

K-Ras is a small GTPase that functions as a molecular switch cycling between inactive (GDP-bound) and active (GTP-bound) states. The conversion of K-Ras-GDP to K-Ras-GTP is the rate-limiting step in the activation of K-Ras and is catalyzed by guanine nucleotide exchange factors such as the son of sevenless (Sos). Mutations in K-Ras fix the protein in the active state and endow cells with capabilities that represent the hallmarks of cancer.[1] These include the ability to proliferate, evade apoptosis, reprogram cell metabolism, induce angiogenesis, activate invasion and metastasis, and escape immune destruction.[2] Indeed, aberrant K-Ras signaling plays a role in 30% of all human cancers, with the highest incidence of activating mutations found in pancreatic (70-90%), colon (30-50%), and lung (20-30%) carcinomas.[3] Downregulation of activated Ras reverses the transformed phenotype of cells and results in the dramatic regression of tumors in murine xenograft models.[4] Thus, K-Ras inhibition represents an attractive therapeutic strategy for many cancers. However, Ras activation and signaling is accomplished primarily through protein-protein interactions. Such protein interfaces typically lack well-defined binding pockets and have been difficult to target with small molecules.[5]
Here we report on the discovery of novel small molecules that bind directly to K-Ras between switch I and switch II and inhibit Sos-catalyzed K-Ras activation. To identify compounds that bind directly to K-Ras, we conducted a fragment screen[6] using uniformly 15N labeled GDP-bound K-Ras (G12D). An NMR-based screen of 11,000 fragments yielded approximately 140 fragments that bind to GDP-bound K-Ras (G12D) (hit rate = 1.3%) as determined by 1H and 15N chemical shift changes in 1H/15N HSQC spectra of uniformly 15N-labeled K-Ras (G12D). Representative examples of some of the hits identified in the fragment-based screen (1, 2, 3) as well as analogs that were synthesized to increase water solubility and binding affinity (4, 5, 6) are depicted in Figure 1. These compounds bind to K-Ras (G12D) with affinities of 1.3-2 mM. The fragment hits identified in the screen were found not only to bind the G12D mutant of K-Ras but also bind to wild-type K-Ras, K-Ras (G12V), and H-Ras (data not shown), indicating that these compounds bind to a site that is conserved among Ras isoforms and different K-Ras mutants.
Figure 1.
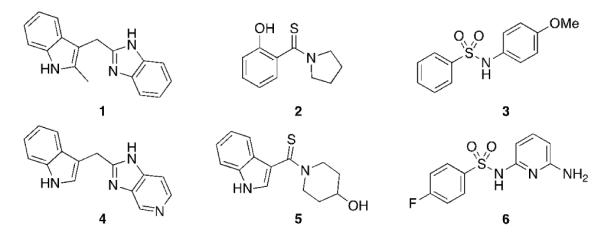
Multiple chemotypes were identified in the fragment-based screen, including indoles (1), phenols (2), and sulfonamides (3). Analogs of these compounds (4, 5, 6) were synthesized to increase their water solubility and binding affinity.
To determine how the fragment hits and analogs bind to K-Ras, we obtained X-ray co-crystal structures. Initial attempts to co-crystallize K-Ras (G12D) with these compounds failed to produce suitable crystals. Due to the limited number of space groups available to this mutant form of the protein, we performed crystallization screens of both wild-type and G12V mutant K-Ras. Both proteins crystallized across a broad range of conditions in multiple space groups and yielded high resolution co-crystal structures. In all 20 co-crystal structures obtained thus far, the compounds occupy a hydrophobic pocket located between the α2 helix of switch II (60-74) and the central β sheet of the protein. Figure 2a depicts a high resolution structure of 4, an analog of a screening hit (1), complexed to the GDP bound form of wild-type K-Ras. The indole of 4 binds into a hydrophobic pocket formed by Val-7, Leu-56, and Tyr-71, as well as the aliphatic portion of the side chains of Lys-5 and Thr-74. The imidazopyridine portion of the molecule lies flat in an adjacent binding cleft formed on one side by the side chain of Tyr-71. The nitrogen at the 1-position of the imidazopyridine is involved in a water-mediated interaction with Ser-39, and the indole NH group forms a hydrogen bond with Asp-54. Another member of the indole series (5) uses a thiocarbonyl instead of a methylene linker to access the secondary binding cleft. The binding mode of the indole moiety in this instance is rotated towards the α2 helix and the switch II loop region, forming a hydrogen bond with Ser-39 instead of Asp-54 and positioning the piperidine ring closer to the helix (Figure 2b). In addition to an indole, a phenol moiety is also able to bind into this hydrophobic pocket, as demonstrated by the X-ray structure of compound 2 bound to K-Ras (Figure 2c). The hydroxyl group of the phenol forms a hydrogen bond with Asp-54 while the pyrrolidine moiety forms stacking interactions with Tyr-71. The binding mode of a member of the sulfonamides series is shown in Figure 2d. The pyridine nitrogen of 6 forms three water mediated hydrogen bonds to Asp-54, Arg-41 and Ser-39, and the ortho amino group interacts with the Asp-54 side chain and a backbone carbonyl. From these structures, it appears that a hydrogen bond donor, such as the -NH on the indole or the -OH on the phenol is important for binding. This is supported by the lack of binding of analogs containing substituents at these positions that occlude hydrogen bond formation.
Figure 2.
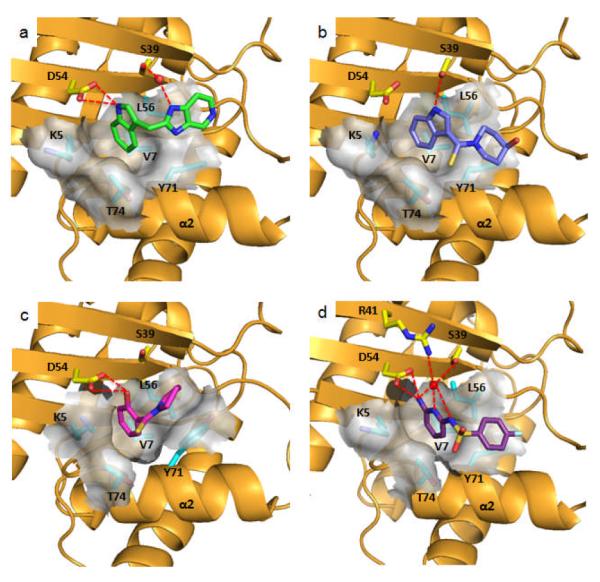
Ribbon and molecular surface representations of the X-ray co-crystal structures of GDP-K-Ras complexed to: a) 4 (PDB code 4EPV), b) 5 (PDB code 4EPW), c) 2 (PDB code 4EPT) and d) 6 (PDB code 4EPX).
Analysis of the ligand-protein co-crystal structures reveal that all the compounds bind to a pocket that is not readily observed in the ligand-free form (Figure 3a) but in an “open” form of the protein (Figure 3b). The pocket is created by a conformational change (Figure 3c) in which the α2 helix moves away from the central β sheet, and the side chain of Tyr-71 breaks the hydrogen bond network present in the ligand-free form. In addition, the side chain of Met-67 rotates out of the way to form a secondary binding cleft. This creates a new binding site for small molecules that is not present in the “closed” form. In subsequent X-ray structures obtained of ligand free K-Ras under different experimental conditions as well as recently published molecular dynamics simulations, the “open” form has been observed, suggesting that the “open” and “closed” conformations are present in equilibrium.[7]
Figure 3.
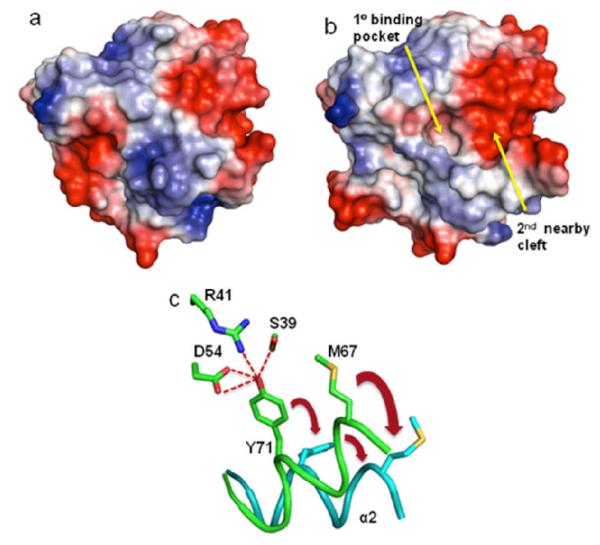
Electrostatic surface representations of GDP-K-Ras a) in the absence of a ligand (PDB code 4EPR) and b) in the “open” form showing the primary hydrophobic binding pocket and the adjacent electronegative cleft. c) Schematic representation of the transition of GDP-K-Ras from the “closed” form (green) to the “open” form (cyan).
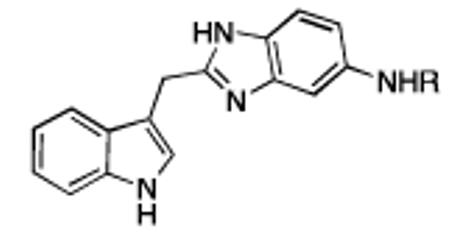
The secondary binding cleft is electronegative in character (Figure 3b) and contains two acidic residues, Glu-37 and Asp-38. To bind to this region of the protein, we synthesized amide-linked amino acid analogs of the indole-benzimidazole fragment (7) containing positively charged amines (Table 1). Improved binding to K-Ras was observed for several of these analogs. The best compound in this series contains an isoleucine (12) which binds to K-Ras with an affinity of 190 μM, an improvement of roughly 10 fold over the unsubstituted analog 7. We were able to obtain a high resolution X-ray co-crystal structure of one of these analogs (13) complexed to GDP-bound K-Ras (G12V). (Figure 4a) As designed, the indole is located in the primary binding pocket, and the positively charged amino group of the amino acid interacts with the carboxylic acid side chain of Asp-38 in the secondary binding cleft.
Table 1.
Compound binding affinity with GDP K-Ras (G12D) and functional activity in a Sos-catalyzed nucleotide exchange assay.
| Cmpd | R | KD (μM) [a] | % Inh. [b] |
|---|---|---|---|
| 7 | -H | ~1300 | no inh. |
| 8 | -Gly | 420 | 27 ±9 |
| 9 | -Ala | 350 | 51 ±4 |
| 10 | -β-Ala | 340 | 32 ±10 |
| 11 | -Val | 240 | 73 ±12 |
| 12 | -Ile | 190 | 78 ±8 |
| 13 | -Pro | 340 | 58 ±8 |
KD values were measured from the chemical shift changes observed in HSQC spectra of uniformly 15N -labeled protein as a function of added compound.
The % inhibition of Sos-catalyzed nucleotide exchange observed using a 1 mM concentration of compound.
Figure 4.
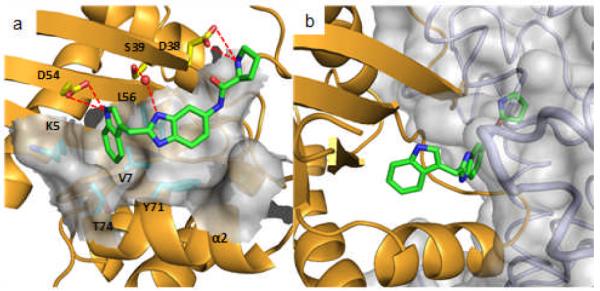
a) Ribbon and molecular surface representations of GDP-bound K-Ras complexed to 13 (PDB code 4EPY). b) K-Ras/13 X-ray structure overlaid with a previously reported[9] Ras-Sos complex crystal structure (PDB code 1BKD).
To examine the functional consequences of binding to K-Ras, compounds were tested for their ability to inhibit Sos-mediated nucleotide exchange. In this assay, unlabeled GDP is exchanged for BODIPY-GTP which is catalyzed by Sos and results in an increase in fluorescence.[8] As shown in Table 1, the extended analogs with binding affinity <500 μM inhibited the nucleotide exchange process at a concentration of 1mM. For example, an analog containing an isoleucine (12) inhibited nucleotide exchange at 78% (Supporting Information) The inhibition of Sos-mediated nucleotide exchange that we observe can be rationalized from a model prepared by overlaying the X-ray structure of the K-Ras/13 complex (Figure 4b) onto a previously reported structure of H-Ras complexed with Sos.[9] The amino acid of 13 clashes with αH of Sos. The model predicts that Sos would not be able to bind to K-Ras when complexed to a small molecule that extends into this space (e.g., 8-13). This model is supported by NMR experiments in which the 1H/15N HSQC spectrum of 15N-labeled K-Ras (G12D) when complexed to unlabeled Sos dramatically changes upon the addition of compound 13. This new spectrum resembles that of the K-Ras/13 complex without Sos (Supporting Information) Furthermore, in the absence of K-Ras, compound 13 does not bind to Sos, and an analog of 13 with the indole moiety N-methylated does not bind to K-Ras and does not inhibit Sos-mediated nucleotide exchange.
In conclusion, using a fragment-based screen, we have identified small molecules that bind to K-Ras in a hydrophobic pocket that is occupied by Tyr-71 in the apo-Ras crystal structure. Using structure-based design, we obtained analogs of the fragment hits with improved binding affinity as well as functional activity in a Sos-catalyzed nucleotide exchange assay. These compounds bind to K-Ras and block binding to Sos which causes the inhibition of Sos-mediated nucleotide exchange. These molecules represent a starting point for obtaining probe molecules useful in elucidating new insights into Ras signaling and for discovering K-Ras inhibitors for the treatment of cancer.
Experimental Section
Details of the protein purification, the fragment screen, X-ray crystallography, nucleotide exchange, 1H/15N-HSQC spectra of K-Ras with and without ligands and Sos, and the synthesis of compounds are given as Supporting Information.
Supplementary Material
Footnotes
This work was supported by the US National Institutes of Health: 5DP1OD006933 (NIH Director’s Pioneer Award) to S.W.F., an ARRA stimulus grant (5RC2CA148375) to L.J. Marnett, and a NCI SPORE grant in GI cancer (5P50CA095103-09) to R.J. Coffey. This work was also funded by the Lustgarten Foundation grant awarded to S.W.F. and the American Cancer Society (Postdoctoral Fellowship, PF1110501CDD) to J.P.B. We thank Matt Mulder (Craig Lindsley lab, Vanderbilt University) for providing HRMS data.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Qi Sun, Department of Biochemistry Vanderbilt University School of Medicine 2215 Garland Ave., 607 Light Hall, Nashville, TN 37232 (USA).
Jason P. Burke, Department of Biochemistry Vanderbilt University School of Medicine 2215 Garland Ave., 607 Light Hall, Nashville, TN 37232 (USA).
Jason Phan, Department of Biochemistry Vanderbilt University School of Medicine 2215 Garland Ave., 607 Light Hall, Nashville, TN 37232 (USA).
Michael C. Burns, Department of Biochemistry Vanderbilt University School of Medicine 2215 Garland Ave., 607 Light Hall, Nashville, TN 37232 (USA)
Edward T. Olejniczak, Department of Biochemistry Vanderbilt University School of Medicine 2215 Garland Ave., 607 Light Hall, Nashville, TN 37232 (USA)
Alex G. Waterson, Department of Pharmacology Vanderbilt University School of Medicine 2200 Pierce Ave., 442 RRB, Nashville, TN 37232 (USA)
Taekyu Lee, Department of Biochemistry Vanderbilt University School of Medicine 2215 Garland Ave., 607 Light Hall, Nashville, TN 37232 (USA).
Olivia W. Rossanese, Department of Biochemistry Vanderbilt University School of Medicine 2215 Garland Ave., 607 Light Hall, Nashville, TN 37232 (USA)
Stephen W. Fesik, Department of Biochemistry Vanderbilt University School of Medicine 2215 Garland Ave., 607 Light Hall, Nashville, TN 37232 (USA).
References
- [1].Hanahan D, Weinberg RA. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- [2].Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. Nat. Rev. Cancer. 2011;11:761–74. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3]a).Laghi L, Orbetegli O, Bianchi P, Zerbi A, Di Carlo V, Boland CR, Malesci A. Oncogene. 2002;21:4301–6. doi: 10.1038/sj.onc.1205533. [DOI] [PubMed] [Google Scholar]; b) Lau KS, Haigis KM. Mol. Cells. 2009;28:315–20. doi: 10.1007/s10059-009-0143-7. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Riely GJ, Marks J, Pao W. Proc. Am. Thorac. Soc. 2009;6:201–5. doi: 10.1513/pats.200809-107LC. [DOI] [PubMed] [Google Scholar]; d) Bos JL. Cancer Res. 1989;49:4682–9. [PubMed] [Google Scholar]; e) Yen LC, Uen YH, Wu DC, Lu CY, Yu FJ, Wu IC, Lin SR, Wang JY. Ann. Surg. 2010;251:254–60. doi: 10.1097/SLA.0b013e3181bc9d96. [DOI] [PubMed] [Google Scholar]
- [4]a).Chin L, Tam A, Pomerantz J, Wong M, Holash J, Bardeesy N, Shen Q, O’Hagan R, Pantginis J, Zhou H, Horner JW, Cordon-Cardo C, Yancopoulos GD, DePinho RA. Nature. 1999;400:468–72. doi: 10.1038/22788. [DOI] [PubMed] [Google Scholar]; b) Podsypanina K, Politi K, Beverly LJ, Varmus HE. Proc. Natl. Acad. Sci. 2008;105:5242–7. doi: 10.1073/pnas.0801197105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Arkin MR, Wells JA. Nat. Rev. Drug Discov. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- [6]a).Shuker SB, Hajduk PJ, Meadows RP, Fesik SW. Science. 1996;274:1531–4. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]; b) Hajduk PJ, Greer J. Nat.Rev.Drug Discov. 2007;6:211–9. doi: 10.1038/nrd2220. [DOI] [PubMed] [Google Scholar]; c) Jhoti H, Cleasby A, Verdonk M, Williams G. Curr Opin Chem Biol. 2007;11:485–93. doi: 10.1016/j.cbpa.2007.07.010. [DOI] [PubMed] [Google Scholar]
- [7]a).Kigawa T, Yamaguchi-Nunokawa E, Kodama K, Matsuda T, Yabuki T, Matsuda N, Ishitani R, Nureki O, Yokoyama SJ. Struct. Funct. Genomics. 2002;2:29–35. doi: 10.1023/a:1013203532303. [DOI] [PubMed] [Google Scholar]; b) Ma J, Karplus M. Proc. Natl. Acad. Sci. 1997;94:11905–10. doi: 10.1073/pnas.94.22.11905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Patgiri A, Yadav KK, Arora PS, Bar-Sagi D. Nat. Chem. Biol. 2011;7:585–7. doi: 10.1038/nchembio.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Boriack-Sjodin PA, Margarit SM, Bar-Sagi D, Kuriyan J. Nature. 1998;394:337–43. doi: 10.1038/28548. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.