

Abstract
Targeted therapeutics have emerged in recent years as an attractive approach to treating various types of cancer. One approach is to modify a cytocidal protein toxin to direct its action to a specific population of cancer cells. We created a targeted toxin in which the receptor‐binding and pore‐forming moiety of anthrax toxin, termed Protective Antigen (PA), was modified to redirect its receptor specificity to HER2, a marker expressed at the surface of a significant fraction of breast and ovarian tumors. The resulting fusion protein (mPA‐ZHER2) delivered cytocidal effectors specifically into HER2‐positive tumor cells, including a trastuzumab‐resistant line, causing death of the cells. No off‐target killing of HER2‐negative cells was observed, either with homogeneous populations or with mixtures of HER2‐positive and HER2‐negative cells. A mixture of mPA variants targeting different receptors mediated killing of cells bearing either receptor, without affecting cells devoid of these receptors. Anthrax toxin may serve as an effective platform for developing therapeutics to ablate cells bearing HER2 or other tumor‐specific cell‐surface markers.
Keywords: Anthrax toxin, HER2/neu, Binary toxin, Affibody, Immunotoxin
Highlights
-
►
We created a protective antigen fusion that targets the HER2 receptor (mPA‐ZHER2).
-
►
mPA‐ZHER2 delivered two cytocidal effectors into the cytosol of HER2‐positive cells.
-
►
Killing was specific for HER2‐positive cells in homogeneous or mixed cell populations.
-
►
A trastuzumab‐resistant tumor cell line was killed by the HER2‐targeted toxin.
-
►
Redirected mPA variants can be combined to eliminate diverse tumor cell populations.
1. Introduction
Amplification and/or overexpression of the HER2 gene at the mRNA or protein level occurs in 20–25% of breast, gastric, and ovarian carcinomas (Berchuck et al., 1990; Gravalos and Jimeno, 2008; Arteaga et al., 2012; Slamon et al., 1989). Particularly in breast cancer, increased expression of HER2 is associated with an aggressive form of the disease, which shows signs of increased tumor growth, recurrence, and resistance to therapy, all contributing to decreased patient survival (Arteaga et al., 2012). Although the FDA‐approved monoclonal antibody, trastuzumab (trade name, Herceptin®), is effective at slowing tumor growth, it remains ineffective at tumor elimination. New therapeutics that actively kill tumor cells thus remain a major goal of cancer‐related research. A promising example of this strategy is to target the action of cytocidal protein toxins to specific cancer cells (Pastan et al., 2007).
Recently, we developed a straightforward way to redirect the receptor specificity of anthrax toxin (Mechaly et al., 2012). First we ablated the native receptor‐binding activity of protective antigen (PA), the receptor‐binding/pore‐forming component of anthrax toxin, and then appended a heterologous, receptor‐binding ligand to the C terminus of the mutated protein (mPA). Using this approach we created fusion proteins that direct toxin action specifically to two different receptors: the diphtheria toxin (DT) receptor (HB‐EGF) and the epidermal growth factor receptor (EGFR) (Mechaly et al., 2012). In the current study we used this approach to redirect toxin action to cells bearing the HER2 receptor.
Anthrax toxin is an ensemble of three nontoxic, monomeric proteins (Young and Collier, 2007). Two of them, the Lethal Factor and the Edema Factor (LF and EF), are enzymatic “effector proteins,” which covalently modify molecular targets within the cytosol. LF is a metalloprotease, which inactivates most members of the mitogen‐activated protein kinase kinase (MEK) family (Duesbery et al., 1998; Vitale et al., 1998), and EF is a calmodulin‐ and Ca2+‐dependent adenylate cyclase, which increases the intracellular concentration of cyclic AMP (Leppla, 1982). The third protein, PA, transports LF and EF from the extracellular milieu to the cytosol by a process that begins with its binding to specific cell‐surface receptors and culminates in its forming pores in the endosomal membrane (Collier, 2009).
After binding to either of its two known receptors — ANTXR1 (also called TEM8) and ANTXR2 (also called CMG2) (Scobie, 2003; Bradley et al., 2001) — PA is proteolytically activated by a furin‐family protease (Klimpel et al., 1992). The activated form self‐assembles into heptameric (Milne et al., 1994) or octameric (Kintzer et al., 2009) ring‐shaped oligomers (pore precursors, or “prepores”), which bind effector proteins with high (nM) affinity (Cunningham et al., 2002; Mogridge et al., 2002). The resulting complexes are endocytosed and delivered to the endosomal compartment, where the acidic pH causes a conformation change in the prepores that enables them to form pores in the endosomal membrane (Miller et al., 1999). The pores, in turn, actively unfold the bound effector proteins and transport them across the membrane to the cytosol (Young and Collier, 2007). There they refold into active enzymes and modify their cytosolic substrates, causing major perturbations of cellular processes and, in some cases, cell death.
HER2 is a receptor tyrosine kinase belonging to the same family as EGFR. Unlike EGFR, however, HER2 has no known natural ligand. In the present study we developed a redirected binary toxin by fusing a high‐affinity Affibody specific for the HER2 receptor (ZHER2:342) (Orlova et al., 2006) to the C terminus of receptor recognition‐deficient PA (mPA), creating the fusion mPA‐ZHER2. Affibodies represent a class of targeting polypeptides derived from the Z domain of Staphylococcus aureus protein A. Advantages over other receptor‐targeting ligands derive from the fact that Affibodies are small (58 amino acids; ∼6 kDa), pH‐ and thermo‐stable, lack Cys residues, and fold independently and reversibly (Nord et al., 1997; Löfblom et al., 2010). Further, they may be rapidly evolved in vitro by phage‐display technologies to affinity levels comparable to those observed with monoclonal antibodies.
Our results show that mPA with the ZHER2:342 Affibody fused to the C terminus can direct the action of either of two cytocidal effector proteins to HER2‐positive tumor cells. These cells, including a HER2‐positive trastuzumab‐resistant tumor cell line, were ablated, and specific killing was observed regardless of whether the cultures consisted of a homogeneous population or had been mixed with cells lacking the HER2 marker.
2. Material and methods
2.1. Reagents and chemicals
Oligonucleotides and the ZHER2:342 gene were synthesized by Integrated DNA Technologies (Coralville, IA). The ZHER2:4 and ZHER2:342 expression plasmids were kindly provided by Dr. Gregory Poon (Washington State University, Pullman, WA). All chemicals were purchased from Sigma–Aldrich (St. Louis, MO), unless noted otherwise.
2.2. Generation of LFN‐RTA expression plasmid
The A chain of ricin (RTA) was fused to the C terminus of the N‐terminal PA‐binding domain of LF (LFN) by overlap extension PCR and cloned into the pet‐SUMO expression vector (Invitrogen, Carlsbad, CA). The first PCR step consisted of two reactions (i) using a forward primer for LFN (LFN FOR – GCGGGCGGTCATGGTGATGTAGGT) and a reverse primer for LFN containing a GS spacer (in bold) and an overlap sequence for RTA (underlined) (LFN‐RTAREV – AATTGGGTATTGTTTGGGGAATATACTACCCCGTTGATCTTGAAGTTCTTCCAA), and (ii) using a forward primer for RTA with a GS spacer (bold) and a 5′ overlap region with LFN (underlined) (LFN‐RTAFOR – TTGGAAGAACTTAAAGATCAACGGGGTAGTATATTCCCCAAACAATACCCAATT) and a reverse primer for RTA encoding a double stop codon (in bold) (RTAREV – CTATTAAAACTGTGACGATGGTGGAGGTGC). A final PCR reaction using the two previous templates was performed with primers LFN FOR and RTAREV to combine the two PCR products, which was subsequently ligated into the pet‐SUMO expression vector (Invitrogen).
2.3. Protein expression and purification
Recombinant WT PA, mPA, mPA‐ZHER2, and mPA‐EGF were expressed and purified as described (Miller et al., 1999; Mechaly et al., 2012). Recombinant LFN‐DTA and LFN‐RTA were expressed as hexahistidine‐SUMO fusions for 4 h at 30 °C under the induction of 1 mM Isopropyl β‐D‐1‐thiogalactopyranoside (IPTG) in the BL21 (DE3) Star strain of Escherichia coli (Invitrogen). Cell pellets were suspended in 100 ml of lysis buffer (20 mM Tris–HCl pH 8.0, 150 mM NaCl, 10 mM imidazole, 10 mg lysozyme, 2 mg DNAse I, supplemented with a Roche complete protease inhibitor tablet per 50 ml) and lysed by sonication. Cell lysates were loaded onto a Ni2+‐NTA agarose column, washed with 100 ml of wash buffer (20 mM Tris–HCl pH 8.0, 150 mM NaCl, and 20 mM imidazole), and eluted with wash buffer supplemented with 250 mM imidazole. The resulting purified protein was exchanged into 20 mM Tris–HCl pH 8.0 and 150 mM NaCl and cleaved with SUMO protease overnight at 4 °C to separate the LFN‐DTA/RTA from the His6‐SUMO protein. Cleaved proteins were then subjected to a second Ni2+‐NTA column to bind His6‐SUMO, leaving the protein of interest (LFN‐DTA/RTA) in the flow‐thru fraction.
Affibodies (ZHER2:4 and ZHER2:342) were expressed from the pet15b expression vector (EMD Millipore, Billerica, MA) and purified in the same manner as LFN‐DTA, without the need for a cleavage step.
2.4. Cell lines and maintenance
The A431 (cat no. CCL‐1555) and CHO‐K1 (cat. no. CCL‐61) cell lines were purchased from ATCC (Manassas, VA). BT‐474, MDA‐MB‐468, and SKBR3 cell lines were generously provided by Dr. Jean Zhao (Dana Farber Cancer Institute, Boston, MA). The MDA‐MB‐231 cell line was provided by Dr. Gregory Poon (Washington State University). The JIMT‐1 cell line was purchased from AddexBio (cat. no. C0006005; San Diego, CA).
A431 and JIMT‐1 cells were maintained in DMEM supplemented with 10% FCS, 500 units/ml penicillin G and 500 units/ml streptomycin sulfate (Invitrogen). CHO‐K1 and all other cell lines were grown in Ham's F12 or RPMI medium (Invitrogen), respectively, supplemented with 10% FCS, 500 units/ml penicillin G and 500 units/ml streptomycin sulfate.
Stable cell lines expressing fluorescent proteins were produced by puromycin‐selectable lentiviral particles coding for CFP, RFP, or GFP (GenTarget, San Diego, CA). Lentiviruses were transduced (MOI = 1) into A431 (CFP), SKBR3 (RFP), and MDA‐MB‐468 (GFP) cell lines. At 48 h post‐transduction, the medium was replaced with medium containing 1 μg/ml puromycin to select for fluorescent cells that were puromycin resistant. Cells were passaged three more times in medium containing 1–5 μg/ml puromycin and analyzed by fluorescence‐activated cell sorting (FACS) to ensure a homogenous, fluorescently‐labeled population of cells were selected.
2.5. Quantifying surface HER2 and EGF receptor levels
Cells (1 × 105/experiment) were dissociated using a non‐enzymatic reagent (Cellstripper™, Cellgro, Herndon, VA) to eliminate the potential for receptor cleavage. Cells were re‐suspended in either 200 μl of PBS or PBS with 1 μg/ml FITC‐labeled anti‐EGFR (cat. no. ab81872; Abcam, Cambridge, MA) or 2 μg/ml FITC‐labeled anti‐HER2 (cat. no. ab31891; Abcam) Affibodies. Cells were incubated for 1‐h at 4 °C, washed twice with 200 μl of PBS, and re‐suspended in PBS. FACS was performed using a BD FACSCalibur flow cytometer. FACS histograms were analyzed using the FlowJo flow cytometry analysis software (Tree Star Inc., Ashland, OR), while mean fluorescence intensity (MFI) was plotted using the GraphPad Prism® software package (GraphPad software Inc., La Jolla, CA). Each plot corresponds to three experiments where 50,000 events/condition were counted.
2.6. Cytotoxicity and competition assays
2.6.1. Protein synthesis inhibition
Cells were plated in appropriate medium at densities of 2.5 × 104 (BT‐474) or 3.5 × 104 cells/well (all other cell lines) in 96‐well plates and incubated overnight at 37 °C. The following day, cells were exposed to ten 10‐fold serial dilutions of LFN‐DTA or LFN‐RTA (starting with a final concentration of 1 μM) in medium supplemented with 20 nM mPA variant. After a 4‐h incubation, toxin‐containing medium was removed and replaced with leucine‐deficient medium supplemented with 1 μCi of [3H]‐leucine/ml (PerkinElmer, Billerica, Massachusetts) and incubated for an additional hour. Plates were washed twice with cold PBS (200 μl) prior to the addition of 200 μl of scintillation fluid. The amount of [3H]‐leucine incorporated was determined by scintillation counting using a Wallac MicroBeta TriLux 1450 LSC (PerkinElmer, Waltham, MA). Percent protein synthesis was normalized against cells treated with the mPA variant alone and was plotted versus the concentration of LFN‐DTA or LFN‐RTA in GraphPad Prism, where each point on the curve corresponds to the average of four experiments.
Competition assays were performed as described above with increasing concentrations of free (i) high‐affinity (ZHER2:342) or (ii) lower‐affinity (ZHER2:4) Affibody added to medium containing 20 nM mPA‐ZHER2 and LFN‐DTA. MDA‐MB‐231 cells which express low levels of HER2 had to be challenged with a higher concentration of LFN‐DTA (1 μM), compared to all other cell lines (10 nM). Percent protein synthesis was normalized against cells treated with mPA‐ZHER2 alone and plotted using GraphPad Prism, where each point on the curve corresponds to the average of four experiments.
2.6.2. Cell viability
Cell viability was measured by an XTT assay, following the manufacturers protocol (Biotium, Hayward, CA). Cells (104/well) were plated in the appropriate medium in 96‐well optical bottom plates, incubated overnight at 37 °C, and exposed to ten 10‐fold serial dilutions of LFN‐DTA in medium supplemented with 20 nM mPA‐ZHER2. After 48 or 72 h, 25 μl of XTT (sodium 2,3‐bis(2‐methoxy‐4‐nitro‐5‐sulfophenyl)‐5‐[(phenylamino)‐carbonyl]‐2H‐tetrazolium inner salt) reagent was added to each well, and the absorbance of reduced XTT was measured at 475 nm, using a SpectraMax M2e microplate reader (Molecular Devices, Sunnyvale, CA). Percent cell viability was normalized against cells treated with mPA‐ZHER2 alone and plotted in GraphPad Prism, versus the concentration of LFN‐DTA. Each data point corresponds to the average of measurements performed in quadruplicate.
2.7. Apoptosis assay
A cell‐based apoptosis assay measuring the activation of known apoptotic markers, caspase 3/7, was performed according the supplier's protocol (Caspase‐Glo 3/7 Assay; Promega, Madison, WI). Cells (104/well) were seeded in 96‐well optical bottom plates and exposed to eight 10‐fold serial dilutions of LFN‐DTA in medium supplemented with 20 nM mPA‐ZHER2. After 24 or 48 h, a proluminescent caspase 3/7 substrate was added to each well, followed by incubation at room temperature for 30 min. Luminescence resulting from substrate cleavage by caspase 3/7 was measured with a Wallac MicroBeta TriLux 1450 LSC (PerkinElmer). Relative luminescence was plotted versus the concentration of LFN‐DTA using GraphPad Prism, where each data point represents the average of four independent measurements.
2.8. Microscopy
Fluorescent cells were mixed (2 × 104 cells each) as described above and grown on tissue culture treated coverslips overnight at 37 °C. Coverslips were exposed to 10 nM LFN‐DTA and mPA, mPA‐ZHER2, mPA‐EGF, or mPA‐ZHER2 and mPA‐EGF (20 nM each). After 24 h, cells were washed twice with PBS, fixed with 4% formaldehyde, and mounted on glass slides. Images were taken with a Nikon Eclipse TE2000‐U fluorescence inverted microscope and analyzed using the MetaMorph software package (Molecular Devices, Sunnyvale, CA).
2.9. Co‐culture cytotoxicity assay
2.9.1. Fluorescence
Fluorescent cell lines (A431CFP, MDA‐MB‐468GFP, and SKBR3RFP) were mixed equally (105 cells each) with unlabeled CHO‐K1 cells, seeded into 6‐well tissue culture dishes in RPMI medium, and incubated overnight at 37 °C. The next day, cells were treated with 10 nM LFN‐DTA and either mPA, mPA‐ZHER2, mPA‐EGF, or mPA‐ZHER2 and mPA‐EGF (20 nM each). Cells were incubated an additional 24 h, washed 2 times with PBS, and detached with trypsin. Cell populations were washed again in PBS and sorted based on fluorescence using a BD FACSCalibur flow cytometer (BD Biosciences, San Jose, CA). Each bar on the graphs corresponds to three experiments where at least 75,000 events were counted. FACS data was analyzed using the FlowJo analysis software and plotted using the GraphPad Prism® software package.
2.9.2. Protein synthesis
A panel of cancer cell lines and CHO‐K1 cells were seeded (3.5 × 104 cells/well) in partitioned sections of a chambered tissue culture slide. After an overnight incubation, the medium was removed, and the partitioning element was discarded. The slides were washed twice with PBS and incubated for 4 h with RPMI containing 10 nM LFN‐DTA with 20 nM of either (i) mPA, (ii) mPA‐ZHER2, (iii) mPA‐EGF, or (iv) a mixture of both mPA variants. Slides were removed from the toxin‐containing medium, washed with 15 ml of PBS, and incubated for an additional hour in leucine‐deficient medium supplemented with 1 μCi of [3H]‐leucine/ml (PerkinElmer). Slides were removed from the medium, washed with 30 ml of PBS, and dried. Individual cell populations were dissolved in 6 M Guanidine–HCl (75 μl) and added to scintillation fluid. The amount of [3H]‐leucine incorporated was determined by scintillation counting. The percent of protein synthesis was normalized against cells treated with mPA and LFN‐DTA and plotted using the GraphPad Prism software package.
3. Results
3.1. mPA‐ZHER2 mediates the killing of HER2‐positive cells
We fused a high‐affinity, 58‐residue Affibody, ZHER2:342, to the C terminus of mPA, a mutated, receptor recognition‐deficient form of PA. The resulting fusion protein (mPA‐ZHER2) was tested in combination with the LFN‐DTA effector protein for ability to kill cancer cell lines displaying various levels of the HER2 receptor. Because LF and EF are not cytocidal for most cells, we used LFN‐DTA, a fusion of the N‐terminal PA‐binding domain of LF (LFN) to the catalytic domain of diphtheria toxin (DTA), as intracellular effector. DTA ADP‐ribosylates eukaryotic elongation factor 2 (eEF‐2) in the cytosol, blocking protein synthesis and causing cell death (Collier and Cole, 1969; Collier, 1967; Honjo et al., 1968).
Various cell lines were incubated 4 h with a constant concentration of mPA‐ZHER2 (20 nM) plus various concentrations of LFN‐DTA, after which protein synthesis over a 1‐h period was measured. The BT‐474 cell line, which expressed the highest level of HER2 among the cell lines tested, was also the most sensitive; that is, it required the lowest concentration (EC50) of LFN‐DTA for 50% inhibition of protein synthesis (Figure 1A). Two cell lines expressing moderate levels of HER2 (SKBR3 and A431) showed intermediate levels of sensitivity; a line with a low level of HER2 (MDA‐MB‐231) showed low sensitivity (EC50 ∼10 nM); and two lines with no detectable HER2 (CHO‐K1, MDA‐MB‐468) were unaffected, even at the highest concentrations of LFN‐DTA tested. Thus, EC50 was inversely related to the level of HER2 on the cell surface (Figure 1B). Levels of HER2 on the various cell lines were determined by FACS analysis after incubation with a fluorescently‐labeled anti‐HER2 Affibody (SFigure 1).
Figure 1.
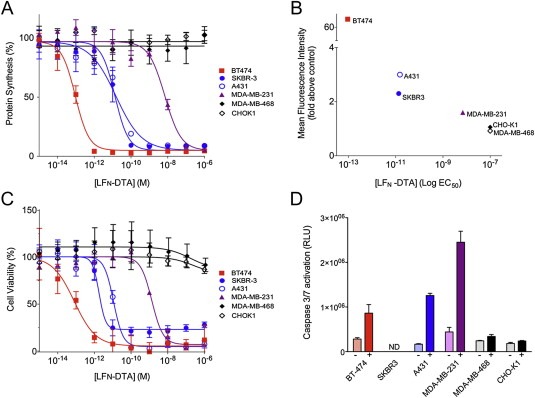
HER2‐dependent killing of cell lines by mPA‐ZHER2 plus LFN‐DTA. (A) Cells were incubated with a fixed concentration of mPA‐ZHER2 (20 nM) plus various concentrations of LFN‐DTA for 4 h and then with medium containing [3H]‐leucine for 1 h. Protein synthesis was measured by scintillation counting and normalized against cells treated with mPA‐ZHER2 alone. (B) HER2 receptor levels were determined by flow cytometry with an FITC‐labeled anti‐HER2 Affibody. Mean fluorescence intensity was calculated using the FloJo software package and plotted versus the logEC50 for [LFN‐DTA]. (C) Cells were exposed to the same conditions as panel A. After 48 h, cell viability was measured by XTT cytotoxicity assay and normalized against cells treated with mPA‐ZHER2 alone. (D) Apoptosis was assessed after exposing cells to either mPA‐ZHER2 alone (−; light bars) or mPA‐ZHER2 plus 10 nM LFN‐DTA (+; dark bars) for 24 h and measuring caspase 3/7 activation. Values corresponding to relative light units (RLU), generated by caspase 3/7 cleavage of a pre‐luminescent substrate, were extracted from dose—response curves presented in SFigure 2 (ND = not determined). In all panels, cell lines with high, moderate, low, and no detectable HER2 receptor levels are colored red, blue, purple, and black, respectively. Each point on the graphs represents the average of four experiments.
Cell viability confirmed that inhibition of protein synthesis by LFN‐DTA caused cell death. Cancer cell lines were exposed to mPA‐ZHER2 (20 nM) and LFN‐DTA, at the indicated concentrations. After 48 h, cell viability was quantified by a cytotoxicity assay that quantifies the reduction of XTT reagent by mitochondrial enzymes that are active in live cells. Protein synthesis inhibition and cell death directly correlated (compare Figures 1A and C), with comparable EC50 values that reflect the amount of HER2 present on the cell surface (Table 1; Figure 1B). Activation of known apoptotic markers, caspase 3/7, confirmed that cell death resulted from apoptosis (Figure 1D; SFigure 2). Caspase 3/7 activation did not increase after 24 h (data not shown) and was dose‐dependent; cells expressing higher amounts of HER2 receptor showed caspase 3/7 activation at a lower LFN‐DTA concentration (SFigure 2). The level of caspase 3/7 activation differed among various cell types and could not be confirmed for the SKBR3 cell line.
Table 1.
In vitro activity of mPA‐ZHER2 and LFN‐DTA against various cancer cell lines.
| Assay | Cell line EC50 (M) | ||||||
|---|---|---|---|---|---|---|---|
| BT‐474 | JIMT‐1 | SKBR‐3 | A431 | MDA‐MB‐231 | MDA‐MB‐468 | CHO‐K1 | |
| Protein synthesis inhibitiona | 9.4 × 10−14 | 3.0 × 10−12 | 1.3 × 10−11 | 1.5 × 10−11 | 7.0 × 10−9 | >1 × 10−6 | >1 × 10−6 |
| Cell viabilityb | 8.0 × 10−14 | 2.5 × 10−12 | 1.6 × 10−12 | 4.1 × 10−11 | 1.3 × 10−9 | >1 × 10−6 | >1 × 10−6 |
| Apoptosisc | 7.2 × 10−13 | 5.1 × 10−11 | NDd | 1.6 × 10−11 | 1.1 × 10−9 | >1 × 10−6 | >1 × 10−6 |
Measured by [3H]‐leucine incorporation after 4 h toxin exposure.
Measured by XTT cell viability assay after 48 h toxin exposure.
Measured by caspase 3/7 activation after 24 h toxin exposure.
Not determined.
Free ZHER2:342 Affibody competitively inhibited mPA‐ZHER2‐dependent killing of HER2‐positive cells (Figure 2). BT‐474 cells expressing high levels of HER2 required a higher level of free Affibody (EC50 ∼400 nM) for toxin blockage relative to cell lines expressing low or moderate levels of HER2 (EC50 ∼20 nM) (Figure 2A). A lower‐affinity Affibody (ZHER2:4) (Wikman et al., 2004) was less effective in blocking toxin action than the higher‐affinity ZHER2:342 Affibody (Figure 2B).
Figure 2.
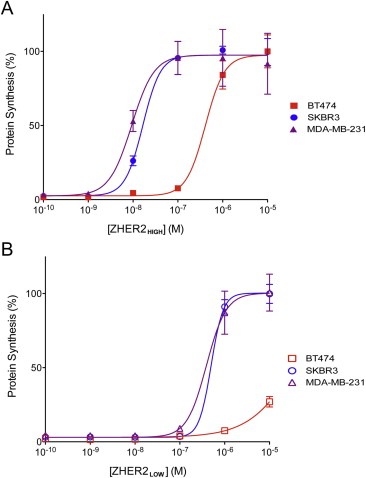
Competition by high‐ and low‐affinity ZHER2 Affibodies for mPA‐ZHER2‐dependent killing. Cells were exposed to a lethal dose of mPA‐ZHER2 and LFN‐DTA in the presence of increasing amounts of a high (ZHER2:342, panel A) or lower (ZHER2:4, panel B) affinity HER2 Affibody for 4 h, and the incorporation of [3H]‐leucine was measured and graphed as described in Figure 1. High, moderate, and low HER2 expressing cell lines are shown in red, blue, and purple, respectively. Each point on the curves represents the average of four experiments.
Bafilomycin A1 protected A431 cells from LFN‐DTA‐dependent killing mediated by either mPA‐ZHER2 or mPA‐EGF (SFigure 3), indicating that translocation of effectors by mPA variants was dependent on the endosomal pH, as is the case with wild‐type PA.
3.2. mPA‐ZHER2 can deliver multiple cytocidal effectors
We tested an analog of LFN‐DTA in which DTA was replaced with the catalytic domain of ricin (RTA). RTA inhibits protein synthesis by a different biochemical mechanism than DTA, namely by depurinating a crucial adenosine residue in the 28S rRNA of the eukaryotic ribosome (Endo and Tsurugi, 1987). LFN‐RTA combined with mPA‐ZHER2 (Figure 3A) or mPA‐EGF (Figure 3B) killed HER2‐positive or EGFR‐positive cells, respectively. Generally LFN‐RTA was 10–100‐fold less efficient than LFN‐DTA in killing the cell lines tested. An exception was the SKBR3 cell line, in which the EC50 values for LFN‐RTA or LFN‐DTA combined with mPA‐ZHER2 were about the same (compare Figures 1A and 3A). Because SKBR3 lacks detectable levels of EGF receptor, it was resistant to mPA‐EGF‐mediated killing.
Figure 3.
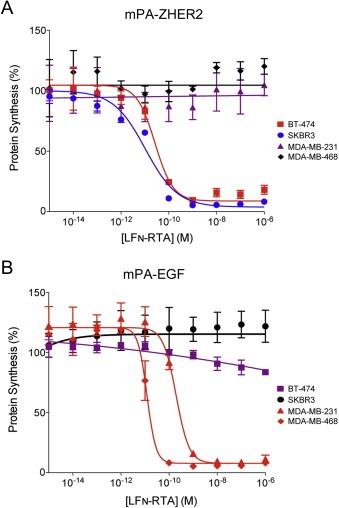
mPA‐ZHER2‐ and mPA‐EGF‐directed killing of cell lines by LFN‐RTA. Cells were exposed to mPA‐ZHER2 (Panel A) or mPA‐EGF (Panel B) in combination with LFN‐RTA, at the indicated concentrations for 4 h, and the level of protein synthesis was measured by scintillation counting. Cells expressing high, moderate, low, or no detectable levels of HER2 or EGFR are colored red, blue, purple, and black, respectively.
3.3. HER2‐targeted anthrax toxin kills a trastuzumab‐resistant tumor cell line
The FDA‐approved monoclonal antibody trastuzumab has been effective in prolonging HER2‐positive patient survival, but not all patients respond, and a large percentage develop therapeutic resistance over time (Arteaga et al., 2012). The JIMT‐1 cell line recently isolated from a patient that had HER2 amplification and clinically resistant to trastuzumab (Tanner et al., 2004). As in other HER2‐positive cell lines we tested, protein synthesis in JIMT‐1 cells was inhibited in response to mPA‐ZHER2 and LFN‐DTA, resulting in cell death by apoptosis (Figure 4). The level of sensitivity was consistent with the HER2 level, and killing mediated by LFN‐RTA was less efficient than by LFN‐DTA (Figure 4A). JIMT‐1 cells required a longer duration of toxin exposure (additional 24 h) to achieve similar cell killing and caspase 3/7 activation, compared to other HER2‐positive cell lines (Figure 4C and D).
Figure 4.
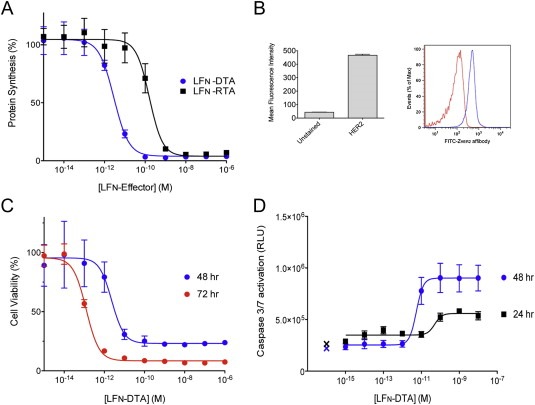
Killing of a HER2‐positive, trastuzumab‐resistant tumor cell line by mPA‐ZHER2 plus LFN‐DTA or LFN‐RTA. (A) The JIMT‐1 tumor cell line was incubated with mPA‐ZHER2 in combination with increasing amounts of LFN‐DTA (blue) or LFN‐RTA (black) for 4 h, and the effects on [3H]‐leucine incorporation were measured as described in Figure 1. (B) FACS analysis using an FITC‐conjugated HER2 Affibody confirms the expression of HER2 on the surface of JIMT‐1 cells. The mean fluorescence was calculated using the FlowJo software package and plotted in the GraphPad Prism® software package (left panel) from the raw data presented in the histogram (right panel), which displays the shift in fluorescence (blue) compared to unstained cells (red). (C) JIMT‐1 cells were exposed to the same conditions as panel A. After 48 or 72 h, cell viability was measured by XTT assay and plotted as percent cell viability normalized against control cells treated with mPA‐ZHER2 alone. (D) Caspase 3/7 activation, an indicator of apoptosis, was measured after a 24 and 48 h exposure to 20 nM mPA‐ZHER2 and LFN‐DTA, at the indicated concentrations. The cleavage of a pre‐luminescent caspase 3/7 substrate generated RLU's that are plotted versus LFN‐DTA concentration. Control cells treated with mPA‐ZHER2 alone are indicated with an X.
3.4. No bystander effect was seen in mixtures of HER2‐positive and HER2‐negative cells
To test for a possible bystander effect, we evaluated the specificity of mPA‐ZHER2 in mixtures of HER2‐positive and HER2‐negative cells. First, to allow individual cell types to be distinguished by fluorescence microscopy or FACS, we labeled selected cell lines by transduction with puromycin‐selectable lentiviruses encoding fluorescent proteins with distinguishable emission properties. Equal numbers of cells from each of 4 cell lines (2 HER2‐positive and 2 HER2‐negative lines) were mixed, and the resulting mixture was incubated 24 h with mPA‐ZHER2 plus LFN‐DTA. Flow cytometry revealed that the HER2‐negative cells, CHO‐K1 (unlabeled) and MDA‐MB‐468GFP (green), now comprised almost the entire population; the HER2‐positive A431CFP (cyan) and SKBR3RFP (red) cells had been reduced from ∼50% to less than 5% of the total (Figure 5A). Fluorescence microscopy of adherent cells gave comparable results (Figure 5B). Because the small remaining population of SKBR3 cells (∼4%) appeared to be dead by microscopy, we believe that flow cytometry may have overestimated this population because of an inherently long half‐life of the fluorescent protein used for labeling. Thus, the mPA‐ZHER2/LFN‐DTA combination was able to kill the HER2‐positive cells in a mixed cell population, with no evident bystander effect on HER2‐negative cells.
Figure 5.
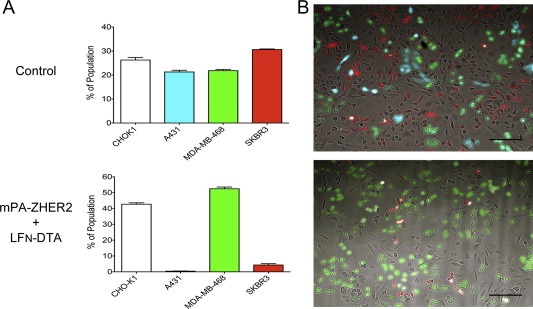
mPA‐ZHER2 mediates specific killing of HER2‐positive cells in a heterogeneous population. Fluorescent cells shown to be sensitive to the actions of mPA‐ZHER2 and LFN‐DTA (A431CFP and SKBR3RFP) were mixed equally with resistant cells (CHO‐K1 and MDA‐MB‐468GFP) and incubated with mPA‐ZHER2 plus LFN‐DTA or with mPA plus LFN‐DTA (control; the control FACS data are identical to those in Figures 7C and 8A, as all of the experiments were conducted simultaneously). After 24 h, cells were detached with trypsin and quantified by FACS (Panel A) or washed with PBS and imaged with a fluorescence microscope (Panel B; scale bar is 100 µm). Each bar represents the average of experiments performed in triplicate.
We also used another approach to test for bystander effects. Multiple cell lines were grown in separate wells of a chambered slide (Figure 6). The partitioning element was removed from the slide, and the slide, containing all cell lines, was then incubated in medium containing mPA‐ZHER2 and LFN‐DTA. After a 4‐h incubation, the slide was washed and transferred to medium supplemented with [3H]‐leucine. After a further 1‐h incubation, the cells were washed, individual cell populations were dissolved with 6 M Guanidine–HCl, and the incorporated radioactive leucine was quantified by scintillation counting. Figure 6 shows that cells expressing high and moderate levels of HER2 were killed, MDA‐MB‐231 cells with low HER2 expression maintained limited resistance, and cells lacking HER2 (CHO‐K1 and MDA‐MB‐468) were unaffected.
Figure 6.
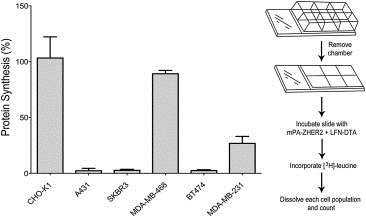
mPA‐ZHER2‐mediated killing in a heterogeneous cell population. Tumor cells were plated in separate compartments of a chambered slide (right panel) and incubated at 37 °C. The following day, the chambers were removed, and the slide was incubated with mPA‐ZHER2 plus LFN‐DTA. After 4 h, cells were incubated with medium supplemented with [3H]‐leucine for 1 h and dissolved in 6 M Guanidine–HCl, and the incorporated radiolabel was quantified by scintillation counting. Percent protein synthesis was normalized against cells treated with mPA + LFN‐DTA.
3.5. Mixing mPA‐ZHER2 and mPA‐EGF allowed killing of both HER2‐ and EGFR‐positive cells in a heterogeneous cell population
Like mPA‐HER2, mPA‐EGF in combination with LFN‐DTA was able to kill cognate cell lines (EGFR‐positive in this case) in both homogeneous (Figure 7A) and heterogeneous cell populations, with no effects on cells lacking the EGF receptor (Figure 7). Killing a mixed population of fluorescent cells by mPA‐EGF presented the same caveats as those described for mPA‐ZHER2, where a small population of EGFR‐positive cells (MDA‐MB‐468, colored green) remained (Figure 7B and C). Once again, a more sensitive assay measuring protein synthesis by incorporation of radioactive leucine showed that EGFR‐positive cells were killed, and cells with very low or no EGFR expression survived (Figure 7D).
Figure 7.
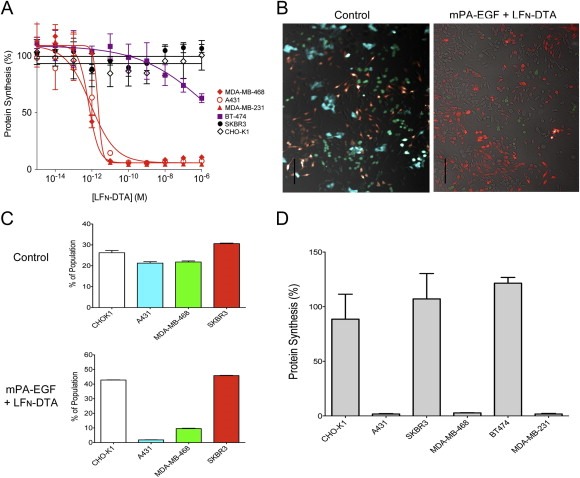
mPA‐EGF specifically kills EGF‐expressing cells in a heterogeneous population. (A) Cells were exposed to 20 nM mPA‐EGF and LFN‐DTA at the concentrations indicated for 4 h and protein synthesis was measured as in experiments described above. Percent protein synthesis was normalized against cells treated with mPA‐EGF alone. Cell lines expressing high, low, and no detectable amounts of EGFR are colored red, purple, and black respectively. Each point on the curves represents the average of four experiments. (B and C) Populations of fluorescent cells were mixed and exposed to a lethal dose of mPA‐EGF and LFN‐DTA or mPA + LFN‐DTA as a control; the control FACS data are identical to those in Figures 5A and 8A, as all of the experiments were conducted simultaneously. After 24 h, cells were washed with PBS and imaged with a fluorescence microscope (B; scale bar is 100 µm) or detached with trypsin and quantified by FACS (C). Each bar represents the average of experiments performed in triplicate. (D) A panel of cancer cell lines was plated in chambered slides overnight. The following day the chambers were removed and cells were exposed to the same treatments as described in panels B and C. Following intoxication for 4 h, cells were processed, and protein synthesis was quantified as described in panel A.
We also tested the ability of a mixture of mPA‐ZHER2 and mPA‐EGF to target specific receptor‐bearing cells in a mixed population of cancer cells. As shown in Figure 8, the combination of mPA‐ZHER2 and mPA‐EGF was able to kill both HER2‐positive and EGFR‐positive cells in the presence of LFN‐DTA, while CHO‐K1 cells, which do not express either receptor, remained unaffected.
Figure 8.
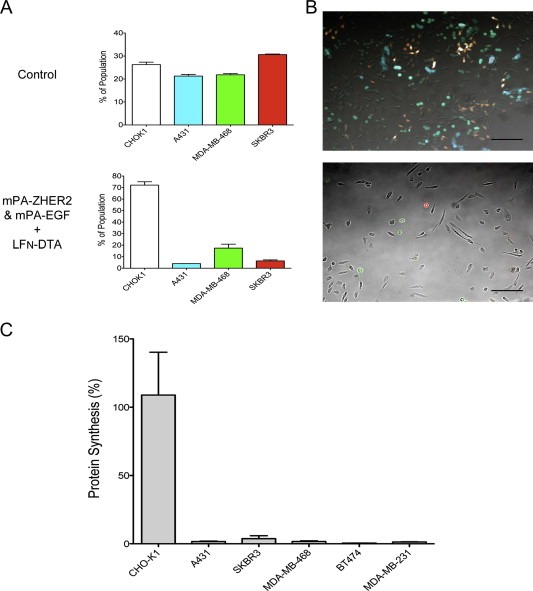
Redirected mPA variants act together to eliminate heterogeneous tumor cell populations. (A and B) Various fluorescent cells were mixed in equal numbers and exposed to LFN‐DTA plus an equimolar mixture of mPA‐ZHER2 and mPA‐EGF. LFN‐DTA plus mPA was used as control (the control FACS data are identical to those in Figures 5A and 7C, as all of the experiments were conducted simultaneously). After 24 h, cell populations were detached with trypsin and quantified by FACS (A) or washed with PBS and imaged with a fluorescence microscope (B; scale bar is 100 µm). Each bar represents the average of experiments performed in triplicate using SKBR3 (red), A431 (cyan), MDA‐MB‐468 (green), and CHO‐K1 (unlabeled) cells. (C) A larger panel of cancer cell lines were plated in separate compartments of a chambered slide overnight. The following day, the partition was removed and cells were exposed to the same treatments as described above. Following intoxication for 4‐h, cells were incubated with medium supplemented with [3H]‐leucine for 1‐h, and protein synthesis was quantified by scintillation counting. Percent protein synthesis was normalized against cells treated with mPA and LFN‐DTA.
4. Discussion
Cell‐surface markers on aggressive forms of certain cancers have been an important focus of efforts to develop targeted anticancer therapies. A prominent example is the monoclonal anti‐HER2 antibody trastuzumab, which is effective in slowing tumor growth and prolonging patient survival (Vogel et al., 2002). However, most patients develop resistance to this antibody over time due to its ineffectiveness in eliminating tumors (Arteaga et al., 2012). Antibody therapies have been combined with conventional chemotherapy or radiation to circumvent such resistance, and antibody‐drug conjugates (ADC's), which kill cells through the action of a linked cytotoxic small molecule compound (“payload”), have recently emerged as an alternative mode of targeted therapy (Carter and Senter, 2008).
Modifying intracellularly acting toxins to direct their actions to tumor cells represents an attractive approach to targeted therapy, in part because the catalytic mode of action of the effector moieties renders these toxins so potent. Replacing the native receptor‐binding domain of toxins such as DT or Pseudomonas exotoxin A (ETA) with a heterologous receptor‐binding protein has been employed effectively to target the cytocidal actions of these toxins (Pastan et al., 2007). This line of investigation has led to a licensed treatment for cutaneous T‐cell lymphomas, termed denileukin diftitox (trade name, Ontak®) (Foss, 2000; Williams et al., 1987), and other targeted protein toxins are currently under investigation (Madhumathi J and Verma, 2012). Ontak is a fusion protein created by replacing the receptor‐binding domain of DT with interleukin‐2 (IL‐2). The IL‐2 moiety binds the fusion toxin to high‐affinity IL‐2 receptors on tumor cells, and the catalytic moiety of DT (DTA) is transported to the cytosol, where it blocks protein synthesis and causes cell death (Collier and Cole, 1969; Collier, 1967).
Elucidation of the structure and activities of anthrax toxin in recent years has led to experiments to explore its potential as a platform for developing anticancer chemotherapeutics. In one study the furin site of PA was mutated to prevent activation of the protein, and the native receptor‐binding activity of the modified PA was exploited to inhibit vascular endothelial growth factor‐induced and basic fibroblast growth factor‐induced angiogenesis (Rogers et al., 2007). In other studies lethal factor combined with native PA was found to induce apoptosis in human melanoma cells, suggesting possible applications for this and other cancers in which disease progression is due in part to constitutive activation of MAPK signaling (Duesbery et al., 2001; Koo et al., 2002). An elegant approach to targeting PA has been to replace its furin cleavage site with a site selective for a different protease — metalloproteinase or urokinase plasminogen activator — that is overexpressed on the surface of cancer cells (Abi‐Habib et al., 2006; Liu et al., 2000).
In the current work we changed the receptor recognition specificity of PA as an approach to using the protein as a vehicle for introducing cytotoxic effectors specifically into HER2‐positive cells. mPA‐ZHER2 proved to be a highly selective mediator of the entry of LFN‐DTA and LFN‐RTA into HER2‐positive cells. The EC50 of LFN‐DTA showed an inverse relationship to the level of HER2 on the cell lines tested (Figure 1). Why LFN‐RTA was 10‐ to 100‐fold less potent than LFN‐DTA (Figure 3) is unclear, but may be related to differences in stability of the effectors in the cytosol, the kinetics of inactivation of target molecules, or any of a number of other factors.
The specificity of mPA‐ZHER2 for cells bearing the HER2 receptor was shown by competition assays (Figure 2) and by its ability to target only HER2‐positive tumor cells in a mixed cell population (Figures 5 and 6). No off‐target effects were observed when HER2‐negative cells were mixed with HER2‐positive cells before treatment with LFN‐DTA plus mPA‐ZHER2. The affinity of monomeric ZHER2 Affibody for the HER2 marker rivals that of the best antibodies (∼20 pM) (Orlova et al., 2006), and the natural oligomerization properties of mPA‐ZHER2 presumably increase the avidity of the interaction of the complex for the HER2 receptor on cells. Once oligomerization takes place, the avidity for the receptor would be such that effectively no dissociation of toxic complexes from cells would occur.
The entry of the cytocidal effectors mediated by either mPA‐ZHER2 or mPA‐EGF was pH‐dependent, as is the case for wild‐type PA (SFigure 3A). The difference in the inhibitory concentration of BFA for the different PA variants is likely due to their respective receptor abundances', where EGFR > ANTXR1/2 > HER2 (SFigure 3B). Alternatively the pH threshold of PA pore formation may vary for the three receptors, as the pH threshold of WT PA bound to ANTXR1 is a full unit higher than when it is bound to ANTXR2, which has higher‐affinity (Lacy et al., 2004; Rainey et al., 2005).
A HER2‐positive, trastuzumab‐resistant tumor cell line (JIMT‐1) was also susceptible to toxin action (Figure 4). The JIMT‐1 cell line, isolated from a patient clinically resistant to trastuzumab, displays properties thought to be associated with the development of HER2‐targeted antibody resistance, including low expression of HER2 (despite gene amplification), receptor masking by other cell‐surface proteins (an event which can mask up to 80% of the trastuzumab‐binding sites), low PTEN expression, activation of the PIK3CA gene, and high expression of neuregulin‐1 (NRG‐1) (Tanner et al., 2004; Nagy et al., 2005; Köninki et al., 2010). The EC50 in relation to HER2 level was consistent with our data on trastuzumab‐sensitive HER2‐positive cell lines. LFN‐RTA was also effective in killing JIMT‐1 cells, but higher concentrations than LFN‐DTA were needed (Figure 4A). The delivery of LFN‐DTA, into the cytosol of JIMT‐1 cells, led to apoptotic cell death, as assessed by an XTT cytotoxicity assay and caspase 3/7 activation (Figure 4C and D). The redirected toxin was able to kill most cells (>75%) after 48 h and achieved almost complete elimination (∼95%) after 72 h (Figure 4C). The greater exposure required to achieve complete cell killing could have resulted from any of a number of differences that increased the time to reach caspase 3/7 activation (48 h versus 24 h; Figure 4D).
The elimination of a trastuzumab‐resistant cell line by anthrax toxin represents a potential advantage over current antibody therapies. Some ADC's have been shown to kill trastuzumab‐resistant tumor cell lines (such as JIMT‐1), but are significantly less effective than LFN‐DTA plus mPA‐ZHER2, and require high doses (μg/ml versus pg/ml) to achieve moderate killing (∼25% cell death) (Lewis Phillips et al., 2008; Köninki et al., 2010). This difference in potency (∼5000‐fold) could result from efficient delivery of a cytocidal enzyme into the cytosol, reflecting the strength of the interaction between mPA‐ZHER2 and the HER2 receptor, as well as the catalytic inactivation of the cytosolic substrate. The accessibility of the mPA‐ZHER2‐binding site on the surface of JIMT‐1 cells compared to the antibody‐binding site, estimated to be 20% available, could also be a factor (Nagy et al., 2005).
Because tumors are composed of a heterogeneous population of cells that have different receptor expression levels, it is unlikely that any single anticancer therapy can achieve complete tumor elimination. Combinations of small molecules, antibodies, and radiation have been used with some success. The binary nature of anthrax toxin and the ability of mPA to oligomerize also suggests that one may be able to combine mPA‐ZHER2 with other forms of mPA targeted to different overexpressed surface tumor markers to eliminate heterogeneous populations of cells. Consistent with this notion, as mPA‐ZHER2 and mPA‐EGF, in combination with LFN‐DTA completely eliminated a panel of tumor cells with different HER2 and EGF receptor expression levels (Figure 8).
The ability of mPA‐ZHER2 to act cooperatively with an analogous mPA variant targeting a different tumor marker highlights the adaptability of targeting with mPA. In addition to combining mPA variants, the ability of the PA pore to translocate any of a variety of intracellular effector enzymes allows the possibility of using combinations of effectors that kill by different biochemical mechanisms. The enzymatic destruction of targeted cells from within by multiple effectors should minimize the likelihood of resistant escape mutants arising, a universal problem in chemotherapy.
In summary, our in vitro data indicate that the targeting of the HER2 receptor by modified, receptor‐targeted anthrax toxin is specific and potent, and displays no off‐target toxicity towards HER2‐negative cell lines. The susceptibility of a HER2‐positive trastuzumab‐resistant tumor cell line to toxin action highlights a significant potential advantage of our system over current FDA‐approved antibody therapies. For these reasons and the advantages described above, the PA‐based targeting of distinct populations of cancer cells represents a promising therapeutic strategy for cancer treatment.
Supporting information
Supplementary data
Acknowledgements
This research was supported by NIAID grant AI022021 to RJC and NIAID grant AI062827 to MNS. We thank Dr. Robin Ross, Ben Seiler, and Erica Gardner from the NERCE Biomolecule Production Core, supported by NIH grant AI057159, for helping with the production of proteins used in this study. The authors would also like to thank Drs. Tom Roberts and Jean Zhao (Dana Farber Cancer Institute) for their critical review of the manuscript. The authors would also like to thank Dr. Jean Zhao again for providing cell lines and Dr. Gregory Poon (Washington State Univeristy, Pullman, WA) for the expression plasmids coding for the ZHER2 proteins and the MDA‐MB‐231 cell line.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2012.12.003.
McCluskey Andrew J., Olive Andrew J., Starnbach Michael N., Collier R. John, (2013), Targeting HER2‐positive cancer cells with receptor‐redirected anthrax protective antigen, Molecular Oncology, 7, doi: 10.1016/j.molonc.2012.12.003.
References
- Abi-Habib, R.J. , 2006. A urokinase-activated recombinant anthrax toxin is selectively cytotoxic to many human tumor cell types. Molecular Cancer Therapeutics 5, (10) 2556–2562. [DOI] [PubMed] [Google Scholar]
- Arteaga, C.L. , 2012. Treatment of HER2-positive breast cancer: current status and future perspectives. Nature Reviews Clinical Oncology 9, (1) 16–32. [DOI] [PubMed] [Google Scholar]
- Berchuck, A. , 1990. Overexpression of HER-2/neu is associated with poor survival in advanced epithelial ovarian cancer. Cancer Research 50, (13) 4087–4091. [PubMed] [Google Scholar]
- Bradley, K.A. , 2001. Identification of the cellular receptor for anthrax toxin. Nature 414, (6860) 225–229. [DOI] [PubMed] [Google Scholar]
- Carter, P.J. , Senter, P.D. , 2008. Antibody-drug conjugates for cancer therapy. Cancer Journal (Sudbury, Mass.) 14, (3) 154–169. [DOI] [PubMed] [Google Scholar]
- Collier, R.J. , Cole, H.A. , 1969. Diphtheria toxin subunit active in vitro. Science 164, (884) 1179–1181. [DOI] [PubMed] [Google Scholar]
- Collier, R.J. , 1967. Effect of diphtheria toxin on protein synthesis: inactivation of one of the transfer factors. Journal of Molecular Biology 25, (1) 83–98. [DOI] [PubMed] [Google Scholar]
- Collier, R.J. , 2009. Membrane translocation by anthrax toxin. Molecular Aspects of Medicine 30, (6) 413–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham, K. , 2002. Mapping the lethal factor and edema factor binding sites on oligomeric anthrax protective antigen. Proceedings of the National Academy of Sciences of the United States of America 99, (10) 7049–7053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duesbery, N.S. , 1998. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 280, (5364) 734–737. [DOI] [PubMed] [Google Scholar]
- Duesbery, N.S. , 2001. Suppression of ras-mediated transformation and inhibition of tumor growth and angiogenesis by anthrax lethal factor, a proteolytic inhibitor of multiple MEK pathways. Proceedings of the National Academy of Sciences of the United States of America 98, (7) 4089–4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo, Y. , Tsurugi, K. , 1987. RNA N-glycosidase activity of ricin A-chain. Mechanism of action of the toxic lectin ricin on eukaryotic ribosomes. The Journal of Biological Chemistry 262, (17) 8128–8130. [PubMed] [Google Scholar]
- Foss, F.M. , 2000. DAB(389)IL-2 (denileukin diftitox, ONTAK): a new fusion protein technology. Clinical Lymphoma 1, (Suppl. 1) S27–S31. [PubMed] [Google Scholar]
- Gravalos, C. , Jimeno, A. , 2008. HER2 in gastric cancer: a new prognostic factor and a novel therapeutic target. Annals of Oncology: Official Journal of the European Society for Medical Oncology/ESMO 19, (9) 1523–1529. [DOI] [PubMed] [Google Scholar]
- Honjo, T. , Nishizuka, Y. , Hayaishi, O. , 1968. Diphtheria toxin-dependent adenosine diphosphate ribosylation of aminoacyl transferase II and inhibition of protein synthesis. The Journal of Biological Chemistry 243, (12) 3553–3555. [PubMed] [Google Scholar]
- Kintzer, A.F. , 2009. The protective antigen component of anthrax toxin forms functional octameric complexes. Journal of Molecular Biology 392, (3) 614–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimpel, K.R. , 1992. Anthrax toxin protective antigen is activated by a cell surface protease with the sequence specificity and catalytic properties of furin. Proceedings of the National Academy of Sciences of the United States of America 89, (21) 10277–10281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köninki, K. , 2010. Multiple molecular mechanisms underlying trastuzumab and lapatinib resistance in JIMT-1 breast cancer cells. Cancer Letters 294, (2) 211–219. [DOI] [PubMed] [Google Scholar]
- Koo, H.-M. , 2002. Apoptosis and melanogenesis in human melanoma cells induced by anthrax lethal factor inactivation of mitogen-activated protein kinase kinase. Proceedings of the National Academy of Sciences of the United States of America 99, (5) 3052–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacy, D.B. , 2004. Structure of heptameric protective antigen bound to an anthrax toxin receptor: a role for receptor in pH-dependent pore formation. Proceedings of the National Academy of Sciences of the United States of America 101, (36) 13147–13151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppla, S.H. , 1982. Anthrax toxin edema factor: a bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proceedings of the National Academy of Sciences of the United States of America 79, (10) 3162–3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis Phillips, G.D. , 2008. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Research 68, (22) 9280–9290. [DOI] [PubMed] [Google Scholar]
- Liu, S. , 2000. Tumor cell-selective cytotoxicity of matrix metalloproteinase-activated anthrax toxin. Cancer Research 60, (21) 6061–6067. [PubMed] [Google Scholar]
- Löfblom, J. , 2010. Affibody molecules: engineered proteins for therapeutic, diagnostic and biotechnological applications. FEBS Letters 584, (12) 2670–2680. [DOI] [PubMed] [Google Scholar]
- Madhumathi, J. , Verma, R.S. , 2012. Therapeutic targets and recent advances in protein immunotoxins. Current Opinion in Microbiology [DOI] [PubMed] [Google Scholar]
- Mechaly, A. , McCluskey, A.J. , Collier, R.J. , 2012. Changing the receptor specificity of anthrax toxin. pp. e00088–12 mBio 3, (3) Available at: http://mbio.asm.org/content/3/3/e00088-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, C.J. , Elliott, J.L. , Collier, R.J. , 1999. Anthrax protective antigen: prepore-to-pore conversion. Biochemistry 38, (32) 10432–10441. [DOI] [PubMed] [Google Scholar]
- Milne, J.C. , 1994. Anthrax protective antigen forms oligomers during intoxication of mammalian cells. The Journal of Biological Chemistry 269, (32) 20607–20612. [PubMed] [Google Scholar]
- Mogridge, J. , 2002. The lethal and edema factors of anthrax toxin bind only to oligomeric forms of the protective antigen. Proceedings of the National Academy of Sciences of the United States of America 99, (10) 7045–7048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy, P. , 2005. Decreased accessibility and lack of activation of ErbB2 in JIMT-1, a herceptin-resistant, MUC4-expressing breast cancer cell line. Cancer Research 65, (2) 473–482. [PubMed] [Google Scholar]
- Nord, K. , 1997. Binding proteins selected from combinatorial libraries of an alpha-helical bacterial receptor domain. Nature Biotechnology 15, (8) 772–777. [DOI] [PubMed] [Google Scholar]
- Orlova, A. , 2006. Tumor imaging using a picomolar affinity HER2 binding affibody molecule. Cancer Research 66, (8) 4339–4348. [DOI] [PubMed] [Google Scholar]
- Pastan, I. , 2007. Immunotoxin treatment of cancer. Annual Review of Medicine 58, 221–237. [DOI] [PubMed] [Google Scholar]
- Rainey, G.J.A. , 2005. Receptor-specific requirements for anthrax toxin delivery into cells. Proceedings of the National Academy of Sciences of the United States of America 102, (37) 13278–13283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers, M.S. , 2007. Mutant anthrax toxin B moiety (protective antigen) inhibits angiogenesis and tumor growth. Cancer Research 67, (20) 9980–9985. [DOI] [PubMed] [Google Scholar]
- Scobie, H.M. , 2003. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proceedings of the National Academy of Sciences 100, (9) 5170–5174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slamon, D.J. , 1989. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 244, (4905) 707–712. [DOI] [PubMed] [Google Scholar]
- Tanner, M. , 2004. Characterization of a novel cell line established from a patient with herceptin-resistant breast cancer. Molecular Cancer Therapeutics 3, (12) 1585–1592. [PubMed] [Google Scholar]
- Vitale, G. , 1998. Anthrax lethal factor cleaves the N-terminus of MAPKKs and induces tyrosine/threonine phosphorylation of MAPKs in cultured macrophages. Biochemical and Biophysical Research Communications 248, (3) 706–711. [DOI] [PubMed] [Google Scholar]
- Vogel, C.L. , 2002. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology 20, (3) 719–726. [DOI] [PubMed] [Google Scholar]
- Wikman, M. , 2004. Selection and characterization of HER2/neu-binding affibody ligands. Protein Engineering, Design & Selection: PEDS 17, (5) 455–462. [DOI] [PubMed] [Google Scholar]
- Williams, D.P. , 1987. Diphtheria toxin receptor binding domain substitution with interleukin-2: genetic construction and properties of a diphtheria toxin-related interleukin-2 fusion protein. Protein Engineering 1, (6) 493–498. [DOI] [PubMed] [Google Scholar]
- Young, J.A.T. , Collier, R.J. , 2007. Anthrax toxin: receptor binding, internalization, pore formation, and translocation. Annual Review of Biochemistry 76, (1) 243–265. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data