

Abstract
Periodontitis is one of the most prevalent human inflammatory diseases. The major clinical phenotypes of this polymicrobial, biofilm-mediated disease are chronic and aggressive periodontitis, the latter being characterized by a rapid course of destruction that is generally attributed to an altered immune-inflammatory response against periodontal pathogens. Still, the biological basis for the pathophysiological distinction of the two disease categories has not been well documented yet.
Type I natural killer T (NKT) cells are a lymphocyte subset with important roles in regulating immune responses to either tolerance or immunity, including immune responses against bacterial pathogens. Here, we delineate the mechanisms of NKT cell activation in periodontal infections.
We show an infiltration of type I NKT cells in aggressive, but not chronic periodontitis lesions in vivo. Murine DCs infected with aggressive periodontitis-associated Aggregatibacter actinomycetemcomitans triggered a type I interferon response followed by type I NKT cell activation. In contrast, infection with Porphyromonas gingivalis, a principal pathogen in chronic periodontitis, did not induce NKT cell activation. This difference could be explained by the absence of a type I interferon response to P. gingivalis infection. We found these interferons to be critical for NKT cell activation.
Our study provides a conceivable biological distinction between the two periodontitis subforms and identifies factors required for the activation of the immune system in response to periodontal bacteria.
Keywords: natural killer T cells, immune evasion, bacterial pathogenesis, Porphyromonas gingivalis, Aggregatibacter actinomycetemcomitans
Introduction
Periodontitis is one of the most prevalent chronic inflammatory diseases in humans, with a prevalence of 30% in the United States population (1). Untreated periodontitis leads to destruction of periodontal tissues and supporting bone, ultimately resulting in tooth loss (2, 3). In addition to its detrimental role in the oral cavity, periodontal infections have been causally linked to atherosclerosis and other systemic diseases (4). Periodontitis is a biofilm-mediated disease, and several oral bacteria that colonize the tooth surfaces, including gram-negative anaerobic species, have been identified as causative agents (3). The two main categories of destructive periodontal disease are chronic and aggressive periodontitis (5, 6). Aggressive periodontitis is characterized by rapid attachment loss and destruction, often at young age and with familiar predisposition (7, 8). Infections with certain serotypes of the facultative aerobic gram-negative pathogen Aggregatibacter actinomycetemcomitans (A.a.) have been causally linked to aggressive periodontitis (9, 10). Chronic periodontitis on the other hand is described as slowly progressing inflammatory loss of periodontal tissues associated with moderate to heavy deposits of bacterial plaque and calculus (7). A principal pathogen in chronic periodontitis is the anaerobic, gram-negative Porpyromonas gingivalis (P.g.)(11). Specifically, no histopathological differences between these two chronic inflammatory subforms of periodontal disease are available to date (12). Importantly, no histological distinction between these two subforms of periodontal disease are available to date. In this study, we assessed the role of type I Natural Killer T (NKT) cells, a cell population with critical properties in guiding immune responses against infection, in both forms of periodontitis, and delineate the mechanisms of their activation.
Natural killer T (NKT) cells are a population of lymphocytes with unique activation and effector properties, which bridge innate and adaptive immunity. The majority of NKT cells, termed type I or invariant NKT cells, are CD1d restricted and express a semi-invariant T cell receptor (TCR) using the segments Vα14 and Jα18 in mice and Vα24 and Jα18 in humans. Type I NKT cells recognize lipid antigens presented in non-polymorphic CD1d molecules, which are predominantly expressed on antigen-presenting cells (DC, macrophages, B cells) (13).
Interactions between DCs, expressing CD1d molecules, and type I NKT cells have intensively been studied (14, 15). Presentation of CD1d-lipid complexes by DCs initiates a positive feedback. In particular, stimulation of DCs by interactions between CD40L (CD154) expressed on type I NKT cells and CD40 molecules on DCs leads to functional maturation and interleukin-12 (IL-12) production in DCs (16-18). This in turn induces the secretion of pro-inflammatory cytokines, including IFN-γ, by type I NKT cells. The secretion of IL-4, which is used as read-out for an anti-inflammatory cytokine profile of NKT cells, is independent of the costimulatory axis between NKT cells and DC (18). Hence, type I NKT cells contribute to host defence against viral and bacterial pathogens.
Lipid antigens derived from certain bacteria, e.g. Sphingomonas and Borrelia burgdorferi (19-21), have been described. However, other pathogens, e.g. viruses do not even contain lipids, or conceivably do not contain CD1d-presentable lipids and thus might not be recognized by NKT cells. Nature has evolved different receptors, including the group of toll-like receptors (TLR), to detect conserved pathogen-associated molecular patterns (PAMPs). Upon ligation of the pattern recognition receptors TLR4 or TLR9, that recognize lipopolysaccharide of gram-negative bacteria and unmethylated CpG DNA sequences, respectively, endogenous glyolipids are generated and loaded onto CD1d molecules in DCs, which then trigger the secretion of IFN-γ by type I NKT cells (22). This process requires the expression of type I interferons, IFN-α o r I F N-β by activated DC. Under normal conditions the glycosphingolipid isoglobotrihexosylceramide (iGb3) is constantly degraded in lysosomes. TLR ligation inhibits activity of the rate-limiting enzyme in iGb3 turnover, α-galactosidase A (α-GalA), and permits the intracellular accumulation and CD1d binding of iGb3. Thus, TLR9-stimulated DC trigger IFN-γ production in type I NKT cells (23).
In this work, we show a pronounced infiltration of type I natural killer T cells in aggressive, but not chronic periodontitis lesions in vivo, and DC-mediated activation of these cells in vitro by aggressive periodontitis-associated A.a., but not by P.g. Furthermore, we demonstrate that in contrast to A.a. infection, P.g. challenge does not result in a type I interferon response or presentation of endogenous glycolipids, thereby preventing the activation of type I NKT cells by bacterial-challenged DCs. Addition of exogenous interferons to DCs challenged with P.g. rescued the production of pro-inflammatory cytokines by type I NKT cells and thus may circumvent the apparent immune evasion by P.g..
Material and Methods
Ethics Statement
All animal experiments were covered by approvals from the Government of the State of Nordrhine-Westphalia (permit no. 8.87-51.04.20.10.042). Design and procedures of the study involving human biopsies were approved by the Columbia University Medical Center Institutional Review Board (IRB – AAAB0869).
Mice
C57BL/6J wildtype (WT) mice were purchased from Jackson Laboratories (Bar Harbor, MA, USA), MyD88-deficient mice were a kind gift of Heike Weighardt (University of Düsseldorf, Germany). Mice were housed under specific pathogen-free conditions and used for experiments at ages of six to eight weeks. Mice lacking the type I interferon receptor (IFNAR) were a kind gift of Beatrix Schumak (University of Bonn, Germany). Animal experiments were performed in accordance to protocols approved by Institutional Animal Care and Use Committee.
Bacteria
Aggregatibacter actinomycetemcomitans Y4 (8) was cultured on blood-agar plates at 37°C/5% CO2. Porphyromonas gingivalis strain 381 (24) was cultured under anaerobic conditions on blood agar plates. Bacterial suspensions in RPMI medium without antibiotics were adjusted to 109 CFU/ml using a spectrophotometer at 600 nm and established growth curves.
Bone-marrow derived DC
DCs were generated from mouse bone-marrow (BM) in the presence of GM-CSF, as described (25). In brief, BM cells were cultured in complete DMEM medium supplemented with 20 ng/mL recombinant GM-CSF (Immunotools, Friesoythe, Germany), replaced every other day. On day 6, BMDC were detached with Trypsin-EDTA (PAA Laboratories, Pasching, Austria) and washed in DMEM/5% FCS without antibiotics. Where indicated, dendritic cells from BMDC cultures further enriched by MACS using pan-DC microbeads (Miltenyi Biotech. Bergisch-Gladbach, Germany) to a purity >90%. BMDC were pulsed with α-GalCer (200 ng/ml, Kirin Ltd., Gunma, Japan) or vehicle (Tween-20) in medium for 3 h at 37°C. For infections, BMDC were cultured in DMEM/5%FCS without antibiotics in the presence of A.a. or P.g. at indicated MOI. After 24 hrs of culture, DCs were washed three times in complete medium before responder NKT cells were added.
Organ preparations
Single cell suspensions from spleens were prepared by standard techniques. Liver MNC were isolated as previously described (26). Briefly, livers were perfused with PBS, minced and iNKT cells were enriched by centrifugation in a two-step Percoll gradient. Enriched populations typically contained 20-30% iNKT cells.
Where indicated, CD1d-restricted NKT cells were further enriched by MACS. Hepatic MNC suspensions were stained with PBS57-loaded, PE-conjugated CD1d-tetramers NIH tetramer facility, Emory University Vaccine Center, Atlanta, GA, USA), washed, and stained with anti-PE microbeads, before selection on miniMACS columns (Miltenyi Biotech, Bergisch-Gladbach, Germany). Enriched preparations contained >70% CD1d-restricted NKT cells and <3% conventional T cells as determined by flow cytometry (Suppl. Fig 1A). Type NKT cells were stimulated in the presence of either bacterial infected or α-GalCer-pulsed BMDC at a DC:NKT ratio of 1:10. After 24 and 48 hours, cell culture supernatants were harvested and used for cytokine analysis. Cytokine-specific ELISA assays (IL-4, IL-17/A, IFNγ, IL-12, eBioscience, San Diego, CA, USA; mouse IFN-β ELISA, Hoelzel Diagnostika, Cologne, Germany) of cell culture supernatants were performed following the manufacturers’ instructions.
alpha-Galactosidase enzyme activity assay
α-GalA enzyme activity in BMDC was determined as described previously (27). In brief, following treatment, DCs were lysed in lysosomal assay buffer (LAB). Protein concentrations were measured using Lowry method. 10 μg of lysates were incubated in 96-well black flat bottom plates in a total volume of 100 μL supplemented with fluorogenic α-GalA substrate 3-methylumbelliferyl-α-D-galactospyranoside (5 mM) and N-acetylgalactosamine (10 mM) for 2 hrs at 37°C. Decreases in fluorescence intensities were assessed at 360 nm and 455 nm for excitation and emission (Infinite M200 fluorometer; Tecan, Crailsheim, Germany). Enzyme activities were normalized to values of untreated BMDC.
Clinical examination and procedures
A total of 15 subjects with moderate to severe generalized chronic periodontitis and ten subjects with generalized aggressive periodontitis were recruited among the patients referred for periodontal therapy to the Clinic for Post-doctoral Periodontics, Columbia University College of Dental Medicine. Eligible patients (i) were at least 25 years old; (ii) had a minimum of 22 teeth present; (iii) had no past history of systematic periodontal therapy other than occasional prophylaxis provided by the referring general dentist, (iv) had received no systemic antibiotics or anti-inflammatory drugs for at least 6 months, (v) harboured a minimum of 4 teeth with radiographic bone loss, (vi) did not suffer from diabetes mellitus, (vii) did not suffer from any of the systemic conditions or genetic disorders that entail a diagnosis of “periodontitis as a manifestation of systemic diseases”, (viii) were not pregnant, and (ix) were not current users of tobacco products or of nicotine replacement medication. Signed informed consent was obtained prior to enrolment. Design and procedures of the study were approved by the Columbia University Medical Center Institutional Review Board (IRB – AAAB0869).
The diagnosis of generalized chronic or aggressive periodontitis was independently performed by two periodontists.
Identification of donor sites and harvesting of gingival tissue samples was performed as earlier described [13]. In brief, a ‘diseased’ interproximal papilla showed bleeding on probing (BoP), probing pocket depth (PPD) ≥ 4mm, and clinical attachment level (CAL) ≥ 3mm, whilst a ‘healthy’ papilla demonstrated no BoP, PPD ≤ 4mm and CAL ≤ 2mm. All tissue specimens were collected during periodontal surgery. In a paired design, each study subject contributed with one ‘diseased’ tissue sample and one ‘healthy’ tissue sample.
Processing of biopsies and quantification of type I NKT cells
Total RNA was extracted from the gingival tissue biopsies, as described previously (28). In brief, the biopsies were homogenized in Trizol reagent (Invitrogen, Carlsbad, CA, USA), and further subjected to a spin-column cleanup (RNeasy Mini Kit, Qiagen, Hilden, Germany). RNA quantity and quality was assessed by Nanodrop spectrophotometer and Agilent Bioanalyzer. Total RNA was reverse-transcribed using Superscript III (Invitrogen).
Aliquots of cDNA reactions were amplified using Sensimix SYBR No-Rox master mix (Bioline, Eberswalde, Germany; Eppendorf RealPlex S machine, Hamburg, Germany) using primers specific for the Vα24Jα18 junction (sense 5′- GAT ATA CAG CAA CTC TGG ATG -3′: antisense 5′- GAG TTC CTC TTC CAA AGT ATA GCC-3′) previously described (14).
cDNA prepared from an human iNKT cell line was amplified using Vα24Jα18 primers using Phusion polymerase (Fermentas, St.Leon-Roth, Germany) and cloned into pGemTeasy vector (Promega, Gainsville, FL, USA). Dilutions of the sequence-verified cloned plasmid were included in qPCR runs as standard curve. Copy numbers were calculated of sample values normalized against the housekeeping gene beta-actin (sense 5′- ACA GAG CCT CGC CTT TGC CG -3′: antisense 5′- TGG GCC TCG TCG CCC ACA TA-3′).
Transcriptomic profiling of periodontal tissue biopsies
Whole-genome gene expression profiles from all biopsies were analyzed using Affymetrix HG-U133plus 2.0 microarrays. In brief, 10 μg of total RNA was reverse transcribed and biotin labeled, fragmented and hybridized to arrays following the manufacturer’s instructions. Data were analyzed using R/Bioconductor and limma. Data were background-corrected, RMA normalized, log transformed and assessed for differentially expressed probes using paired statistics. Probes differentially regulated in aggressive and chronic periodontitis (affected versus clinically healthy biopsies) were identified using a Venn diagram. Experimental details and results following the MIAME standards are available at the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE16134) under the accession number GSE16134.
Gene expression analysis
mRNA expression was analyzed by real-time PCR using primers specific for IFN-α (sense 5′- CTT CCA CAG GAT CAC TGT GTA CCT -3′; antisense 5′-TTC TGC TCT GAC CAC CTC CC-3′), IFN-β (sense 5′- CTG GAG CAG CTG AAT GGA AAG -3′; antisense 5′-CTT CTC CGT CAT CTC CAT AGG G -3′), GNPAT (sense 5- CCA TGG ACG TTC CTA GCT CC -3′; antisense 5′- ACT TGA TGT CCC CTG GCT TG -3′), UGCG (sense 5-′ CCG TAT AGC AAG CTC CCT GG -3′; antisense 5′- AAG CCT TGT CTG TCG GCT AC -3′) and normalized against GAPDH (sense 5′- AAC TTT GGC ATT GTG GAA GG -3′: antisense 5′- ACA CAT TGG GGG TAG GAA CA -3′) as housekeeping gene.
Transcriptomic profiling of A.a. and P.g. infected DCs
100 ng of TRIzol isolated and spin-column cleaned total RNA was subjected to a single round of in vitro transcription and biotin labeling (Total Prep RNA amplification kit, Ambion, Austin, TX, USA). cRNA was hybridized on Mouse WG-6 v2 Expression BeadChips (Illumina, San Diego, CA, USA) according to standard protocols.
Expression data were exported as unnormalized sample and control probe profiles from the Illumina BeadStudio software and analyzed using R/Bioconductor and limma. Data were quality weighed [16], background-corrected, quantile normalized, log transformed [17], and explored for differentially expressed genes with a false discovery rate (FDR) < 0.05 [18] using Bayesian statistics. Transcriptomic datasets were assessed for similarity using hierarchical clustering. A heatmap illustrating the differential regulation of probes in A.a. and P.g. infected DCs versus untreated controls was constructed using the CIMminer program at http://discover.nci.nih.gov/, a development of the Genomics and Bioinformatics Group, Laboratory of Molecular Pharmacology, Center for Cancer Research, National Cancer Institute [19]. Probes differentially regulated by A.a. and P.g. infection in DCs were identified using a Venn diagram. Differential regulation of signaling pathways was performed using the signaling pathway impact analysis algorithm [20]. Interferon-stimulated transcripts in the different experimental groups were identified using the Interferome database (29). Experimental details and results following the MIAME standards are available at the Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=vtkvuiesiyiapi&acc=GSE41383) under accession number GSE41383.
Statistics
Statistical analyses not involving microarrays were performed using GraphPad Prism V (GraphPad Software, San Diego, CA, USA). Parametric testing for two groups was performed using unpaired, two-tailed t-tests, three or more groups were analyzed using one-way ANOVA with post-hoc Newman-Keuls tests. Non-parametric testing was performed using Wilcoxon signed rank test (two groups) and Kruskal-Wallis tests (three or more groups). Values of p<0.05 were considered significant.
Results
Differential infiltration of type I NKT cells in aggressive and chronic periodontitis lesions
Given the proposed role of type I NKT cells in bacterial infections, we asked whether type I NKT cells infiltrate periodontal lesions. We extracted RNA from paired ‘diseased’ and clinically ‘healthy’ gingival tissue specimens (average periodontal probing depth ‘healthy’ sites 2.25 ± 0.058 mm, ‘diseased’ sites 7.67 ± 1.78 mm, average probing depth difference 5.44 ± 1.81 mm) obtained from patients diagnosed with aggressive and chronic periodontitis, respectively. cDNAs were analyzed for the presence of type I NKT cells. To this end, from paired biopsies we amplified transcripts of the Vα24-Jα18 rearranged TCR which is selectively carried on type I NKT cells. Enumeration of type I NKT cells according to TCR mRNA transcripts has been described previously (30, 31).
Numbers of type I NKT cells present in unaffected (‘healthy’) periodontal tissue were not significantly different between both patient groups (p=0.11).
8 of 10 patients with aggressive periodontitis showed increasing numbers of type I NKT cells in diseased compared to healthy biopsies. Overall, in patients with aggressive periodontitis type I NKT cells infiltrated the tissue lesions at significantly higher levels (p<0.01, mean fold influx of Vα24-Jα18+ cells: 41.59) (Fig.1A). In contrast, in only 5 of 15 patients with chronic periodontitis we found type I NKT increases greater than 5-fold, thus, the overall type I NKT cell infiltration in this group did not reach statistical significance (Fig.1B, p<0.09, mean fold influx of Vα24-Jα18+ cells=3.73).
Fig. 1. Differential type I NKT cell infiltration in aggressive and chronic periodontitis lesions.
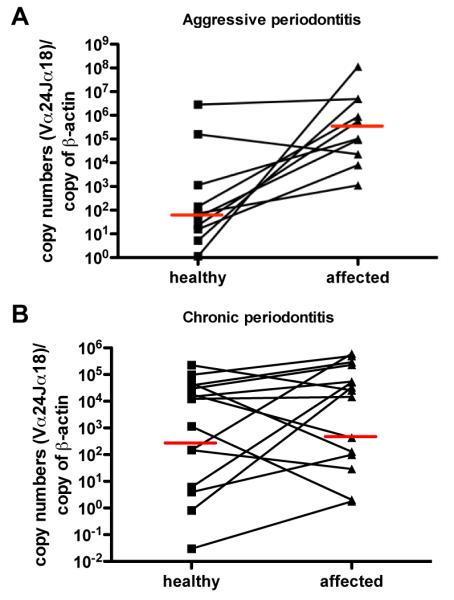
RNA of biopsies extracted of aggressive (A) and chronic (B) periodontal lesions and respective contralateral healthy biopsies were transcribed into cDNA. Numbers of iNKT cells expressing the rearranged Vα24Jα18 TCR was determined by SYBR green qPCR, normalized against β-actin levels and calculated using standard curves of known copy numbers. n=10, Wilcoxons paired t-test. Red bars indicate median values.
To establish the determinants and consequences of increased NKT cell influx in aggressive periodontitis lesions, we then assessed the whole-genome transcriptomic profiles of the utilized biopsies for transcripts related to NKT cell biology. We found increased expression of several chemokines in aggressive periodontitis lesions, as compared to the clinically healthy pairs, that may explain the increased influx of type I NKT cells in these lesions (Supplemental table 1, panels A-C). Specifically, we observed a significant induction of the chemokine CCL8 and its receptor CCR5 (Supplemental table 2) that is well documented to be strongly expressed on type I NKT cell surfaces (32). When assessing the transcriptomic data for evidence of NKT cell activation, we found the pro-inflammatory product of activated type I NKT cells, IFN-γ, to be significantly induced in aggressive, but not chronic lesions. Furthermore, IL-17A and -B mRNA was significantly induced in aggressive, but not chronic periodontitis lesions. Since type I NKT cells require for activation mature DCs presenting CD1d molecules, we subsequently assessed the expression of the maturation marker CD86 and CD1d, and found both to be significantly induced in aggressive, but not chronic periodontitis (Supplemental table 1,). Based on these data, we then proceeded to further characterize the activation of type I NKT cells by periodontal pathogens in vitro.
A.a.-infected DCs activate type I NKT cells
We assessed whether A.a., causally related to aggressive periodontitis, and P.g., a major pathogen in chronic periodontitis, were able to stimulate DCs to activate type I NKT cells in vitro. DC differentiated from BM of healthy WT mice were infected with either A.a. or P.g. at different MOI for 24 hrs. Cells were washed and used as stimulator cells for type I NKT cells enriched from livers of WT mice. DCs infected with A.a. stimulated the secretion of pro-inflammatory cytokine IFN-γ (48 hrs.) in a MOI-dependent manner. Similar results were seen in response to other A.a. strains (data not shown). In contrast, levels of the anti-inflammatory cytokine IL-4 in culture supernatants did not increase above background levels (Fig.2A, black bars). Contrary to this, DCs infected with P.g. neither stimulated IL-4 nor IFN-γ release in type I NKT cells (Fig.2A, right panels). Furthermore, infection of purified DCs with A.a. but not P.g. stimulated significantly elevated levels of IL-17A protein in purified type I NKT cells (Figure S1). To test whether activation of type I NKT cells by bacteria-infected DCs is mediated by CD1d, groups of DCs were incubated with CD1d blocking antibodies. Blockade of CD1d molecules at MOI 50 and 100 significantly abrogated the production of IFN-γ by NKT cells (Fig.2A, shaded bars). This indicates that the activation of type I NKT cells by A.a.-infected DCs requires expression of CD1d molecules.
Fig. 2. Type I NKT cells are activated by A.a.-infected DCs.

Murine BMDCs were infected with A.a. (panel A) or P.g. at indicated MOI for 24 hrs in the presence (shaded bars) of blocking CD1d antibodies (clone 1B1, 20 μg/mL) or isotype controls (black bars). Liver MNC were added as responder cells and co-cultured with DCs for further 48 hrs. IL-4 and IFNγ levels in supernatants were determined 24 and 48h later, respectively, by ELISA. C. Murine BMDCs were infected with A.a. or P.g. or cultured in the presence or absence of LPS for 24 hrs. Histograms show CD86 expression on MHC-II+CD11c+ cells. Data shown are representative of three experiments. n=3; *, p<0.05; **, p <0.01; ***, p<0.001.
Furthermore, we explored whether A.a. and P.g. differ in the ability to induce functional maturation of DCs. To this end, BMDCs were infected with A.a. or P.g. at MOI of 10 or cultured in the presence of LPS used as positive control. As shown in Fig.2C, LPS up-regulates the expression of the maturation marker CD86 compared to unstimulated controls (34.2% vs. 23.5%). Levels of CD86 expression as well as the frequency of CD86-expressing DCs after A.a. infection (35.9%) were comparable to values of LPS control. In contrast - and corroborating the lack of type I NKT cell activation - infection with P.g. did not up-regulate CD86 on BMDCs (Fig.2B). These data demonstrate a clear lack of activation of DCs and type I NKT cells in response to challenge with P.g.
A.a.-mediated type I NKT cell activation requires TLR signaling
To gain further insight into the mechanisms of type I NKT cell activation in response to periodontal bacteria, we asked whether toll-like receptor signaling in DCs is required to promote type I NKT cell activation. BMDCs generated from WT mice and mice homo- and heterozygous for the adaptor molecule MyD88, involved in signaling by most toll-like receptors, were infected with A.a. and subsequently used as stimulators for freshly isolated type I NKT cells. WT DCs infected with A.a. stimulated a strong IFN-γ secretion in type I NKT cells. In contrast, IFN-γ levels to DCs hemizygous for MyD88 were reduced to ~50% of those induced by WT DC. DCs lacking both MyD88 alleles were unable to activate type I NKT cells in response A.a. challenge (Fig.3A).
Fig. 3. NKT cell activation by A.a.-infected DCs depends on Toll-like receptor signaling.

Murine BMDCs generated from WT (closed bars), MyD88 hemizygous (gray bars) or MyD88−/− (open bars) mice were infected with A.a. Y4 at MOI 100. 24 hrs later, BMDC cultures were washed and liver MNC added with or without rIL-12. IFNγ levels in supernatants were determined 48h later by ELISA. Data shown are representative of three experiments. n=3,*, p<0.05 ***, p<0.001. Liver MNC were exposed to WT or MyD88−/− DC infected with A.a. at indicated MOI. 24 hrs. later cultures were stained with mAbs specific for NK1.1, CD3, CD25, and α-GC-loaded CD1d tetramers and analyzed by flow cytometry. Graph shows CD25 expression on α-GC:CD1d-tetramer+ NK1.1+CD3+ type I NKT cells, relative to untreated controls. Data shown are representative of three experiments. *, p<0.05; **, p <0.01; n.s., not significant.
Furthermore, we stained type I NKT cells (gated as CD3+ α-GC-loaded CD1d-tetramer+ NK1.1+) for the expression of CD25 protein. CD25 is expressed by type I NKT cells and further upregulated upon activation. Type I NKT cells stimulated with A.a.-infected WT DCs MOI-dependently up-regulated the expression of CD25 (Fig.3B, triangles). In contrast, no up-regulation of CD25 was detectable on type I NKT cells co-cultured with infected MyD88-deficient DCs (Fig. 3B, squares). Similar results were observed using Dc generated from MyD88 hemizygous mice (Fig. 3B, circles)
These data indicate that TLR signaling is required for the activation of type I NKT cells by DCs challenged with A.a.
Transcriptomic profiling of infected DCs reveals lacking type I interferon response to P.g. challenge
The observation of the apparent inability of P.g. to activate type I NKT cells via DCs (in contrast to A.a.) prompted us to evaluate whether this effect was reflected in genome-wide transcriptomic profiles of dendritic cells infected with P.g. or A.a., as compared to uninfected controls.
Indeed, the genes differentially regulated by P.g. infection were markedly different from those regulated in A.a. infections (Fig.4A, supplemental table 1, panel A-C). Unsupervised hierarchical clustering revealed that expression profiles of P.g. infected DCs were in fact more similar to uninfected controls than A.a. infected DCs that cluster completely differently (Fig.4B). A Venn diagram of genes differentially regulated in P.g. and A.a. infected cells showed that both pathogens share a certain proportion of commonly regulated genes. Still, P.g. challenge leads to an apparently dampened inflammatory reaction, as indicated by the far less pronounced induction of interleukins and interferons (table I). However, and in line with the aforementioned analyses, A.a. infection triggered exclusive differential regulation of 3830 genes, whilst P.g. challenge only leads to the exclusive regulation of less than one tenth, 336 genes (Fig.4C, supplemental table 1, panel D-L).
Fig. 4. Transcriptomic profiling of infected DCs reveals lacking type I interferon response to P.g. challenge.

BMDCs were infected with A.a. or P.g. at MOI 10 for 24h. Full-genome transcriptomic profiles were explored for differential regulation (FDR < 0.05). Expression profiles of A.a. and P.g. infected DCs are markedly different (heatmap, panel A). Unbiased hierarchial clustering showed a higher similarity of expression profiles of P.g.-infected DCs to uninfected controls than to A.a.-infected cells (panel B). Both pathogens share a certain proportion of commonly regulated genes. A.a. infection triggered exclusive differential regulation of 3830 genes, whilst P.g. challenge only lead to the exclusive regulation of less than one tenth, 336 genes at FDR < 0.05 (panel C). Using the Interferome database, we identified 450 and 34 genes to be interferon-regulated by A.a. and P.g. challenge, respectively (panel D). The transcripts induced or repressed by A.a. challenge indicate a type I interferon response (panel E).
Table 1.
Selected genes that were regulated by Aggregatibacter actinomycetemcomitans or Porphyromonas gingivalis.
| Gene | Fold change A.a. (MOI 10) |
p/q value | Fold change P.g. (MOI 10) |
p/q value |
|---|---|---|---|---|
| Cytokines | ||||
| Interleukin-1 alpha | 32.0±0.16a) | <1x10−5 | 9.4±0.15a) | <1×10−5 |
| Interleukin-1 beta | 18.0±0.26a) | <1×10−5 | 9.0±0.25a) | <1×10−5 |
| Interferon-alpha | 5.4±2.16b) | <1×10−5,a) / <0.05b) |
1.1±0.89b) | <1×10−5,a) / 0.42b) |
| Interferon-beta | 9.9±2.57b) | <1×10−5,a) / 0.01b) |
0.04±0.1b) | <1×10−5,a) / <0.05b) |
| Interferon regulatory factor 7a) | 1.3±0.21a) | <1×10−5 | 6.2±0.2a) | 0.033 |
| IL-6 | 30.4±0.21a) | <1×10−5 | 10.8±0.21a) | 0.53 |
| IL-10 | 42.0±0.3a) | <1×10−5 | 3.26±0.3a) | <1×10−5 |
| IL-12 | 20.0±0.19a) | <1×10−5 | 12.0±0.18a) | <1×10−5 |
| IL-23, p19 | 18.8±0.15a) | <1×10−5 | 1.1±0.14a) | <1×10−5 |
| Surface receptors | ||||
| Interleukin-2R *** chain (CD25) | 4.16±0.16a) | <1×10−5 | 1.5±0.16a) | 0.0015 |
| Antigen presentation | ||||
| CD1d | 1.7±0.15a) | 0.995 | 1.2±0.15a) | <1×10−5 |
| CD40 | <1×10−5 | 1.5±0.22a) | <1×10−5 | |
| histocompatibility 2, class II Mb2 |
4.0±0.14a) | <1×10−5 | 1.4±0.14a) | 0.0077 |
| histocompatibility 2, D region, locus 1 |
2.0±0.22a) | <9×10−5 | 1.5±0.22a) | 0.0074 |
| histocompatibility 2, Q region locus 8 |
3.4±0.1a) | <1×10−5 | 1.4±0.09a) | 0.0035 |
microarray;
Q-PCR. Data are displayed as means plus/minus SEM. Note that the standard errors for array data tend to be relatively small due to the pre-normalization and the linear modeling.
Statistical analysis of the data was performed using unpaired t-tests (qPCR data) or Bayesian statistics and linear modeling using Bioconductor/limma (microarray data). p values (qPCR) and q values (array data) are displayed.
Interferome database analysis of the differentially regulated genes demonstrated that A.a. infection lead to the exclusive differential regulation (FDR < 0.05) of 450 transcripts that are known to be triggered by interferon stimulation, whereas P.g. challenge merely elicited differential regulation of 34 interferon-regulated genes (Fig 4D). Importantly, most of the genes triggered by A.a. are known to be regulated by type I interferons (Fig 4E). When evaluating the impact of P.g. and A.a. infection on signaling pathways in dendritic cells, we found that A.a. lead to a highly significant activation of the ‘antigen processing and presentation’ pathway, whilst this pathway was not significantly regulated by P.g. (supplemental table 1, panels M and N).
These data indicate that challenge with P.g., in contrast does not trigger a type I interferon response, leading to an impaired interleukin to induction in BMDCs. Therefore, the antigen processing machinery necessary for activation A.a., of NKT cells is seemingly not activated in response to P.g. infection.
P.g. does not stimulate type I interferon production
Based on our microarray data showing a lack of a type I interferon response in P.g.- infected versus A.a.-infected DCs, we subsequently sought to further characterize the production of type I interferons. First, we stimulated DCs with A.a. or P.g and analyzed IFN-α and IFN-β mRNA expression. IFN-α mRNA expression levels did not change significantly between the tested groups (Fig.5A). In contrast, infection with A.a. induced a significant induction of IFN-β mRNA expression in DCs (Fig. 5B). Consistent to this, we observed a significant induction if IFN-β protein secretion by DCs infected with A.a. (Fig. 5C). However, no significant increase of IFN-β protein nor its mRNA level was detectable after P.g. infection (Fig. 5C and B).
Fig.5. A.a. and P.g. differ in the ability to stimulate IFN-β production and disrupt GSL turnover.

A. Murine BMDCs were infected with A.a. or P.g. (MOI 10) or left unstimulated for 24 hrs. A./B. Graphs show IFN-α and IFN-β mRNA expression relative to untreated DCs, obtained from at least three independent experiments and analyzed by real-time PCR. C. IFN-β concentrations in cell culture supernatants of BM-DCs infected as in A. D. Enzyme activity of α-GalA in DCs after infection with A.a. or P.g. (MOI 10), or culture in the presence of endotoxin-free IFN-α or IFN-β (both at 1 U/ml), or LPS. Enzymatic activity was measured in DC lysates using a fluorogenic α-GalA substrate. Graph shows relative activity compared to untreated DCs (n=3). E-G. mRNA expression of mRNA transcripts (E: α-GalA, F: glyceronephosphate O-acyltransferase, G: ceramide glucosyltransferase) in DCs after infection with A.a. or P.g. (MOI 10), or culture in the presence of IFN-α, IFN-β, or LPS, respectively. *, p<0.05; **, p<0.01; n.s, not significant.
In response to TLR ligation, the endosomal glycosphingolipid (GSL) turnover in DCs is disrupted, leading to their accumulation and presentation in CD1d molecules (23). We explored whether A.a. and P.g. were able to downregulate the rate-limiting enzyme of GSL turnover, α-galactosidase A, in infected DCs. DCs were infected with A.a. or P.g. or were incubated with recombinant IFN-α or IFN-β. First, we evaluated the enzymatic activity of α-GalA in DCs after 6 hours of incubation. Compared to α-GalA activity in uninfected BMDC, A.a. infection lead to a 50% decrease in α-GalA activity. In contrast, P.g. infected DC showed slightly elevated activity of this enzyme compared to untreated DCs (Fig. 5D). Notably, the type I interferons IFN-α and IFN-β as well as LPS were also able to reduce the activity of α-GalA in DCs (Fig.5D).
We next investigated whether type I interferons which are up-regulated upon bacterial infection directly inhibited α-galactosidase A as analyzed by its mRNA expression in DCs after 24 hours of infection. Corroborating the marked differences in α-GalA enzymatic activity observed between A.a. and P.g. infection, A.a. challenge led to a down-regulation of α-GalA mRNA to approximately 40%. In contrast, P.g infection did not induce a clear change in α -GalA mRNA levels, nor did IFN-α or IFN-β significantly change α-GalA mRNA levels (Fig. 5E).
We extended this analysis on genes, namely glyceronephosphate O-acyltransferase (α -GNPAT) and ceramide glucosyltransferase (UGCG), which had previously been reported in the context of lipid loading on CD1d (33, 34). Neither infection with A.a. or P.g. nor stimulation with LPS regulated the expression of these two genes (Fig.5F,G), indicating the effect of A.a. infection to be specific for α -GalA expression and activity.
These data indicate that A.a. infection leads to an induction of IFN-β in DCs. Corroborating our finding that the activation of type I NKT cells by A.a. is TLR-dependent, we observed a disruption of GSL turnover in A.a. infected DCs. Infection with P.g., however, did not induce type I interferons in DCs. The TLR-dependent GSL turnover disruption could not be observed in P.g. infected DCs.
Autocrine type I interferons on A.a.-infected DCs
Whether type I interferons are acting on DCs in an autocrine fashion or are necessary for the activation of type I NKT cells is unknown. To address this question, we performed co-culture experiments in which DCs were infected in the presence or absence of a blocking antibody against the type I interferon receptor (IFNaR) before type I NKT cells were added as responder cells. Parallel to this, blocking antibodies were added during the stimulation of responder type I NKT cells. DCs infected with A.a. for 24 hrs in the absence of antibodies (Fig. 6A, bar 2) or in presence of isotype controls (Fig. 6A, bar 3) triggered significant amounts of IFN-γ released from responding type I NKT cells. Addition of IFNaR-blocking antibodies with A.a. abrogated the ability of DC to stimulate type I NKT cells (Fig. 6A, bar 4). However, when IFNaR-neutralizing antibodies were added simultaneously to type I NKT cells the IFN-γ release remained unaffected (Fig.6A, bar 5). Corroborating these results, DCs generated from BM of mice lacking the type I interferon receptor and infected with A.a. failed to stimulate type I NKT cells to secrete IFN-γ (Fig.6B). These data suggest that type I interferons (IFN-α, IFN-β) secreted from bacterial-infected DCs, rather than acting on type I NKT cells, are necessary to achieve DC stimulation of type I NKT cells.
Fig.6. Autocrine type I interferons are required for the ability of A.a.-infected DCs to stimulate type I NKT cells.

A. Murine BMDCs were infected with A.a. (MOI 10) with or without blocking mAb against IFNaR, or mouse IgG1 isotype control, or left uninfected. 24 h later, liver MNCs were added with or without anti-IFNaR mAb or isotype controls. Data shown are representative of three experiments. B. BM-DC of WT and IFNAR knockout mice were infected with A.a. (MOI 10) or left untreated (n=3). IFN-γ levels in supernatants (48h) were assessed by ELISA. * p<0.05;**, p<0.01; ***, p<0.001; n.d, not detectable; n.s, not significant.
Exogenous type I interferon rescues NKT cell stimulation by P.g.-infected DCs
To confirm that the absence of type I interferon production in P.g.-infected DCs is in fact responsible for the observed inability of P.g.-infected DCs to activate type I NKT cells, we performed ‘rescue’-experiments.
First, we tested whether exogenous type I interferons added during the infection of DCs would rescue the ability of P.g.-infected DCs to stimulate type I NKT cells. BMDCs were infected with either A.a. or P.g. at MOI of 10 in the presence or absence of recombinant IFN-α in the culture medium. After 24 hours, DCs were washed and type I NKT cells were added as responder cells. Confirming data shown in Figs. 2 and 4, A.a.-infected DCs triggered the production of IFN-γ in type I NKT cells, whereas DCs infected with P.g. failed to do so (Fig.7A, bars 1,2). Importantly, addition of exogenous type I interferons (IFN-α, IFN-β) during the infection with P.g. was sufficient to promote the downstream activation of type I NKT cells (Fig.7, bars 3+4 and 7+8). Addition of exogenous type I interferons simultaneously to type I NKT cells comparably ‘rescued’ the IFNg production by type I NKT cells.
Fig.7. Exogenous type I interferon restores the lack of IFN-α induction and NKT cell activation by P.g-infected DCs.
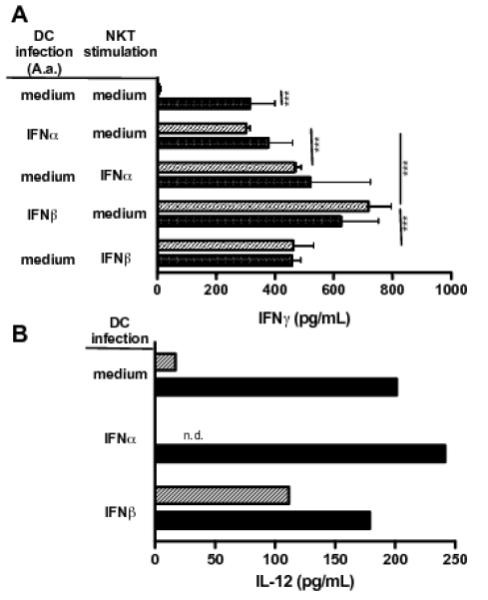
Murine BMDCs were infected (MOI 10, 24h) with A.a. (closed bars) or P.g (shaded bars) in the presence or absence of recombinant IFN-α or IFN-β (both 10 U/mL). Supernatants were tested for IL-12 release (panel A, n=2) and liver MNC were added to remaining DCs with or without addition of cytokines (48h). IFN-γ levels in supernatants were assessed by ELISA (panel B, n=3). Data shown are representative of three experiments. *, p<0.05;**, p<0.01; ***, p<0.001; n.d, not detectable; n.s, not significant.
To test whether the inability of P.g.-infected DCs is due to a lack of IL-12 secretion cell culture supernatants were tested for their IL-12 content. As expected from maturation data shown in Fig.2C, infection with A.a. results in the release of IL-12 by DCs (Fig.7B). Addition of exogenous IFN-α to DC cultures resulted in reduced release of IL-12, probably due to DC exhaustion. In contrast, infection with P.g did not result in IL-12 release (Fig.7B, grey bars). The addition of IFN-α to P.g./DC cultures significantly increased the IL-12 secretion of DC to levels comparable to those elicited by A.a. infections.
The addition of exogenous interferons was sufficient to restore the immunogenic cytokine secretion of type NKT cells in response to challenge with P.g.
Discussion
In this study we describe clear differences in type I NKT cell infiltration in chronic and aggressive periodontitis lesions (Fig.1). Type I NKT cells specifically infiltrated lesions of the aggressive subtype. Albeit numbers of paired periodontal biopsies available to this study were limited due to the restrictions imposed on the resection of clinically healthy gingiva, significant differences could be observed in the ‘disease’-‘health’ comparison in these cohorts. We furthermore observed a trend (p=0.11) towards lower type I NKT cell numbers in clinically ‘healthy’ aggressive versus ‘chronic’ lesions. If confirmed, this finding could demonstrate an inherent difference in innate immune cell repertoire even in unchallenged gingiva from both disease entities. We analyzed the frequency of type I NKT cells measuring the presence of Va24-Ja18 TCR transcripts, selectively expressed by type I NKT cells. To date, no working antibody specifically staining type I NKT cells is available, which conceivably might provide additional insight into the localization of NKT in periodontal lesions. Furthermore, we also found significant differences in transcripts linked to DC-mediated NKT cell activation. Specifically, chemokines that bind chemokine receptors expressed by type I NKT cells were exclusively significantly regulated in aggressive periodontitis lesions in vivo, possibly explaining the increased influx of type I NKT cells in this disease subtype. Furthermore, markers of DC-mediated activation, such as CD86 and CD1d were induced in aggressive periodontitis lesions only.
Therefore, we investigated the DC-mediated type I NKT cell responses to the periodontal pathogens A.a. and P.g. in vitro. DCs infected with A.a at different MOI dose-dependently stimulated a vigorous secretion of the proinflammatory cytokine IFN-γ in type I NKT cells (Fig.2A). Strikingly, DCs infected with P.g did not elicit any detectable cytokine secretion in type I NKT cells (Fig. 2). We did not detect elevated concentrations of anti-inflammatory IL-4 in co-cultures of infected DC and type I NKT cells, corroborating earlier data showing that upon TLR ligation, DCs selectively stimulate the production of IFN-γ in type I NKT cells (16, 22, 23, 35, 36).
The cytokine secretion by type I NKT cells depended on the expression of CD1d molecules on the surface of infected DCs. This indicates that the impact of DCs that were inherently, albeit in small numbers, present in our type I NKT cell preparations was negligible in these experiments. In line with the aforementioned in vivo and in vitro results, we found that infection of DCs with A.a., but not P.g, upregulated the expression of CD86 costimulatory molecules on DC surfaces. Whereas A.a. stimulated strong IL-12 secretion by DCs, challenge with P.g only led to minimal IL-12 protein production (Fig.7). Whole-genome expression analyses of DCs infected with A.a. and P.g. indicate that P.g. infection, in contrast to A.a., resulted in a lacking type I interferon response.
The observed mild induction of IL-12 mRNA in P.g.-infected DCs did apparently not result in protein translation. Thus, our data are not in contrast to results by Zhou and coworkers who demonstrated human macrophage activation by live P.g. and isolated fimbriae, as assessed by TNF-α secretion and upregulation of IL-12 and NFkB mRNA (37). Importantly, and in line with our data, neither live P.g nor isolated fimbriae stimulated IFN-γ secretion by macrophages. Other studies only analyzed the effects of cell wall extracts on human monocyte-derived DCs (38-40). To our knowledge, our study is first to show a lack of DC maturation in response to infection with live P.g.
Recognition of conserved pathogen-associated molecular patterns (PAMPs) by toll-like receptors is a key element of innate immunity. Several lines of evidence exist for activation of NKT cells by glycolipids presented in DCs recognizing PAMPs via their TLRs (16, 22, 23, 35, 36, 41). Ligation of TLR9 on DCs was demonstrated to result in production of IFN-γ by NKT cells in vitro (22). Type I interferon signalling and presentation of de novo synthesized charged glycolipids on CD1d molecules was required for this process (22). TLR activation and downstream NKT cell activation within mononuclear cell preparations containing both cell types required secretion of IFN-β by DCs and functional expression of the type I interferon receptor IFNaR on type I NKT cells (22). However, the question whether infected DCs require IFNaR expression has not been analysed. In addition, for a number of bacterial species lipid antigens have been described that are able to bind CD1d molecules and stimulate type I NKT cells, including Salmonella and Borrelia (19-21). Our result strongly suggest that disruption of the glycolipid turnover by α-GalA is critical to the ability of A.a.-infected DCs to stimulate type I NKT cells. In view of this requirement, it appears unlikely that A.a. contains lipid stimulating type I NKT cells.
We analyzed the requirements of TLR and type I interferon signalling in response to infection with periodontal pathogens. Our data indicate that the stimulatory capacity of A.a.-infected DCs depends on MyD88 expression, which is the central signalling molecule downstream of most TLR family members, including TLR2, 4, and 9 (42). MyD88 gene-deficient DCs exhibited a gene dose-dependent lack of stimulatory capacity for type I NKT cells (Fig. 3). Corroborating our data, Iweala and coworkers recently found that Salmonella vaccination in MyD88-deficient mice skewed the effector functions of type I NKT cells (43).
P.g expresses several cell wall components mediating its virulence in vivo (44). In particular, P.g fimbriae and lipopolysaccharide possess immune evasive properties. Hajishengalis and coworkers showed that P.g. fimbriae bind to the chemokine receptor CXCR4 expressed on mature DCs, leading to elevated intracellular cAMP levels that antagonize the TLR-mediated NF-κB activation in DCs (44). This constitutes one of conceivably several non-exclusive mechanisms which enable P.g. to evade the immune system. Indeed, we show a notably reduced inflammatory response in P.g. versus A.a.-infected DCs that could be explained by active immune evasion by P.g..
We investigated whether the lack of NKT cell stimulation by P.g-infected DCs is due to a lack of type I interferon expression in infected DCs. Infection of murine DC with A.a induced type I interferons (Fig. 5C) and several transcripts involved in type I interferon signaling and DC maturation (Table I, Fig 4D). In stark contrast, P.g was unable to upregulate the expression of type I interferons in DCs (Fig.5). The activation of type I NKT cells by A.a. infection required the expression of the IFNaR and functional TLR signalling in DCs. Interestingly, A.a. infection led to the strong upregulation of IFN-β production whilst transcription levels of IFN-α remained unchanged. The molecular basis of this observation is still elusive. However, A.a. is known to secrete the ‘immune-suppressive factor’ (ISF) and thereby inhibit protein synthesis in leukocytes, as shown for inhibition of IgG and IgM production in B cells as well as IL-2 translation in T cells (45). Along this line, Gobl et al. using human monocytes showed that interferon-alpha but not -beta secretion requires de novo protein synthesis. (46). To our knowledge their are no data available as to whether this is also holds true for murine DCs. However, since murine DCs retain the ability to up-regulate other proteins (i.e. IL-12, CD86) this remains further investigation.
Recently, it was demonstrated that combinations of IFN-β and presentation of self-glycolipids from TLR9-stimulated DCs were required for the downstream activation of type I NKT cells (22). It was, however, still unclear whether type I interferons exert additional autocrine effects on DC. To address this question, we blocked the IFNaR during the infection of DCs and whilst co-incubating DCs and NKT cells. Using this experimental approach, we clearly demonstrate that expression of the IFNaR is required on the surface of infected DC and indispensable for responding type I NKT cells. This in turn suggests, the stimulatory capacity of A.a.-infected DCs to type I NKT cell is attributed to the ability of A.a. to signal via toll-like receptors and elicit the production of both IL-12 and type I interferons by DCs. Darmoise et al. previously showed that the activation of type I NKT cells by α-GalA-deficient DCs despite their enhanced basal stimulatory capacity still depends on their IL-12 production (23). Whether autocrine IFN-β secretion and IFNAR signalling besides downmodulating of endosomal glycolipid turnover also acts on the production of IL-12 remains further investigation.
Taken together, we show that infiltration by type I NKT cells is a feature of aggressive, but not chronic periodontitis. In line with these in vivo observations, we found that the aggressive periodontitis-associated pathogen A.a. triggered type I NKT cell activation and proinflammatory IFN-γ secretion via DCs in vitro. In contrast, P.g., a principal pathogen in chronic periodontitis, results in a dampened immune response in DCs characterized by a lack of type I interferon production and a subsequent lack of type I NKT cell activation. Strikingly, the ability of P.g. to accumulate endogenous glycosphingolipids in DCs and leading to the activation of type I NKT cells was restored by addition of exogenous type I interferons. This indicates that immune evasion actively mediated by P.g. may be responsible for the less pronounced activation of the inflammatory repertoire in chronic than in aggressive periodontitis. The activation of type I NKT cells with subsequent production of abundant IFN-γ triggered by A.a. possibly aggravates tissue destruction in aggressive periodontitis (47).
Supplementary Material
Acknowledgements
The authors thank Ryan T. Demmer (Columbia University, New York, NY, USA) for logistical help with patient samples, Heike Weighardt (University of Düsseldorf, Germany) for providing MyD88 knockout mice, and Beatrix Schumak (University of Bonn,Germany) for IFNAR kncout mice. We thank Bettina Jux for critically reading the manuscript.
Research in the authors’ laboratories was supported by the German Research Foundation (DFG KFO208), NIH/NIDCR, DGP, DGZMK.
Glossary
- A.a.
Aggregatibacter actinomycetemcomitans
- α-GalCer
alpha-galactosylceramide
- BMDC
bone marrow-derived dendritic cells
- FDR
false discovery rate
- GSL
glycosphingolipid
- MNC
mononuclear cells
- MOI
multiplicity of infection
- NKT
Natural Killer T cells
- P.g
Porphyromonas gingivalis
References
- 1.Dye BA, Barker LK, Li X, Lewis BG, Beltran-Aguilar ED. Overview and quality assurance for the oral health component of the National Health and Nutrition Examination Survey (NHANES), 2005-08. J Public Health Dent. 2011;71:54–61. doi: 10.1111/j.1752-7325.2010.00202.x. [DOI] [PubMed] [Google Scholar]
- 2.Darveau RP. The oral microbial consortium’s interaction with the periodontal innate defense system. DNA Cell Biol. 2009;28:389–395. doi: 10.1089/dna.2009.0864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Darveau RP. Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Microbiol. 2010;8:481–490. doi: 10.1038/nrmicro2337. [DOI] [PubMed] [Google Scholar]
- 4.Kebschull M, Demmer RT, Papanou PN. “Gum bug, leave my heart alone!”-- epidemiologic and mechanistic evidence linking periodontal infectiond atherosclerosis. J Dent Res. 2010;89:879–902. doi: 10.1177/0022034510375281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Armitage GC. Development of a classification system for periodontal diseases and conditions. Ann Periodontol. 1999;4:1–6. doi: 10.1902/annals.1999.4.1.1. [DOI] [PubMed] [Google Scholar]
- 6.Demmer RT, Papapanou PN. Epidemiologic patterns of chronic and aggressive periodontitis. Periodontol 2000. 2010;53:28–44. doi: 10.1111/j.1600-0757.2009.00326.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Armitage GC, Cullinan MP. Comparison of the clinical features of chronic and aggressive periodontitis. Periodontol 2000. 2010;53:12–27. doi: 10.1111/j.1600-0757.2010.00353.x. [DOI] [PubMed] [Google Scholar]
- 8.Haffajee AD, Socransky SS. Microbial etiological agents of destructive periodontal diseases. Periodontol 2000. 1994;5:78–111. doi: 10.1111/j.1600-0757.1994.tb00020.x. [DOI] [PubMed] [Google Scholar]
- 9.Schacher B, Baron F, Rossberg M, Wohlfeil M, Arndt R, Eickholz P. Aggregatibacter actinomycetemcomitans as indicator for aggressive periodontitis by two analysing strategies. J Clin Periodontol. 2007;34:566–573. doi: 10.1111/j.1600-051X.2007.01080.x. [DOI] [PubMed] [Google Scholar]
- 10.Dhillon PK, Barry M, Stampfer MJ, Perner S, Fiorentino M, Fornari A, Ma J, Fleet J, Kurth T, Rubin MA, Mucci LA. Aberrant cytoplasmic expression of p63 and prostate cancer mortality. Cancer Epidemiol Biomarkers Prev. 2009;18:595–600. doi: 10.1158/1055-9965.EPI-08-0785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Q, Zhou XD, Zheng QH, Wang Y, Tang L, Huang DM. Distribution of Porphyromonas gingivalis fimA genotypes in chronic apical periodontitis associated with symptoms. J Endod. 2010;36:1790–1795. doi: 10.1016/j.joen.2010.08.018. [DOI] [PubMed] [Google Scholar]
- 12.Smith M, Seymour GJ, Cullinan MP. Histopathological features of chronic and aggressive periodontitis. Periodontol 2000. 2010;53:45–54. doi: 10.1111/j.1600-0757.2010.00354.x. [DOI] [PubMed] [Google Scholar]
- 13.Kronenberg M. Toward an understanding of NKT cell biology: progress and paradoxes. Annu Rev Immunol. 2005;23:877–900. doi: 10.1146/annurev.immunol.23.021704.115742. [DOI] [PubMed] [Google Scholar]
- 14.Fujii S, Shimizu K, Kronenberg M, Steinman RM. Prolonged IFN-gamma-producing NKT response induced with alpha-galactosylceramide-loaded DCs. Nat.Immunol. 2002;3:867–874. doi: 10.1038/ni827. [DOI] [PubMed] [Google Scholar]
- 15.Fujii S, Shimizu K, Smith C, Bonifaz L, Steinman RM. Activation of natural killer T cells by alpha-galactosylceramide rapidly induces the full maturation of dendritic cells in vivo and thereby acts as an adjuvant for combined CD4 and CD8 T cell immunity to a coadministered protein. J.Exp.Med. 2003;198:267–279. doi: 10.1084/jem.20030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brigl M, Bry L, Kent SC, Gumperz JE, Brenner MB. Mechanism of CD1d-restricted natural killer T cell activation during microbial infection. Nat.Immunol. 2003;4:1230–1237. doi: 10.1038/ni1002. [DOI] [PubMed] [Google Scholar]
- 17.Tomura M, Yu WG, Ahn HJ, Yamashita M, Yang YF, Ono S, Hamaoka T, Kawano T, Taniguchi M, Koezuka Y, Fujiwara H. A novel function of Valpha14 + CD4 + NKT cells: stimulation of IL-12 production by antigen-presenting cells in the innate immune system. J.Immunol. 1999;163:93–101. [PubMed] [Google Scholar]
- 18.Hayakawa Y, Takeda K, Yagita H, Van Kaer L, Saiki I, Okumura K. Differential regulation of Th1 and Th2 functions of NKT cells by CD28 and CD40 costimulatory pathways. J.Immunol. 2001;166:6012–6018. doi: 10.4049/jimmunol.166.10.6012. [DOI] [PubMed] [Google Scholar]
- 19.Kinjo Y, Tupin E, Wu D, Fujio M, Garcia-Navarro R, Benhnia MR, Zajonc DM, Ben-Menachem G, Ainge GD, Painter GF, Khurana A, Hoebe K, Behar SM, Beutler B, Wilson IA, Tsuji M, Sellati TJ, Wong CH, Kronenberg M. Natural killer T cells recognize diacylglycerol antigens from pathogenic bacteria. Nat Immunol. 2006;7:978–986. doi: 10.1038/ni1380. [DOI] [PubMed] [Google Scholar]
- 20.Kinjo Y, Wu D, Kim G, Xing GW, Poles MA, Ho DD, Tsuji M, Kawahara K, Wong CH, Kronenberg M. Recognition of bacterial glycosphingolipids by natural killer T cells. Nature. 2005;434:520–525. doi: 10.1038/nature03407. [DOI] [PubMed] [Google Scholar]
- 21.Sriram V, Du W, Gervay-Hague J, Brutkiewicz RR. Cell wall glycosphingolipids of Sphingomonas paucimobilis are CD1d-specific ligands for NKT cells. Eur J Immunol. 2005;35:1692–1701. doi: 10.1002/eji.200526157. [DOI] [PubMed] [Google Scholar]
- 22.Paget C, Mallevaey T, Speak AO, Torres D, Fontaine J, Sheehan KC, Capron M, Ryffel B, Faveeuw C, Leite de Moraes M, Platt F, Trottein F. Activation of invariant NKT cells by toll-like receptor 9-stimulated dendritic cells requires type I interferon and charged glycosphingolipids. Immunity. 2007;27:597–609. doi: 10.1016/j.immuni.2007.08.017. [DOI] [PubMed] [Google Scholar]
- 23.Darmoise A, Teneberg S, Bouzonville L, Brady RO, Beck M, Kaufmann SH, Winau F. Lysosomal alpha-galactosidase controls the generation of self lipid antigens for natural killer T cells. Immunity. 2010;33:216–228. doi: 10.1016/j.immuni.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ono M, Okuda K, Takazoe I. Purification and characterization of a thiol-protease from Bacteroides gingivalis strain 381. Oral Microbiol Immunol. 1987;2:77–81. doi: 10.1111/j.1399-302x.1987.tb00294.x. [DOI] [PubMed] [Google Scholar]
- 25.Nowak M, Lynch L, Yue S, Ohta A, Sitkovsky M, Balk SP, Exley MA. The A2aR adenosine receptor controls cytokine production in iNKT cells. Eur J Immunol. 2010;40:682–687. doi: 10.1002/eji.200939897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nowak M, Arredouani MS, Tun-Kyi A, Schmidt-Wolf I, Sanda MG, Balk SP, Exley MA. Defective NKT cell activation by CD1d+ TRAMP prostate tumor cells is corrected by interleukin-12 with alpha-galactosylceramide. PLoS One. 2010;5:e11311. doi: 10.1371/journal.pone.0011311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ford PJ, Gamonal J, Seymour GJ. Immunological differences and similarities between chronic periodontitis and aggressive periodontitis. Periodontol 2000. 2010;53:111–123. doi: 10.1111/j.1600-0757.2010.00349.x. [DOI] [PubMed] [Google Scholar]
- 28.Kornman KS, Page RC, Tonetti MS. The host response to the microbial challenge in periodontitis: assembling the players. Periodontol 2000. 1997;14:33–53. doi: 10.1111/j.1600-0757.1997.tb00191.x. [DOI] [PubMed] [Google Scholar]
- 29.Samarajiwa SA, Forster S, Auchettl K, Hertzog PJ. INTERFEROME: the database of interferon regulated genes. Nucleic Acids Res. 2009;37:D852–857. doi: 10.1093/nar/gkn732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grose RH, Thompson FM, Cummins AG. Deficiency of 6B11+ Invariant NK T-Cells in Celiac Disease. Dig Dis Sci. 2008;53:1846–1851. doi: 10.1007/s10620-007-0093-x. [DOI] [PubMed] [Google Scholar]
- 31.Prussin C, Foster B. TCR V alpha 24 and V beta 11 coexpression defines a human NK1 T cell analog containing a unique Th0 subpopulation. J Immunol. 1997;159:5862–5870. [PubMed] [Google Scholar]
- 32.Thomas SY, Hou R, Boyson JE, Means TK, Hess C, Olson DP, Strominger JL, Brenner MB, Gumperz JE, Wilson SB, Luster AD. CD1d-restricted NKT cells express a chemokine receptor profile indicative of Th1-type inflammatory homing cells. J. Immunol. 2003;171:2571–2580. doi: 10.4049/jimmunol.171.5.2571. [DOI] [PubMed] [Google Scholar]
- 33.Brennan PJ, Tatituri RV, Brigl M, Kim EY, Tuli A, Sanderson JP, Gadola SD, Hsu FF, Besra GS, Brenner MB. Invariant natural killer T cells recognize lipid self antigen induced by microbial danger signals. Nat Immunol. 2011;12:1202–1211. doi: 10.1038/ni.2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Facciotti F, Ramanjaneyulu GS, Lepore M, Sansano S, Cavallari M, Kistowska M, Forss-Petter S, Ni G, Colone A, Singhal A, Berger J, Xia C, Mori L, De Libero G. Peroxisome-derived lipids are self antigens that stimulate invariant natural killer T cells in the thymus. Nat Immunol. 2012;13:474–480. doi: 10.1038/ni.2245. [DOI] [PubMed] [Google Scholar]
- 35.Marschner A, Rothenfusser S, Hornung V, Prell D, Krug A, Kerkmann M, Wellisch D, Poeck H, Greinacher A, Giese T, Endres S, Hartmann G. CpG ODN enhance antigen-specific NKT cell activation via plasmacytoid dendritic cells. Eur J Immunol. 2005;35:2347–2357. doi: 10.1002/eji.200425721. [DOI] [PubMed] [Google Scholar]
- 36.Mattner J, Debord KL, Ismail N, Goff RD, Cantu C, III, Zhou D, Saint-Mezard P, Wang V, Gao Y, Yin N, Hoebe K, Schneewind O, Walker D, Beutler B, Teyton L, Savage PB, Bendelac A. Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature. 2005;434:525–529. doi: 10.1038/nature03408. [DOI] [PubMed] [Google Scholar]
- 37.Zhou Q, Desta T, Fenton M, Graves DT, Amar S. Cytokine profiling of macrophages exposed to Porphyromonas gingivalis, its lipopolysaccharide, or its FimA protein. Infect Immun. 2005;73:935–943. doi: 10.1128/IAI.73.2.935-943.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chino T, Santer DM, Giordano D, Chen C, Li C, Chen CH, Darveau RP, Clark EA. Effects of oral commensal and pathogenic bacteria on human dendritic cells. Oral Microbiol Immunol. 2009;24:96–103. doi: 10.1111/j.1399-302X.2008.00478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kanaya S, Nemoto E, Ogawa T, Shimauchi H. Porphyromonas gingivalis fimbriae induce unique dendritic cell subsets via Toll-like receptor 2. J Periodontal Res. 2009;44:543–549. doi: 10.1111/j.1600-0765.2008.01149.x. [DOI] [PubMed] [Google Scholar]
- 40.Vernal R, Leon R, Herrera D, Garcia-Sanz JA, Silva, Sanz M. Variability in the response of human dendritic cells stimulated with Porphyromonas gingivalis or Aggregatibacter actinomycetemcomitans. J Periodontal Res. 2008;43:689–697. doi: 10.1111/j.1600-0765.2007.01073.x. [DOI] [PubMed] [Google Scholar]
- 41.Askenase PW, Itakura A, Leite-de-Moraes MC, Lisbonne M, Roongapinun S, Goldstein DR, Szczepanik M. TLR-dependent IL-4 production by invariant Valpha14+Jalpha18+ NKT cells to initiate contact sensitivity in vivo. The Journal of Immunology. 2005;175:6390–6401. doi: 10.4049/jimmunol.175.10.6390. [DOI] [PubMed] [Google Scholar]
- 42.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 43.Iweala OI, Smith DW, Matharu KS, Sada-Ovalle I, Nguyen DD, Dekruyff RH, Umetsu DT, Behar SM, Nagler CR. Vaccine-induced antibody isotypes are skewed by impaired CD4 T cell and invariant NKT cell effector responses in MyD88-deficient mice. J Immunol. 2009;183:2252–2260. doi: 10.4049/jimmunol.0804011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hajishengallis G, Wang M, Liang S, Triantafilou M, Triantafilou K. Pathogen induction of CXCR4/TLR2 cross-talk impairs host defense function. Proc Natl Acad Sci U S A. 2008;105:13532–13537. doi: 10.1073/pnas.0803852105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shenker BJ, Vitale LA, Welham DA. Immune suppression induced by Actinobacillus actinomycetemcomitans: effects on immunoglobulin production by human B cells. Infect Immun. 1990;58:3856–3862. doi: 10.1128/iai.58.12.3856-3862.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gobl AE, Cederblad B, Sandberg K, Alm GV. Interferon-alpha but not -beta genes require de novo protein synthesis for efficient expression in human monocytes. Scand J Immunol. 1992;35:177–185. doi: 10.1111/j.1365-3083.1992.tb02848.x. [DOI] [PubMed] [Google Scholar]
- 47.Garlet GP. Destructive and protective roles of cytokines in periodontitis: a re-appraisal from host defense and tissue destruction viewpoints. J Dent Res. 2010;89:1349–1363. doi: 10.1177/0022034510376402. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.