

Abstract
Inhibitors of the PI3K/mTOR signaling network are under development as novel cancer therapies. However, these compounds do not cause robust cytotoxic responses in tumor cells unless combined with other agents. Rational combinations with other targeted therapies will likely be necessary to achieve the potential of PI3K/mTOR inhibitors in oncology.
Introduction
Phosphoinositide 3-kinase (PI3K) and the mammalian target of rapamycin (mTOR) participate in a signaling network that drives cell proliferation and survival in response to growth factors or oncogenic changes.[1, 2] Development of small molecule inhibitors of PI3K and/or mTOR has been an active area of drug discovery for cancer, and several compounds are in clinical trials. However, PI3K/mTOR inhibitors show limited ability to cause cancer cell death when used as single agents. In preclinical models, PI3K/mTOR inhibitors achieve tumor regression most effectively when used on combination with other targeted therapies.[3–5] Here we discuss models to explain cancer cell resistance to apoptosis, and speculate on rational combinations to unleash the pro-apoptotic potential of PI3K/mTOR inhibitors.
PI3K/mTOR signaling and cell survival
PI3Ks are a family of broadly expressed enzymes that produce 3-phosphorylated inositol lipids to promote membrane recruitment of specific effectors.[6] Among the different PI3K catalytic isoforms, the class I PI3Ks (PI3Kα, PI3Kβ, PI3Kγ, PI3Kδ) are mainly responsible for signals leading to cell growth, proliferation and survival. Activating mutations in the gene encoding PI3Kα (PIK3CA) are frequent in human cancer and the other three class I isoforms are thought to promote cancer in certain contexts.[1] A key downstream effector of PI3K signaling is mTOR, a serine/threonine kinase encoded by a single gene, MTOR. The mTOR kinase is the catalytic subunit of two multiprotein complexes, TORC1 and TORC2, with different substrates and functions.[2] TORC1 and TORC2 together control a large fraction of the cellular phosphorylation responses to growth factors and mTOR substrate phosphorylation is elevated in most cancer cells.
Class I PI3K and TORC2 provide essential inputs to activate AKT (also known as PKB), a multi-functional serine/threonine kinase with many cellular substrates.[2, 6] A large body of literature supports the idea that AKT promotes cell survival through phosphorylation of many proteins, including BAD, MDM2, and FOXO transcription factors.[7] However, much of the evidence for PI3K/AKT-mediated survival signaling came from early studies using non-specific inhibitors such as wortmannin and LY294002. Development of compounds with selectivity towards class I PI3Ks and/or mTOR, or AKT, has prompted reconsideration of earlier models. Namely, most cell types (cancer or normal) do not display a dramatic cell death response when treated with inhibitors of PI3K, AKT or mTOR as single agents. In our experience, ATP-competitive mTOR inhibitors or dual PI3K/mTOR inhibitors can induce significant apoptosis in murine bone marrow cells transformed with the human oncogene BCR-ABL.[5] However, in bona fide human leukemia cells these compounds are more effective when used in combination with inhibitors of the BCR-ABL tyrosine kinase.[5, 8] For scientists working in the PI3K/mTOR field, early optimism about single agent efficacy is evolving into the realization that effective application of PI3K/mTOR inhibitorsin oncology will usually require rational combinations. Even in the case of CAL-101, a PI3Kδ inhibitor showing promising results in B cell malignancies, clinical trials suggest greater efficacy when CAL-101 is combined with rituximab or bendamustine.[1] How can we apply existing knowledge to design the best rational combinations?
Resistance model #1: compensatory signaling pathways
In most cancer cells, elevated PI3K/mTOR activity is not the only signaling mechanism that feeds into pro-survival mechanisms. The RAS/RAF/MEK/ERK pathway is activated in most cancer cells and acts in parallel to promote cell survival, sometimes converging on shared targets with PI3K/mTOR (Fig. 1A). Combinations of MEK inhibitors with PI3K/mTOR inhibitors have proved to be more effective than single agents in mouse models of KRAS-driven lung cancer[3] and other models. In many cases, the PI3K and RAS pathways are downstream of activated receptor tyrosine kinases (e.g. EGFR) or non-receptor tyrosine kinases (e.g. BCR-ABL). Combining mTOR kinase inhibitors with tyrosine kinase inhibitors (TKIs) has shown promise in BCR-ABL–dependent leukemias, as mentioned above[5, 6], and in EGFR-dependent breast cancer.[4] Therefore, a general strategy for tumors with known lesions in tyrosine kinases might be to combine PI3K/mTOR inhibitors with TKIs or with MEK inhibitors (Fig. 1A).
Figure 1.
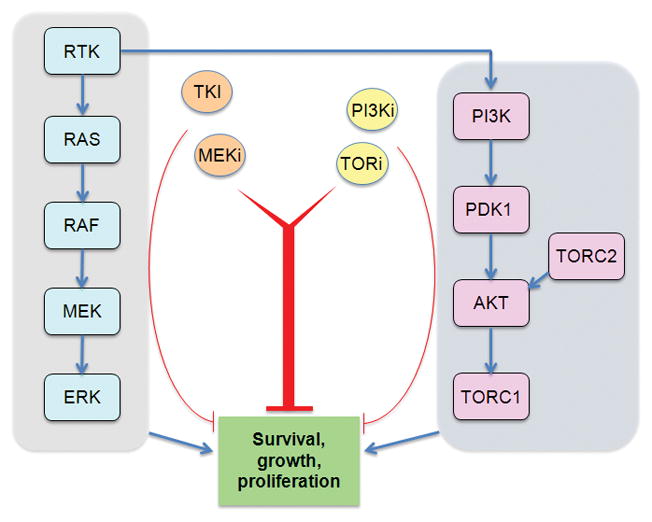
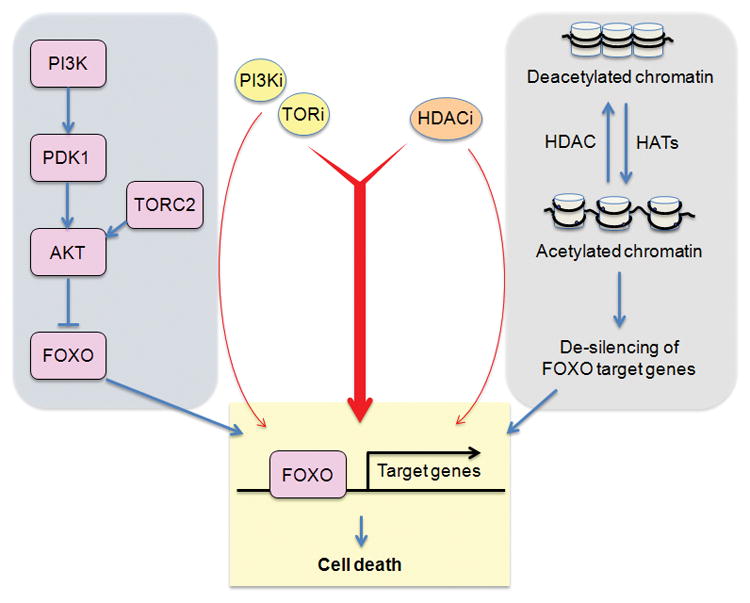
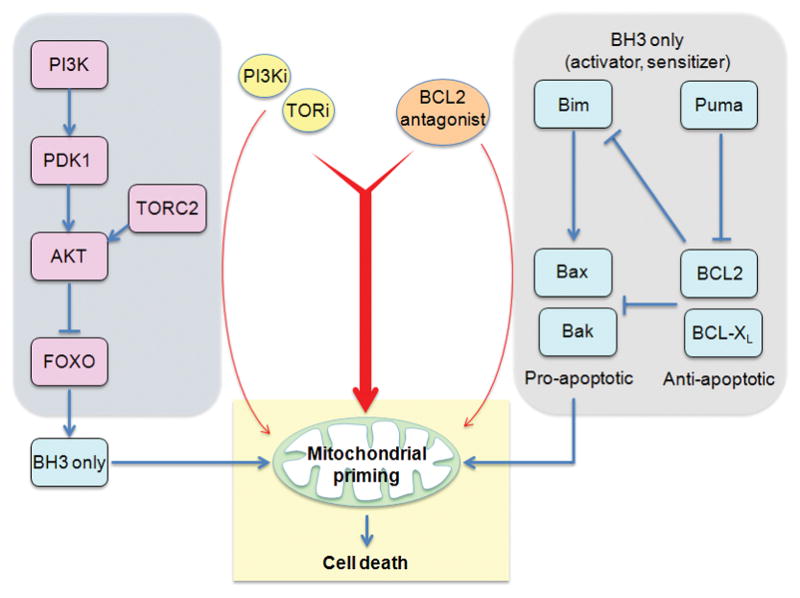
Pharmacological strategies to overcome resistance to apoptosis in cancer cells treated with PI3K/mTOR inhibitors. (A) Many tumor cells have activated receptor tyrosine kinases (RTKs), or cytoplasmic tyrosine kinases, and/or activation of the RAS/RAF/MEK/ERK pathway. Combining TKIs or MEK inhibitors with PI3K/mTOR inhibitors can induce cancer cell death. (B) Pro-apoptotic genes are often silenced epigenetically in tumor cells. HDAC inhibition leads to increased histone acetylation and more open chromatin state at gene regulatory elements. PI3K/mTOR inhibition triggers nuclear accumulation of FOXO transcription factors, which can access pro-apoptotic gene promoters when cells are treated with HDAC inhibitors. (C) Low mitochondrial priming is a significant barrier to apoptotic induction in some cancer cells. BCL2 antagonists can increase priming and lower the threshold for apoptotic induction by PI3K/mTOR inhibitors.
Resistance model #2: epigenetic status
It is becoming accepted that epigenetic changes play a major role in maintaining the transformed state of cancer cells, and providing resistance to cancer therapies.[9] A simple model is that most apoptotic stimuli act by induction of pro-death genes, and such genes may be silenced epigenetically in cancer cells. This could occur by promoter methylation or through various post-translational modifications of histones. Reversing the suppressive epigenetic marks might potentiate the apoptotic program.
One of the mechanisms for gene silencing is histone deacetylation (Fig. 1B). The concept of treating cancer with histone deacetylase inhibitors (HDACi) has gained acceptance with the approval of vorinostat for the treatment of cutaneous T cell lymphoma. A number of studies have reported synergistic killing of tumor cells with HDACi combined with inhibitors of the PI3K/mTOR network. Our laboratory has observed synergy of vorinostat with mTOR kinase inhibitors in B cell acute lymphoblastic leukemia (B-ALL) cells (D.A. Fruman, unpublished data). A possible mechanism is the de-silencing of genes induced by FOXO transcription factors, which enter the nucleus when TORC2 and AKT are suppressed. Hence, we propose that HDACi should be explored more broadly for their ability to potentiate FOXO-dependent death when the PI3K/mTOR network is suppressed (Fig. 1B). Other drugs targeting the epigenome are also worth investigating if a molecular rationale exists.
Resistance model #3: mitochondrial priming
Apoptosis is an orderly process whose rate-limiting step is loss of mitochondrial membrane potential. Ultimately, the ability of drugs to induce cell death is determined by the balance of pro-survival and pro-apoptotic factors at the mitochondrial membrane. The closeness of cells to the threshold for apoptosis has been termed mitochondrial “priming”.[10] Recent studies have shown that the mitochondrial priming status is a major determinant of response to chemotherapy in a number of different malignancies. It is also likely that priming controls the response to targeted therapies. Consistent with this, CAL-101 treatment was shown to increase priming of CLL cells, potentiating apoptosis especially in the presence of chemotherapeutic agents.[10]
The main determinant of mitochondrial priming is the BCL2 family of apoptotic regulators (Fig. 1C). Elevated priming can be achieved by increased function of BH3-only family members, or by decreased function of pro-survival members BCL2, BCL-XL, MCL-1, and others. Based on this knowledge, BCL2 antagonists (e.g. ABT-263) represent a rational strategy to increase the pro-apoptotic effect of various cancer drugs including PI3K/mTOR inhibitors (Fig. 1C). For example, PI3K/mTOR inhibitors can increase expression of FOXO targets such as Bim and Puma, whose pro-apoptotic effects would be potentiated by increased priming through BCL2 antagonists.
Summary
Great progress has been achieved in PI3K/mTOR drug discovery. Small molecules with high selectivity and good drug properties have entered oncology trials after demonstrating proof of principle in animal models of cancer. However, optimism about this drug class has been tempered by the evidence that single agent PI3K/mTOR inhibitors are generally more cytostatic than cytotoxic. For these compounds to become a broadly used tool to eradicate cancer, it is essential to unleash the pro-apoptotic potential of inhibiting this pathway. A common strategy is to combine PI3K/mTOR inhibitors with compounds targeting other oncogenic signaling kinases, such as tyrosine kinases or MEK. Two other strategies discussed herein are to combine PI3K/mTOR inhibitors with agents that alter the cancer epigenome or that increase mitochondrial priming. Many other strategies exist and deserve consideration.
On a cautionary note, it is likely that combinations that augment killing of cancer cells will also have greater toxicity to normal tissues. Identifying drug combinations with an acceptable therapeutic window will be a continuing challenge, but is hoped to eventually improve healthy survival of patients afflicted with cancer.
Acknowledgments
S.S.Y. was supported by the Inje Research and Scholarship Foundation in, 2011. D.A.F was supported by NIH R01-CA158383
References
- 1.Fruman DA, Rommel C. PI3Kdelta Inhibitors in Cancer: Rationale and Serendipity Merge in the Clinic. Cancer Discov. 2011;1:562–572. doi: 10.1158/2159-8290.CD-11-0249. [DOI] [PubMed] [Google Scholar]
- 2.Laplante M, Sabatini DM. mTOR Signaling in Growth Control and Disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Engelman JA, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351–1356. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garcia-Garcia C, et al. Dual mTORC1/2 and HER2 blockade results in antitumor activity in preclinical models of breast cancer resistant to anti-HER2 therapy. Clin Cancer Res. 2012;18:2603–2612. doi: 10.1158/1078-0432.CCR-11-2750. [DOI] [PubMed] [Google Scholar]
- 5.Janes MR, et al. Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor. Nat Med. 2010;16:205–213. doi: 10.1038/nm.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vanhaesebroeck B, et al. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;11:329–341. doi: 10.1038/nrm2882. [DOI] [PubMed] [Google Scholar]
- 7.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Janes MR, et al. Efficacy of the investigational mTOR kinase inhibitor MLN0128/INK128 in models of B-cell acute lymphoblastic leukemia. Leukemia. 2012 doi: 10.1038/leu.2012.276. Epub Oct. 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Popovic R, Licht JD. Emerging epigenetic targets and therapies in cancer medicine. Cancer Discov. 2012;2:405–413. doi: 10.1158/2159-8290.CD-12-0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davids MS, et al. Decreased mitochondrial apoptotic priming underlies stroma-mediated treatment resistance in chronic lymphocytic leukemia. Blood. 2012;120:3501–3509. doi: 10.1182/blood-2012-02-414060. [DOI] [PMC free article] [PubMed] [Google Scholar]