

Abstract
Retinoids (vitamin A and its analogs) are highly potent regulators of cell differentiation, cell proliferation, and apoptosis. Because of these activities, retinoids have been most extensively studied in the contexts of embryonic development and of proliferative diseases, especially cancer and skin disease. Recently, there has been considerable new research interest focused on gaining understanding of the roles that retinoids and/or retinoid-related proteins may have in the development of metabolic diseases, primarily obesity, diabetes, and dyslipidemia. This review will summarize recent advances that have been made in these areas, focusing on the role of retinoids in modulating adipogenesis, the roles of retinoids and retinoid-related proteins as signaling molecules linking obesity with the development of type II diabetes, the roles of retinoids in pancreatic β-cell biology/insulin secretion, and the actions of retinoids in hepatic steatosis.
1. Introduction
The term retinoid is used to refer collectively to compounds that have a structural resemblance to all-trans-retinol (which by definition is vitamin A), with or without the biological activity of vitamin A (1,2). Thus, retinoids are both natural substances found in the diet at low levels and synthetic compounds designed as pharmacologic agents. It has long been established that 11-cis-retinaldehyde is the visual chromophore (3). Retinoids, acting as ligands for ligand-dependent transcription factors, are potent regulators of cellular proliferation, differentiation and apoptosis (4–6). Because of these effects on cells, retinoids are required for maintaining immunity, barrier function, bone health, embryogenesis, and reproduction. Recently, there is growing research interest focused on retinoid actions in metabolism and metabolic disease. The goal of this article is to review briefly retinoid biology and to consider in more depth some recent advances in understanding retinoid actions in metabolism and metabolic disease.
2. Retinoid Biology
A. Intake, Storage and Metabolism
Mammals obtain retinoid from the diet either in the form of provitamin A carotenoids or as preformed vitamin A, primarily retinol and retinyl esters. In the intestine, retinyl esters must be hydrolyzed to retinol before uptake by enterocytes; studies utilizing non-hydrolyzable ether analogs of retinyl ester have demonstrated the necessity of hydrolysis prior to absorption (7). This involves the catalytic activities of intestinal retinyl ester hydrolases (REH), including the bile salt-dependent cholesterol ester lipase and pancreatic triglyceride lipase, which together account for 50% of intestinal REH activities (8).
Within enterocytes, retinol is re-esterified to retinyl ester predominantly by lecithin:retinol acyltransferase (LRAT) and, to a very small extent, by acyl-CoA:retinol acyltransferases (ARATs) (9). Diacylglycerol acyltransferase 1 (DGAT1) has been shown to be a physiologically significant intestinal ARAT (10). The intestinal mucosa is also the major site of carotenoid conversion to retinoid. In humans, up to 90% of absorbed all-trans-β-carotene is enzymatically cleaved by β-carotene 15,15′-monooxygenase 1 (BCMO1) to form retinaldehyde (11), which is then reduced to retinol by retinaldehyde reductases and esterified by LRAT (12). Retinyl esters are then incorporated into nascent chylomicrons, which enter the circulation via the mesenteric lymph. Approximately 25–33% of chylomicron retinyl esters are hydrolyzed by lipoprotein lipase (LPL) in the circulation and delivered to extrahepatic tissues, while the rest is taken up by the liver as a component of chylomicron remnants (CRs) (13). For a detailed account of chylomicron metabolism and the steps leading to hepatic clearance, the reader is referred to two excellent reviews on the topic of CR uptake by Cooper and colleagues (14,15).
The liver is the most important tissue in the body for retinoid uptake, storage and mobilization. There are two major hepatic cells types involved in retinoid metabolism, hepatocytes and hepatic stellate cells (HSCs). Hepatocytes play an indispensable role in uptake and processing of dietary retinoid into the liver, and in synthesis and secretion of retinol-binding protein (RBP), which is required for mobilizing hepatic retinoid stores in times of dietary vitamin A-insufficiency. Cellular retinol-binding protein I (CRBPI) binds newly absorbed retinol and serves as an intracellular transporter of retinol, linking and facilitating the processes of retinol uptake, metabolism and mobilization. From hepatocytes, newly absorbed retinol is transferred to HSCs for re-esterification and storage. The molecular mechanisms for the transfer of retinol from hepatocyte to HSCs, or inversely from HSCs to hepatocyte for mobilization remain to be clearly established. HSCs are the central cellular site for retinoid storage in the healthy animal, accounting for as much as 50–60% of the total retinoid present in the entire body, mostly in the form of retinyl esters. LRAT, which is highly expressed in HSCs, is the sole enzyme responsible for retinyl ester synthesis in liver. For more on hepatic retinoid metabolism, the reader is referred to two recent reviews (16,17).
A generalized scheme for the metabolism of retinoids is provided in Figure 1. It should be noted that all of these metabolic steps may not occur in every cell type. Moreover, these processes may be more complex than depicted, involving multiple binding proteins and mutiple enzyme species.
Figure 1. General scheme of cellular retinol metabolism.
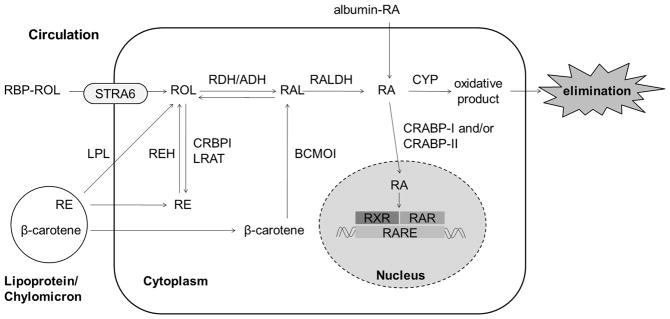
Cellular uptake of retinol (ROL) is mediated by STRA6 in tissues that express this surface receptor. This uptake process requires the presence of CRBPI and depends on the activity of LRAT in esterifying ROL to retinol ester (RE). STRA6 is, however, not universally expressed and is not expressed in liver and heart. Thus, it has been proposed that ROL can diffuse across the plasma membrane down a concentration gradient. RE associated with lipoproteins or chylomicrons can be hydrolyzed to ROH by LPL and taken up by this “flip-flop” mechanism across the plasma membrane. Alternatively, the lipoproteins can be taken up whole by endocytosis. Within the cell, RE is hydrolyzed by REHs to ROH, which is then either oxidized to retinaldehyde (RAL) by one of the short chain dehydrogenase/reductases reported to have retinol dehydrogenase (RDH) activity or by medium chain alcohol dehydrogenases (ADH) able to use retinol as a substrate, or esterified to RE by LRAT for storage in lipid droplets. β-carotene is enzymatically cleaved by BCMO1 in an irreversible step to form two molecules of RAL, which can be reduced to ROH by RDH or oxidized to RA by RALDH. RA can also be taken up from the circulation where it is found bound to albumin. From the cytoplasm, RA is transferred to the nucleus, by CRABPI and/or CRABPII, where it binds with RARs and RXRs to induce transcription of target genes. Alternatively, RA can be catabolized by the cytochrome enzymes (CYP) and the oxidative products are eliminated from the cell.
B. Retinoid Actions – Genomic Regulation
Retinoids have pleiotropic actions in all vertebrate tissues, affecting cellular proliferation, differentiation, and apoptosis. As discussed below, retinoids also regulate metabolism. These bioactivities are due largely to the ability of retinoids to regulate expression levels of target genes. Representative genes involved in metabolism and metabolic disease reported to be regulated by retinoic acid (RA) are summarized in Table I. RA is the major transcriptionally active retinoid species, regulating gene expression of over 500 genes (4) through binding to retinoic acid receptors (RARs) and retinoid X receptors (RXRs) (18). Each of these families comprises three isotypes (α, β and γ) with additional isoforms generated by alternative splicing and differential promoter usage (5). RARs and RXRs, which work as RXR/RAR heterodimers, mediate the ligand-dependent regulation of target gene transcription through cis interactions with retinoic acid response element (RARE) and trans interactions with other transcription factors and coregulators. RARs are activated by all-trans-retinoic acid (atRA) and its 9-cis-isomer (9cRA), while RXRs are only activated by one natural retinoid, 9cRA. RARs and RXRs share 6 conserved modular structures designated A–F, which are characterized by a variable NH2-terminal region (A/B) that has a ligand-independent activation function (AF1); a conserved DNA binding domain (C); a hinge region (D); and a multi-functional ligand-binding domain (LBD) (E/F) consisting of a conserved carboxy terminal motif required for ligand-dependent transactivation (AF2), homo- and heterodimerization, and the association of coactivator and corepressor proteins (19–22).
Table I.
Some Representative Genes Involved in Metabolism Which Are Reported to be regulated by RA
| Symbol (Gene) | Names in Refences | Direction | Function | |
|---|---|---|---|---|
| Acadm | MCAD | Acyl-Coenzyme A dehydrogenase or medium-chain acyl-Coenzyme A dehydrogenase. | Up | Fatty acid metabolism |
| ApoA1 and 2 | Apo A-I and-II | Apolipoprotein A-I and-II | Up | Lipid metabolism |
| Cetp | CETP | Cholesteryl ester transfer protein | Up | Lipid metabolism |
| Cyp7a1 | CYP7A1 | Cholesterol 7 alpha-hydroxylase or cholesterol 7-alpha-monooxygenase or cytochrom P450 7A1 | - | Bile acid synthesis, lipid metabolism |
| Facl2 | Acyl-CoA Synthase, ACS | Fatty-acyl-CoA synthase | Up | Fatty acid biosynthesis |
| Fasn | FAS | Fatty acid synthase | - | Fatty acid biosynthesis |
| Fbp1 | Fru-1,6-P2ase, FBPase | Fructose bisphosphatase | Up | Gluconeogenesis |
| Hnf4A | HNF4α | Hepatocyte nuclear factor 4 alpha or NR2A1 (nuclear receptor subfamily 2, group A, member 1) | - | Transcription factor controlling expression of several hepatic genes; non-insulin-dependent diabetes type II in mutation |
| Gck | Glucokinase | Up | Glucose homeostasis | |
| Glut4 | GLUT4 | Glucose transporter type 4 | Up | Insulin-regulated glucose transporter found in adipose tissues and skeletal muscle |
| Il6 | IL6 | Interleukin-6 | Down | A pro-inflammatory and anti- inflammatory cytokine; energy mobilization in muscle and fatty tissue |
| Ins | Proinsulin, insulin | Up | Regulates carbohydrate and fat metabolism | |
| Lep, ob | Leptin | Down | Adipose derived hormone to regulate energy intake and energy expenditure, including appetite and metabolism | |
| Nr2f1 | COUP-TF1 | COUP transcription factor 1 or nuclear receptor subfamily 2, group F, member 1 | Up | A member of nuclear hormone receptor family of transcription factors |
| Pck1 | PEPCK | Phosphoenolpyruvatecarboxykinase 1 | Up | Gluconeogenesis |
| Ppara | PPAR-α or NR1C1 | Peroxisome proliferator-activated receptor alpha or nuclear receptor subfamily 1, group C, member 1 | Up | A transcription factor and major regulator of lipid metabolism in the liver |
| Pparg | PPAR-γ or NR1C3 | Peroxisome proliferator-activated receptor gamma or glitazone receptor or nuclear receptor subfamily 1, group C, member 3 | Up | Fatty acid storage and glucose metabolism; the genes activated by PPARγ stimulate lipid uptake and adipogenesis by fat cells |
| Slc2a2 | GLUT2 | Solute carrier family 2 (facilitated glucose transporter), member 2 or glucose transporter 2 | Up | A transporter for transfer of glucose between liver, pancreas and blood, and for renal glucose reabsorption |
| Ucp1 | Ucp, ucp-1 | Uncoupling protein 1 or thermogenin | Up | An uncoupling protein found in the brown adipose tissue, responsible for thermogenesis |
Retinoid receptors bind as asymmetrically oriented RXR/RAR heterodimers to specific promoter sequences composed typically of two direct repeats (referred to as a DR) of a core hexameric motif, PuG(G/T)TCA (23), separated most commonly by 5 bases (a DR5 is considered a canonical RARE). RAREs have been identified in the promoters of a large number of retinoid-target genes implicated in a wide variety of functions. Unliganded retinoid receptors bound to DNA response elements located in the promoter of target genes repress transcription through the recruitment of the corepressor NCoR (nuclear receptor corepressor) and SMRT (silencing mediator for retinoid and thyroid hormone receptor) (21). The corepressors reside in high molecular weight complexes endowed with histone deacetylase activity which increase the interaction of the N-terminal histone tails with the nucleosomal DNA. Thus, to activate gene expression, retinoid receptors must modify repressive chromatin structures in order to allow for the recruitment of the transcription machinery. Ligand-induced conformational changes in the receptors cause dissociation of corepressors and the coordinated and/or combinatorial recruitment of coactivators associated with complexes displaying histone acetyltransferase, methyltransferase, kinase or ATP-dependent remodeling activities that decompact repressive chromatin (24).
RXRs serve as obligate heterodimeric partners not only with RARs, but also with several other nuclear receptors, including but not limited to peroxisome proliferator-activated receptors (PPARs), liver X receptors (LXRs), vitamin D receptors (VDRs), farnesoid X receptor (FXR), and thyroid hormone receptors (TRs). Thus, it is clear that retinoids can influence transcription of a wider set of hormone-responsive genes (5,23), suggesting a high degree of cross talk and the potential for shared ligand activation and reciprocal signaling effects.
C. Retinoid Actions--Non-Genomic Processes
There is growing evidence that retinoid actions in the body do not solely depend on the transcriptional activity of RA. Protein retinoylation is proposed to be responsible for mediating some known actions of retinoids, including cellular differentiation, cell growth and possibly steroidogenesis (25,26). Retinoylation is a cell-specific, post-transcriptional modification of proteins, which is described for a number of tissues including rat liver, kidney, testis and brain, and for different cell lines. RA has been shown to bind covalently, via a thioester bond, to proteins such as serum albumin and cyclic adenosine monophosphate (cAMP)-binding proteins, and this reaction is believed to occur enzymatically (27). A listing of some proteins that are reported to be retinoylated is provided in Table II. Because retinoylation in vitamin A-deficient rats occurs at a greater rate than that in vitamin A-sufficient rats, protein retinolyation has been proposed to be an essential process (25). Age, diet and cellular RA concentrations are factors proposed to influence protein retinoylation (28).
Table II.
Some Proteins Reported in the Literature to Undergo Retinoylation
| Retinoylated Proteins | Tissues/Cell lines | Species | Implicated Diseases/Possible Roles | Citations |
|---|---|---|---|---|
|
| ||||
| Nuclear matrix proteins including p51 and p55 | Bone marrow | Rat | Aging | (26,28) |
|
| ||||
| Oxoglutarate carrier | Testes mitochondria | Rat | Testosterone biosynthesis/Steroidogenesis | (94) |
|
| ||||
| Thioredoxin reductase | Keratinocytes | Human | Inhibition of thioredoxin reductase | (95) |
|
| ||||
| Protein kinase (PKA) types I and II cyclic AMP-binding regulatory subunits | Human acute myeloid leukemia cell line (HL60) | Human | RA induced differentiation of HL60 cells to granulocyte-like cells | (96) |
| MCF-7 breast cancer cell line | Cell growth | (97) | ||
| Fibroblasts | Psoriasis | (98) | ||
|
| ||||
| Vimentin | Human acute myeloid leukemia cell line (HL60) and HL60 mutants | Human | RA induced differentiation of HL60 cells to granulocyte-like cells | (99,100) |
|
| ||||
| Actin binding protein (α-actinin) | Human acute myeloid leukemia cell line (HL60) | Human | RA induced differentiation of HL60 cells to granulocyte-like cells | (25) |
|
| ||||
| Basic proteins (Histone) | Human acute myeloid leukemia cell line (HL60) | Human | RA induced differentiation of HL60 cells to granulocyte-like cells | (101) |
|
| ||||
| Cytokeratins 16 and 10 | Skin | Mice | Normal skin function; Epidermal differentiation | (27) |
|
| ||||
| Serum albumin | Skin | Mice | Mediate in vivo response of RA | (27) |
It is also clear that there are other non-genomic mechanisms that directly influence directly cell signaling pathways. The best characterized of these is the demonstration that retinoids can mediate cellular apoptosis through RXR-mediated interactions with the nerve growth factor IB, also known as Nur77, a nuclear receptor which promotes cell growth when localized in the nucleus, and apoptosis when present in mitochondria (29). Specifically, RXR contains a nuclear export sequence that is used to shuttle Nur77 out of the nucleus and into the mitochondria upon RXR/Nur77 heterodimerization and in response to extracellular stimuli (30). Various RXR ligands including 9cRA can influence RXR binding to Nur77 and consequently affect Nur77 translocation and cellular apoptosis.
Presently, there is considerable research activity focused on identifying how retinoids act non-genomically to influence cellular processes. At this early stage, it is clear that retinoids have both genomic and non-genomic roles within cells. However, specific mechanisms regarding the non-genomic actions of retinoids still need to be elucidated.
3. Retinoid Regulation of Adipocyte Biology
A. Vitamin A uptake, storage and mobilization in white adipose tissue
White adipose tissue (WAT) plays an important role in retinoid storage and metabolism. It has been estimated that as much as 20% of the total retinoid present in a vitamin A-sufficient rat is stored in adipose tissue, mostly in the form of retinyl ester (31). WAT can acquire retinoids from the circulation as retinol bound to RBP or from postprandial CRs facilitated by LPL, synthesized and secreted by adipocytes (32). BCMO1 is expressed in WAT, so β-carotene present in CRs is also a potential source of adipocyte retinol or RA (33). Adipocytes express RARs and RXRs as well as genes necessary to store, mobilize, and oxidize retinol to RA (34); thus retinoid metabolism and actions are important to adipose tissue biology.
When dietary retinol intake is insufficient, retinol can be mobilized from WAT bound to RBP to maintain retinoid-dependent functions in other tissues. This was established from the observation that WAT retinyl ester stores decrease in wild type (WT) and Lrat−/−mice fed a retinol-deficient diet (9). The molecular identity of the enzyme(s) responsible for synthesis of retinyl esters in adipocytes remains to be established since relatively high levels of retinyl esters are found in adipose tissue of mice which lack both Lrat and Dgat1 (10). The enzyme responsible for the hydrolysis of WAT retinyl esters is hormone sensitive lipase (HSL). Wei et al. showed that HSL displays robust REH activity in cultured adipocytes treated with dibutyryl-cAMP, which activates HSL and stimulates retinol release into the culture media at the expense of cellular retinyl ester stores (35). Subsequent studies of Hsl-null mice showed that these mutant mice have elevated WAT retinyl ester levels and decreased REH activity in WAT, supporting the proposal that HSL is the major REH in WAT (36).
B. Retinoid Effects on Adipogenesis
The literature on the possible link between dietary retinoid intake and adiposity is contradictory. High dietary retinol intake has been reported by Jeyakumar et al. to decrease body weight and WAT mass in both lean and obese rats (37,38) but other investigations involving mice showed no change in body weight and WAT mass upon high dietary retinol intake for 18 weeks (39). These differences may arise due to differences in the species studied. Findings from studies employing retinol-deficient diets are also contradictory, one study reported increased adiposity (40), whereas another reported decreased adiposity (41). This discrepancy may be due to differences in the degree of retinol-deficiency, with more severe deficiency inhibiting body fat formation, whereas less severe deficiency increasing adiposity. Results from RA-supplementation studies are more consistent. Several studies from Palou and colleagues have shown a decrease in body weight and WAT mass upon RA-treatment (39,42). However, it is difficult to attach substantial physiological significance to these studies since atRA was administered to the mice at doses of 10, 50 and 100 mg/kg body weight (50,53). At the highest dose, where the largest effect on body weight was observed, mice would be receiving 3–4 mg atRA per day. Since the total quantity of atRA present in the body of a mouse receiving the dose would normally be less than 200 ng, the doses employed in these studies are very large. This raises concerns regarding the physiologic significance of the work. Interestingly, a recent study by Berry et al. reported that obese mice showed a decrease in body weight attributed to reduced abdominal and epididymal WAT mass and improved insulin sensitivity after systemic atRA treatment using subcutaneous slow-release RA pellets (15 mg, 90-day release) (43).
Many published studies have shown that atRA treatment can inhibit early events of adipocyte differentiation in 3T3-L1 preadipocytes and in primary rat adipocytes (44,45), most notably by blocking the transcriptional activity of CCAAT-enhancer-binding protein (C/EBP) β. C/EBPβ regulates expression of PPARγ and C/EBPα, two transcription factors required for adipocyte differentiation (46). In the 3T3-L1 adipocyte model, it was shown that atRA treatment of mature adipocytes did not affect lipid accumulation (45), even though RARα was expressed in these cells, albeit at a relatively low level. This was taken as evidence that the inhibitory effect of atRA on adipogenesis is limited to early differentiation events and that mature adipocytes become insensitive to the atRA-induced inhibition of adipogenesis. Moreover, this work sheds some light on why RA has an inhibitory role at early but not at later stages by showing that cellular retinoic acid-binding protein II (CRABPII) and RARα, -β and -γ expression are all decreased in adipocytes compared to preadipocytes. Furthermore, fatty acid binding protein 5 (FABP5), an intracellular lipid-binding protein (iLBP) capable of binding and transporting atRA to the nucleus, and PPARβ/δ are increased in mature adipocytes. atRA has been characterized as a high affinity ligand for PPARβ/δ (47), although this is not universally accepted (48,49). An increase in the FABP5/CRABPII ratio, as seen during adipose differentiation, is proposed to channel atRA away from the RAR-mediated inhibition of adipogenesis leading to increased PPARβ/δ activation (43). Noy and colleagues have emphasized the importance of CRABPII repression for adipocyte differentiation (50). These investigators proposed that CRABPII expression is critical for the ability of atRA to inhibit adipogenesis and that adipocyte differentiation is accompanied by a down-regulation of CRABPII expression. Furthermore, these investigators demonstrated that many hormonal factors responsible for inducing preadipocyte differentiation also repress Crabp2 gene expression. Importantly, five C/EBPα response elements are present in the Crabp2 promoter. In mature adipocytes, C/EBPα binds the CrabpII promoter and suppresses its expression, thus maintaining adipocytes in the differentiated state by inhibiting atRA transfer to RARs in the nucleus. Recently, Noy and colleagues provided further evidence supporting this mechanism of atRA-mediated inhibition of adipogenesis by showing that the activation of the CRABPII/RAR pathway results in diminished adipogenesis through inhibition of preadipocyte differentiation (51).
A study by Lobo et al. showed that β-carotene treatment of 3T3-L1 adipocytes, which express BCMO1, decreased PPARγ, C/EBPα and aP2/FABP4 mRNA levels, and reduced triglyceride content of the cells (33). This did not occur when mature adipocytes were treated with retinol or atRA. Surprisingly, atRA levels were increased more in mature adipocytes when treated with β-carotene than with atRA itself. These effects of β-carotene treatment were reversed when the 3T3-L1 adipocytes were cotreated with a RAR pan-antagonist. Thus, Lobo et al. concluded that the inhibitory effect of β-carotene on mature adipocytes is dependent on the activation of the RAR, and atRA can have an inhibitory effect on adipogenesis even in mature adipocyte, but only when the atRA originates from β-carotene. This is consistent with the observation of increased adiposity and increased PPARγ expression in adipose tissue of Bcmo1−/−mice.
It has also been proposed that retinaldehyde can inhibit adipogenesis (52). Retinaldehydrogenase dehydrogenase 1 (RALDH1)-deficient (Raldh1−/−) mice are reported to have increased adipose retinaldehyde levels and decreased adiposity. Specifically, the literature reports that retinaldehyde, acting as a transcriptional repressor, binds both PPARγ and RXR, thereby inhibiting PPARγ activity and consequently adipocyte differentiation (52). However, this work is controversial since other laboratories have not been able to detect high tissue levels of retinaldehyde (53) like the ones reported in this study (52). More recent studies by Plutzky and colleagues exploring the role of RALDH1 in mice have established a role for this enzyme and its substrate, retinaldehyde, as determinants of adipocyte plasticity and adaptive thermogenesis in mice (54). These investigators showed that RALDH1 deficiency induced a brown adipose tissue-like transcriptional program in WAT and drove uncoupled respiration and adaptive thermogenesis in WAT.
C. RBP as novel adipokine: effects on insulin sensitivity
The established physiologic function of RBP is to mobilize cellular retinoid stores and transport retinol to extrahepatic tissues where it is used for RA synthesis. In 2005, Yang et al. reported a study linking increased adipose-derived RBP to insulin resistance. (This literature employs the gene nomenclature for RBP, i.e. RBP4. The abbreviations RBP and RBP4 refer to the same protein.) These investigators reported elevated levels of serum RBP in mouse models of insulin resistance and in humans with obesity and type II diabetes (55). They further showed that increased serum RBP, in RBP-overexpressing mice or upon RBP injection into the circulation, caused insulin resistance, whereas decreased serum RBP, in Rbp−/− mice or upon pharmacologic reduction of RBP levels, improved insulin action. The effects of elevated serum RBP on insulin resistance were characterized in muscle, where RBP impairs insulin signaling and in the liver where it increases Pepck gene expression and glucose output. Taken together, this work suggests that adipose-derived RBP is a novel adipokine, which contributes to the development of insulin resistance and a potential therapeutic target for the treatment of insulin resistance in type II diabetes.
Subsequent clinical studies have assessed in humans possible associations between elevated plasma RBP concentrations and individual components of the metabolic syndrome, especially insulin resistance. This concept was later extended and it was proposed that the ratio of RBP to retinol is more strongly correlated with insulin resistance, suggesting that apo-RBP may play the predominant role and that these effects could be independent of retinoid metabolism (56). The results from these clinical studies have been conflicting; a considerable number of clinical studies have shown a correlation between elevated RBP levels and at least one component of the metabolic syndrome. But a nearly equal number of other studies were unable to detect an association. Thus, the role of RBP on insulin resistance is still very much controversial. A good recent review from Kotnik et al. considers extensively the conflicting results from clinical studies and proposes confounding factors that may explain some differences (57). Additional clinical studies will be needed to clarify the link between elevated serum RBP levels and insulin resistance in humans.
The relationship between serum RBP levels and insulin resistance remains controversial but molecular mechanisms that underlie this relationship are now being actively explored. Work by Berry et al. gives some insights into RBP’s action on insulin response in WAT and muscle (58). A protein termed “stimulated by retinoic acid 6” (STRA6) has been proposed to be a cell-surface receptor for RBP that mediates the uptake of retinol into cells (59). However, Berry et al. showed that STRA6 induces an intracellular signaling cascade which impairs insulin signaling. Binding of retinol-RBP induces STRA6 phosphorylation which activates the Janus kinase 2 (JAK2)/Signal transducer and activator of transcription 5 (STAT5) signaling pathway. This signaling pathway contributes to activation of STAT target genes, including Pparγ and suppressor of cytokine signaling 3 (Socs3), which is a known inhibitor of insulin signaling. The retinol-RBP/STRA6/JAK2/STAT5 mediated induction of the insulin signaling inhibitor Socs3 provides a potential molecular mechanism for how RBP inhibits insulin action in tissues that express STRA6, including WAT and muscle.
Kahn and colleagues have provided a different mechanistic explanation for how RBP modulates insulin responsiveness (60). These investigators showed that RBP induces expression of proinflammatory cytokines in mouse and human macrophages, which indirectly inhibit insulin signaling in cocultured adipocytes. This effect was reported to involve activation of c-Jun-N-terminal protein kinase (JNK) and Toll-like-receptor 4 (TLR4) pathways. Activation was independent of STRA6, and apo-RBP was much more potent in inducing proinflammatory cytokines than retinol-RBP. This is very different from what was reported by Berry et al. (72) but the two mechanisms are not mutually exclusive and may both act in regulating insulin responsiveness.
4. Retinoids in Pancreatic Islet Biology
A. Retinoids are required for normal endocrine function of pancreatic β-cells
Early evidence for a role of retinoids in pancreatic islet biology came from Kato et al. (61) who localized relatively high levels of RBP, transthyretin (TTR), CRBPI, and CRABPI proteins to pancreatic islet cells. The presence of these retinoid-related proteins in islets suggests that retinoids may have an important role in pancreatic islet function (61). In vivo and in vitro work by Chertow and colleagues showed that islets obtained from retinoid-deficient rats displayed impaired glucose-induced insulin release, but this effect was reversed upon treatment of the rats with either retinyl palmitate or RA prior to islet isolation (62). Insulin release remained impaired upon repletion with low levels of dietary RA (2 μg/g diet) but were partially normalized at higher levels (8 μg/g diet). In the fed state, retinoid-deficient rats displayed increased plasma glucose levels and decreased insulin levels, impaired glucose-induced acute insulin release, and glucose intolerance. The impaired insulin secretion was reversed upon dietary retinoid-repletion. Interestingly, islets obtained from control rats had significantly greater concentrations of CRBPI than islets obtained from retinoid-deficient rats, suggesting a possible relationship between CRBPI expression and insulin secretion. It should be noted that differences in plasma insulin or insulin secretion were only observed in the fed state or when islets were stimulated with high glucose concentrations, suggesting a role for retinoids in regulating insulin secretion from pancreatic β-cells. Subsequent studies showed a protective effect of retinyl palmitate on streptozotocin- and alloxan-induced β-cell loss and diabetes development, supporting the idea that retinoids are beneficial or even protective to endocrine β-cell function by delaying drug-induced diabetes and a decline in β-cells numbers, as opposed to a decline in β-cell insulin secreting capacity seen in vitamin A-deficiency (63).
Results from cell culture studies by Chertow and colleagues are in agreement with the observed relationships regarding retinoid nutritional status and CRBPI and CRABPI expression and insulin secretion (64). In rat insulinoma RINm5F cells, these investigators observed increases in CRBPI and CRABPI as well as KCl-induced insulin release in cells treated with either retinol or RA. An independent study from Fernandez-Mejia et al. showed in the same cell line that atRA treatment increased not only insulin secretion, but also proinsulin mRNA levels and glucokinase mRNA levels and activity (65). It should be noted that the atRA doses required for increasing proinsulin and glucokinase mRNA levels correspond to physiologic levels (1–10 nM). atRA treatment also increased insulin secretion independently at both low and high glucose concentrations. These effects of atRA on insulin secretion and glucokinase expression were later confirmed in primary cultures of pancreatic islets obtained from both adult and fetal rats (66). A number of studies have explored expression of RARs and RXRs in β-cell lines and primary islets (67–70). Expression of particular subtypes of RARs and RXRs varies depending on the cell line or the species studied. RARα is abundant in human and rat pancreatic islets but is relatively poorly expressed in mice. In mouse islets, RARγ and RXRβ are the most abundant retinoid nuclear receptors, but RARβ is also expressed at low levels (69). The identification and characterization of a functional RARE in the promoter of the human insulin gene further strengthens the idea that the retinoid nuclear receptors help mediate insulin output from β-cells (71). Moreover, atRA treatment of isolated human islets increased insulin mRNA levels, suggesting that atRA plays a direct role in regulating insulin gene transcription in humans (71).
Several other studies have confirmed the importance of retinoids for maintaining normal endocrine functions of β-cells by linking atRA to the expression of genes indispensable to regulated insulin secretion. An upregulation in the expression of glucokinase (65,66) and glucose transporter Glut2, both of which are essential for glucose entry and sensing by β-cells, has been observed in INS1 cells upon atRA treatment (72).
In summary, the literature compellingly shows that retinoids play an important role for islet endocrine function. This conclusion is based on expression data which indicate that RBP, CRBPI, CRABPI, RARs and RXRs, as well as enzymes involved in RA synthesis, are found in pancreatic islets and/or β-cells (61,62,64,68,69,73). Other β-cell genes that are essential to the regulation of insulin secretion, including glucokinase and Glut2, appear to be regulated by atRA. But the direct relevance of RA/RAR signaling in the regulation of insulin secretion by β-cells has yet to be unequivocally demonstrated.
B. Retinoids are required for normal endocrine function of pancreatic α-cells
Chertow and colleagues also investigated the role of retinoids in glucagon secretion by pancreatic α-cells in rats (74). These investigators showed that vitamin A-deficiency is associated with defects in glucagon secretion. Unlike for insulin secretion however, this defect was not reversed upon repletion with retinyl palmitate or atRA. Arginine-stimulated glucagon secretion was decreased in islets from vitamin A-deficient rats. The detection of CRBPI and CRABPI in glucagon-secreting α-cell lines and α-cells of intact islets suggested a role for retinoids in helping mediate α-cell actions. Other studies by these investigators showed that retinol and atRA treatment of cultured rat islets and glucagon-secreting cell lines inhibited glucagon secretion in a dose-dependent manner (67). RARα and RARγ mRNAs have been reported to be present in α-cells, suggesting that atRA-induced inhibition of glucagon secretion in α-cells may be mediated through effects on transcription.
C. Endogenous 9cRA and RXR functions in pancreas
Several recent studies have suggested direct actions of retinoids in insulin secretion. Kane et al. proposed that 9cRA is an endogenous pancreas-specific autacoid, which attenuates glucose-stimulated insulin secretion (75). Using liquid chromatography tandem mass spectrometry, these investigators reported the presence of 9cRA in pancreas at concentrations ranging from 20–30 pmol/g tissue, well above those of atRA. Pancreatic 9cRA levels varied with fasting, feeding and glucose challenge and were inversely correlated with insulin levels. Kane et al. also showed that 9cRA treatment promoted glucose intolerance and rapidly attenuated glucose sensing and insulin secretion in mice. Treatment of 832/13 β-cells with 9-cis-retinol induced the synthesis of 9cRA. This, along with the presence of 9-cis-retinol in pancreatic microsomes, was taken to suggest that β-cells have the capacity and the available substrate to synthesize 9cRA locally in vivo. Moreover, these investigators found elevated levels of 9cRA in pancreas of several mouse models of glucose intolerance, including mice experiencing diet-induced obesity, ob/ob mice, and db/db mice.
Miyazaki et al. reported that ablation of RXR signaling by a dominant-negative RXR-mutant form, specifically expressed in pancreatic β-cells of mice, improved glucose tolerance and increased glucose-stimulated insulin secretion in islets isolated from those mice (76). These investigators then further showed that treatment of isolated mouse islets with 10 μM 9cRA decreased glucose-stimulated insulin secretion at high glucose concentrations. This study indicates that RXR signaling in β-cells may inhibit excessive insulin release under conditions when glucose concentrations are high. It is not clear whether RXR predominantly functions as a homodimer or as a heterodimer associated with other nuclear receptors such as the RARs, VDRs, PPARs, TRs, LXRs, FXR and/or others, to mediate this effect. This is important since several studies have shown that combined overexpression of PPARγ and RXR in INS1 cells as well as treatment with rosiglitazone (a PPARγ agonist) inhibits glucose-stimulated insulin secretion (72,77).
Taken together, the studies of Kane et al. and Miyazaki et al. suggest mechanistic roles for 9cRA and RXRs in preventing excessive insulin secretion in condition of high glucose. More research is needed to fully understand the mechanism of action (genomic, non-genomic, or both) of 9cRA in pancreatic β-cells. It is important to stress several differences between the findings reported by these groups. Kane et al. proposed a non-genomic effect of 9cRA on insulin secretion in β-cells, whereas Miyazaki et al. proposed a RXR-mediated effect on gene transcription. 9cRA was not identified to be present in any other tissues in the body in earlier studies by Kane et al. (90). Thus, the pancreas, or more specifically β-cells, may be the only tissue in the body where 9cRA is synthesized at high levels and where it plays a physiologic role acting in a non-genomic manner (87). Furthermore, 9cRA has only been indirectly determined to be present in β-cells; Kane et al. observed a decrease in pancreatic 9cRA levels in mouse models of decreased β-cell numbers (Ins2Akita mice and mice treated with streptozotocin). This was taken to suggest that β-cells serve as the cellular site where 9cRA is localized in the pancreas, but this is an inference and 9cRA concentrations remain to be directly measured in primary islet isolates. As pointed out by Kane in a review (78), 9cRA concentrations are approximately 20 pmol/g for whole pancreas and vary with fasting/refeeding. Yet, pancreatic islets represent about 1 to 5% of cells in the pancreas and in mice around 80% of those are β-cells (79). This implies the presence of 20- to 100-fold greater concentrations of 9cRA in β-cells than in the whole pancreas. These concentrations would be in great excess of those of atRA.
Interestingly, a study by Shimamura et al., published at around the same time as the studies of Kane et al. and Miyazaki et al., reported that the expression of retinaldehyde dehydrogenase 3 (RALDH3), which synthesizes atRA from all-trans-retinaldehyde (80) but which cannot catalyze the synthesis of 9cRA from 9-cis-retinaldehyde, is highly upregulated in pancreatic islets obtained from mouse models of diabetes, including high fat-fed BDF1 mice and db/db mice (81). Shimamura et al. also showed that overexpression of Raldh3 in high glucose conditions increased glucagon secretion from alpha TC1 clone 9 cells and decreased insulin secretion from MIN6 β-cells. These data are consistent with the notion that altered retinoid metabolism in islet cells is associated with type 2 diabetes and that accumulated atRA in the pancreatic islets may precede islet dysfunction.
A study of Rbp1−/− mice, which lack CRBPI, by Kane et al. confirmed the importance of CRBPI in glucose homeostasis (82). Rbp1−/− mice fed a diet providing a relatively large amount of retinol (30 IU/g of diet) showed abnormally elevated pancreatic 9cRA levels and low insulin levels in the fed state, hyperglycemia, and marked glucose intolerance. Surprisingly, Rbp1−/−mice fed a high fat diet showed improved glucose tolerance compared to WT mice (83). According to Kane et al., Rbp1−/− mice are resistant to high fat diet-induced alterations in glucose tolerance. These findings might be explained by a study from Zizola et al. (83), which showed a similar decrease in insulin secretion in Rbp1−/− mice fed a high fat diet, resulting from a marked improvement in whole body insulin sensitivity and glucose tolerance, along with increased adiposity. Further investigations of glucose-stimulated insulin secretion involving the use of islets isolated from Rbp1−/− and WT mice will be needed to clearly establish whether the decreased plasma insulin originates from improved insulin sensitivity or impaired glucose-stimulated insulin secretion. Interestingly, Kane et al. also showed an increase in Crbp2 expression in isolated islets of Rbp1−/− mice. These investigators hypothesized that CRBPII functionally replaces CRBPI in islets, possibly increasing the rate of 9-cis-retinol oxidation to 9cRA, thereby increasing the 9cRA levels in β-cells and impairing insulin secretion.
Thus, the literature establishes an important role for retinoids in mediating pancreatic β-cell function. This is summarized in Figure 2. The recent literature has proposed both a direct non-genomic role for 9cRA and one for RXR signaling in β-cell function, both of which prevent over-secretion of insulin. However, this does not explain older observations regarding the activation of insulin transcription and secretion upon atRA treatment or the reported regulation by atRA of the insulin sensing genes glucokinase and Glut2 in β-cells. Those two seemingly opposing roles proposed for retinoids in β-cell function (enhancement of insulin secretion by atRA and inhibition of insulin secretion by 9cRA) are not mechanistically conflicted. atRA activation of the RAR nuclear receptors may be important to insulin secretion, whereas 9cRA and RXR activation, perhaps through its action with one or more of the RXR-associated nuclear hormone receptors, may play a role in preventing excessive insulin secretion in times of already high circulating insulin concentrations. The precise mechanism directly responsible for retinoid actions in β-cells within the adult pancreas, involving RAR and/or RXR activation, has yet to be clearly identified and systematically explored.
Figure 2. Retinoid regulation of insulin secretion in pancreatic β-cells.
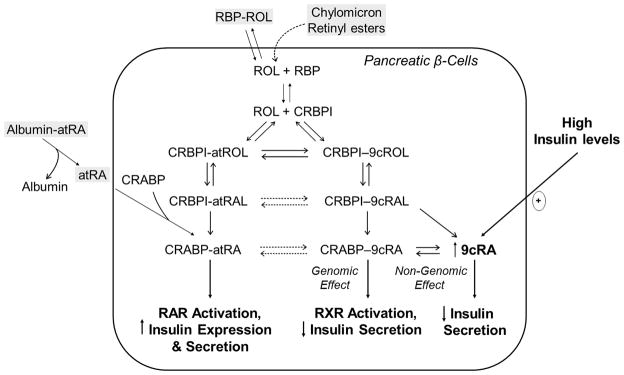
RA has been proposed to regulate insulin secretion from β-cells in three ways: (1) atRA stimulates insulin secretion through RAR activation, (2) 9cRA inhibits insulin secretion through RXR activation, (3) 9cRA inhibits insulin secretion through non-genomic effects. Abbreviations: atROH, all-trans-retinol; 9cROH, 9-cis-retinol; RAL, retinaldehyde; RA, retinoic acid; CRBPI, cellular retinol-binding protein I; CRABP, cellular retinoic acid-binding proteins I and/or II; RAR, retinoic acid receptor; RXR, retinoid X receptor; and RBP, retinol-binding protein.
5. Retinoid Regulation of Hepatic Lipid Metabolism
There is accumulating evidence in the literature suggesting a role for retinoids in the regulation of lipid metabolism in the liver. Data from a number of human studies strongly support this conclusion. For a cross-sectional study of 91 children, a trend was observed between low serum retinol levels and an ultrasound-determined risk of hepatic steatosis development (84). For a case-control study involving 138 adults, low dietary intake of retinoids was one of only two differing features between control subjects and those with non-alcoholic fatty liver disease (NAFLD) (85). Another study of 145 obese adults reported that subjects with NAFLD had significantly lower serum retinol and β-carotene levels than healthy subjects (86). These human population studies suggest an inverse relationship between dietary retinoid and carotenoid intake and the development of NAFLD.
Animal studies exploring the effects of both deficiency or excess of dietary retinoids on hepatic lipids fail to reach a consensus regarding the role of retinoids in fatty liver development. Mice fed a retinol-deficient diet starting at day 10 of gestation by McGrane and colleagues exhibited decreased expression of genes involved in hepatic mitochondrial and peroxisomal fatty acid oxidation, including fatty acyl-CoA ligase 2, carnitine palmitoyltransferase-1 (CPT-1), medium chain acyl CoA dehydrogenase and peroxisomal acyl CoA oxidase 1 (87). These mice developed fatty liver, characterized by an excessive accumulation of hepatic triglycerides as early as 9 weeks of age. Treatment with a gavage dose of atRA (10 mg/kg body weight) resulted in increased expression of these downregulated genes. While these observations are consistent with the human data, other investigators have observed opposite effects in rats fed a retinol-deficient diet. Oliveros et al. reported decreases in [14C]choline incorporation into phosphatidylcholine and [14C]acetate incorporation into saponifiable lipids in livers of male rats fed a retinol-deficient diet for 3 months (88). Acetyl-CoA carboxylase activity was reported to be decreased, while CPT-I activity increased. Repletion with retinol reversed the decrease in synthesis of phosphatidylcholine and fatty acids, and increased mitochondrial fatty acid oxidation. Liver microarray studies by McClintick et al. carried out in rats fed a retinol-deficient diet for 53 days, are consistent with these later findings, showing decreases in expression of enzymes involved in fatty acid synthesis and increases in expression for those involved in fatty acid oxidation (89).
Inconsistent results from the animal studies of diet-induced retinol deficiency have led researchers to study mice with genetic alterations in retinoid-related proteins. One group explored the effects of inhibiting retinoid signaling in the liver by generating transgenic mice expressing a dominant-negative form of RARα in hepatocytes (90). These mice developed microvesicular steatosis, with reduced mitochondrial β-oxidation activity, along with decreased expression of fatty acid oxidation-related enzymes, including CPT-I. These findings are similar to those reported by McGrane and colleagues in mice fed a retinol-deficient diet (87). Expression of enzymes involved in peroxisomal β-oxidation, however, was upregulated, and this is inconsistent with data reported by McGrane and colleagues obtained upon feeding of a retinol-deficient diet. Feeding the dominant-negative RAR transgenic mice a diet containing excessive atRA reversed some of the observed changes, which these investigators took as confirming that the observed differences seen for the transgenic mice resulted from obstruction of RA signaling.
Investigations into the effects of excessive retinoid intake on hepatic lipid metabolism have been carried out using rodents fed either excessive retinoid-supplementation or with agonists of RAR signaling. Amengual et al. injected mice subcutaneously with very large daily doses of atRA at 10 or 100 mg/kg body weight for four days and observed increased hepatic expression of CPT-1, carnitine/acylcarnitine carrier, and PPARα, along with decreased hepatic expression of sterol regulatory element binding protein 1c and fatty acid synthase, and a dose-dependent decrease in hepatic triglyceride content (91). Surprisingly, lipogenic genes including PPARβ and PPARγ mRNA levels were observed to decrease while hepatic expression of acetyl-CoA carboxylase-1 mRNA increased in these same mice. Although the results from this study seem to support those from McGrane’s group and also the published human findings, this study needs to be interpreted with caution because of the large pharmacological doses of atRA employed.
The cannabinoid type 1 receptor (CB1R) has been implicated in alcoholic fatty liver disease, where it increases lipogenesis and decreases fatty acid oxidation (92). Mukhopadhyay et al. demonstrated that treatment of hepatocytes with either 1 or 5 μM atRA or CD437, a RARγ agonist, led to increases in CB1R mRNA and protein levels. Furthermore, siRNA knockdown of RARγ mRNA decreased the ability of a RAR pan-agonist to induce CB1R expression. Both ethanol-containing and high fat diets increased RARγ and CB1R protein in mouse liver, along with hepatic triglyceride levels. Chromatin immunoprecipitation studies showed that RAR agonists increased the occupancy of RARγ on the CB1R gene promoter. Thus, atRA, through actions mediated by RARγ, may be responsible for the effects of chronic alcohol consumption on the development of hepatic steatosis. Administration of a RXR agonist was also reported to result in an increase in the size and number of fat droplets in the livers of genetically diabetic mice (93). Surprisingly, this increase was greater than that produced by PPARγ agonists.
Collectively, these studies suggest that retinoids may be importantly involved in the regulation of hepatic lipid metabolism. Inconsistencies reported by different groups may arise from differences in study designs, since the majority of these studies were not designed to directly address the effects of retinoids on hepatic lipids, but rather to produce liver effects secondary to systemic lipid changes, such as increased circulating free fatty acid levels. These may be responsible for increases in hepatic uptake of fatty acids. For this reason, no unequivocal conclusions regarding the effects of retinoids on hepatic lipid metabolism can be established at this point. Further investigations will be necessary to elucidate the molecular events that underlie these relationships.
Acknowledgments
The work cited in this review from the authors’ laboratory was supported by grants R01 DK068437, R01 DK079221, and RC2 AA019413 from the National Institutes of Health.
References
- 1.Goodman DS. Vitamin A and retinoids in health and disease. N Engl J Med. 1984;310:1023–1031. doi: 10.1056/NEJM198404193101605. [DOI] [PubMed] [Google Scholar]
- 2.Goodman DS. Vitamin A metabolism. Fed Proc. 1980;39:2716–2722. [PubMed] [Google Scholar]
- 3.Wald G. Molecular basis of visual excitation. Science. 1968;162:230–239. doi: 10.1126/science.162.3850.230. [DOI] [PubMed] [Google Scholar]
- 4.Balmer JE, Blomhoff R. Gene expression regulation by retinoic acid. J Lipid Res. 2002;43:1773–1808. doi: 10.1194/jlr.r100015-jlr200. [DOI] [PubMed] [Google Scholar]
- 5.Chambon P. A decade of molecular biology of retinoic acid receptors. FASEB J. 1996;10:940–954. [PubMed] [Google Scholar]
- 6.Mangelsdorf DJ, Umesono K, Evans RM. The retinoid receptors. In: Sporn MB, Roberts AB, Goodman DS, editors. The Retinoids, Biology, Chemistry and Medicine. 2. Raven Press Ltd; New York: 1994. [Google Scholar]
- 7.Weng W, Li L, van Bennekum AM, Potter SH, Harrison EH, Blaner WS, Breslow JL, Fisher EA. Intestinal absorption of dietary cholesteryl ester is decreased but retinyl ester absorption is normal in carboxyl ester lipase knockout mice. Biochemistry. 1999;38:4143–4149. doi: 10.1021/bi981679a. [DOI] [PubMed] [Google Scholar]
- 8.Gilham D, Labonte ED, Rojas JC, Jandacek RJ, Howles PN, Hui DY. Carboxyl ester lipase deficiency exacerbates dietary lipid absorption abnormalities and resistance to diet-induced obesity in pancreatic triglyceride lipase knockout mice. J Biol Chem. 2007;282:24642–24649. doi: 10.1074/jbc.M702530200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Byrne SM, Wongsiriroj N, Libien J, Vogel S, Goldberg IJ, Baehr W, Palczewski K, Blaner WS. Retinoid absorption and storage is impaired in mice lacking lecithin:retinol acyltransferase (LRAT) J Biol Chem. 2005;280:35647–35657. doi: 10.1074/jbc.M507924200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wongsiriroj N, Piantedosi R, Palczewski K, Goldberg IJ, Johnston TP, Li E, Blaner WS. The molecular basis of retinoid absorption: a genetic dissection. J Biol Chem. 2008;283:13510–13519. doi: 10.1074/jbc.M800777200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lietz G, Lange J, Rimbach G. Molecular and dietary regulation of beta,beta-carotene 15,15′-monooxygenase 1 (BCMO1) Arch Biochem Biophys. 2010;502:8–16. doi: 10.1016/j.abb.2010.06.032. [DOI] [PubMed] [Google Scholar]
- 12.Herr FM, Wardlaw SA, Kakkad B, Albrecht A, Quick TC, Ong DE. Intestinal vitamin A metabolism: coordinate distribution of enzymes and CRBP(II) J Lipid Res. 1993;34:1545–1554. [PubMed] [Google Scholar]
- 13.Blaner WS, Obunike JC, Kurlandsky SB, al-Haideri M, Piantedosi R, Deckelbaum RJ, Goldberg IJ. Lipoprotein lipase hydrolysis of retinyl ester. Possible implications for retinoid uptake by cells. J Biol Chem. 1994;269:16559–16565. [PubMed] [Google Scholar]
- 14.Cooper AD. Hepatic uptake of chylomicron remnants. J Lipid Res. 1997;38:2173–2192. [PubMed] [Google Scholar]
- 15.Yu KC, Cooper AD. Postprandial lipoproteins and atherosclerosis. Front Biosci. 2001;6:D332–354. doi: 10.2741/yu. [DOI] [PubMed] [Google Scholar]
- 16.Shirakami Y, Lee SA, Clugston RD, Blaner WS. Hepatic metabolism of retinoids and disease associations. Biochim Biophys Acta. 2012;1821:124–136. doi: 10.1016/j.bbalip.2011.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.D’Ambrosio DN, Clugston RD, Blaner WS. Vitamin A metabolism: an update. Nutrients. 2011;3:63–103. doi: 10.3390/nu3010063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kastner P, Mark M, Ghyselinck N, Krezel W, Dupe V, Grondona JM, Chambon P. Genetic evidence that the retinoid signal is transduced by heterodimeric RXR/RAR functional units during mouse development. Development. 1997;124:313–326. doi: 10.1242/dev.124.2.313. [DOI] [PubMed] [Google Scholar]
- 19.Lee MS, Kliewer SA, Provencal J, Wright PE, Evans RM. Structure of the retinoid X receptor alpha DNA binding domain: a helix required for homodimeric DNA binding. Science. 1993;260:1117–1121. doi: 10.1126/science.8388124. [DOI] [PubMed] [Google Scholar]
- 20.Schwabe JW, Chapman L, Finch JT, Rhodes D. The crystal structure of the estrogen receptor DNA-binding domain bound to DNA: how receptors discriminate between their response elements. Cell. 1993;75:567–578. doi: 10.1016/0092-8674(93)90390-c. [DOI] [PubMed] [Google Scholar]
- 21.Glass CK, Rosenfeld MG. The coregulator exchange in transcriptional functions of nuclear receptors. Genes & development. 2000;14:121–141. [PubMed] [Google Scholar]
- 22.Wurtz JM, Bourguet W, Renaud JP, Vivat V, Chambon P, Moras D, Gronemeyer H. A canonical structure for the ligand-binding domain of nuclear receptors. Nature structural biology. 1996;3:206. doi: 10.1038/nsb0296-206. [DOI] [PubMed] [Google Scholar]
- 23.Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83:841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- 24.Bastien J, Rochette-Egly C. Nuclear retinoid receptors and the transcription of retinoid-target genes. Gene. 2004;328:1–16. doi: 10.1016/j.gene.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 25.Kubo Y, Wada M, Ohba T, Takahashi N. Formation of retinoylated proteins from retinoyl-CoA in rat tissues. J Biochem. 2005;138:493–500. doi: 10.1093/jb/mvi145. [DOI] [PubMed] [Google Scholar]
- 26.Pingitore A, Cione E, Senatore V, Genchi G. Adrenal glands and testes as steroidogenic tissue are affected by retinoylation reaction. Journal of bioenergetics and biomembranes. 2009;41:215–221. doi: 10.1007/s10863-009-9220-z. [DOI] [PubMed] [Google Scholar]
- 27.Takahashi N, Fujiu Y. Cytokeratins 16 and 10 bind to retinoic acid covalently in skin tissue of mice. Br J Dermatol. 2010;162:974–979. doi: 10.1111/j.1365-2133.2009.09592.x. [DOI] [PubMed] [Google Scholar]
- 28.Pipkin JL, Hinson W, Lyn-Cook LE, Duffy PH, Feuers RJ, Leakey JE, Aly KB, Hart RW, Casciano DA. The effect of aging and dietary restriction on the retinoylation of nuclear matrix proteins in rats. Aging. 1996;8:263–270. doi: 10.1007/BF03339577. [DOI] [PubMed] [Google Scholar]
- 29.Zhang XK. Targeting Nur77 translocation. Expert opinion on therapeutic targets. 2007;11:69–79. doi: 10.1517/14728222.11.1.69. [DOI] [PubMed] [Google Scholar]
- 30.Cao X, Liu W, Lin F, Li H, Kolluri SK, Lin B, Han YH, Dawson MI, Zhang XK. Retinoid X receptor regulates Nur77/TR3-dependent apoptosis [corrected] by modulating its nuclear export and mitochondrial targeting. Molecular and cellular biology. 2004;24:9705–9725. doi: 10.1128/MCB.24.22.9705-9725.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsutsumi C, Okuno M, Tannous L, Piantedosi R, Allan M, Goodman DS, Blaner WS. Retinoids and retinoid-binding protein expression in rat adipocytes. J Biol Chem. 1992;267:1805–1810. [PubMed] [Google Scholar]
- 32.van Bennekum AM, Kako Y, Weinstock PH, Harrison EH, Deckelbaum RJ, Goldberg IJ, Blaner WS. Lipoprotein lipase expression level influences tissue clearance of chylomicron retinyl ester. J Lipid Res. 1999;40:565–574. [PubMed] [Google Scholar]
- 33.Lobo GP, Amengual J, Li HN, Golczak M, Bonet ML, Palczewski K, von Lintig J. Beta,beta-carotene decreases peroxisome proliferator receptor gamma activity and reduces lipid storage capacity of adipocytes in a beta,beta-carotene oxygenase 1-dependent manner. J Biol Chem. 2010;285:27891–27899. doi: 10.1074/jbc.M110.132571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bonet ML, Ribot J, Felipe F, Palou A. Vitamin A and the regulation of fat reserves. Cell Mol Life Sci. 2003;60:1311–1321. doi: 10.1007/s00018-003-2290-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wei S, Lai K, Patel S, Piantedosi R, Shen H, Colantuoni V, Kraemer FB, Blaner WS. Retinyl ester hydrolysis and retinol efflux from BFC-1beta adipocytes. J Biol Chem. 1997;272:14159–14165. doi: 10.1074/jbc.272.22.14159. [DOI] [PubMed] [Google Scholar]
- 36.Strom K, Gundersen TE, Hansson O, Lucas S, Fernandez C, Blomhoff R, Holm C. Hormone-sensitive lipase (HSL) is also a retinyl ester hydrolase: evidence from mice lacking HSL. FASEB J. 2009;23:2307–2316. doi: 10.1096/fj.08-120923. [DOI] [PubMed] [Google Scholar]
- 37.Jeyakumar SM, Vajreswari A, Giridharan NV. Vitamin A regulates obesity in WNIN/Ob obese rat; independent of stearoyl-CoA desaturase-1. Biochem Biophys Res Commun. 2008;370:243–247. doi: 10.1016/j.bbrc.2008.03.073. [DOI] [PubMed] [Google Scholar]
- 38.Jeyakumar SM, Vajreswari A, Sesikeran B, Giridharan NV. Vitamin A supplementation induces adipose tissue loss through apoptosis in lean but not in obese rats of the WNIN/Ob strain. J Mol Endocrinol. 2005;35:391–398. doi: 10.1677/jme.1.01838. [DOI] [PubMed] [Google Scholar]
- 39.Felipe F, Mercader J, Ribot J, Palou A, Bonet ML. Effects of retinoic acid administration and dietary vitamin A supplementation on leptin expression in mice: lack of correlation with changes of adipose tissue mass and food intake. Biochim Biophys Acta. 2005;1740:258–265. doi: 10.1016/j.bbadis.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 40.Ribot J, Felipe F, Bonet ML, Palou A. Changes of adiposity in response to vitamin A status correlate with changes of PPAR gamma 2 expression. Obes Res. 2001;9:500–509. doi: 10.1038/oby.2001.65. [DOI] [PubMed] [Google Scholar]
- 41.Sagazio A, Piantedosi R, Alba M, Blaner WS, Salvatori R. Vitamin A deficiency does not influence longitudinal growth in mice. Nutrition. 2007;23:483–488. doi: 10.1016/j.nut.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 42.Mercader J, Ribot J, Murano I, Felipe F, Cinti S, Bonet ML, Palou A. Remodeling of white adipose tissue after retinoic acid administration in mice. Endocrinology. 2006;147:5325–5332. doi: 10.1210/en.2006-0760. [DOI] [PubMed] [Google Scholar]
- 43.Berry DC, Noy N. All-trans-retinoic acid represses obesity and insulin resistance by activating both peroxisome proliferation-activated receptor beta/delta and retinoic acid receptor. Mol Cell Biol. 2009;29:3286–3296. doi: 10.1128/MCB.01742-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suryawan A, Hu CY. Effect of retinoic acid on differentiation of cultured pig preadipocytes. J Anim Sci. 1997;75:112–117. doi: 10.2527/1997.751112x. [DOI] [PubMed] [Google Scholar]
- 45.Xue JC, Schwarz EJ, Chawla A, Lazar MA. Distinct stages in adipogenesis revealed by retinoid inhibition of differentiation after induction of PPARgamma. Mol Cell Biol. 1996;16:1567–1575. doi: 10.1128/mcb.16.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schwarz EJ, Reginato MJ, Shao D, Krakow SL, Lazar MA. Retinoic acid blocks adipogenesis by inhibiting C/EBP beta-mediated transcription. Mol Cell Biol. 1997;17:1552–1561. doi: 10.1128/mcb.17.3.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shaw N, Elholm M, Noy N. Retinoic acid is a high affinity selective ligand for the peroxisome proliferator-activated receptor beta/delta. J Biol Chem. 2003;278:41589–41592. doi: 10.1074/jbc.C300368200. [DOI] [PubMed] [Google Scholar]
- 48.Rieck M, Meissner W, Ries S, Muller-Brusselbach S, Muller R. Ligand-mediated regulation of peroxisome proliferator-activated receptor (PPAR) beta/delta: a comparative analysis of PPAR-selective agonists and all-trans retinoic acid. Mol Pharmacol. 2008;74:1269–1277. doi: 10.1124/mol.108.050625. [DOI] [PubMed] [Google Scholar]
- 49.Borland MG, Foreman JE, Girroir EE, Zolfaghari R, Sharma AK, Amin S, Gonzalez FJ, Ross AC, Peters JM. Ligand activation of peroxisome proliferator-activated receptor-beta/delta inhibits cell proliferation in human HaCaT keratinocytes. Mol Pharmacol. 2008;74:1429–1442. doi: 10.1124/mol.108.050609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Berry DC, Soltanian H, Noy N. Repression of cellular retinoic acid-binding protein II during adipocyte differentiation. J Biol Chem. 2010;285:15324–15332. doi: 10.1074/jbc.M110.110635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Berry DC, Desantis D, Soltanian H, Croniger CM, Noy N. Retinoic Acid Upregulates Preadipocyte Genes to Block Adipogenesis and Suppress Diet-Induced Obesity. Diabetes. 2012 doi: 10.2337/db11-1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ziouzenkova O, Orasanu G, Sharlach M, Akiyama TE, Berger JP, Viereck J, Hamilton JA, Tang G, Dolnikowski GG, Vogel S, Duester G, Plutzky J. Retinaldehyde represses adipogenesis and diet-induced obesity. Nat Med. 2007;13:695–702. doi: 10.1038/nm1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kane MA, Folias AE, Napoli JL. HPLC/UV quantitation of retinal, retinol, and retinyl esters in serum and tissues. Anal Biochem. 2008;378:71–79. doi: 10.1016/j.ab.2008.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kiefer FW, Vernochet C, O’Brien P, Spoerl S, Brown JD, Nallamshetty S, Zeyda M, Stulnig TM, Cohen DE, Kahn CR, Plutzky J. Retinaldehyde dehydrogenase 1 regulates a thermogenic program in white adipose tissue. Nat Med. 2012 doi: 10.1038/nm.2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang Q, Graham TE, Mody N, Preitner F, Peroni OD, Zabolotny JM, Kotani K, Quadro L, Kahn BB. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature. 2005;436:356–362. doi: 10.1038/nature03711. [DOI] [PubMed] [Google Scholar]
- 56.Aeberli I, Biebinger R, Lehmann R, L’Allemand D, Spinas GA, Zimmermann MB. Serum retinol-binding protein 4 concentration and its ratio to serum retinol are associated with obesity and metabolic syndrome components in children. J Clin Endocrinol Metab. 2007;92:4359–4365. doi: 10.1210/jc.2007-0468. [DOI] [PubMed] [Google Scholar]
- 57.Kotnik P, Fischer-Posovszky P, Wabitsch M. RBP4: a controversial adipokine. European journal of endocrinology/European Federation of Endocrine Societies. 2011;165:703–711. doi: 10.1530/EJE-11-0431. [DOI] [PubMed] [Google Scholar]
- 58.Berry DC, Jin H, Majumdar A, Noy N. Signaling by vitamin A and retinol-binding protein regulates gene expression to inhibit insulin responses. Proc Natl Acad Sci U S A. 2011;108:4340–4345. doi: 10.1073/pnas.1011115108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kawaguchi R, Yu J, Honda J, Hu J, Whitelegge J, Ping P, Wiita P, Bok D, Sun H. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science. 2007;315:820–825. doi: 10.1126/science.1136244. [DOI] [PubMed] [Google Scholar]
- 60.Norseen J, Hosooka T, Hammarstedt A, Yore MM, Kant S, Aryal P, Kiernan UA, Phillips DA, Maruyama H, Kraus BJ, Usheva A, Davis RJ, Smith U, Kahn BB. Retinol-binding protein 4 inhibits insulin signaling in adipocytes by inducing proinflammatory cytokines in macrophages through a c-Jun N-terminal kinase- and toll-like receptor 4-dependent and retinol-independent mechanism. Mol Cell Biol. 2012;32:2010–2019. doi: 10.1128/MCB.06193-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kato M, Kato K, Blaner WS, Chertow BS, Goodman DS. Plasma and cellular retinoid-binding proteins and transthyretin (prealbumin) are all localized in the islets of Langerhans in the rat. Proc Natl Acad Sci U S A. 1985;82:2488–2492. doi: 10.1073/pnas.82.8.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chertow BS, Blaner WS, Baranetsky NG, Sivitz WI, Cordle MB, Thompson D, Meda P. Effects of vitamin A deficiency and repletion on rat insulin secretion in vivo and in vitro from isolated islets. J Clin Invest. 1987;79:163–169. doi: 10.1172/JCI112778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chertow BS, Webb MD, Leidy JW, Jr, Cordle MB. Protective effects of retinyl palmitate on streptozotocin- and alloxan-induced beta cell toxicity and diabetes in the rat. Res Commun Chem Pathol Pharmacol. 1989;63:27–44. [PubMed] [Google Scholar]
- 64.Chertow BS, Moore MR, Blaner WS, Wilford MR, Cordle MB. Cytoplasmic retinoid-binding proteins and retinoid effects on insulin release in RINm5F beta-cells. Diabetes. 1989;38:1544–1548. doi: 10.2337/diab.38.12.1544. [DOI] [PubMed] [Google Scholar]
- 65.Fernandez-Mejia C, Davidson MB. Regulation of glucokinase and proinsulin gene expression and insulin secretion in RIN-m5F cells by dexamethasone, retinoic acid, and thyroid hormone. Endocrinology. 1992;130:1660–1668. doi: 10.1210/endo.130.3.1537314. [DOI] [PubMed] [Google Scholar]
- 66.Cabrera-Valladares G, German MS, Matschinsky FM, Wang J, Fernandez-Mejia C. Effect of retinoic acid on glucokinase activity and gene expression and on insulin secretion in primary cultures of pancreatic islets. Endocrinology. 1999;140:3091–3096. doi: 10.1210/endo.140.7.6765. [DOI] [PubMed] [Google Scholar]
- 67.Chertow BS, Driscoll HK, Primerano DA, Cordle MB, Matthews KA. Retinoic acid receptor transcripts and effects of retinol and retinoic acid on glucagon secretion from rat islets and glucagon-secreting cell lines. Metabolism. 1996;45:300–305. doi: 10.1016/s0026-0495(96)90282-6. [DOI] [PubMed] [Google Scholar]
- 68.Chertow BS, Blaner WS, Rajan N, Primerano DA, Meda P, Cirulli V, Krozowski Z, Smith R, Cordle MB. Retinoic acid receptor, cytosolic retinol-binding and retinoic acid-binding protein mRNA transcripts and proteins in rat insulin-secreting cells. Diabetes. 1993;42:1109–1114. doi: 10.2337/diab.42.8.1109. [DOI] [PubMed] [Google Scholar]
- 69.Kutlu B, Burdick D, Baxter D, Rasschaert J, Flamez D, Eizirik DL, Welsh N, Goodman N, Hood L. Detailed transcriptome atlas of the pancreatic beta cell. BMC Med Genomics. 2009;2:3. doi: 10.1186/1755-8794-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chertow BS, Driscoll HK, Goking NQ, Primerano D, Cordle MB, Matthews KA. Retinoid-X receptors and the effects of 9-cis-retinoic acid on insulin secretion from RINm5F cells. Metabolism. 1997;46:656–660. doi: 10.1016/s0026-0495(97)90009-3. [DOI] [PubMed] [Google Scholar]
- 71.Clark AR, Wilson ME, London NJ, James RF, Docherty K. Identification and characterization of a functional retinoic acid/thyroid hormone-response element upstream of the human insulin gene enhancer. Biochem J. 1995;309 ( Pt 3):863–870. doi: 10.1042/bj3090863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Blumentrath J, Neye H, Verspohl EJ. Effects of retinoids and thiazolidinediones on proliferation, insulin release, insulin mRNA, GLUT 2 transporter protein and mRNA of INS-1 cells. Cell Biochem Funct. 2001;19:159–169. doi: 10.1002/cbf.907. [DOI] [PubMed] [Google Scholar]
- 73.Chertow BS, Goking NQ, Driscoll HK, Primerano DA, Matthews KA. Effects of all-trans-retinoic acid (ATRA) and retinoic acid receptor (RAR) expression on secretion, growth, and apoptosis of insulin-secreting RINm5F cells. Pancreas. 1997;15:122–131. doi: 10.1097/00006676-199708000-00003. [DOI] [PubMed] [Google Scholar]
- 74.Chertow BS, Driscoll HK, Blaner WS, Meda P, Cordle MB, Matthews KA. Effects of vitamin A deficiency and repletion on rat glucagon secretion. Pancreas. 1994;9:475–484. doi: 10.1097/00006676-199407000-00010. [DOI] [PubMed] [Google Scholar]
- 75.Kane MA, Folias AE, Pingitore A, Perri M, Obrochta KM, Krois CR, Cione E, Ryu JY, Napoli JL. Identification of 9-cis-retinoic acid as a pancreas-specific autacoid that attenuates glucose-stimulated insulin secretion. Proc Natl Acad Sci U S A. 2010;107:21884–21889. doi: 10.1073/pnas.1008859107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Miyazaki S, Taniguchi H, Moritoh Y, Tashiro F, Yamamoto T, Yamato E, Ikegami H, Ozato K, Miyazaki J. Nuclear hormone retinoid X receptor (RXR) negatively regulates the glucose-stimulated insulin secretion of pancreatic ss-cells. Diabetes. 2010;59:2854–2861. doi: 10.2337/db09-1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ravnskjaer K, Boergesen M, Rubi B, Larsen JK, Nielsen T, Fridriksson J, Maechler P, Mandrup S. Peroxisome proliferator-activated receptor alpha (PPARalpha) potentiates, whereas PPARgamma attenuates, glucose-stimulated insulin secretion in pancreatic beta-cells. Endocrinology. 2005;146:3266–3276. doi: 10.1210/en.2004-1430. [DOI] [PubMed] [Google Scholar]
- 78.Kane MA. Analysis, occurrence, and function of 9-cis-retinoic acid. Biochim Biophys Acta. 2012;1821:10–20. doi: 10.1016/j.bbalip.2011.09.012. [DOI] [PubMed] [Google Scholar]
- 79.Jain R, Lammert E. Cell-cell interactions in the endocrine pancreas. Diabetes, obesity & metabolism. 2009;11(Suppl 4):159–167. doi: 10.1111/j.1463-1326.2009.01102.x. [DOI] [PubMed] [Google Scholar]
- 80.Sima A, Parisotto M, Mader S, Bhat PV. Kinetic characterization of recombinant mouse retinal dehydrogenase types 3 and 4 for retinal substrates. Biochim Biophys Acta. 2009;1790:1660–1664. doi: 10.1016/j.bbagen.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 81.Shimamura M, Karasawa H, Sakakibara S, Shinagawa A. Raldh3 expression in diabetic islets reciprocally regulates secretion of insulin and glucagon from pancreatic islets. Biochem Biophys Res Commun. 2010;401:79–84. doi: 10.1016/j.bbrc.2010.09.013. [DOI] [PubMed] [Google Scholar]
- 82.Kane MA, Folias AE, Pingitore A, Perri M, Krois CR, Ryu JY, Cione E, Napoli JL. CrbpI modulates glucose homeostasis and pancreas 9-cis-retinoic acid concentrations. Mol Cell Biol. 2011;31:3277–3285. doi: 10.1128/MCB.05516-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zizola CF, Frey SK, Jitngarmkusol S, Kadereit B, Yan N, Vogel S. Cellular retinol-binding protein type I (CRBP-I) regulates adipogenesis. Mol Cell Biol. 2010;30:3412–3420. doi: 10.1128/MCB.00014-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Suano de Souza FI, Silverio Amancio OM, Saccardo Sarni RO, Sacchi Pitta T, Fernandes AP, Affonso Fonseca FL, Hix S, Ramalho RA. Non-alcoholic fatty liver disease in overweight children and its relationship with retinol serum levels. Int J Vitam Nutr Res. 2008;78:27–32. doi: 10.1024/0300-9831.78.1.27. [DOI] [PubMed] [Google Scholar]
- 85.Musso G, Gambino R, De Michieli F, Biroli G, Premoli A, Pagano G, Bo S, Durazzo M, Cassader M. Nitrosative stress predicts the presence and severity of nonalcoholic fatty liver at different stages of the development of insulin resistance and metabolic syndrome: possible role of vitamin A intake. Am J Clin Nutr. 2007;86:661–671. doi: 10.1093/ajcn/86.3.661. [DOI] [PubMed] [Google Scholar]
- 86.Villaca Chaves G, Pereira SE, Saboya CJ, Ramalho A. Non-alcoholic fatty liver disease and its relationship with the nutritional status of vitamin A in individuals with class III obesity. Obes Surg. 2008;18:378–385. doi: 10.1007/s11695-007-9361-2. [DOI] [PubMed] [Google Scholar]
- 87.Kang HW, Bhimidi GR, Odom DP, Brun PJ, Fernandez ML, McGrane MM. Altered lipid catabolism in the vitamin A deficient liver. Mol Cell Endocrinol. 2007;271:18–27. doi: 10.1016/j.mce.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 88.Oliveros LB, Domeniconi MA, Vega VA, Gatica LV, Brigada AM, Gimenez MS. Vitamin A deficiency modifies lipid metabolism in rat liver. Br J Nutr. 2007;97:263–272. doi: 10.1017/S0007114507182659. [DOI] [PubMed] [Google Scholar]
- 89.McClintick JN, Crabb DW, Tian H, Pinaire J, Smith JR, Jerome RE, Edenberg HJ. Global effects of vitamin A deficiency on gene expression in rat liver: evidence for hypoandrogenism. J Nutr Biochem. 2006;17:345–355. doi: 10.1016/j.jnutbio.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 90.Shiota G. Role of retinoic acid receptor in steatohepatitis-related tumor formation. J Gastroenterol Hepatol. 2007;22(Suppl 1):S101–107. doi: 10.1111/j.1440-1746.2006.04668.x. [DOI] [PubMed] [Google Scholar]
- 91.Amengual J, Ribot J, Bonet ML, Palou A. Retinoic acid treatment enhances lipid oxidation and inhibits lipid biosynthesis capacities in the liver of mice. Cell Physiol Biochem. 2010;25:657–666. doi: 10.1159/000315085. [DOI] [PubMed] [Google Scholar]
- 92.Mukhopadhyay B, Liu J, Osei-Hyiaman D, Godlewski G, Mukhopadhyay P, Wang L, Jeong WI, Gao B, Duester G, Mackie K, Kojima S, Kunos G. Transcriptional regulation of cannabinoid receptor-1 expression in the liver by retinoic acid acting via retinoic acid receptor-gamma. J Biol Chem. 2010;285:19002–19011. doi: 10.1074/jbc.M109.068460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lenhard JM, Lancaster ME, Paulik MA, Weiel JE, Binz JG, Sundseth SS, Gaskill BA, Lightfoot RM, Brown HR. The RXR agonist LG100268 causes hepatomegaly, improves glycaemic control and decreases cardiovascular risk and cachexia in diabetic mice suffering from pancreatic beta-cell dysfunction. Diabetologia. 1999;42:545–554. doi: 10.1007/s001250051193. [DOI] [PubMed] [Google Scholar]
- 94.Cione E, Pingitore A, Perri M, Genchi G. Influence of all-trans-retinoic acid on oxoglutarate carrier via retinoylation reaction. Biochim Biophys Acta. 2009;1791:3–7. doi: 10.1016/j.bbalip.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 95.Schallreuter KU, Janner M, Mensing H, Breitbart EW, Berger J, Wood JM. Thioredoxin reductase activity at the surface of human primary cutaneous melanomas and their surrounding skin. Int J Cancer. 1991;48:15–19. doi: 10.1002/ijc.2910480104. [DOI] [PubMed] [Google Scholar]
- 96.Takahashi N, Liapi C, Anderson WB, Breitman TR. Retinoylation of the cAMP-binding regulatory subunits of type I and type II cAMP-dependent protein kinases in HL60 cells. Arch Biochem Biophys. 1991;290:293–302. doi: 10.1016/0003-9861(91)90544-s. [DOI] [PubMed] [Google Scholar]
- 97.Takahashi N, Breitman TR. The covalent labeling of proteins by 17 beta-estradiol, retinoic acid, and progesterone in the human breast cancer cell lines MCF-7 and MCF-7/AdrR. The Journal of steroid biochemistry and molecular biology. 1992;43:489–497. doi: 10.1016/0960-0760(92)90235-b. [DOI] [PubMed] [Google Scholar]
- 98.Tournier S, Raynaud F, Gerbaud P, Lohmann SM, Anderson WB, Evain-Brion D. Retinoylation of the type II cAMP-binding regulatory subunit of cAMP-dependent protein kinase is increased in psoriatic human fibroblasts. J Cell Physiol. 1996;167:196–203. doi: 10.1002/(SICI)1097-4652(199605)167:2<196::AID-JCP2>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 99.Takahashi N, Breitman TR. Retinoylation of vimentin in the human myeloid leukemia cell line HL60. J Biol Chem. 1994;269:5913–5917. [PubMed] [Google Scholar]
- 100.Takahashi N, Breitman TR. Retinoylation of proteins in leukemia, embryonal carcinoma, and normal kidney cell lines: differences associated with differential responses to retinoic acid. Arch Biochem Biophys. 1991;285:105–110. doi: 10.1016/0003-9861(91)90334-f. [DOI] [PubMed] [Google Scholar]
- 101.Takahashi N, Ohba T. Demonstration of basic proteins that bind retinoic acid in the human myeloid leukemia cell line HL60. Biol Pharm Bull. 2009;32:1943–1946. doi: 10.1248/bpb.32.1943. [DOI] [PubMed] [Google Scholar]