

Abstract
Rationale
The intracellular trafficking of connexin 43 (Cx43) hemichannels presents opportunities to regulate cardiomyocyte gap junction coupling. Although it is known that Cx43 hemichannels are transported along microtubules to the plasma membrane, the role of actin in Cx43 forward trafficking is unknown.
Objective
We explored whether the actin cytoskeleton is involved in Cx43 forward trafficking.
Methods and Results
High-resolution imaging reveals that Cx43 vesicles colocalize with nonsarcomeric actin in adult cardiomyocytes. Live-cell fluorescence imaging reveals Cx43 vesicles as stationary or traveling slowly (average speed 0.09 μm/s) when associated with actin. At any time, the majority (81.7%) of vesicles travel at subkinesin rates, suggesting that actin is important for Cx43 transport. Using Cx43 containing a hemagglutinin tag in the second extracellular loop, we developed an assay to detect transport of de novo Cx43 hemichannels to the plasma membrane after release from Brefeldin A-induced endoplasmic reticulum/Golgi vesicular transport block. Latrunculin A (for specific interference of actin) was used as an intervention after reinitiation of vesicular transport. Disruption of actin inhibits delivery of Cx43 to the cell surface. Moreover, using the assay in primary cardiomyocytes, actin inhibition causes an 82% decrease (P<0.01) in de novo endogenous Cx43 delivery to cell–cell borders. In Langendorff-perfused mouse heart preparations, Cx43/β-actin complexing is disrupted during acute ischemia, and inhibition of actin polymerization is sufficient to reduce levels of Cx43 gap junctions at intercalated discs.
Conclusions
Actin is a necessary component of the cytoskeleton-based forward trafficking apparatus for Cx43. In cardiomyocytes, Cx43 vesicles spend a majority of their time pausing at nonsarcomeric actin rest stops when not undergoing microtubule-based transport to the plasma membrane. Deleterious effects on this interaction between Cx43 and the actin cytoskeleton during acute ischemia contribute to losses in Cx43 localization at intercalated discs.
Keywords: actin, connexin 43, cytoskeletal dynamics, gap junctions, trafficking
Gap junctions permit the direct electric coupling of cardiomyocytes, providing rapid and well-orchestrated propagation of action potentials through the myocardium, which is necessary for each heartbeat.1 Remodeling of gap junction expression, either through their downregulation or through altered membrane localization, results in pathological electric defects of the myocardium and leads to malignant arrhythmias.2,3 Ventricular gap junctions primarily comprise connexin 43 (Cx43), a short-lived protein with a half-life of several hours.4 The rapid turnover and specific plasma membrane localization of Cx43 at intercalated discs suggest that the channels’ intracellular movements are highly regulated. Coordination of these intracellular movements is not well-understood.
Groups of six de novo Cx43 molecules oligomerize into hemichannels at the trans-Golgi network (TGN).5 These gap junction hemichannels are packaged into vesicles that, on exiting the TGN, are transported by motor proteins along microtubules to the plasma membrane.6 Interactions between microtubule plus-end binding proteins and membrane scaffolding proteins can effect specificity in targeting of vesicular cargo to distinct plasma membrane subdomains such as the intercalated discs (for Cx43) and T-tubules (for voltage-gated calcium channels) of cardiomyocytes.7,8 In addition, we have found that oxidative stress can affect the microtubule trafficking machinery through displacement of the plus-end binding protein EB1 and subsequent perturbation of Cx43 delivery to the plasma membrane in human and mouse myocardium.9 This finding highlights the importance of understanding the cytoskeletal trafficking mechanisms of ion channels and how this machinery is affected in disease.
The cellular cytoskeleton comprises microtubules, actin filaments, and intermediate filaments. Of these three components, microtubules and actin are understood to regulate intracellular vesicular transport both through motor protein transport and through regulation of events such as vesicular fusion with the plasma membrane.10,11 Actin micro-filaments provide the cellular architecture important for maintenance of cell shape but are highly dynamic and essential to enable cell motility, division, and morphogenesis. In muscle cells, such as cardiomyocytes, actin has an additional role in the form of sarcomeric thin filaments.12 Three isoforms of actin have been described; α, β, and γ. The α-actin is preferentially incorporated into sarcomeric thin filaments in cardiomyocytes, whereas β-actin and γ-actin are understood to predominate in the F-actin responsible for nonsarcomeric cellular functions.13 The formation and maintenance of specialized cardiomyocyte plasma membrane subdomains such as intercalated discs, caveolae, and T-tubules are also dependent on F-actin.14,15 The role of nonsarcomeric actin filaments in cardiomyocyte vesicular transport from the TGN to the plasma membrane, however, remains poorly defined.
In this study, we explored the role of actin in forward trafficking of Cx43-containing vesicles. We find that Cx43 colocalizes with nonsarcomeric actin structures in cardiomyocytes and filamentous actin structures in nonmyocyte cells. Live-cell imaging studies reveal that Cx43 vesicles surprisingly spend a majority of their time in the cytoplasm associated with actin structures. We then timed forward trafficking of de novo Cx43 hemichannels by utilizing the reversible endoplasmic reticulum (ER)/Golgi transport inhibitor Brefeldin A. Replacing a segment of the second extracellular loop of Cx43 with a hemagglutinin (HA) epitope tag permitted specific antibody-mediated biochemical and immunofluorescence detection of surface channels. Disruption of actin polymerization after release of Brefeldin A block revealed the actin cytoskeleton as necessary for forward trafficking of de novo Cx43 hemichannels to the plasma membrane. Formation of gap junction plaques with endogenous de novo Cx43 was also perturbed in similar experiments using primary cardiomyocytes. Moreover, we used Langendorff-perfused mouse hearts to investigate the effects of ischemic stress on actin-based Cx43 trafficking. Biochemical assays revealed disruption of Cx43/β-actin complexing during ischemia, and immunofluorescence studies show interference of actin polymerization using latrunculin A was sufficient to reduce Cx43 expression at intercalated discs to comparable levels of that found in ischemic hearts.
Methods
Mice
C57BL/6 mice used for primary cardiomyocyte isolation were maintained under sterile barrier conditions. All procedures were reviewed and approved by the University of California Institutional Animal Care and Use Committee. Detailed Methods are provided in the online-only Data Supplement.
Molecular Biology
Human Gja1 for Cx43 expression was cloned using Gateway technology (Invitrogen) as previously described.9 The LifeAct ENTR clone was constructed using DNA oligos encoding LifeAct (BamHI sense: GATCATGGGCGTGGCCGACCTGATCAAGAAGTTCGAGAGCATCAGCAAGGAGGAG, XhoI antisense: TCGACTCCTCCTTGCTGATGCTCTCGAACTTCTTGATCAGGTCGGCCACGCCCAT), which were annealed at 95°C and phosphorylated using T4 PNK (New England Biolabs [NEB]) before ligation into pENTR1a (Invitrogen) using BamHI and XhoI sites. Destination clones expressing Cx43-eGFP and LifeAct-mCherry were generated using the converted vectors pDEST-eGFP-N1 and pDEST-mCherry-N1 as previously described.8
We targeted the relatively variable region between the two peripheral cysteines at the apex of E2 for tag insertion with a glycine–proline-linked nine residue HA sequence via site-directed mutagenesis, restriction enzyme digestion, and ligation. Two unique restriction sites (XhoI sense primer: GTTTACACTTGCACTCGAGATCCCTG; AgeI antisense primer: GAACAGTCCACCGGTTGTGGGCAGG) were mutated into the E2 loop coding region of a human pENTR221-Cx43-stop construct using megaprimer mutagenesis polymerase chain reaction with the Quikchange II kit (Stratagene). As a result of this mutation, the nonconserved lysine at 188 was mutated to a threonine. To insert the HA tag into the modified E2 loop of Cx43, the mutated pENTR221-Cx43*-stop construct was codigested with XhoI and AgeI-HF (NEB). Digested DNA was dephosphorylated using calf intestinal alkaline phosphatase (NEB) and purified using the QIAquick polymerase chain reaction Purification Kit (Qiagen). Sense (TCGAGGCCCGGGCGGCTATCCGTATGATGTGCCGGATTATGCGGGCGGCGGC) and antisense (CCGGGCCGCCGCCCGCATAATCCGGCACATCATACGGATAGCCGCCCGGGCC) oligos coding for gly-prohemagluttinin with XhoI and AgeI overhangs (Integrated DNA Technologies) were annealed using touchdown polymerase chain reaction. Annealed oligos were then phosphorylated with T4 polynucleotide kinase (NEB) at 37°C for 30 minutes before inactivation at 65°C for 20 minutes. Annealed phosphorylated HA oligo was ligated into pENTR221-Cx43*-stop using T4 ligase (NEB) according to manufacturer’s instructions. The Cx43-E2HA-stop expression clone was generated using Gateway LR cloning (Invitrogen) into the pcDNA3.2-V5/Dest destination vector.
To generate HA-tagged transferrin receptor (TfR), a human TfR entry clone was obtained from the DF/HCC DNA Resource Core (clone ID: HsCD00041847). The N-terminal HA-tagged TfR expression clone was generated via Gateway LR cloning with a gateway-converted pCAN-HA-Dest plasmid (kind gift from F. McCormick, University of California San Francisco). For C-terminal tagging, pcDNA3.2-V5/Dest was converted to pcDNA3.2-HA/Dest by first introducing unique NotI and XhoI sites by sire-directed mutagenesis (Not I sense: GGCCTACCCATACGACGTCCCAGATTACGCG, XhoI sense: CCCTCTCCTCGGTCTCGAGTCTACGCGTACCGG), followed by ligation of annealed HA-stop peptide as described (sense primer: GGCCCGTACCCATACGACGTCCCAGATTACGCGCGTACCGGTTAGTAATGAC, antisense: TCGAGTCATTACTAACCGGTACGCGCGTAATCTGGGACGTCGTATGGGTACG).
The R202E mutation was generated in the pENTR221-Cx43-stop clone using the sense (GACTGTTTCCTCTCTGAGCCCACGGAGAAAACC) and antisense (GGTTTTCTCCGTGGGCTCAGAGAGGAAACAGTC) primers and the with the Quikchange II kit (Stratagene) according to manufacturer’s instructions. Gateway LR cloning was then used with pCAN-HA-Dest to generate the N-terminally tagged expression clone pCAN-HA-Cx43R202E-stop.
Cell Culture
Primary adult and P3 neonatal mouse cardiomyocytes were isolated and either fixed or maintained in culture as previously described.8,9 HaCaT cells were a kind gift from R. Derynck (University of California San Francisco) and were maintained in DMEM supplemented with 10% fetal bovine serum, nonessential amino acids, sodium pyruvate (Invitrogen), and Mycozap (Lonza). Stable overexpression of Cx43 in HaCaT cells was achieved through transduction with pLenti6.3-hCx43-stop as previously described.9 HeLa cells were obtained from ATCC and maintained as previously described.16
Immunofluorescence
Cells were fixed with 4% paraformaldehyde (Electron Microscopy Services) in phosphate-buffered saline (PBS) for 30 minutes and immunostained for Cx43 (rabbit polyclonal, 1/3000; Sigma) and α-actinin (mouse monoclonal, 1/500; Abcam) as previously described.8,9 F-actin and cell membranes were identified using phalloidin-AlexaFluor555 and wheat germ agglutinin AlexaFluor647 (Invitrogen), respectively, according to manufacturer’s instructions. ProLong gold-containing DAPI (Invitrogen) was used to mount slides for image acquisition.
Live Cell Imaging
HaCaT cells were plated on glass-bottom dishes (MatTek) and cotransfected with pDEST-Cx43-eGFP-N1 and pDEST-LifeAct-mCherry-N1 using lipofectamine 2000 (Invitrogen) according to manufacturer’s instructions. Sixteen hours after transfection, medium was replaced with warm Hanks balanced salts solution (HBSS) containing 10% fetal bovine serum and live-cell spinning-disk confocal microscopy performed at 37°C with acquisitions every 2.5 seconds for 3 minutes as previously described8,9 using a Yokogowa CSU-X1 spinning disk confocal unit with 486 and 561 nm laser sources and Coolsnap HQ2 camera controlled by NIS Elements software.
Surface Detection of HA-Tagged Cx43 Hemichannels
Transfected cells were washed twice briefly with HBSS (Invitrogen) and blocked for 5 minutes on ice in HBSS with 5% bovine serum albumin (Sigma). Supernatant was removed and monoclonal mouse anti-HA antibody (Sigma) was added in HBSS at a concentration of 1 μg/mL. Cells were incubated for another 5 minutes on ice before three brief washes with HBSS. For surface immunoprecipitations, cells were lysed in RIPA buffer at this point. For immunofluorescence, AlexaFluor555 goat antimouse (Invitrogen) was diluted 1/1000 in HBSS and added to cells. After 5 minutes of incubation on ice, cells were washed five times with HBSS and fixed for 30 minutes in 4% paraformaldehyde. To detect total HA, fixed cells were then permeabilized for 5 minutes with 0.1% Triton-X 100 in PBS and blocked for 1 hour at room temperature with 5% normal goat serum (NGS) in PBS. Cells were then incubated for 1 hour with mouse anti-HA antibody diluted 1/20 000 in 5% NGS in PBS before several PBS washes and incubation for another hour in AlexaFluor488 goat antimouse (Invitrogen) diluted 1/500 in 5% NGS in PBS. Unbound antibodies were removed with several PBS washes and cells were mounted in ProLong gold-containing DAPI.
Immunoprecipitation of Surface Cx43-E2HA
For surface protein immunoprecipitation, RIPA lysates were sonicated and clarified by centrifugation before protein concentration determination (using the BioRad DC protein assay) and normalization; 1 mg of protein was then nutated for 30 minutes with 20 μL Dynabeads ProteinG (Invitrogen) at 4°C. Beads were washed five times with 1 mL RIPA buffer and bound protein was denatured and resolved by SDS-PAGE and Western blot as previously described.9
Brefeldin A Trafficking Block and Release Assay
Cells were treated with 2.5 μg/mL Brefeldin A (Sigma) or DMSO vehicle control for 16 hours. Drugs were removed with three washes of warm PBS, medium was replaced, and cells were incubated for another 2 hours to allow proteins to move into the Golgi apparatus. Latrunculin A (Sigma) was added or not to a final concentration of 2 μg/mL and cells were incubated for another 2 hours. Cells were then fixed for immunofluorescence studies or surface-labeled live for HA-tag surface immunoprecipitation or immunofluorescence.
Langendorff-Perfused Mouse Heart Preparation
Hearts from 6- to 8-week-old C57BL/6 mice were cannulated and maintained on a Langendorff perfusion apparatus as previously described.9 Normal perfusion was maintained for 30 minutes, followed by 15 minutes of perfusion with 1 μmol/L latrunculin A (Sigma) or DMSO. Hearts were then either perfused for another 30 minutes for control conditions or perfusion was terminated and hearts were exposed to ischemia for 30 minutes. During no-flow ischemia, the heart was immersed in warm K-H buffer to maintain warmth and moisture. Immediately after Langendorff procedure, hearts were placed in cryovials and snap-frozen in liquid nitrogen for biochemical studies. For cryosectioning, hearts were embedded in OCT (Sakura Finotek) and snap-frozen by immersing in liquid nitrogen-chilled isopentane to snap-freeze before storage at −80°C.
Coimmunoprecipitation of Cx43/β-Actin From Primary Adult Mouse Cardiomyocytes and Mouse Heart Tissue
To enrich cytoplasmic Cx43 pools, a low-detergent coimmunoprecipitation lysis buffer was prepared (0.1% Nonidet P-40; Roche; 50 mmol/L Hepes, pH 7.4; 150 mmol/L KCl; 1 mmol/L EDTA; 1 mmol/L EGTA; 1 mmol/L DTT; 1 mmol/L NaF; 0.1 mmol/L Na3VO4) supplemented with HALT protease and phosphatase inhibitors (Pierce). Tissue was weighed and homogenized for 10 seconds in lysis buffer and immunoprecipitation was performed as previously described.9 For pull-downs, 2 μg of antibody was used (mouse anti-β-actin, Sigma; mouse anti-GST as IgG control, SantaCruz). Freshly isolated primary adult mouse cardiomyocytes were plated and maintained in the presence or absence of 1 μmol/L latrunculin A for 2 hours before lysis and immunoprecipitation.
Cryosectioning and Immunofluorescence of Mouse Heart Tissue
Cryosections (8 μm) were prepared and positioned on poly-l-lysine–coated slides before acetone fixation and air drying. Sections were rehydrated for 10 minutes in PBS, followed by 1 hour of blocking at room temperature with 5% NGS in PBS. After blocking, sections were incubated overnight at 4°C with primary antibodies diluted in 5% NGS; rabbit anti-Cx43 (1:3000; Sigma) and mouse anti-N-cadherin (1:200; BD Biosciences). After several PBS washes, cells were incubated for an additional hour at room temperature with goat secondary antibodies conjugated to Alexa Fluor 488 or 555. Slides were then washed with PBS, briefly washed with dH2O, and coverslips were mounted using ProLong gold antifade reagent containing DAPI. Slides were dried overnight and imaged using a Nikon Ti microscope with a ×60/1.49 Apo TIRF objective, Yokogowa CSU-X1 spinning disk confocal unit with 486 and 561 nm laser sources, and Coolsnap HQ2 camera controlled by NIS Elements software.
Image Analysis
Colors in Figures are chosen for clarity and may not correlate to the emission spectra of the actual fluorophore used. ImageJ software (NIH) was used for all image analysis. To determine vesicle velocities in live-cell acquisitions (Figures 2 and 3), the MTrackJ plugin was used to track individual vesicles. Only vesicles that traversed more than 10 μm were tracked. Actin images were thresholded and colocalization of Cx43 track and actin were scored at each point for each vesicle being tracked (n=14 cells, 50 vesicles).
Figure 2. Connexin 43 (Cx43) vesicles experience slower velocities when associated with actin structures.
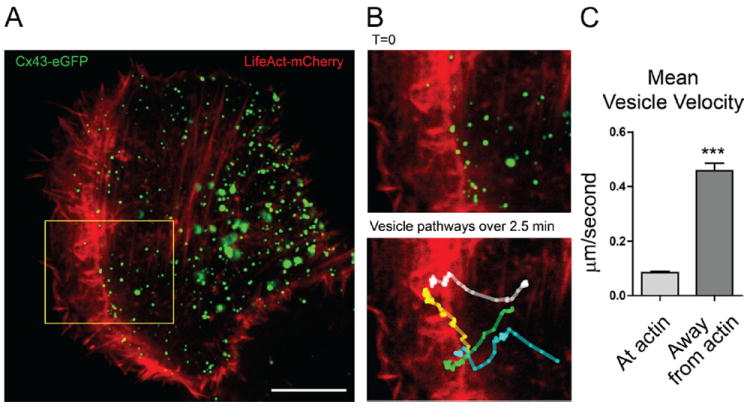
HaCaT cells transiently expressing Cx43-eGFP (green) and LifeAct-mCherry (red) were imaged by spinning-disk confocal microscopy. A, Representative frame of entire cell from which individual Cx43 vesicles were tracked. B, Enlarged region and colored vesicle tracks (bottom frame has full move collapsed into a single image). C, Average Cx43 velocities when colocalized with or without actin. Data are from 14 individual cells from which 50 vesicles were tracked, generating a total 2663 data points. n=2274 for “at actin” and n=389 for “away from actin.” Original magnification: ×100; scale bar: 10 μm.
Figure 3. Distribution of intracellular connexin 43 (Cx43) particle velocities.
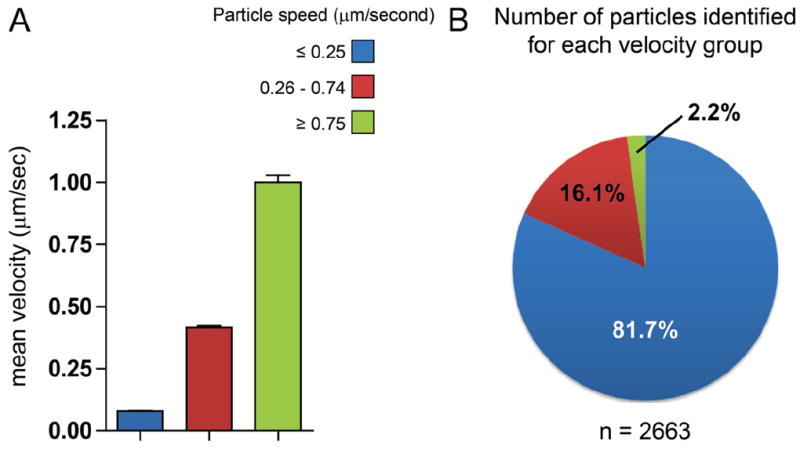
HaCaT cells were cotransfected with Cx43-eGFP and LifeAct-mCherry. Individual Cx43 vesicle dynamics were tracked over time using the MTrackJ plugin for ImageJ. Particles were arbitrarily separated into three groups based on speed: ≥0.25 μm/s (blue), 0.26 to 0.74 μm/s (red), and ≥0.75 μm/s (green). Group velocities are plotted in (A) and proportional distribution of groups is represented by a pie chart in (B). Data are from 14 individual cells from which 50 vesicles were tracked, generating a total 2663 data points.
To quantify surface HA expression (Figure 5), maximum intensity projections were generated for confocal z-stacks of whole cells. A region of interest was defined by outlining the entire cell and fluorescence intensity within this region of interest measured for total and surface HA images. Background was subtracted and surface HA values were divided by total HA (n=10 cells per condition).
Figure 5. Actin disruption perturbs delivery of de novo connexin 43 (Cx43) to the cell surface.
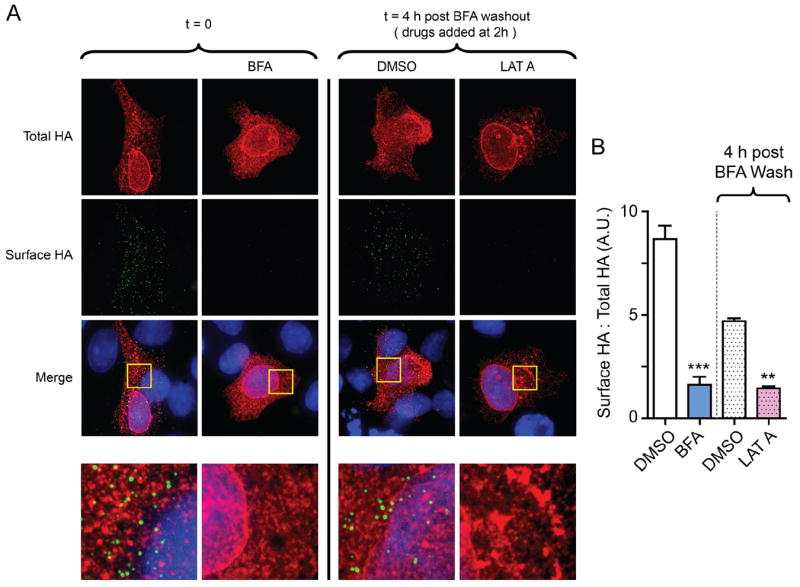
HaCaT cells were transfected with Cx43-E2HA and treated with Brefeldin A for 16 hours to block endoplasmic reticulum (ER)/Golgi transport. A, DMSO or latrunculin A was added to cells 2 hours after Brefeldin A washout and surface immunofluorescence was performed 2 hours after addition of drug. Surface hemagglutinin (HA) is presented in green, and total HA is presented in red (100×). Bottom panels are zoomed-in squares from merge images. B, Ratiometric quantification of surface HA expression in (A), relative to total HA (n=10 cells per condition).
For quantification of Cx43 expression at cell–cell borders (Figures 6 and 7), wheat germ agglutinin images were used to identify cell border regions. These were then applied to maximum intensity projections of 7.5 μm confocal z-stacks of Cx43 signal. Ten 10-μm lines were drawn perpendicular to cell–cell borders and the plot profile function in ImageJ was used to quantify Cx43 levels. Finally, a 150×150-pixel square was used to quantify background in a cell-free region of the image for background subtraction (n=10 cell pairs for each condition).
Figure 6. Actin structures are necessary for forward trafficking of de novo connexin 43 (Cx43) hemichannels from the Golgi apparatus to the plasma membrane in HaCaT cells.
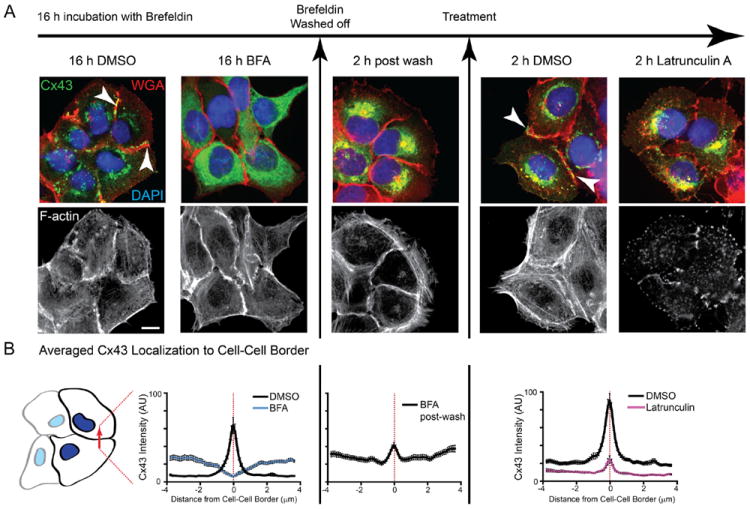
HaCaT cells were treated with Brefeldin A (BFA) to inhibit vesicular transport and released after 16 hours. A, Fixed cells were immunostained for Cx43 (green), cell membranes were labeled with wheat germ agglutinin (red), and nuclei were counterstained with DAPI (blue). Filamentous actin was labeled with phalloidin (black and white panels). B, Fluorescence intensity profiles of Cx43 expression at cell–cell borders. Original magnification: ×60. Scale bar: 10 μm.
Figure 7. Actin structures are necessary for forward trafficking of de novo connexin 43 (Cx43) hemichannels from the Golgi apparatus to the plasma membrane in neonatal cardiomyocytes.
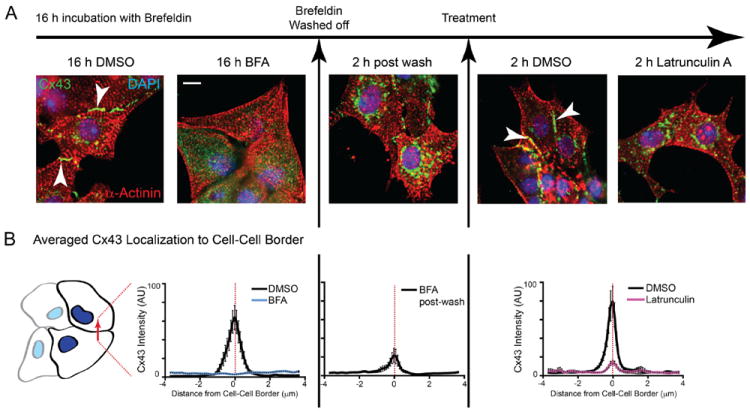
Neonatal mouse cardiomyocytes were treated with Brefeldin A (BFA) to inhibit vesicular transport and released after 16 hours. A, Fixed cells were immunostained for Cx43 (green) and α-actinin (red) and nuclei were counterstained with DAPI (blue). B, Fluorescence intensity profiles of Cx43 expression at cell–cell borders. Original magnification: ×60; scale bar: 10 μm.
Quantification of Cx43 expression at intercalated discs (Figure 8) was based on colocalization with N-cadherin. Using ImageJ software, background was subtracted from N-cadherin images, to which equal thresholds were applied, generating binary masks of intercalated disc regions. Within this mask image, intercalated disc regions had a value of 1 and all remaining pixels had a value of 0. Masks were image-multiplied by corresponding Cx43 images, and fluorescence intensity was subsequently measured. For each sample, six randomly selected images from three separate hearts were analyzed per condition.
Figure 8. Acute ischemia or actin disruption alone limits connexin 43 (Cx43) expression at the intercalated disc in Langendorff-perfused mouse hearts.
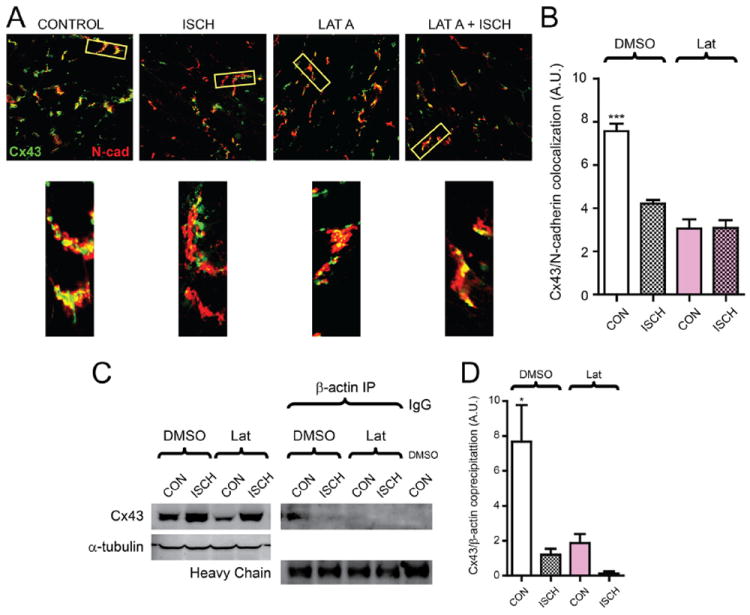
Hearts from 6- to 8-week-old C57BL/6 mice were maintained using a Langendorff perfusion apparatus for 15 minutes, followed either by 15 minutes of normal perfusion (control) or with 1 μmol/L latrunculin A. Perfusion was then continued for another 30 minutes, or hearts were subjected to no-flow ischemia for 30 minutes. A, Immunofluorescence labeling of Cx43 (green) and N-cadherin (red) in cryosections from snap-frozen hearts. Bottom panels are zoomed-in regions to highlight Cx43/N-cadherin colocalization. B, Quantification of Cx43/N-cadherin colocalization in (A). C, Coimmunoprecipitation of Cx43 with β-actin from mouse heart tissue lysates. D, Densitometry of precipitated Cx43 relative to total Cx43 in lysate for (C). n=3 hearts for each condition.
Results
Cx43 Vesicles Colocalize With Actin in Cardiomyocytes
Cx43 vesicles are transported along the microtubule cytoskeleton from the TGN to their destination in the plasma membrane. We were interested in the distribution of nonsarcomeric actin in adult cardiomyocytes and the spatial relationship between actin and Cx43 vesicles. Adult mouse cardiomyocytes were isolated and incubated on glass-bottom chamber slides for 2 hours before fixation and high-resolution imaging. We found regions of Cx43 enrichment, as seen in confocal slices presented in Figure 1A, to be perinuclear, which is their trafficking origin, and at intercalated discs, which is their destination. Also, between the Golgi apparatus and intercalated discs are multiple vesicles of Cx43. These cytoplasmic localizations of Cx43 often colocalized with nonsarcomeric actin structures, visualized as the yellow punctae between blue myofibrils of α-actinin in the overlay image of Figure 1A, which is a combination of Cx43 in green and actin in red.
Figure 1. Intracellular connexin 43 (Cx43) colocalizes with nonsarcomeric actin structures in adult mouse cardiomyocytes.
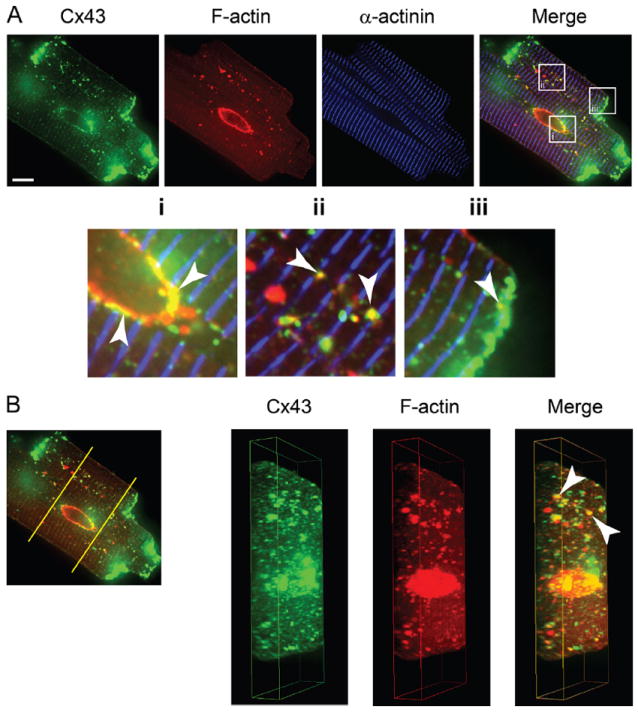
Freshly isolated adult mouse cardiomyocytes immunolabeled for Cx43 (green), filamentous actin (red), and α-actinin (blue). A, A 0.5-μm slice from spinning-disk confocal z-stack. Enlarged regions indicate colocalization of Cx43 and nonsarcomeric F-actin (arrows). B, Three-dimensional reconstruction and cross-section of cardiomyocyte from confocal z-stack. Original magnification: ×100; scale bars: 10 μm.
To better-visualize the cellular distribution of cytoplasmic Cx43 vesicles and actin, we generated three-dimensional reconstructions of cardiomyocytes from confocal Z-stacks (Figure 1B; full 360-degree view in Supplemental Movie I). These reconstructions confirm that cytoplasmic Cx43 and nonsarcomeric actin colocalize. Visually, it is apparent that these collections are not associated with the sarcolemma (Figure 1B, arrows). Moreover, using a low-detergent buffer to biochemically enrich nonsarcolemmal Cx43, it was possible to coprecipitate Cx43 with (nonsarcomeric) β-actin from isolated adult mouse cardiomyocytes. Complexing of Cx43 and β-actin was ablated with 2 hours of treatment with the actin-disrupting drug latrunculin A (Supplemental Figure I). Given the role of Cx43 as a plasma membrane protein, the actin-based collections of cytoplasmic Cx43 are likely to be in the process of trafficking. Thus, we investigated the role of actin in Cx43 vesicular transport.
Cx43-Containing Vesicles Pause at Actin Filaments En Route to the Plasma Membrane
Live-cell imaging permits exploration of the time course and directionality of Cx43 movement in relation to actin structures. HaCaT cells, which express low levels of endogenous Cx43, were transiently transfected with Cx43-eGFP and LifeAct-mCherry. LifeAct associates with filamentous actin in live cells, with minimal interference of actin dynamics.17 In these cells, Cx43 and actin were imaged at the same time points, for a total imaging time of 3 minutes and sampling rate of 2.5 seconds. Within each movie, the movements of individual Cx43 vesicles were identified and tracked using the MTrackJ plugin for ImageJ. A representative frame from a movie is shown in Figure 2A. We focused on dynamic vesicles for this study, and only those that traversed at least 10 μm during the time course acquired were tracked. Four representative vesicles are tracked in Supplemental Movie II and presented in the still-frame of Figure 2B. From the movies, a consistent pattern emerged that rapid vesicular movements occur when the vesicles are not associated with actin structures. The corollary is also true. Significantly slower Cx43 vesicle velocities occur when the vesicles are coincident with actin fibers, with average velocities of 0.09 μm/s in comparison to 0.46 μm/s for vesicles away from actin (Figure 2C).
Anterograde vesicular transport along cytoskeleton structures is dependent on kinesin transporters for microtubule-based transport and the myosin family for actin-based transport. It previously has been found that connexin transport can occur along microtubules at speeds consistent with kinesin.6,7 We arbitrarily grouped particles into three populations based on velocity to examine the distribution of “fast” (≥0.75 μm/s) versus “slow” (≤0.25 μm/s) vesicular transport, with the middle range accounting for transitioning particles (Figure 3). These values were designated based on previous studies that report, depending on the cell system and cargo, kinesin-based vesicular transport has reported speeds up to 1.8 μm/s in vivo,18 whereas myosin-based transport along actin is much slower, with average travel velocities of 0.2 μm/s.19 Average velocities of each group are presented in Figure 3A, whereas the proportional distribution of each group within the population of 2663 points is plotted in Figure 3B. The striking result of these data are that the majority of particles (81.7%) travel at speeds of 0.25 μm/s or slower, with only 2.2% traveling at 0.75 μm/s or higher. Speeds lower than 0.25 μm/s are more consistent with myosin-based rather than kinesin-based transport. More generally, at any given point in time, the majority of Cx43 vesicles are not undergoing microtube-based transport.
Actin Is Necessary for Transport of Cx43 to the Cell Surface
The dynamically tracked Cx43 vesicles of Figures 2 and 3 were selected for those involved in forward trafficking (membrane delivery) and not retrograde trafficking (internalization). To rigorously test the involvement of actin in forward trafficking, we introduced Brefeldin A, which reversibly blocks ER/Golgi transport of vesicles and, in time, causes accumulation of proteins within the ER.20,21 We previously timed forward trafficking of de novo Cx43 hemichannels in HeLa cells using a tetracycline-inducible Cx43-eYFP fusion.16 Brefeldin A block permits the timed release of endogenous membrane proteins from the ER, isolating forward trafficking for study.
To establish the timing of Cx43 forward trafficking after Brefeldin A block, we developed a Cx43 mutant containing an extracellular epitope tag for reliable surface detection. The variable region between the two peripheral cysteines at the apex of the second extracellular loop of Cx43, E2, was replaced with a glycine–proline-linked nine residue HA sequence via site-directed mutagenesis (Figure 4A). To establish that this modified Cx43 protein (Cx43-E2HA) could be detected on the cell surface in HeLa cells, we performed surface protein immunoprecipitation and immunofluorescence labeling in transiently transfected cells. For surface immunoprecipitation experiments, live cells were incubated with anti-HA antibody before lysis and pull-down (Figure 4B). Cx43-E2HA and extracellularly tagged transferrin receptor (TfR-HA, positive control) were detectable by Western blot analysis of HA-immunoprecipitated surface protein, whereas intracellularly HA-tagged variants of TfR (HA-TfR) were not and the trafficking mutant Cx43 (HA-Cx43R202E) showed very low signal (Supplemental Figure II). Similar data were obtained using HaCaT cells (Figure 4B). Cx43R202E is a previously described trafficking mutant22 that remains trapped in the ER and served as a negative control in these experiments. We chose to tag this Cx43 construct with the HA epitope at the N-terminus to further abrogate forward trafficking and to limit rescue of forward transport by endogenous Cx43. Note that this fusion protein migrates at a higher molecular weight than Cx43-E2HA because it is an addition of the HA epitope to the Cx43 N-terminus rather than a substitution of part of the E2 region. A faint surface band was observed for HA-Cx43R202E in both cell lines, however, so some trafficking to the cell surface may have occurred, although these bands were relatively faint considering comparable input levels to Cx43-E2HA and IgG background signal. To visualize both extracellular and intracellular HA, live cells were incubated with primary anti-HA antibody, followed by fluorophore-conjugated secondary antibody. Cells were then fixed, permeabilized, and immunofluorescently labeled for intracellular HA before confocal image acquisition. All HA-tagged constructs displayed intracellular HA labeling, but only TfR-HA or Cx43-E2HA was detectable by extracellular HA staining (Supplemental Figure IA). Ratiometric analysis of extracellular to intracellular HA signal was significant only for cells expressing TfR-HA or Cx43-E2HA (Supplemental Figure III).
Figure 4. Detection of surface connexin 43 (Cx43) expression by insertion of a hemagglutinin (HA) epitope tag in the second extracellular loop.
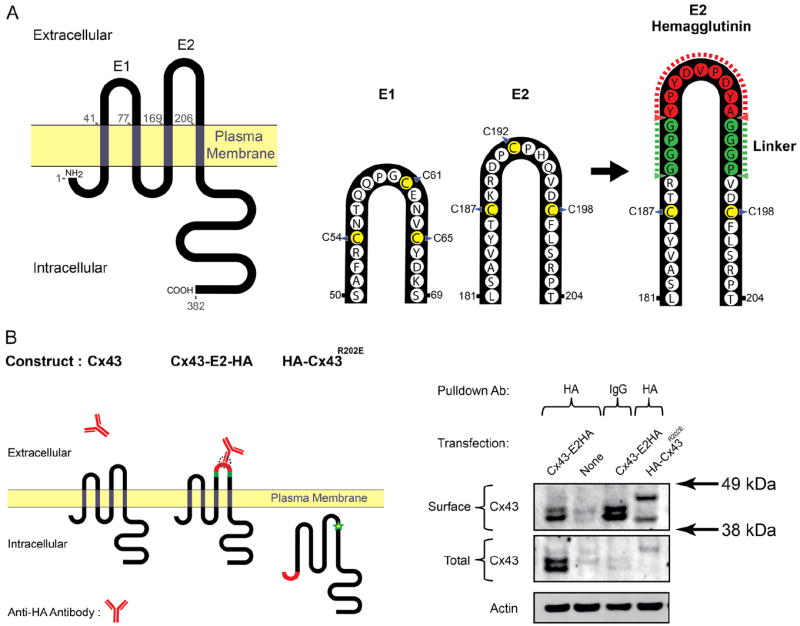
By site-directed mutagenesis of unique restriction sites and subsequent ligation, a HA epitope tag was inserted into the second extracellular loop of Cx43 (E2) together with a Pro-Gly linker sequence (A). B, Cx43-E2HA was transfected into HaCaT cells and surface expression was confirmed by incubation of live cells with anti-HA antibody and immunoprecipitation.
We used Cx43-E2HA to time forward transport of de novo Cx43 from the ER after Brefeldin A treatment. Transiently transfected HaCaT cells were pretreated for 16 hours with Brefeldin A and surface HA immunoprecipitation was performed at 0, 2, and 4 hours after Brefeldin A washout. No surface Cx43 was detected at 0 or 2 hours, but a strong signal was present by 4 hours after wash (Supplemental Figure IV, top row). From these data, we conclude that de novo Cx43 hemichannels traffic to the plasma membrane within 4 hours of release from Brefeldin A-mediated vesicular transport block. To then test the requirement of actin structures in delivery of Cx43 to the plasma membrane, we introduced an actin polymerization inhibitor, latrunculin A, 2 hours after Brefeldin A washout. By surface immunofluorescence, we found that disruption of actin by latrunculin A perturbed delivery of Cx43-E2HA to the surface (Figure 5A, zoomed-in merged images are shown in bottom panels). Ratiometric quantification of surface expression with and without latrunculin A treatment is presented in Figure 5B. These studies support a role for actin in the anterograde transport of de novo Cx43 hemichannels to the plasma membrane.
Actin Structures Are Necessary for Cx43 Forward Trafficking and Gap Junction Plaque Formation
Having found that Cx43 vesicles colocalize and dynamically interact with actin filaments en route to the plasma membrane (Figures 1, 2, 3), and that filamentous actin is important for delivery of Cx43 to the plasma membrane (Figure 5), we were interested in the role of actin in wild-type Cx43 gap junction plaque formation. To first test this in a cell line, we used lentiviral transduction to generate a HaCaT cell line stably expressing Cx43 and performed timed trafficking experiments using Brefeldin A as in Figure 5.
As seen in the first two columns of Figure 6A, after 16 hours of exposure to Brefeldin A, treated cells no longer have Cx43 at cell–cell borders with the majority of Cx43 signal in a reticular pattern consistent with the ER. By 2 hours after Brefeldin A washout, Cx43 is enriched at the perinuclear Golgi apparatus (Figure 6A, middle column). At this point, with an enrichment of Cx43 hemichannels in the Golgi apparatus awaiting transport to the membrane, the cells were treated with either latrunculin A to inhibit actin polymerization or DMSO as vehicle control. DMSO-treated cells developed rich deposits of de novo Cx43 gap junctions at cell–cell borders within the next 2 hours. Disruption of actin using lantrunculin A, however, was sufficient to prevent delivery of these de novo Cx43 channels to the plasma membrane (Figure 6A, right columns). To quantify levels of Cx43 localization at cell–cell borders, 10-μm lines were drawn perpendicular to, and bisecting, cell–cell borders. Averaged fluorescence intensity profiles of these lines are presented in Figure 6B. As seen in the last panel of Figure 6, latrunculin A limits Cx43 delivery to cell–cell borders by 75%.
Comparable data were obtained in neonatal cardiomyocytes and are presented in Figure 7. Inhibiting actin with latrunculin A results in 82% decrease of Cx43 at the cell–cell border of neonatal cardiomyocytes. Based on the HaCaT cells studies, as well as those in primary neonatal cardiomyocytes, it appears that actin dependence on Cx43 forward trafficking is generalizable to multiple cell types.
Ischemic Stress Disrupts Cx43/β-Actin Interaction in Langendorff-Perfused Mouse Hearts
To investigate the potential pathophysiological role of actin-based Cx43 transport, we used Langendorff-perfused mouse hearts subjected to acute no-flow ischemia, latrunculin A, or both. Ischemic stress resulted in altered localization of Cx43 (green) as determined by immunofluorescence analysis with N-cadherin (red) as a marker of the intercalated disc. Latrunculin A treatment was sufficient to reduce Cx43 levels at the intercalated disc in a comparable manner to ischemia, but these values did not decrease further after ischemia combined with latrunculin A treatment (Figure 8A, B). Using a low-detergent buffer to enrich cytoplasmic Cx43, we analyzed levels of Cx43 coprecipitating with β-actin, which is understood to be predominantly nonsarcomeric.13 Total lysates revealed increases in these soluble Cx43 levels during ischemia in comparison to control lysates, consistent with our previous studies in human tissue.9 Despite an enrichment in total Cx43 in these soluble fractions, ischemia, latrunculin A, and ischemic latrunculin A hearts all showed almost a complete lack of detectable complexing of Cx43 and β-actin, whereas control hearts displayed robust complexing (Figure 8C, D). These findings suggest actin is necessary for transport and maintenance of Cx43 at intercalated discs, and actin-Cx43 interaction is limited during ischemic stress, inhibiting Cx43 plaque size.
Discussion
The Cx43 life cycle consists of forward/anterograde trafficking, movements within the plasma membrane, and retrograde trafficking.23,24 It is well-established that Cx43 vesicles are transported from the TGN along microtubules to the plasma membrane.7,9,25,26 Furthermore, specific plus-end tracking proteins and membrane anchors allow microtubule-based directed delivery of channels to the intercalated disc.7,9 The actin cytoskeleton previously has been identified to be involved in melanosome movements,27 and “actin barriers” have been described in impeding endocytosis.28 There is increasing evidence for cooperation between the actin and microtubule cytoskeletal systems in active transport to vesicles to the plasma membrane29 and, more recently, long-range vesicular transport solely along actin filaments to the plasma membrane has been demonstrated in mouse oocytes.30 Interestingly, disruption of the actin cytoskeleton has been reported to affect Cx43 gap junction coupling in astrocytes31 and the assembly and maintenance of connexin 30 gap junctions in HeLa cells.32 In this study, we provide direct evidence that, in addition to microtubules, the actin cytoskeleton is a necessary component of the Cx43 forward trafficking machinery in cardiomyocytes.
One of the key findings in this study is that Cx43 vesicles pause, slow down, and generally exist in the cytoplasm for a substantial amount of time before plasma membrane delivery. A recent report by Fort et al6 on connexin 32 transport in liver cells also reports vesicles pausing en route. It is not clear why Cx43 vesicle transport involves such a substantial time in the cytoplasm. A possibility is that Cx43 trafficking requires multiple microtubules to traffic from the TGN to the plasma membrane. Microtubules are highly dynamic structures whose plus-ends exist in a state of dynamic instability and last from seconds to minutes before undergoing catastrophe.33,7 Therefore, actin filaments could act as a transition point for Cx43 en route to the plasma membrane. Interestingly, a close relationship between actin transport machinery and the microtubule plus-end binding protein EB1 has been observed and proposed as a mechanism of transfer of vesicles between microtubules and actin.29 The microtubule that carries the Cx43 vesicle from the TGN may not necessarily be the microtubule that effects the final delivery at the plasma membrane. There are several advantages for a staccato type of delivery event. First, premade Cx43 hemichannels can be stored near, but not in, the plasma membrane, providing a source of channels that could be quickly accessed and delivered to the membrane should the need arise, such as during an acute ischemic event. Second, membrane targeting need not occur on the initial microtubule that transports that Cx43 vesicle from the TGN. Cx43 vesicles could be positioned at actin-based intracellular reserves near the membrane, awaiting a microtubule with the appropriate plus-end-tracking protein to provide the final delivery event.
Many unconventional myosins, such as myosin V, are now known to be involved in vesicular transport along actin and are distinct from those of the sarcomere. Myosin Va previously has been implicated in tethering melanosomes near the cell periphery,34 as well as in effecting actual transport along actin structures.35 When associated with actin, the relatively slow Cx43 vesicle velocities reported here are consistent with myosin-based vesicular transport.19 We therefore can speculate that Cx43 vesicles can be transferred from kinesin-based transport machinery on microtubules to unconventional myosins on actin filaments as they are trafficked through the cell interior. This would also account for the dramatic differences in velocity that occur within individual Cx43 vesicle trafficking paths. Identification of the motor proteins responsible for actin-based Cx43 trafficking in cardiomyocytes and how transfer to and from microtubules occurs will represent a key step in understanding how these processes are regulated.
We previously have described disruption of the microtubule Cx43 trafficking apparatus occurs in stressed mouse and human myocardium, whereby displacement of EB1 from microtubule plus-ends perturbs delivery of Cx43 to the plasma membrane.9 Susceptibility of the actin cytoskeleton to cellular stresses such as ischemia is known to occur in many cell types, and activation of pathways leading to disruption of actin structures has been demonstrated in renal smooth muscle cells.36 Although disruption of myofibrillar structures is well-described in stressed and failing hearts,37-39 the Langendorff perfusion experiments in this study provide novel insight into the fate of nonsarcomeric actin during ischemic stress in cardiomyocytes. Abrogation of Cx43 complexing with β-actin structures during ischemia supports a necessary role for the actin cytoskeleton in gap junction formation. Consistent with our studies in isolated cardiomyocytes and cell lines, disruption of actin polymerization is sufficient to reduce Cx43 gap junction levels. It is worth noting that exposure of hearts to ischemia in addition to latrunculin A-mediated actin disruption did not lead to additive reductions in Cx43 remodeling, suggesting a common mechanism. In addition to a direct role in vesicular trafficking, it is possible that actin lays the scaffold by which microtubules are organized, laying the foundation for the path Cx43 vesicles take to the plasma membrane.40,41
Cardiomyocytes are highly structured, with different ion channels existing in distinct subdomains of the cardiomyocyte sarcolemma.16,42 Recently, explorations have begun to uncover the importance and mechanisms of trafficking in determining ion channel density on the cell surface. In an analysis of 34 KV11.1 (hERG) mutations, Anderson et al43 determined that most (82%) of these mutations led to defective trafficking rather than defective channel kinetics. In the atria, a deletion mutation in the Cx43 c-terminus has been identified that disrupts hemichannel trafficking and is associated with atrial fibrillation.44 The adherens junction structure provides an anchor for EB1 and p150GLUED working in concert to effect targeted microtubule-based delivery of gap junctions to intercalated discs in the ventricle7 in a process that is disrupted under conditions of oxidative stress.9 More aspects of the ion channel-targeting complexes are being uncovered, with the ankyrin family of scaffolding proteins now known to localize NaV1.5 to intercalated discs and T-tubules (ankyrin-G)45 and NCX and sodium–potassium ATPase to T-tubules (ankyrin-B).46 Ankyrin-G may have multiple roles at the intercalated disc organizing gap junctions, desmosomes, and sodium channels.47 We also recently determined that the L-type calcium channels can be directly delivered to T-tubule membrane by microtubules via the BIN1 membrane scaffolding protein acting as an anchor for calcium channel delivery much in the way adherens junctions anchor Cx43-based delivery.8
Cx43 exists within the cytoplasm in many membranous compartments of the cardiomyocyte, from the ER through the Golgi, within individual transport vesicles, and, finally, endosomal and lysosomal structures. Another nonsarcolemmal population of cytoplasmic Cx43 has been identified within the mitochondrial membrane, and although this phenomenon appears closely linked to cell survival and stress pathways, the model of delivery of Cx43 to the mitochondrial membrane remains elusive. In the future it will be interesting to explore cytoskeleton-based trafficking of Cx43 to nonsarcolemmal membrane.
The findings that actin is involved in Cx43-based forward delivery and that this process is perturbed during ischemia introduce interesting complexity to the forward trafficking paradigm. Cx43, and presumably other ion channel-containing vesicles, need not be restricted to a single microtubule for their membrane delivery event. It will be interesting to determine whether actin serves to maintain intermediate pools for reserves of channel or for allowing specific membrane targeting. We are finding that a complex interplay between different members of the cytoskeleton exists that regulates ion channel trafficking to the plasma membrane. Further understanding the role of actin in gap junction regulation increases opportunity for preservation or enhancement of channel density by manipulating cytoskeleton-based vesicular transport in cardiomyocytes.
Supplementary Material
Novelty and Significance.
What Is known?
Alterations in connexin 43 (Cx43) gap junctional coupling occur in many forms of heart disease and can contribute to the arrhythmias of sudden cardiac death.
The microtubule cytoskeleton transports newly synthesized Cx43 hemichannels from the cardiomyocyte interior to specific subregions of the cardiomyocyte plasma membrane.
What New Information Does This Article Contribute?
The majority of intracellular Cx43 is associated with nonsarcomeric actin; in this state, Cx43 channels are either stationary or move at very slow velocities.
Actin is necessary for Cx43 to be delivered to the plasma membrane and likely cooperates with microtubules for organized Cx43 trafficking.
Acute ischemia disrupts actin interaction with Cx43, thus actin preservation may be an option for improving gap junctional coupling in the ischemic heart.
Ventricular gap junctions comprise Cx43, a protein with a half-life of hours. Remodeling of gap junctions occurs rapidly after cardiac insult and contributes to the arrhythmias of sudden cardiac death. The dynamic microtubule cytoskeleton is well-established as necessary in effecting the delivery of Cx43 to the intercalated disc, and we previously have found this process to be perturbed during stress. However, the role of nonsarcomeric actin in Cx43 vesicular transport has not been explored. Here, we examine how the actin cytoskeleton participates in Cx43 trafficking to the plasma membrane. In this study, we find that Cx43 vesicles colocalize with nonsarcomeric actin structures in cardiomyocytes and, at any one time, most Cx43 vesicles are either stationary or moving at relatively slow velocities consistent with myosin-based transport. Moreover, we demonstrate that the actin cytoskeleton is necessary for delivery of Cx43 vesicles to the cell surface and that the interaction between Cx43 and actin is ablated during ischemic insult. These findings highlight a novel role for nonsarcomeric actin in the regulation of cardiomyocyte gap junctional coupling. A complex interplay between microtubules and actin therefore exists in intracellular ion channel trafficking. Further exploration of this process will contribute to the development of therapies aimed at preserving normal cardiac electric function.
Acknowledgments
The authors are grateful to Samy Lamouille (University of California San Francisco) for critical review of this manuscript.
Sources of Funding
This work was supported in part by an American Heart Association Scientist Development grant 10SDG3429942 (J.W.S.) and National Institutes of Health grant HL094414 (R.M.S.).
Nonstandard Abbreviations and Acronyms
- BFA
Brefeldin A
- Cx43
connexin 43
- ER
endoplasmic reticulum
- HA
hemagglutinin
- hERG
human ether-a-go-go-related gene
- IP
immunoprecipitation
- ISCH
ischemia
- LAT A
latrunculin A
- TfR
transferrin receptor
- TGN
trans-Golgi network
Footnotes
The online-only Data Supplement is available with this article at http://circres.ahajournals.org/lookup/suppl/doi:10.1161/CIRCRESAHA.111.257964/-/DC1
Disclosures
None.
References
- 1.Shaw RM, Rudy Y. Ionic mechanisms of propagation in cardiac tissue. Roles of the sodium and L-type calcium currents during reduced excitability and decreased gap junction coupling. Circ Res. 1997;81:727–741. doi: 10.1161/01.res.81.5.727. [DOI] [PubMed] [Google Scholar]
- 2.Smith JH, Green CR, Peters NS, Rothery S, Severs NJ. Altered patterns of gap junction distribution in ischemic heart disease. An immunohistochemical study of human myocardium using laser scanning confocal microscopy. Am J Pathol. 1991;139:801–821. [PMC free article] [PubMed] [Google Scholar]
- 3.Peters NS, Coromilas J, Severs NJ, Wit AL. Disturbed connexin43 gap junction distribution correlates with the location of reentrant circuits in the epicardial border zone of healing canine infarcts that cause ventricular tachycardia. Circulation. 1997;95:988–996. doi: 10.1161/01.cir.95.4.988. [DOI] [PubMed] [Google Scholar]
- 4.Beardslee MA, Laing JG, Beyer EC, Saffitz JE. Rapid turnover of connexin43 in the adult rat heart. Circ Res. 1998;83:629–635. doi: 10.1161/01.res.83.6.629. [DOI] [PubMed] [Google Scholar]
- 5.Musil LS, Goodenough DA. Multisubunit assembly of an integral plasma membrane channel protein, gap junction connexin43, occurs after exit from the ER. Cell. 1993;74:1065–1077. doi: 10.1016/0092-8674(93)90728-9. [DOI] [PubMed] [Google Scholar]
- 6.Fort AG, Murray JW, Dandachi N, Davidson MW, Dermietzel R, Wolkoff AW, Spray DC. In vitro motility of liver connexin vesicles along microtubules utilizes kinesin motors. J Biol Chem. 2011;286:22875–22885. doi: 10.1074/jbc.M111.219709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shaw RM, Fay AJ, Puthenveedu MA, von Zastrow M, Jan YN, Jan LY. Microtubule plus-end-tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell. 2007;128:547–560. doi: 10.1016/j.cell.2006.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hong TT, Smyth JW, Gao D, Chu KY, Vogan JM, Fong TS, Jensen BC, Colecraft HM, Shaw RM. Bin1 localizes the l-type calcium channel to cardiac t-tubules. PLoS Biol. 2010;8:e1000312. doi: 10.1371/journal.pbio.1000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smyth JW, Hong TT, Gao D, Vogan JM, Jensen BC, Fong TS, Simpson PC, Stainier DY, Chi NC, Shaw RM. Limited forward trafficking of connexin 43 reduces cell-cell coupling in stressed human and mouse myocardium. J Clin Invest. 2010;120:266–279. doi: 10.1172/JCI39740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rogers SL, Gelfand VI. Membrane trafficking, organelle transport, and the cytoskeleton. Curr Opin Cell Biol. 2000;12:57–62. doi: 10.1016/s0955-0674(99)00057-5. [DOI] [PubMed] [Google Scholar]
- 11.Jaiswal JK, Rivera VM, Simon SM. Exocytosis of post-Golgi vesicles is regulated by components of the endocytic machinery. Cell. 2009;137:1308–1319. doi: 10.1016/j.cell.2009.04.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boateng SY, Goldspink PH. Assembly and maintenance of the sarcomere night and day. Cardiovasc Res. 2008;77:667–675. doi: 10.1093/cvr/cvm048. [DOI] [PubMed] [Google Scholar]
- 13.Hayakawa K, Ono S, Nagaoka R, Saitoh O, Obinata T. Differential assembly of cytoskeletal and sarcomeric actins in developing skeletal muscle cells in vitro. Zoolog Sci. 1996;13:509–517. doi: 10.2108/zsj.13.509. [DOI] [PubMed] [Google Scholar]
- 14.Noorman M, van der Heyden MA, van Veen TA, Cox MG, Hauer RN, de Bakker JM, van Rijen HV. Cardiac cell-cell junctions in health and disease: Electrical versus mechanical coupling. J Mol Cell Cardiol. 2009;47:23–31. doi: 10.1016/j.yjmcc.2009.03.016. [DOI] [PubMed] [Google Scholar]
- 15.Itoh T, Erdmann KS, Roux A, Habermann B, Werner H, De Camilli P. Dynamin and the actin cytoskeleton cooperatively regulate plasma membrane invagination by BAR and F-BAR proteins. Dev Cell. 2005;9:791–804. doi: 10.1016/j.devcel.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 16.Smyth JW, Shaw RM. Forward trafficking of ion channels: what the clinician needs to know. Heart Rhythm. 2010;7:1135–1140. doi: 10.1016/j.hrthm.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke F, Jenne D, Holak TA, Werb Z, Sixt M, Wedlich-Soldner R. Lifeact: a versatile marker to visualize F-actin. Nat Methods. 2008;5:605–607. doi: 10.1038/nmeth.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brady ST, Pfister KK, Bloom GS. A monoclonal antibody against kinesin inhibits both anterograde and retrograde fast axonal transport in squid axoplasm. Proc Natl Acad Sci U S A. 1990;87:1061–1065. doi: 10.1073/pnas.87.3.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Howard J. Molecular motors: structural adaptations to cellular functions. Nature. 1997;389:561–567. doi: 10.1038/39247. [DOI] [PubMed] [Google Scholar]
- 20.Klausner RD, Donaldson JG, Lippincott-Schwartz J. Brefeldin a: insights into the control of membrane traffic and organelle structure. J Cell Biol. 1992;116:1071–1080. doi: 10.1083/jcb.116.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Misumi Y, Misumi Y, Miki K, Takatsuki A, Tamura G, Ikehara Y. Novel blockade by brefeldin A of intracellular transport of secretory proteins in cultured rat hepatocytes. J Biol Chem. 1986;261:11398–11403. [PubMed] [Google Scholar]
- 22.Olbina G, Eckhart W. Mutations in the second extracellular region of connexin 43 prevent localization to the plasma membrane, but do not affect its ability to suppress cell growth. Mol Cancer Res. 2003;1:690–700. [PubMed] [Google Scholar]
- 23.Hesketh GG, Van Eyk JE, Tomaselli GF. Mechanisms of gap junction traffic in health and disease. J Cardiovasc Pharmacol. 2009;54:263–272. doi: 10.1097/FJC.0b013e3181ba0811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smyth JW, Shaw RM. The gap junction life cycle. Heart Rhythm. 2012;9:151–153. doi: 10.1016/j.hrthm.2011.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lauf U, Giepmans BN, Lopez P, Braconnot S, Chen SC, Falk MM. Dynamic trafficking and delivery of connexons to the plasma membrane and accretion to gap junctions in living cells. Proc Natl Acad Sci U S A. 2002;99:10446–10451. doi: 10.1073/pnas.162055899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomas T, Jordan K, Laird DW. Role of cytoskeletal elements in the recruitment of Cx43-GFP and Cx26-YFP into gap junctions. Cell Commun Adhes. 2001;8:231–236. doi: 10.3109/15419060109080729. [DOI] [PubMed] [Google Scholar]
- 27.Ross JL, Ali MY, Warshaw DM. Cargo transport: molecular motors navigate a complex cytoskeleton. Curr Opin Cell Biol. 2008;20:41–47. doi: 10.1016/j.ceb.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aschenbrenner L, Naccache SN, Hasson T. Uncoated endocytic vesicles require the unconventional myosin, Myo6, for rapid transport through actin barriers. Mol Biol Cell. 2004;15:2253–2263. doi: 10.1091/mbc.E04-01-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu XS, Tsan GL, Hammer JA., III Melanophilin and myosin Va track the microtubule plus end on EB1. J Cell Biol. 2005;171:201–207. doi: 10.1083/jcb.200503028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schuh M. An actin-dependent mechanism for long-range vesicle transport. Nat Cell Biol. 2011;13:1431–1436. doi: 10.1038/ncb2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Theiss C, Meller K. Microinjected anti-actin antibodies decrease gap junctional intercellular commmunication in cultured astrocytes. Exp Cell Res. 2002;281:197–204. doi: 10.1006/excr.2002.5652. [DOI] [PubMed] [Google Scholar]
- 32.Qu C, Gardner P, Schrijver I. The role of the cytoskeleton in the formation of gap junctions by Connexin 30. Exp Cell Res. 2009;315:1683–1692. doi: 10.1016/j.yexcr.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 33.Saxton WM, Stemple DL, Leslie RJ, Salmon ED, Zavortink M, McIntosh JR. Tubulin dynamics in cultured mammalian cells. J Cell Biol. 1984;99:2175–2186. doi: 10.1083/jcb.99.6.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu X, Bowers B, Rao K, Wei Q, Hammer JA., III Visualization of melanosome dynamics within wild-type and dilute melanocytes suggests a paradigm for myosin V function in vivo. J Cell Biol. 1998;143:1899–1918. doi: 10.1083/jcb.143.7.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chabrillat ML, Wilhelm C, Wasmeier C, Sviderskaya EV, Louvard D, Coudrier E. Rab8 regulates the actin-based movement of melanosomes. Mol Biol Cell. 2005;16:1640–1650. doi: 10.1091/mbc.E04-09-0770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kwon O, Phillips CL, Molitoris BA. Ischemia induces alterations in actin filaments in renal vascular smooth muscle cells. Am J Physiol Renal Physiol. 2002;282:F1012–F1019. doi: 10.1152/ajprenal.00294.2001. [DOI] [PubMed] [Google Scholar]
- 37.Oxford EM, Danko CG, Kornreich BG, Maass K, Hemsley SA, Raskolnikov D, Fox PR, Delmar M, Moise NS. Ultrastructural changes in cardiac myocytes from Boxer dogs with arrhythmogenic right ventricular cardiomyopathy. J Vet Cardiol. 2011;13:101–113. doi: 10.1016/j.jvc.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dhalla NS, Rangi S, Babick AP, Zieroth S, Elimban V. Cardiac remodeling and subcellular defects in heart failure due to myocardial infarction and aging. Heart Fail Rev. 2011 Aug 18; doi: 10.1007/s10741-011-9278-7. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 39.Decker RS, Decker ML, Kulikovskaya I, Nakamura S, Lee DC, Harris K, Klocke FJ, Winegrad S. Myosin-binding protein C phosphorylation, myofibril structure, and contractile function during low-flow ischemia. Circulation. 2005;111:906–912. doi: 10.1161/01.CIR.0000155609.95618.75. [DOI] [PubMed] [Google Scholar]
- 40.Sainsbury F, Collings DA, Mackun K, Gardiner J, Harper JD, Marc J. Developmental reorientation of transverse cortical microtubules to longitudinal directions: a role for actomyosin-based streaming and partial microtubule-membrane detachment. Plant J. 2008;56:116–131. doi: 10.1111/j.1365-313X.2008.03574.x. [DOI] [PubMed] [Google Scholar]
- 41.Schaefer AW, Schoonderwoert VT, Ji L, Mederios N, Danuser G, Forscher P. Coordination of actin filament and microtubule dynamics during neurite outgrowth. Dev Cell. 2008;15:146–162. doi: 10.1016/j.devcel.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin X, Liu N, Lu J, Zhang J, Anumonwo JM, Isom LL, Fishman GI, Delmar M. Subcellular heterogeneity of sodium current properties in adult cardiac ventricular myocytes. Heart Rhythm. 2011;8:1923–1930. doi: 10.1016/j.hrthm.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anderson CL, Delisle BP, Anson BD, Kilby JA, Will ML, Tester DJ, Gong Q, Zhou Z, Ackerman MJ, January CT. Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation. 2006;113:365–373. doi: 10.1161/CIRCULATIONAHA.105.570200. [DOI] [PubMed] [Google Scholar]
- 44.Thibodeau IL, Xu J, Li Q, Liu G, Lam K, Veinot JP, Birnie DH, Jones DL, Krahn AD, Lemery R, Nicholson BJ, Gollob MH. Paradigm of genetic mosaicism and lone atrial fibrillation: physiological characterization of a connexin 43-deletion mutant identified from atrial tissue. Circulation. 2010;122:236–244. doi: 10.1161/CIRCULATIONAHA.110.961227. [DOI] [PubMed] [Google Scholar]
- 45.Mohler PJ, Rivolta I, Napolitano C, LeMaillet G, Lambert S, Priori SG, Bennett V. Nav1.5 E1053K mutation causing brugada syndrome blocks binding to ankyrin-g and expression of nav1.5 on the surface of cardiomyocytes. Proc Natl Acad Sci U S A. 2004;101:17533–17538. doi: 10.1073/pnas.0403711101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mohler PJ, Davis JQ, Bennett V. Ankyrin-b coordinates the na/k atpase, na/ca exchanger, and insp3 receptor in a cardiac t-tubule/sr microdomain. PLoS Biol. 2005;3:e423. doi: 10.1371/journal.pbio.0030423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sato PY, Coombs W, Lin X, Nekrasova O, Green KJ, Isom LL, Taffet SM, Delmar M. Interactions between ankyrin-G, Plakophilin-2, and Connexin43 at the cardiac intercalated disc. Circ Res. 2011;109:193–201. doi: 10.1161/CIRCRESAHA.111.247023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.