

Abstract
Humans have had a significant impact on the distribution and abundance of Saccharomyces cerevisiae through its widespread use in beer, bread and wine production. Yet, similar to other Saccharomyces species, S. cerevisiae has also been isolated from habitats unrelated to fermentations. Strains of S. cerevisiae isolated from grapes, wine must and vineyards worldwide are genetically differentiated from strains isolated from oak-tree bark, exudate and associated soil in North America. However, the causes and consequences of this differentiation have not yet been resolved. Historical differentiation of these two groups may have been influenced by geographic, ecological or human-associated barriers to gene flow. Here, we make use of the relatively recent establishment of vineyards across North America to identify and characterize any active barriers to gene flow between these two groups. We examined S. cerevisiae strains isolated from grapes and oak-trees within three North American vineyards and compared them to those isolated from oak-trees outside of vineyards. Within vineyards we found evidence of migration between grapes and oak-trees and potential gene flow between the divergent oak-tree and vineyard groups. Yet, we found no vineyard genotypes on oak-trees outside of vineyards. In contrast, S. paradoxus isolated from the same sources showed population structure characterized by isolation by distance. The apparent absence of ecological or genetic barriers between sympatric vineyard and oak-tree populations of S. cerevisiae implies that vineyards play an important role in the mixing between these two groups.
Keywords: yeast, S. cerevisiae, gene flow, population genetics, vineyard, oak
Introduction
Most species exhibit some degree of population differentiation. This genetic differentiation is often a consequence of physical barriers to migration, but can also arise through local adaptation. The budding yeast Saccharomyces cerevisiae is widely distributed and consistently associated with two distinct habitats: human related fermentations (Fay and Benavides, 2005; Legras et al., 2007; Liti et al., 2009) and oak-trees and their associated substrates (Naumov et al., 1998; Sniegowski et al., 2002; Zhang et al., 2010). Previous studies have demonstrated that the population genetic structure of S. cerevisiae is correlated with this ecological differentiation rather than geographical distance (Fay and Benavides, 2005; Legras et al., 2007; Diezmann and Dietrich, 2009; Liti et al., 2009; Schacherer et al., 2009). In contrast, the population structure of Saccharomyces paradoxus, the closest relative of S. cerevisiae, seems to be driven by geographical distance (Koufopanou et al., 2006; Liti et al., 2009). Because S. paradoxus is predominantly associated with oak-tree habitats and rarely associated with human related fermentations, the difference in population genetic structure between S. cerevisiae and S. paradoxus could simply be a consequence of S. cerevisiae’s association with humans, either through human-associated dispersal (Legras et al., 2007) or through artificial selection in the form of domestication (Fay and Benavides, 2005; Liti et al., 2009).
In S. cerevisiae, population structure is largely defined by multiple strains isolated from a variety of human-related fermentations. Genetically differentiated groups have been identified in association with the production of beer, bread, grape wine, sake wine, palm wine and various food products (Sicard and Legras, 2011). One of the best characterized groups includes strains isolated from grapes and grape must as well as those used in commercial grape wine production. These strains have been isolated from vineyards around the world and form a genetically homogeneous group (Fay and Benavides, 2005; Liti et al., 2009), which we refer to as vineyard strains. Strains unrelated to human fermentations have primarily been isolated from oak-tree exudate, bark and associated soil as well as clinical samples from immunocompromised patients (Liti et al., 2009). These strains are more diverse and unless isolated from the same location do not form a well-defined group. Given the widespread occurrence and co-occurrence of Saccharomyces species on oak-trees in the Northern Hemisphere (Sniegowski et al., 2002; Sampaio and Gonçalves, 2008) and beech trees in the Southern Hemisphere (Libkind et al., 2011), the tree habitat is thought to represent the wild source from which many human associated strains were derived. This has been confirmed in at least once case: the non-S. cerevisiae contribution to the alloploid lager-brewing S. pastorianus genome has recently been identified as S. eubayanus, a species isolated from trees in Patagonia (Libkind et al., 2011).
The underlying cause of genetic differentiation between vineyard and non-vineyard strains of S. cerevisiae has been difficult to determine. Differentiation of vineyard and non-vineyard strains could be the result of historical patterns of migration, in which case the genetic similarity of vineyard strains isolated from around the world is a consequence of human-assisted migration, either intentional or not, from European vineyards to those in other locations. It is also important to consider the possibility that historical population structure contributed to the genetic differentiation of vineyard and non-vineyard strains. The majority of oak-tree and clinical strains have been isolated from North America whereas the vineyard strains are thought to have a European origin (Liti et al., 2009). Most oak-tree samples from Europe yield other Saccharomyces species (Johnson et al., 2004; Koufopanou et al., 2006; Sampaio and Gonçalves, 2008), and the few S. cerevisiae strains that have been obtained have not yet been genetically characterized. Furthermore, of the few clinical isolates and wild isolates from Europe, most group with vineyard strains (Liti et al., 2009). However, without a well-defined group of non-vineyard strains from Europe it has been difficult to understand the multitude of factors responsible for genetic differentiation of vineyard and non-vineyard groups. Local adaptation to the vineyard environment and/or human selection could also help maintain differentiation between these groups, an idea that is supported by a variety of phenotypes that have been associated with vineyard strains. Divergent phenotypes include resistance to copper (Fay et al., 2004; Liti et al., 2009) and sulfite (Park and Bakalinsky, 2000), two chemicals used in vineyards and for wine production, growth and fermentation parameters (Spor et al., 2009), freeze/thaw tolerance (Will et al., 2010), sporulation efficiency (Gerke et al., 2006), and wine aroma and flavor (Hyma et al., 2011).
The relatively recent establishment of vineyards outside of Europe presents the opportunity to examine recent migration and recombination between vineyard and oak-tree populations of S. cerevisiae. In New Zealand, isolates from natural sources and vineyard strains migrate between habitats and interbreed (Zhang et al., 2010). However, the extent to which this occurs in other locations is not known. The degree of migration and genetic exchange between vineyards and other natural habitats, such as oak trees, is important to knowing whether vineyard strains are adapted to vineyard environment, whether oak-tree strains can invade vineyards and contribute to wine fermentations, and whether recent genetic exchange is breaking down the differentiation between these two groups.
In order to better understand the evolutionary forces that contribute to population structure in S. cerevisiae, we examined local population structure between oak-tree and vineyard isolates in North America. Because samples from a single location are often very closely related to one another we used a recently developed genome-sequencing approach that enabled us to interrogate ~200kb of sequence distributed across S. cerevisiae’s 12.5 Mbp genome. By comparing this sequence to other sequenced S. cerevisiae genomes we found strains that clearly fall within previously defined vineyard and oak-tree groups. We isolated strains from both of these genetically defined groups from both the grape and oak-tree substrates within vineyards, but not from oak-trees sampled outside of vineyards. In comparison, samples of S. paradoxus, which can also be isolated both within and outside of vineyards, exhibit a genetic pattern of isolation by distance. Our results provide insight into population structure in North America and demonstrate that migration and potential genetic exchange between vineyard and oak-tree habitats has occurred over a short time-scale.
Materials and Methods
Strains
S. cerevisiae and S. paradoxus strains were collected from a total of eight study sites. Two different vineyard and two different non-vineyard locations were sampled from the states of Missouri and Oregon, USA. In Missouri, vineyard sites were located in Ste. Genevieve County (Chaumette Vineyards) and St. Charles County (Mount Pleasant Winery), 106 km apart, and non-vineyard sites were in St. Louis County (Tyson Research Center), >28 km from either vineyard, and Washington County (L. Watrud, personal property), >50 km from either vineyard. In Oregon, vineyard sites were located in Polk County (Whistling Dog Cellars) and Benton County (Tyee Wine Cellars), 58 km apart, and non-vineyard sites were in Benton County (Chip Ross State Park and M. Bollman, personal property), >17 km from either vineyard.
Sampling and Enrichment
Samples were collected from two different environments at vineyard locations; from damaged grapes and from adjacent, vineyard associated oak-trees. At non-vineyard locations samples were collected from oak-trees. Damaged grapes and oak-trees were chosen for sampling based on previously published studies showing high recovery rates (Naumov et al., 1998; Mortimer and Polsinelli, 1999; Sniegowski et al., 2002; Sampaio and Gonçalves, 2008). Damaged grapes were removed from the vine using ethanol sterilized forceps and macerated using an ethanol sterilized metal rod. Oak-tree samples were taken from bark, twig and surrounding soil found at the base of established trees > 8.9 cm (3.5 inches) in diameter. Oak bark samples were scraped from the tree using ethanol sterilized knives, twigs were cut using ethanol sterilized scissors, and soil was collected using ethanol sterilized spatulas. All types of samples were placed into sterile plastic 15 ml screw cap conical vials. In addition to grape and oak samples, samples from dejuiced grape mash and from a spontaneously generated wine fermentation were collected at Chaumette Vineyards. Samples were collected in 2008 from all 8 locations during the harvest season for vineyards in Missouri (September), and Oregon (October). Additional samples were collected from the Missouri Chaumette Vineyard and Tyson sampling locations in 2009.
Samples were enriched for S. cerevisiae and other yeasts species that favor similar growing conditions by adding 6 mL of sterile enrichment media to the sample, closing the tube and allowing it to ferment. Two different types of enrichment media were used, a high sugar medium (H), YPD containing 10% dextrose and 5% ethanol, adjusted to pH 5.3 (Mortimer and Polsinelli, 1999) and a low sugar medium (L) containing 6.7 g/L yeast nitrogen base, 1% w/v glucose, and 8% v/v ethanol, an adaptation from (Sampaio and Gonçalves, 2008) in order to determine which enrichments increase the recovery of S. cerevisiae. After 7 days of fermentation, a 200 μl sample was transferred into a new 15 ml vial with 6 mL of fresh sterile enrichment media, and allowed to ferment for an additional 4 days. Following the second fermentation, 2 μl of enriched media was plated onto YPD plates, and incubated at 30°C for 2 days. One to six colonies from each plate was re-streaked for purity and frozen stock cultures of an overnight (YPD) culture were prepared in 15% glycerol at −80°C. For samples collected in 2009, only the high sugar enrichment medium was used for both stages of enrichment, and only colonies that resembled S. cerevisiae were re-streaked and frozen.
Isolate screening and species identification
Colonies that resembled bacteria were tested on YPD agar containing 10 mg/L chrloramphenicol and 100 mg/L ampicillin, bacterial specific antibiotics. If colonies failed to survive antibiotic screening (indicating likely bacterial species) they were excluded from the study. Remaining “yeast-like” colonies were further screened with molecular methods to identify isolates belonging to the Saccharomyces sensu stricto group. DNA was purified from each isolate by resuspending a colony grown on YPD in 100 μl of10 mg/ml lyticase with a small amount of glass beads in a 96 well PCR plate. Plates were sealed and incubated at 37 °C for 15 minutes, followed by a brief vortexing for 2-3 seconds and incubation at 95 °C for 10 minutes. The resulting DNA was then used as a template for a multiplex PCR assay (Nardi et al., 2006). The assay included two primer pairs, one specific to the Saccharomyces sensu stricto group, and the other which acts as a universal fungal primer (Table S1). Amplification of two PCR products indicated presence of Saccharomyces sensu stricto specific priming, and thus identification of Saccharomyces species. PCR reactions were carried out in a 25 μl reaction using 3 μl of DNA template, 0.5 μl of each primer at 10 μM concentration, 1 μl Taq polymerase, 1.2 mM DNTPs, and 4 mM MgCl2. PCR reactions were incubated at 94 °C for 2 minutes followed by 35 cycles of 94 °C for 30 seconds, 51 °C for 30 seconds and 72 °C for 2 minutes, followed by a final incubation at 72 °C for 7 minutes.
Isolates that were identified as Saccharomyces sensu stricto using this method were further classified using ribotyping; restriction digests of the intergenic transcribed spacer region (ITS) (McCullough et al., 1998). An initial digestion by the restriction enzyme HaeIII was first used to differentiate S. cerevisiae and S. paradoxus from S. mikatae, S. bayanus, and S. kudriavzevii. A second digestion by either BfaI or MwoI was used to further differentiate species within these two groups, respectively (Table S2).
A large number of oak isolates were obtained and a subset was selected for sequence analysis with preference given to those from different trees. A total of 49 S. cerevisiae and 28 S. paradoxus strains were selected for analysis (Table 1). A single S. cerevisiae isolate from a spontaneous fermentation, two isolated from macerated grapes near the winery. Four additional strains isolated in Wisconsin, two from cherries and two from oak-trees, were provided by Audrey Gasch. See tables S3 and S4 for a description of S. cerevisiae and S. paradoxus strains used in this study.
Table 1.
Saccharomyces isolates analyzed by substrate
| substrate | S. cerevisiae | S. paradoxus |
|---|---|---|
| grape | 17 | 3 |
| macerated grapes | 2 | 0 |
| spontaneous fermentation | 1 | 0 |
| vineyard oak | 10 | 10 |
| non-vineyard oak | 17 | 15 |
| cherry | 2 | 0 |
| Total | 49 | 28 |
Genotyping
Restriction-site associated DNA tags (RAD tags) were sequenced using a protocol based on (Baird et al., 2008). Genomic DNA was isolated using ArchivePure DNA Yeast & Gram −+ Kits (5 Prime, Inc.), quantified using the Quant-it™ dsDNA HS Assay (Invitrogen Corporation), adjusted to a standard concentration, and digested for 60 minutes at 37°C in a 50 μl reaction with 5 units (U) each of MfeI and MboI (New England Biolabs, Inc.), followed by heat inactivation for 20 minutes at 65°C. Digested genomic DNA was ligated to P1 adaptor, a modified Solexa® adaptor (2006 Illumina, Inc., all rights reserved; top: 5′ –ACA CTC TTT CCC TAC ACG ACG CTC TTC CGA TCT xxxx – 3′ [x = barcode], bottom: 5′- Phos – AATT xxxx AGA TCG GAA GAG CGT CGT GTA GGG AAA GAG TGT - 3′, and P2 adaptor, a modified Solexa® adaptor (2006 Illumina, Inc., all rights reserved; top: 5′ - Phos – GAT CCT CAG GCA TCA CTC GAT TCC TCC GAG AAC AA – 3′ : bottom: 5′ - CAA GCA GAA GAC GGC ATA CGA CGG AGG AAT CGA GTG ATG CCT GAG – 3′) with 1000 U concentrated T4 DNA ligase (New England Biolabs, Inc.) at room temperature for 20 minutes, followed by heat inactivation at 65°C for 20 minutes. Ligated and digested DNA was pooled and purified using a QIAquick PCR Purification Kit (Qiagen, Inc.). Fragments from 150-500 bp were isolated using a QIAquick Gel Extraction kit (Qiagen, Inc.). Fragments were then PCR amplified using 5-10 ng DNA, 25 μl Phusion High-Fidelity PCR Master Mix (New England Biolabs,Inc.), 0.5 μM of each modified Solexa® pcr primer: (solexa pcr forward P1 5′ - AAT GAT ACG GCG ACC ACC GAG ATC TAC ACT CTT TCC CTA CAC GAC GCT CT - 3′ and solexa pcr reverse P2 5′ - CAA GCA GAA GAC GGC ATA CGA - 3′), and water to a final volume of 50 μl. Cycling conditions were 98°C for 1 minute followed by 14-18 cycles of 98°C for 10 seconds, 60°C for 30 seconds, 72°C for 30 seconds, and a final extension at 72°C for 4 minutes. The resulting PCR product was purified using a QIAquick PCR Purification Kit (Qiagen, Inc.) and adjusted to 10 nm. Illumina Solexa protocols were followed for sequencing.
Sequence Analysis
Raw sequence reads were processed to reduce sequencing artifacts within the data using custom Perl scripts. First, reads were separated by barcodes, which were examined for quality and trimmed from reads prior to mapping. Reads with a Phred-scaled sequence quality score of less than 20 for any bp within the barcode, as well as reads with an unknown barcode sequence, were removed. For S. cerevisiae, reads were aligned to the Saccharomyces genome resequencing project (SGRP) reference genome (available at http://www.sanger.ac.uk/research/projects/genomeinformatics/sgrp.html) (Liti et al., 2009) using the short read alignment program Bowtie (Langmead et al., 2009). Reads that aligned to more than one location were suppressed (option –m 1), two mismatches were allowed in the 28 bp seed (options –n 2, −l 28), and the –tryhard option was enabled. Reads that lacked a MfeI restriction site or did not align adjacent to an MfeI restriction site (AATG), allowing for a one bp mismatch from the reference sequence within the restriction site, were filtered from the data set.
Alignment rates to the SGRP S. paradoxus reference genome were low (< 50%) for most S. paradoxus strains, including the control strain YPS138, likely due to the large amount of sequence divergence between North American isolates and the European isolates used to generate the reference genome (Liti et al., 2009). A new assembly was created using the SGRP genome sequences of North American S. paradoxus strains UFRJ50791, UFRJ50816, A12, A4, YPS138 and DBVPG6304, that resulted in 5-6x coverage and was used for alignment.
After alignment, the first four and last four base pairs of each read were discarded. Any position in an aligned read with a Phred-scaled sequence quality score of less than 15 was masked by converting that position to an ‘n,’ changing its quality score to 0, and removing it from the calculation of sequence coverage at that position. Consensus pileups for each strain were generated using Samtools (Li et al., 2009). Sequenced positions with a consensus quality score of less than 40 or with less than 3x coverage were filtered out of the data set. Single nucleotide polymorphisms (SNPs) in the data set were retained if the SNP quality score was greater than or equal to 20, and there were no more than 2 SNPs in a 10 bp window.
During each run we included two control strains with independent genome sequence data in order to estimate the false positive rate for SNPs resulting from Solexa sequencing. The expected number of false positives was calculated for each control strain as FP*T, where T is the total number of Solexa sequenced positions for the strain and FP is the false positive rate estimated by the number of SNPs found by Solexa sequencing but not found in the previously sequenced genome. To exclude any errors present in the previously sequenced genomes we estimated the rate of false positives from sites that were the same in both the M22 and YPS163 reference genomes. False discovery rate estimates are found in Table S7.
The average number of sequenced positions for 51 S. cerevisiae and 40 S. paradoxus isolates, was 462,972 and 284,334 bp respectively. Several S. paradoxus isolates had very low sequence coverage (10,000 bp or less), and were excluded from analysis. After removing those isolates, the average number of sequenced positions for S. paradoxus was 292,572 bp.
Statistical Analysis
Population differentiation between vineyard and oak-tree populations of S. cerevisiae was characterized using 49 S. cerevisiae isolates including 17 grape isolates (MO, OR), 10 oak isolates recovered within or adjacent to vineyards (MO, OR), 15 oak isolates recovered outside of vineyards (MO, OR), along with a single isolate from a spontaneous fermentation, 2 isolates from macerated grapes near a winery, 2 oak and 2 cherry isolates from Wisconsin. Population differentiation within S. paradoxus samples was characterized using 28 isolates including 3 grape isolates (MO, OR), 10 vineyard oak isolates (MO, OR) and 15 non-vineyard oak isolates (MO, OR) (Table 1).
Due to the properties of RAD tagging and Solexa sequencing, certain regions of the genome may not be sequenced in every isolate. To adjust for this possibility, the sequence data set was compiled for RAD genotyped S. cerevisiae strains, and any position that was sequenced for at least 48 of the 49 strains was retained. After filtering, the data set included 215,395 base pairs, representing about 1.7% of the S. cerevisiae genome. Orthologous sequences were obtained from a set of 38 S. cerevisiae strains with sequenced genomes (Liti et al., 2009). Genotypes for these strains were extracted from the alignments available at http://www.sanger.ac.uk/research/projects/genomeinformatics/sgrp.html. See Table S5 for a list of strains. Sequenced positions with a Phred score of less than 20 were converted to “N”s. Orthologous sequences were also obtained from a set of 25 newly sequenced S. cerevisiae strains (Table S6), available at http://www.genetics.wustl.edu/jflab/data4.html. Genotype information for these strains was obtained using BLAST (Altschul et al., 1990), using the reference sequence at RAD genotyped positions as a query against nucleotide blast databases created from genome assemblies. After the addition of the previously sequenced strains, the dataset was further restricted to positions for which sequence data was available for at least 80% of strains (including both RAD genotyped and previously sequenced strains). After filtering, our data set included 5,425 variable positions (SNPs).
The sequence data were compiled separately for RAD genotyped S. paradoxus strains. Filters were similar to those used for S. cerevisiae except that we included positions that were sequenced for at least 24 of the 40 strains. The filtered data set included 281,944 base pairs, representing approximately 2.4% of the S. paradoxus genome. Additional genome sequences for a diverse set of 37 S. paradoxus strains have been described previously (Liti et al., 2009). Genotypes for these strains were extracted using BLAST (Altschul et al., 1990). See Table S6 for a list of strains. The assemblies of these strains were downloaded from http://www.sanger.ac.uk/research/projects/genomeinformatics/sgrp.html. After filtering, our data set included 9,809 variable SNPs.
Sequence diversity was estimated for noncoding regions, coding regions, two-fold, four-fold and non-degenerate sites based on the SGRP reference genome annotation (Liti et al., 2009) for S. cerevisiae. Sequence diversity was estimated as the number of nucleotide substitutions per site (π) using MEGA4 (Tamura et al., 2007). All positions containing alignment gaps, missing or ambiguous data were eliminated only in pairwise sequence comparisons. The ratio πN/πS was estimated based on substitutions per non-degenerate site/ substitutions per four-fold degenerate site, and two-fold degenerate sites were excluded from the calculation. Minor allele frequencies (MAF) were calculated for biallelic sites using PLINK (Purcell et al., 2007). For minor allele frequencies in S. cerevisiae, only one isolate from each clonal group, defined as a single clade in which the pairwise nucleotide p-value between any two strains within the group is less than 0.0002 was included in the analysis. The neutral expectation for minor allele frequencies was calculated using Watterson’s θ (Watterson, 1975) following (Lu et al., 2006).
Phylogenetic Analysis and Population Structure
Phylogenetic trees were inferred with MEGA4 (Tamura et al., 2007) using the Neighbor-Joining method based on pairwise distances (measured as nucleotide substitutions per site) with 1,000 bootstrap replicates. All positions containing alignment gaps and missing data were eliminated only in pairwise sequence comparisons.
Population structure was examined using the model-based program STRUCTURE (Pritchard et al., 2000). For S. cerevisiae, population structure was inferred from 3,087 parsimony informative loci assuming the admixture model with uncorrelated allele frequencies and no linkage. Three initial simulations at K =1 were used to infer lambda, which parameterizes the allele frequency prior, and based on these initial simulations lamda was set at 0.4912 for subsequent simulations. Three replicate simulations were performed for each inferred number of populations (K), for K = 2 through K = 10 with a burn-in period of 10,000, followed by 10,000 additional Markov Chain Monte Carlo replications. An additional 7 replicate simulations were run for K=6 and K=7. CLUMPP (Jakobsson and Rosenberg, 2007) was used to assess the similarity between replicate STRUCTURE results (G’) in order to determine the relative likelihood of multimodality of the inferred population structure. For K=6 and 7, the LargeKGreedy algorithm was used with 10,000 random permutations. DISTRUCT (Rosenberg, 2003) was used to visualize the results. The STRUCTURE simulation at k=7 with the highest estimated Ln probably of the data was used for population assignment and inferences of admixture.
Population structure was also examined using a similar model-based program, InStruct,(Gao, Williamson and Bustamante, 2007), that accounts for inbreeding. Biallelic SNPs were filtered to remove SNPs that were in approximate linkage equilibrium with each other in a 50 base pair window using PLINK (Purcell et al., 2007) before randomly selecting 100 SNPs in order to reduce runtime. Five chains for each of K=2 through K=10 were run with a burn-in period of 100,000 followed by 2,000,000 additional Markov Chain Monte Carlo replications. The 20 stored iteration results after burn-in were used to calculated the Gelman-Rubin statistic. DISTRUCT (Rosenberg, 2003) was used to visualize the results.
Multidimensional scaling was use to graph genetic similarity among strains. To focus on alleles relevant to admixture a subset of 285 biallellic SNPs segregating within both the wine-clade and oak-clade were mean centered and variance normalized. Multidimensional scaling was then applied to the euclidean distance matrix of the scaled genotypes.
Population structure in S. paradoxus was examined for 7,063 parsimony informative SNPs as in S. cerevisiae, with 10 replications each of K=2 through K=5 with lambda equal to 1. Similarity was assessed using the Fullsearch algorithm to compare 10 permutations for each inferred number of populations.
Results
Sampling of yeast from vineyard and oak substrates
S. cerevisiae and S. paradoxus were isolated from grapes and oak-trees in Missouri and Oregon. While the isolation rates of S. cerevisiae and S. paradoxus from oak samples were similar to previous studies (Sampaio and Gonçalves, 2008), 14% and 28% in 2008, respectively, the isolation rate of S. cerevisiae from grape samples (2%) was very low compared to isolations rates from vineyards in Italy (20%), (Mortimer and Polsinelli, 1999). A total of 49 S. cerevisiae and 28 S. paradoxus strains were selected for analysis, including all of the grape isolates and a subset of the oak isolates with preference given to those from different trees (Table 1). A single S. cerevisiae isolate from a spontaneous fermentation, two isolated from macerated grapes near the winery, as well as two cherry and two oak-tree strains sampled from Wisconsin were also included.
Rates of polymorphism and heterozygosity based on restriction site associated sequencing
Restriction site associated (RAD) sequencing identified 5,425 polymorphic sites in S. cerevisiae and 9,809 polymorphic sites in S. paradoxus. Based on RAD sequence of four previously sequenced strains, we estimated false positive rates of 2.0×10−5 and 1.2×10−5 for two S. cerevisiae strains and 4.1×10-4 and 1.6×10-4 for two S. paradoxus strains (Table S7). The higher estimated rate of false positives for S. paradoxus may be due to divergence between stocks of the same strain; the S. paradoxus sequences were generated from different stocks whereas the S. cerevisiae sequences were generated from the same stock.
Most strains exhibited low levels of heterozygosity, fewer than 2% of SNPs were called heterozygous based on the total number of differences between the sequenced strain and reference genome. High rates of heterozygosity were found in two cherry strains, DCM6 and DCM21 (52.7% and 47.4% of SNPs, respectively), a wine strain isolated from a vineyard grape (KEH000415, 12.2%), and a wine strain isolated from a spontaneous wine fermentation (KEH02575, 22.5%).
Phylogenetic Analysis of S. cerevisiae
A bootstrap consensus phylogeny was constructed for all S. cerevisiae samples including 17 grape samples (MO, OR), 10 oak samples located within or adjacent to vineyards (MO, OR), 15 oak samples located outside of vineyards (MO, OR), a single strain from a spontaneous fermentation, 2 strains from macerated grapes near a winery, 2 oak and 2 cherry samples from Wisconsin. The neighbor-joining tree (Figure 1) shows that with the exception of the two cherry isolates, all of the isolates group with previously described European/wine/vineyard or North American oak populations (Fay and Benavides, 2005; Aa et al., 2006; Legras et al., 2007; Liti et al., 2009; Schacherer et al., 2009) with high bootstrap support. The North American oak clade includes all 6 previously sequenced oak-tree isolates. The vineyard clade includes seven previously sequenced vineyard isolates as well as seven non-vineyard strains, only one of which (CLIB215, a bakery strain) was isolated outside of Europe. Of the remaining previously sequenced strains, many are consistent with mating between distinct groups. Included within these are five (I14, T73, YIIc17-E5, Y55, YJM269) vineyard strains that fall outside of the vineyard clade (Figure 1).
Figure 1.

Evolutionary relationships of S. cerevisiae strains. The bootstrap consensus tree based on 197,125 bp of sequence data was inferred from 1000 replicates using pairwise distances (substitutions per site) between strains. Positions containing alignment gaps and missing data were eliminated only in pairwise sequence comparisons. Branches with bootstrap values of less than 50% have been collapsed. The tree is drawn to scale. Label colors correspond to their isolation source, and previously described European/vineyard (A) and N. American/oak lineages (B) are noted on the tree. Bootstrap support for nodes A and B is 100%.
The clear genetic differentiation between strains within the wine/European clade and North American clade make it possible to test for migration between sympatric oak and grape populations in N. America. Out of the 19 grape and macerated grape isolates, 11 fall within the vineyard clade and 8 fall within the oak-tree clade (Figure 1). Out of the 10 vineyard oak isolates, 6 fall within the vineyard clade and 4 fall within the oak-tree clade. In contrast, all 17 of the 17 non-vineyard oak isolates fall within the oak clade.
Our study sites included two vineyard locations each in both Missouri (MO) and Oregon (OR). Sample sizes in one MO vineyard (Chaumette) allowed for finer grained analysis of the distribution of wine and oak strains. Within this vineyard, both wine and oak strains are present, but their distribution is not associated with sample substrate, i.e. wine strains are found on both grapes and oak-trees and oak strains are also found on both grapes and oak-trees. Thus, over small geographic distances there is no evidence for restricted migration between oak-trees and grapes. In the other three vineyards isolation rates of S. cerevisiae from grapes was too low to infer genetic patterns. From the second MO winery (Mount Pleasant), four strains (2 from grapes and 2 oaks) were all of the oak genotype, whereas no strains were isolated from grapes in OR, and both strains isolated from oaks in OR vineyards were of the vineyard genotype.
Strains of the oak genotype exhibit a clonal population structure; many of the strains isolated have nearly identical genotypes with no strong geographic pattern. Within oak-tree genotype strains, two clonal subpopulations (pairwise distances < 0.0002) contain 24 of the 27 strains in the group (Figure 1). The dominant genotype (KEH00729, 20 isolates) is widespread, found in both vineyards and non-vineyard locations in Missouri and Oregon. The oak strains from Wisconsin (DY8 and DY9), as well as another US oak-tree strain (T7) are also very closely related to the dominant clone KEH00729 (nucleotide p-distances are 0.0009, 0.0005 and 0.0.0012 respectively). The second subpopulation (KEH00411, 4 isolates) was found at both a vineyard and a non-vineyard location in Missouri (Figure 1). Vineyard genotype strains, in contrast to the oak genotype strains, have a less clonal structure with the exception of one group of strains (KEH02580, 8 isolates). The clonal structure of this group of wine strains shows the frequent occurrence of a single genotype within the Chaumette vineyard during the 2009 harvest season.
Patterns and levels of nucleotide diversity also differ between the vineyard and oak clades. Despite the smaller number of clones within the vineyard clade, the overall nucleotide diversity (π*100) within the vineyard clade (0.063) is 46% that of the oak clade (0.137)(Table 2). As observed in previous studies, the vineyard clade shows lower levels synonymous polymorphism (4-fold degenerate sites) but a higher ratio of nonsynonymous to synonymous polymorphism (4-fold to 0-fold degenerate sites, Chi-square test, p < 0.001, Table 2), compared to the oak clade, consistent with a bottleneck in the lineage leading to the vineyard clade. The vineyard clade also has a higher proportion of rare alleles to high frequency alleles relative to the oak clade (Figure S1), a pattern consistent with either an increase in the number of rare alleles in the vineyard clade during the recovery from a population bottleneck or a decrease in the number of rare alleles in the oak clade due to clonal expansion or a reduction in population size.
Table 2.
Nucleotide diversity (π) within and between strains of S. cerevisiae.
| comparison | # strains | all sites | non-coding | 4fold | non-degenerate | πN / πS |
|---|---|---|---|---|---|---|
| all S. cerevisiae | 112 | 0.297 | 0.455 | 0.680 | 0.120 | 0.176 |
| wine/vineyard lineage | 32 | 0.063 | 0.085 | 0.109 | 0.040 | 0.367 |
| N. American/oak lineage | 35 | 0.137 | 0.185 | 0.370 | 0.044 | 0.119 |
Nucleotide diversity is the number of substitutions per site (π) * 100, calculated using MEGA4.0 (Tamura et al., 2007) based on pairwise comparisons of nucleotide substitutions per site. πN / πS is the ratio of nucleotide diversity at nondegenerate (N) to 4-fold degenerate (S) sites, 2-fold degenerate sites were excluded from the analysis.
Gene flow between differentiated populations of S. cerevisiae
To determine whether there has been any historical gene flow between vineyard and oak strains we examine the genome distribution of oak and vineyard-clade genotypes using STRUCTURE. Congruent with a previous inference of population structure (Liti et al., 2009), both the wine/European lineage and the North American oak lineage were clearly differentiated, both for previously characterized isolates as well as isolates collected in this study (Figure 2). As such, our micro-scale sampling of yeast samples from vineyard and non-vineyard locations resolved previously identified subpopulations of S. cerevisiae. Replicate simulations at a given K value became less consistent above K=3, as indicated with decreased values for G’ (Jakobsson and Rosenberg, 2007), even though likelihood increased. Although values of similarity (G’) between replicate runs at K=8 and K=9 are roughly comparable, our results are congruent with a previous inference of population structure (Liti et al., 2009) at K=7, providing resolution of sake, Malaysian, and West African lineages in addition to the wine/European and North American oak lineages.
Figure 2.

Inferred population structure of S. cerevisiae.
Population structure was examined using the model-based programs STRUCTURE (Pritchard, Stephens, and Donnelly 2000), and Instruct (Gao, Williamson and Bustamante, 2007)(REF!!!). DISTRUCT (Rosenberg 2003) was used to visualize STRUCTURE results for K=5, K=6, K=7 and K=8 with the highest estimated Ln probably of the data, and Instruct results for the K=9 run with the highest posterior likelihood value. H’ G’ values from replicate STRUCTURE runs were calculated using CLUMPP (Jakobsson and Rosenberg, 2007). The Gelman-Rubin statistics (g-r) for K=9, was calculated to test for convergence of multiple Instruct Chains. Populations are labeled by their assignment from the most likely STRUCTURE run at K=7, but colored by inferred clustering at the given K value.
To account for inbreeding, we also examined the genome distribution of oak and vineyard-clade genotypes using InStruct. Although the Gelman-Rubin statistic showed convergence at K=3, K=7, K=8, and K=9 (GR < 1.1), K=9 with a mean posterior likelihood value of −1589.8756 was chosen by the program as the most likely number of subpopulations. For 5 of the 9 inferred subpopulations, no individual had greater than 12% membership. The remaining four subpopulations correspond nearly identically with the vineyard, two different oak and W. African/Malaysian subpopulations inferred by STRUCTURE at K=7 (Figure 2).The sake group clusters with one of the oak groups, and the ‘other’ group is inferred to represent a mostly vineyard genetic background. Both STRUCTURE and InStruct show evidence for admixture between subpopulations.
Potential introgression or admixture between the ‘vineyard and ‘oak’ genotypes was detected by STRUCTURE for several of the strains isolated for this study and fell into two categories, those with predominately vineyard backgrounds, and those with predominately oak backgrounds (Table 4 and Figure 3). Six strains with oak backgrounds (KEH00146, KEH02595, DY8, DY9, KEH00088 and KEH01205) were inferred to have between 0 and 12% vineyard ancestry, (Table 4). Eight strains with vineyard backgrounds (DCM21, DCM6, KEH00221, KEH00415, KEH02575, KEH02714, KEH02809 and KEH02884) are inferred to have between 0 and 2930% oak ancestry. InStruct typically inferred a smaller amount of admixture, yet the results are not not directly comparable to those of STRUCTURE since only a subset of SNPs were used in the InStruct analysis. The relationships among strains are shown in Figure 3 by multidimensional scaling of genotypic distances, which assumes no underlying population genetic model.
Table 4.
Admixture between vineyard and oak-tree strains.
| ID | Primary Population |
Collection Site | Source | STRUCTURE K = 7 | InStruct K = 9 | ||||
|---|---|---|---|---|---|---|---|---|---|
| Vineyard | Oak1 | Oak2 | Vineya rd |
Oak1 | Oak2 | ||||
| DCM21 | vineyard | WI, 2009 | cherry | 0.63 | 0.29 | 0.00 | 0.97 | 0.01 | 0.00 |
| DCM6 | vineyard | WI, 2009 | cherry | 0.60 | 0.23 | 0.00 | 0.95 | 0.00 | 0.01 |
| KEH00221 | vineyard | MO Vineyard 2008 |
oak | 1.00 | 0.00 | 0.00 | 0.94 | 0.05 | 0.00 |
| KEH00415 | vineyard | MO Vineyard 2008 |
grape | 0.91 | 0.09 | 0.00 | 0.96 | 0.02 | 0.02 |
| KEH02575 | vineyard | MO Vineyard 2008 |
fermen t |
0.92 | 0.03 | 0.00 | 0.96 | 0.03 | 0.00 |
| KEH02714 | vineyard | MO Vineyard 2008 |
grape | 1.00 | 0.00 | 0.00 | 0.96 | 0.02 | 0.01 |
| KEH02809 | vineyard | MO Vineyard 2008 |
grape | 0.93 | 0.07 | 0.00 | 0.86 | 0.14 | 0.00 |
| KEH02884 | vineyard | MO Vineyard 2008 |
grape | 0.89 | 0.08 | 0.00 | 0.98 | 0.01 | 0.00 |
| KEH01146 | oak2 | MO 2008 | oak | 0.00 | 0.00 | 1.00 | 0.03 | 0.01 | 0.96 |
| KEH02595 | oak2 | MO Vineyard 2009 |
grape | 0.00 | 0.00 | 1.00 | 0.06 | 0.02 | 0.91 |
| DY8 | oak1 | WI, 2009 | oak | 0.12 | 0.81 | 0.00 | 0.00 | 0.98 | 0.00 |
| DY9 | oak1 | WI, 2009 | oak | 0.06 | 0.94 | 0.00 | 0.01 | 0.98 | 0.00 |
| KEH00088 | oak1 | MO Vineyard 2008 |
oak | 0.04 | 0.53 | 0.00 | 0.59 | 0.01 | 0.35 |
| KEH01205 | oak1 | MO 2008 | oak | 0.00 | 1.00 | 0.00 | 0.06 | 0.93 | 0.00 |
The proportion of membership to vineyard and oak populations inferred by STRUCTURE (Pritchard, Stephens, and Donnelly 2000) and InStruct ,(Gao, Williamson and Bustamante, 2007) is show for each strain isolated for this study for which admixture between vineyard and oak-tree strains was inferred. The primary population listed is the population with greater than 50% membership based on STRUCTURE K=7.
Figure 3.
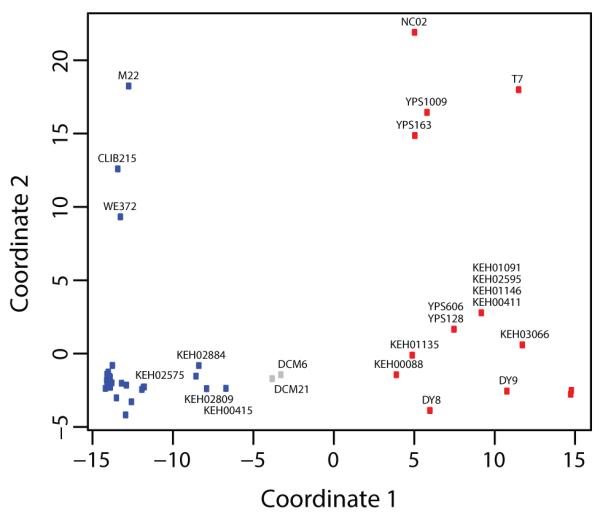
Multidimensional scaling of genotypic distances among strains. The first coordinate (x-axis) differentiates strains within the wine (blue) and oak-tree (red) clades shown in Figure 1. The second coordinate (y-axis) differentiates wine and oak-tree strains alleles showing mixture with non-wine and non-oak populations inferred by STRUCTURE. Only a subset of strains were labeled for clarity.
Phylogenetic Analysis and Distribution of Genetic Diversity in S. paradoxus
The absence of any geographic differentiation in the S. cerevisiae oak clade is notable given that S. paradoxus is geographically differentiated. However, there are few studies of S. paradoxus population structure in North America. To determine if the differences in population structure between S. cerevisiae and S. paradoxus are observed within N. America, we compared population structure between contemporary isolates of S. cerevisiae and S. paradoxus.
We examine 28 S. paradoxus strains collected from MO (14) and OR (14) (Table S4), in addition to 37 previously sequenced S. paradoxus isolates (Liti et al., 2009) (Table S6). Phylogenetic analysis resolved the same four populations that genome resequencing uncovered: American, European, Far Eastern, and Hawaiian (represented by a single strain) (Liti et al., 2009) (Figure 3). Most of the strains isolated from Missouri and Oregon belong to the American clade, with the exception of KEH0229, KEH02271, KEH02530 and KEH00137, which belong to the European clade. Isolates belonging to the American clade show further geographic structure. Strains from Missouri and strains from Oregon form two distinct clades that are highly supported by bootstrap analysis (Figure 1).
Analysis of population structure using the program STRUCTURE is consistent with the phylogenetic analysis, revealing three populations corresponding to the previously described American, European and Far Eastern populations, with strain membership corresponding to the clades described above (Figure S2). The Hawaiian strain appears to show a signal of genetic admixture, although it is likely that this is an artifact of sample size.
The overall nucleotide diversity (π*100) for S. paradoxus (1.413) is nearly five times higher than for S. cerevisiae (0.297). However, most of that diversity is found between the American and European clades. The amount of diversity contained within the American clade of S. paradoxus (0.167) is only slightly higher than for the oak clade of S. cerevisiae (0.134) (Table 3). Minor allele frequencies in S. paradoxus show a significant shift towards higher frequency alleles compared to the neutral expectation, both for the entire population and for the American lineage considered independently (Chi-square test, p < 0.001 and p = 0.017, respectively). However, when the European lineage is considered independently, there is a significant shift towards lower frequency alleles compared to the neutral expectation (Chi-square test p < 0.001) (Figure S3).
Table 3.
Nucleotide diversity in S. paradoxus.
| # of strains | π * 100 | |
|---|---|---|
| American | 30 | 0.167 |
| European | 28 | 0.074 |
| Far Eastern | 4 | 0.057 |
| Total | 63 | 1.413 |
| Within populations | 63 | 0.099 (7%) |
| Between populations | 63 | 1.314 (93%) |
Nucleotide diversity is π * 100, calculated using MEGA4.0 (Tamura et al., 2007) based on pairwise comparisons of nucleotide p-value.
Discussion
S. cerevisiae is characterized by a number of genetically distinct groups. One group includes vineyard strains and other strains of European origin. Another group includes oak-tree strains from North America. In this study we show that distinct wine and oak populations of S. cerevisiae, corresponding to these two groups, occur sympatrically within vineyards in North America. While oak and wine strains are present both on grapes and oak-trees in vineyards, wine stains are not established or do not persist in non-vineyard habitats. These two clades show major differences in population genetic parameters, indicating separate and distinct demographic histories. We provide evidence of genetic exchange between wine and oak yeast populations, documented by heterozygous hybrid wine/oak strains from cherry trees and admixed strains from vineyards. While our results do not exclude adaptive divergence between these two groups, they are consistent with a neutral model of divergence mediated by historical barriers to gene flow, particularly between North American and European populations. However, we also find that the population structure of oak S. cerevisiae strains is dominated by several clones that exhibit no geographical structure, in stark contrast to the geographical separation observed for populations of S. paradoxus isolated from the same sources.
As demonstrated here and in previous studies, S. cerevisiae and S. paradoxus show very different patterns of genetic diversity and population structure (Naumov et al., 1997; Johnson et al., 2004; Koufopanou et al., 2006; Liti et al., 2009). Of particular note is the correlation of genetic diversity with geographic distance observed in S. paradoxus, and the presence of genetic barriers between allopatrically diverged populations (Sniegowski et al., 2002). Similar to previous studies (Liti et al., 2009), the level of genetic diversity we observed within S. paradoxus was approximately 5 times greater than for S. cerevisiae. The pattern of genetic diversity observed in S. paradoxus is congruent with isolation between continents (i.e. North America, Europe, Asia), as previously reported (Johnson et al., 2004; Liti et al., 2009), but this study provides additional evidence demonstrating genetic differentiation in S. paradoxus associated with geographical distance within a continent, specifically North America. Saccharomyces paradoxus isolates from Missouri and Oregon formed well supported groups within North American isolates and there is some support for geographic substructure within Oregon as well.
We also observe a major difference between S. cerevisiae and S. paradoxus regarding the movement of genes between populations. Four of the S. paradoxus strains isolated from Missouri and Oregon were found to cluster with European S. paradoxus, suggesting migration of European isolates into the US. While we observe admixture between the European (wine) and North American (oak) genotypes of S. cerevisiae, we find no evidence for genetic exchange between European and American S. paradoxus genotypes. The migration of European S. paradoxus isolates and their genetic isolation from N. American strains has been observed previously in the North and Eastern US and Canada (Kuehne et al., 2007) and may be indicative of allopatric divergence leading to speciation. Indeed, hybrids between S. paradoxus strains from different geographical origins show a significant decrease in spore viability, indicating partial reproductive isolation (Sniegowski et al., 2002).
The presence of both European (wine) and North American (oak) genotypes of S. cerevisiae on grapes and oak-trees in the vineyard demonstrates that these two groups are sympatric and raises the possibility that mating and genetic exchange occur within vineyards. Previous work has shown that strains from diverse sources appear to be mosaics of other well-defined lineages (Liti et al., 2009). However, because of asexual reproduction it is difficult to know when or where admixed strains arose. We found most admixed strains in vineyards and little evidence for admixed strains from oak-trees outside of vineyards (two isolates with 3-6% vineyard background were identified only using InStruct). While it is possible that these admixed strains were derived from matings within the vineyard, where both genetic backgrounds were isolated, no hybrids were recovered within vineyards and the admixed strains carried only a small portion of genotypes from the other population. The two heterozygous hybrid strains from a cherry orchard in Wisconsin combined with two admixed oak-tree strains from Wisconsin raise the possibility that admixed strains found in the vineyard were migrants from other locations. Interestingly, strains isolated from orchards in China are in some cases most closely related to European/wine strains while in others are more closely related to North American oak-tree strains (Wang et al., 2012). The discovery of multiple arboreal populations in China, distinct from both North American oak-tree and wine populations, demonstrates that not all S. cerevisiae diversity is captured in existing genome data and suggests that other admixed or distinct populations may await discovery (Wagn et al., 2012). With only a limited repertoire of population genetic variation in North America, it is difficult to infer how recent or where any genetic exchange occurred between the oak-tree and vineyard lineages.
What can explain the historical divergence between the vineyard and oak-tree groups? While there are several potential scenarios that could contribute to the population structure we observed, two likely mechanisms include neutral demographic processes, such as recent migration of allopatrically diverged isolates, or selective forces such as postzygotic barriers to gene flow between locally adapted genotypes.
In regard to the neutral demographic scenario, a potential explanation for the presence of distinct vineyard and oak-tree clades is that i) these groups were established by the historical separation of European and North American populations and ii) the dispersal of European vineyard type strains with the establishment of vineyards around the world has been too recent for the observed subdivision between vineyard and any indigenous non-vineyard strains to erode. A number of our results are consistent with the neutral demographic model. The history of US winemaking is relatively recent; commercial vineyards have been established within the last 300 years, and the wineries sampled in this study were established between 150 (Mount Pleasant Winery) and 20 (Chaumette Vineyards) years ago.
Both vineyard and oak genotypes were isolated from vineyard grapes and vineyard oaks, yet only oak genotypes were isolated from non-vineyard locations. The presence of both genotypes in vineyards is most likely due to the migration of commercial wine genotypes out of winery facilities and onto grapes and adjacent oak-trees. The lack of wine genotypes isolated from non-vineyard locations may indicate that S. cerevisiae lacks sufficient dispersal ability to reach oak-trees outside of vineyards and/or that not enough time has passed for this dispersal to occur. The lack of geographic structure within oak strains suggests that either dispersal may not be a limiting factor in S. cerevisiae, or that dispersal events are temporally heterogeneous on a timeframe longer than the time since vineyard establishment in North America. It is also possible that migration ability has diverged between vineyard and oak populations and contributes to the differences in their distribution. Very little is known about the dispersal range and mechanism for the movement of S. cerevisiae strains under normal conditions, although it has been postulated they are primarily transported by insects, although recent work has shown that wasps (Stefanini et al., 2012) and bees (Goddard et al., 2010) may be important vectors within vineyards.
Adaptation and/or domestication could also explain divergence between vineyard and oak clades. Under this scenario vineyard strains have adapted to the vineyard environment, and have potentially become less fit in the oak-tree environment. It is also possible that humans have knowingly or unknowingly propagated yeasts that have desirable enological characteristics. Previous studies have shown differentiation between vineyard and oak strains in wine flavor and aroma (Hyma et al., 2011), freeze-thaw tolerance and other environmental stresses (Kvitek et al., 2008; Will et al., 2010), sporulation, which also indicates a partial loss of out-crossing in vineyard strains(Gerke et al., 2006), and copper and sulfite resistance (Park and Bakalinsky, 2000; Fay et al., 2004; Liti et al., 2009). Interestingly, oak-tree strains tend to grow better than vineyard strains in both grape and oak-tree simulated medium (Hyma, 2010). In either case, the ubiquitous presence of vineyard-type yeast in vineyards from around the world is almost certainly mediated by human-associated migration, which may not be available to strains in the oak-tree environment.
Another explanation for the restricted range of wine strains is that they are introduced seasonally and do not persist in the vineyard year round. Other studies have revealed that S. cerevisiae exists on grapes in high frequency only in the few weeks surrounding the grape harvest season (Valero et al., 2007), and that commercial wine making strains disseminate into the vineyard on a seasonal basis (Valero et al., 2005) which may limit the ability of wine genotypes to migrate to non-vineyard oaks. However, another study reported that commercial strains persist in the vineyard on a perennial basis (Schuller et al., 2005), and there is evidence that S. cerevisiae can colonize wine cellars (Versavaud et al., 1995; Blanco et al., 2011).
This study represents one of the first examinations of genome wide population level differentiation within Saccharomyces species in a single ecological context. Distinct wine and oak populations of S. cerevisiae are observed within vineyards and each population has unique differences in genetic variation and nucleotide diversity. We find evidence for genetic exchange between the populations which may suggest that local adaptation is not the primary driving force of genetic differentiation between the populations. However, wine genotypes are restricted to vineyard locations, which may be a result of neutral demographic processes or fitness differences; it remains to be seen whether gene flow between the populations results in individuals that are less fit. It is clear that S. paradoxus and S. cerevisiae, despite their similarities and isolation from sympatric arboreal habitats even the same substrates (Sniegowski et al., 2002; Sampaio and Gonçalves, 2008) have dramatically different population structure even in the same environment. Future studies of S. cerevisiae including increased global sampling, especially of European populations will be critical to assess the degree to which local adaptation or domestication is responsible for the presence of distinct populations of S. cerevisiae.
Supplementary Material
Figure 4.
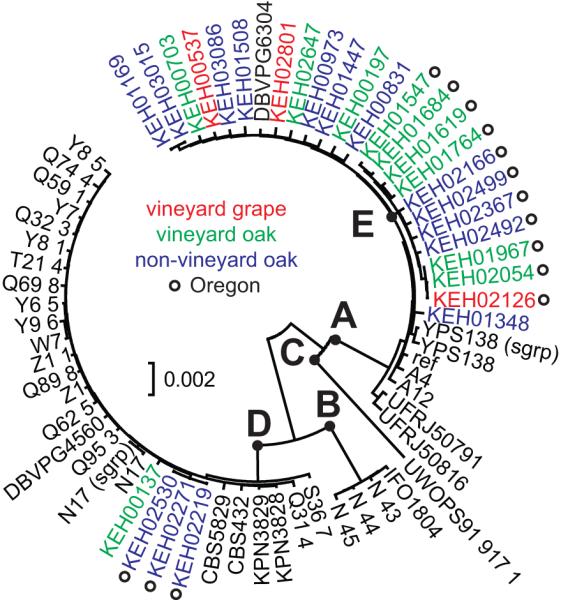
Evolutionary relationships of S. paradoxus strains.
The bootstrap consensus tree (1,000 replicates) of 66 taxa based on pairwise genetic distances (nucleotide substitutions per site) at 96,753 positions. All positions with missing and ambiguous data were removed. The tree is drawn to scale. Branches with bootstrap values of less than 50% have been collapsed. The tree is drawn to scale. Previously described groups (A) American, (B) Far Eastern (C) Hawaiian, and (D) European are noted. The node labeled “E” indicates the separation between Missouri and Oregon isolates from this study. Bootstrap support for nodes A, B, D and E is 100%. Bootstrap support could not be calculated for node C.
Acknowledgements
We would like to thank Jason Londo, Elizabeth Engle, Devjanee Swain, Vitas Wagner, Juyoung Huh and Maia Dorsett for assistance with collections, Kim Lorenz and Barak Cohen for sharing protocols and reagents, the Washington University Genomic Technology Access Center and the Center for Genome Sciences for sequencing services and support, along with all of the property owners and managers who made the collections possible: Lidia Watrud, Mike Bollman, Hank Johnson of Chaumette Vineyard (Ste. Genevieve, MO), Tony Saballa of Charleville Vineyard (Ste. Genevieve, MO), Mark Baehman of Mt. Pleasant Vineyard (Augusta, MO), Tom and Celeste Symonette of Whistling Dog Cellars (Polk Co., OR), and Marilee Buchanan of Tyee Vineyards (Benton Co, OR). This work was supported in part by a NIH Genome Analaysis Training Grant to KEH and a NIH grant to JCF (GM080669).
Footnotes
Data Accessibility:
Raw data (fastq files per individual) and fasta sequence alignment for S. cerevisiae and S. paradoxus, S. paradoxus assembly, and scripts used to generate sequence alignments: Dryad entry doi:10.5061/dryad.58745.
References
- Aa E, Townsend JP, Adams RI, Nielsen KM, Taylor JW. Population structure and gene evolution in Saccharomyces cerevisiae. FEMS Yeast Research. 2006;6:702–715. doi: 10.1111/j.1567-1364.2006.00059.x. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, Selker EU, Cresko WA, Johnson EA. Rapid SNP Discovery and Genetic Mapping Using Sequenced RAD Markers. PLoS ONE. 2008;3:e3376. doi: 10.1371/journal.pone.0003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco P, Orriols I, Losada A. Survival of commercial yeasts in the winery environment and their prevalence during spontaneous fermentations. J Ind Microbiol Biotechnol. 2011;1:235–239. doi: 10.1007/s10295-010-0818-2. [DOI] [PubMed] [Google Scholar]
- Diezmann S, Dietrich FS. Saccharomyces cerevisiae: Population divergence and resistance to oxidative stress in clinical, domesticated and wild isolates. PLoS ONE. 2009;4:e5317. doi: 10.1371/journal.pone.0005317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay JC, Benavides JA. Evidence for domesticated and wild populations of Saccharomyces cerevisiae. PLoS Genet. 2005;1:66–71. doi: 10.1371/journal.pgen.0010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay JC, McCullough HL, Sniegowski PD, Eisen MB. Population genetic variation in gene expression is associated with phenotypic variation in Saccharomyces cerevisiae. Genome Biol. 2004;5:R26–R26. doi: 10.1186/gb-2004-5-4-r26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H, Williamson S, Bustamante CD. An MCMC Approach for Joint Inference of Population Structure and Inbreeding rates from Multi-Locus Genotype Data. Genetics. 2007;176:1635–1651. doi: 10.1534/genetics.107.072371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerke JP, Chen CTL, Cohen BA. Natural Isolates of Saccharomyces cerevisiae display complex genetic variation in sporulation efficiency. Genetics. 2006;174:985–997. doi: 10.1534/genetics.106.058453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard MR, Anfang N, Tang R, Gardner RC, Jun C. A distinct population of Saccharomyces cerevisiae in New Zealand: evidence for local dispersal by insects and human-aided global dispersal in oak barrels. Environmental Microbiology. 2010;12:63–73. doi: 10.1111/j.1462-2920.2009.02035.x. [DOI] [PubMed] [Google Scholar]
- Hyma KE. PhD. Dissertation. Washington University; Saint Louis, Mo: 2010. Genetic and phenotypic differentiation between winemaking and wild strains of Saccharomyces cerevisiae. [Google Scholar]
- Hyma KE, Saerens SM, Verstrepen KJ, Fay JC. Divergence in wine characteristics produced by wild and domesticated strains of Saccharomyces cerevisiae. FEMS Yeast Research. 2011;11:540–551. doi: 10.1111/j.1567-1364.2011.00746.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsson M, Rosenberg NA. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 2007;23:1801–1806. doi: 10.1093/bioinformatics/btm233. [DOI] [PubMed] [Google Scholar]
- Johnson LJ, Koufopanou V, Goddard MR, Hetherington R, Schafer SM, Burt A. Population genetics of the wild yeast Saccharomyces paradoxus. Genetics. 2004;166:43–52. doi: 10.1534/genetics.166.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koufopanou V, Hughes J, Bell G, Burt A. The spatial scale of genetic differentiation in a model organism: the wild yeast Saccharomyces paradoxus. Philosophical Transactions of the Royal Society B: Biological Sciences. 2006;361:1941–1946. doi: 10.1098/rstb.2006.1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehne HA, Murphy HA, Francis CA, Sniegowski PD. Allopatric divergence, secondary contact, and genetic isolation in wild yeast populations. Curr. Biol. 2007;17:407–411. doi: 10.1016/j.cub.2006.12.047. [DOI] [PubMed] [Google Scholar]
- Kvitek DJ, Will JL, Gasch AP. Variations in stress sensitivity and genomic expression in diverse S. cerevisiae isolates. PLoS Genet. 2008;4:e1000223. doi: 10.1371/journal.pgen.1000223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legras JL, Merdinoglu D, Cornuet JM, Karst F. Bread, beer and wine: Saccharomyces cerevisiae diversity reflects human history. Molecular Ecology. 2007;16:2091–2102. doi: 10.1111/j.1365-294X.2007.03266.x. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libkind D, Hittinger CT, Valério E, Gonçalves C, Dover J, Johnston M, Gonçalves P, Sampaio JP. Microbe domestication and the identification of the wild genetic stock of lager-brewing yeast. Proceedings of the National Academy of Sciences. 2011;108:14539–14544. doi: 10.1073/pnas.1105430108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liti G, Carter DM, Moses AM, Warringer J, Parts L, James SA, Davey RP, Roberts IN, Burt A, Koufopanou V, et al. Population genomics of domestic and wild yeasts. Nature. 2009;458:337–341. doi: 10.1038/nature07743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Tang T, Tang H, Huang J, Shi S, Wu C-I. The accumulation of deleterious mutations in rice genomes: a hypothesis on the cost of domestication. Trends in Genetics. 2006;22:126–131. doi: 10.1016/j.tig.2006.01.004. [DOI] [PubMed] [Google Scholar]
- McCullough MJ, Clemons KV, McCusker JH, Stevens DA. Intergenic transcribed spacer PCR ribotyping for differentiation of Saccharomyces species and interspecific hybrids. J. Clin. Microbiol. 1998;36:1035–1038. doi: 10.1128/jcm.36.4.1035-1038.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortimer R, Polsinelli M. On the origins of wine yeast. Research in Microbiology. 1999;150:199–204. doi: 10.1016/s0923-2508(99)80036-9. [DOI] [PubMed] [Google Scholar]
- Nardi T, Carlot M, Bortoli ED, Corich V, Giacomini A. A rapid method for differentiating Saccharomyces sensu stricto strains from other yeast species in an enological environment. FEMS Microbiology Letters. 2006;264:168–173. doi: 10.1111/j.1574-6968.2006.00450.x. [DOI] [PubMed] [Google Scholar]
- Naumov GI, Naumova ES, Sniegowski PD. Differentiation of European and Far East Asian populations of Saccharomyces paradoxus by allozyme Analysis. Int J Syst Bacteriol. 1997;47:341–344. doi: 10.1099/00207713-47-2-341. [DOI] [PubMed] [Google Scholar]
- Naumov GI, Naumova ES, Sniegowski PD. Saccharomyces paradoxus and Saccharomyces cerevisiae are associated with exudates of North American oaks. Canadian Journal of Microbiology. 1998;44:1045–1050. [PubMed] [Google Scholar]
- Park H, Bakalinsky AT. SSU1 mediates sulphite efflux in Saccharomyces cerevisiae. Yeast. 2000;16:881–888. doi: 10.1002/1097-0061(200007)16:10<881::AID-YEA576>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg NA. distruct: a program for the graphical display of population structure. Molecular Ecology Notes. 2003;4:137–138. [Google Scholar]
- Sampaio JP, Gonçalves P. Natural populations of Saccharomyces kudriavzevii in portugal are associated with oak bark and are sympatric with S. cerevisiae and S. paradoxus. Appl Environ Microbiol. 2008;74:2144–2152. doi: 10.1128/AEM.02396-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schacherer J, Shapiro JA, Ruderfer DM, Kruglyak L. Comprehensive polymorphism survey elucidates population structure of Saccharomyces cerevisiae. Nature. 2009;458:342–345. doi: 10.1038/nature07670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuller D, Alves H, Dequin S, Casal M. Ecological survey of Saccharomyces cerevisiae strains from vineyards in the Vinho Verde Region of Portugal. FEMS Microbiology Ecology. 2005;51:167–177. doi: 10.1016/j.femsec.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Sicard D, Legras J-L. Bread, beer and wine: Yeast domestication in the Saccharomyces sensu stricto complex. C. R. Biol. 2011;334:229–236. doi: 10.1016/j.crvi.2010.12.016. [DOI] [PubMed] [Google Scholar]
- Sniegowski PD, Dombrowski PG, Fingerman E. Saccharomyces cerevisiae and Saccharomyces paradoxus coexist in a natural woodland site in North America and display different levels of reproductive isolation from European conspecifics. FEMS Yeast Res. 2002;1:299–306. doi: 10.1111/j.1567-1364.2002.tb00048.x. [DOI] [PubMed] [Google Scholar]
- Spor A, Nidelet T, Simon J, Bourgais A, de Vienne D, Sicard D. Niche-driven evolution of metabolic and life-history strategies in natural and domesticated populations of Saccharomyces cerevisiae. BMC Evolutionary Biology. 2009;9:296. doi: 10.1186/1471-2148-9-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanini I, Dapporto L, Legras J-L, Calabretta A, Paola MD, Filippo CD, Viola R, Capretti P, Polsinelli M, Turillazzi S, et al. Role of social wasps in Saccharomyces cerevisiae ecology and evolution. PNAS. 2012;109:13398–13403. doi: 10.1073/pnas.1208362109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Valero E, Cambon B, Schuller D, Casal M, Dequin S. Biodiversity of Saccharomyces yeast strains from grape berries of wine-producing areas using starter commercial yeasts. FEMS Yeast Research. 2007;7:317–329. doi: 10.1111/j.1567-1364.2006.00161.x. [DOI] [PubMed] [Google Scholar]
- Valero E, Schuller D, Cambon B, Casal M, Dequin S. Dissemination and survival of commercial wine yeast in the vineyard: a large-scale, three-years study. FEMS Yeast Res. 2005;5:959–969. doi: 10.1016/j.femsyr.2005.04.007. [DOI] [PubMed] [Google Scholar]
- Versavaud A, Courcoux P, Roulland C, Dulau L, Hallet JN. Genetic diversity and geographical distribution of wild Saccharomyces cerevisiae strains from the wine-producing area of Charentes, France. Applied and Environmental Microbiology. 1995;61:3521–3529. doi: 10.1128/aem.61.10.3521-3529.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QM, Liu WQ, Liti G, Wang SA, Bai FY. Surprisingly diverged populations of Saccharomyces cerevisiae in natural environments remote from human activity. Mol Ecol. 2012 doi: 10.1111/j.1365-294X.2012.05732.x. (in press) [DOI] [PubMed] [Google Scholar]
- Watterson GA. On the number of segregating sites in genetical models without recombination. Theor Popul Biol. 1975;7:256–276. doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- Will JL, Kim HS, Clarke J, Painter JC, Fay JC, Gasch AP. Incipient balancing selection through adaptive loss of aquaporins in natural Saccharomyces cerevisiae populations. PLoS Genet. 2010;6:e1000893. doi: 10.1371/journal.pgen.1000893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Skelton A, Gardner RC, Goddard MR. S. paradoxus and S. cerevisiae reside on oak trees in New Zealand: evidence for migration from Europe and inter-species hybrids. FEMS Yeast Research. 2010;7:941–947. doi: 10.1111/j.1567-1364.2010.00681.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.