

Abstract
Termination and resolution of inflammation are tightly linked to the inactivation of one of its strongest inducers, NF-κB. While canonical post-stimulus inactivation is achieved by upregulation of inhibitory molecules that relocate NF-κB complexes to the cytoplasm, termination of the NF-κB response can also be accomplished directly in the nucleus by posttranslational modifications, e.g., ubiquitination of the RelA subunit. Here we reveal a functional role for RelA monoubiquitination in regulating NF-κB activity. By employing serine-to-alanine mutants, we found that hypo-phosphorylated nuclear RelA is monoubiquitinated on multiple lysine residues. Ubiquitination was reversed by IκBα expression and was reduced when nuclear translocation was inhibited. RelA monoubiquitination decreased NF-κB transcriptional activity despite prolonged nuclear presence and independently of RelA degradation, possibly through decreased CREB-binding protein (CBP) co-activator binding. Polyubiquitin-triggered proteasomal degradation has been proposed as a model for RelA inactivation. However, here we show that proteasomal inhibition, similar to RelA hypo-phosphorylation, resulted in nuclear translocation and monoubiquitination of RelA. These findings indicate a degradation-independent mechanism for regulating the activity of nuclear RelA by ubiquitination.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-011-0912-2) contains supplementary material, which is available to authorized users.
Keywords: NF-κB, IκBα, Ubiquitination, Transcription
Introduction
The NF-κB/Rel family of dimeric transcription factors is involved in the immediate early transcription of a large array of genes induced by mitogenic or pathogen-associated stimuli. The canonical and most abundant form of NF-κB is composed of a 50-kDa (p50; NFkB1) and a 65-kDa (p65; RelA) subunit. In the resting state NF-κB is retained in the cytoplasm by inhibitory proteins of the IκB family, namely IκBα, IκBβ, IκBε, p100 (IκBδ) and p105 (IκBγ) [1]. Formation of NF-κB/IκB complexes masks the nuclear localization signals present in NF-κB dimers, which prevent nuclear translocation and transcriptional activity. One of the key events in the activation of NF-κB is the liberation of functional NF-κB dimers from IκBs, which is achieved by signal-induced complete (e.g., IκBα, IκBβ, IκBε) or partial (e.g., p100 and p105) proteolytic degradation of the inhibitor involving several pathway-specific protein kinase modules [2]. The transcriptional activator RelA/NF-κB is targeted by multiple post-translational modifications including phosphorylation [3–5], acetylation [6, 7], proline isomerization [8], methylation [9] and ubiquitination [10–12] controlling DNA binding, transcriptional activity, interaction with co-activators and protein stability.
Ubiquitin is covalently linked to proteins due to the sequential action of three types of enzymes known as E1, E2 and E3 with the respective ability to activate, conjugate and transfer the ubiquitin moiety to the ε-amino group of a lysine residue within the target protein. To become eligible for degradation by the 26S proteasome, ubiquitin chains consisting of at least four ubiquitin peptides that are branching of lysine 48 (K48) must be attached to the substrate [13]. Degradation of transcriptional activators by the proteasome machinery is an efficient way to limit transcription to a specific time window. Several mammalian transcription factors, c-Jun [14], c-myc [15] and p53 [16], for example, are short-lived proteins that are destroyed by ubiquitin-mediated proteolysis. While many transactivators are regulated in this way, the traditional view has linked termination of NF-κB-mediated transcription to nuclear expression of IκBα. It is believed that after stimulus-induced degradation newly synthesized IκB proteins enter the nucleus and bind to DNA-resident NF-κB molecules, removing them from their binding sites and resulting in Crm-1-dependent relocation to the cytoplasm [17, 18]. However, this classical model of IκB-dependent termination of NF-κB signaling has been challenged by the fact that NF-κB transcriptional activity is efficiently terminated in the absence of IκBα once the stimulating agent is removed [11]. In this model, nuclear RelA is proposed to be ubiquitinated and degraded, and therefore inhibition of the 26S proteasome results in higher transcriptional activity. The nature of RelA ubiquitination however has not been revealed.
In this study we investigated the functional impact of RelA ubiquitination elicited by either hypo-phosphorylation or proteasomal inhibition on NF-κB activity. We found that mutation of previously described phosphorylation sites within its rel homology domain (RHD) leads to multiple monoubiquitin additions. RelA monoubiquitination inhibited its transcriptional activity without affecting protein stability. Instead we find that RelA monoubiquitination results in decreased binding of the CBP transcriptional co-activator providing a possible mechanism for its reduced transcriptional potential. We further show that nuclear localization is essential for RelA ubiquitination and that proteasomal inhibition leads to RelA monoubiquitination by prolonging RelA nuclear retention. These findings raise the possibility that nuclear activity of RelA is controlled by monoubiquitination.
Materials and methods
Antibodies
The following antibodies were used in this study: β-actin (AC-15, Sigma), CBP (A-22, Santa Cruz Biotechnology), c-myc (9E10), Eps15 (Covance), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Chemicon), hemagglutinin (HA) (12CA5, Roche Applied Science and F-7, Santa Cruz Biotechnology), IκBα (6A920, Imgenex), lamin B (M-20), RelA (C-20, both Santa Cruz Biotechnology), mono/polyubiquitin (P4D1, Covance and Ubi-1, Invitrogen) and polyubiquitin (FK1, Biomol).
Plasmid constructs
pcDNA3 and pLXIH vectors expressing human myc-RelA wt, S205A, S276A and S281A have been described [19]. HA-RelA wt and S276A were cloned into a pcDNA3-HA vector (Invitrogen) by standard cloning procedures. pcDNA3-myc-RelA S205/276/281A was generated by one-step mutagenesis of S276/281A using myc-RelA S205A as a template. pcDNA-myc-RelA S276A was used as template for mutation of all 18 K located in human RelA to R. pcDNA-myc-RelA S276A NLS mutant was obtained by mutagenesis of aa 301–304 from KRKR to DQNQ [20]. pMT107 encoding for wt polyhistidine-tagged ubiquitin was obtained from D. Bohmann (URMC, Rochester, NY). pcDNA3-HA-ubiquitin wt, K29R, K48R, K63R and K29/48/63R (K3xR) were from I. Dikic (Goethe University, Frankfurt am Main, Germany). These constructs were used to clone ubiquitin variants by replacing the HA-tag with a His6-tag via unique BamHI/NheI cloning sites. pcDNA3-his-ubiquitin K7xR was generated out of pcDNA3-his-ubiquitin K3xR by sequentially mutating K6, K11, K27 and K33 to R. To obtain pLXIH-Ub-RelA and pcDNA3-Ub-RelA, HA-tagged ubiquitin was cloned into pLXIH (Clontech) and pcDNA3 (Invitrogen) by PCR amplification with HpaI/XhoI and HindIII restriction sites, respectively. Myc-RelA was placed downstream of the ubiquitin coding sequence by standard cloning techniques. To prevent isopeptidases from cleaving the ubiquitin tag from RelA the carboxyl-terminal GG motif of ubiquitin was mutated to AV [21]. For pLXIH-pLacZ-RelA wt, aa 756–829 of the bacterial LacZ gene were cloned upstream of RelA wt. This stretch was chosen because of its similarity to human ubiquitin in terms of aa composition resulting in a comparable isoelectric point of pLacZ (pI 7.01) and Ub (pI 7.02). The construct was cloned by 2-fragment cloning into XhoI/BamHI-digested pLXIH. pLacZ was amplified from pLacZi (Clontech) with primers containing XhoI and HindIII restriction sites and placed upstream of a myc-RelA wt fragment featuring HindIII/BamHI overhangs. HA-IκBα was obtained by transferring a BamHI/EcoRI fragment from pKSII/ECI-6 (kind gift from R. de Martin, Medical University of Vienna, Vienna, Austria) to a modified pcDNA3 vector harboring an amino-terminal HA-tag. Human p50 was obtained through PCR amplification from HeLa cDNA with primers harboring EcoRI/XhoI restriction sites and cloned into pCMV-myc vector (Clontech). Mutations were performed using the QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions. Plasmid constructs were verified by automated DNA sequencing.
Cell culture reagents and transfections
Human embryonic kidney (HEK) 293T, HeLa, RelA−/− and IκBα−/− 3T3 cells were maintained in DMEM supplemented with 10% FBS, 100 U/ml penicillin G and 100 μg/ml streptomycin B. Retrovirally transduced 3T3 stably expressing myc-RelA wt or mutants, Ub-RelA wt or pLacZ-RelA wt were generated as described [19]. Positive clone pools were cultured in medium containing 250 μg/ml hygromycin B (InvivoGen). Transfection of HEK293T was carried out with calcium phosphate. Where indicated, cells were treated with 5 μM Clasto-Lactacystin β-lactone (Cayman), 50 μM MG132, 1 μM MG262, 100 μg/ml CHX (all Calbiochem), 1 μg/ml LPS (Sigma), 10 ng/ml human recombinant TNF, 100 ng/ml epidermal growth factor (EGF) (both Invitrogen) or 20 ng/ml LMB (Alexis Biochemicals).
Histidine-pull-down ubiquitination assay
Cells were harvested in phosphate-buffered saline (PBS), and 1/20 of samples was removed for expression control. Residual cells were pelleted, lysed in buffer A (6 M guanidine-hydrochloride, 0.1 M Na2HPO4/NaH2PO4, 10 mM imidazole, pH 8) and sonicated for 15 s at 25% amplitude. Then 400 μg of each protein lysate was incubated with Ni-NTA agarose resin (Qiagen). Beads were washed twice in buffer A and twice in buffer A/TI, once in buffer TI (25 mM Tris-HCl, 20 mM imidazole, pH 6.8), and suspended in SDS sample buffer. Ubiquitinated RelA was detected in pull-downs with RelA-specific antibody.
RelA pull-down ubiquitination assay
Cells were harvested in PBS, resuspended in 50 mM Tris-HCl pH 8, 1% SDS and 5 mM DTT, and boiled for 10 min to denature all proteins. For immunoprecipitation, lysates were diluted with 10 vol of 50 mM Tris-HCl pH 8, 150 mM sodium chloride, 1 mM EDTA, 1% Igepal CA-630 and 0.5% sodium-deoxycholate. All buffers were supplemented with protease inhibitors (Roche Applied Science) and 20 mM N-ethylmaleimide (Calbiochem). For endogenous RelA, after pre-clearing, 1/20 of lysates was removed for expression control. RelA was then pulled down with RelA-specific antibody coupled to protein-A Sepharose. Overexpressed RelA from HEK293T was pulled down with anti-c-myc antibody. Precipitates were examined for the presence of ubiquitinated RelA with respective anti-ubiquitin antibodies.
Co-immunoprecipitation assay
For RelA-CBP assay, 3T3 cells were lysed in 50 mM HEPES pH 7.9, 250 mM sodium chloride, 1% Igepal CA-630, 1 mM EDTA, protease inhibitors. For RelA-p50 assay, HEK293T cells co-transfected with HA-RelA and myc-p50 expression plasmids were harvested in lysis buffer containing 50 mM Tris-HCl pH 7.5, 150 mM sodium chloride, 1% Igepal CA-630, 0.5 M sodium-deoxycholate, 0.1% SDS and 1 mM EDTA, supplemented with protease inhibitor. After incubation for 30 min in lysis buffer, lysates were clarified by centrifugation, and supernatants were incubated with CBP-specific antibody coupled to protein-A Sepharose or HA-coupled affinity agarose (Sigma); 1/20 of lysates was kept for expression control. Samples were washed and immunoprecipitated proteins were eluted by addition of SDS-sample buffer. Lysates and precipitated proteins were analyzed by SDS-PAGE and immunoblotting.
Identification of ubiquitinated RelA by nanoLC/MS/MS analysis
Samples obtained by histidine-ubiquitin-pull-down were separated on SDS-PAGE, visualized in gel with SYPRO Ruby (Molecular Probes), and the positive gel protein bands in the range from 80 to 150 kDa found in a separate immunoblotting assay were excised. The SDS-gel slices were subjected to in-gel digestion by trypsin and subsequent extraction as reported previously [22]. The digest was reconstituted in 10 µl of 2% acetonitrile with 0.5% formic acid for nanoLC-ESI-MS/MS analysis, which was carried out using a LTQ-Orbitrap Velos (Thermo-Fisher Scientific) mass spectrometer equipped with a “Plug and Play” nano ion source device (CorSolutions LLC). The nanoLC was carried out by the UltiMate3000 MDLC system (Dionex). A detailed protocol of the MS analysis can be found in the Supplementary Information.
RelA and IκBα immunohistochemistry
Cells seeded on glass cover slips were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100 and blocked in PBS/0.05% Tween20/0.1% bovine serum albumin (Sigma-Aldrich). Cells were incubated with rabbit polyclonal RelA, mouse monoclonal HA (isotype IgG2b) or mouse monoclonal c-myc (isotype IgG1) antibodies in blocking buffer. After incubation with Alexa488-labeled anti-rabbit IgG, Alexa488-labeled anti-mouse IgG1 or Alexa568-labeled anti-mouse IgG2b (all Molecular Probes) in PBS/0.05% Tween20, cover slips were mounted on microscope slides with SlowFade Gold antifade reagent containing 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen). Subcellular distribution of RelA and IκBα was analyzed using a Nikon Eclipse TE2000 microscope equipped with a 40× objective. Pictures were acquired with a charge-coupled device using identical acquisition parameters.
RelA protein stability
Cells were treated with CHX for the indicated time points. Total cell lysates were prepared by lysis in RIPA buffer (50 mM Tris-HCl pH 8, 150 mM sodium chloride, 1 mM EDTA, 1% Igepal CA-630, 0.5% sodium-deoxycholate, 0.1% SDS, protease inhibitors). For cytosolic extracts cells were harvested in PBS, pelleted and lysed in 10 mM Tris-HCl pH 8, 320 mM sucrose, 3 mM CaCl2, 2 mM MgOAc, 0.5% Triton X-100, 1 mM DTT, protease inhibitors). Nuclei were spun down, washed once and sonicated in Laemmli buffer for 10 s at 25% amplitude. Separation of cytosol and nuclei was ensured by exclusive detection of GAPDH in cytosolic and lamin B in nuclear fractions. For pulse chase metabolic labeling cells were washed twice with warm PBS, starved in methionine/cysteine-free DMEM (Invitrogen)/0.5% dialyzed FBS for 30 min and labeled with 200 μCi/ml [35S]-methionine (Perkin Elmer) for 1 h. Cells were washed twice in warm PBS, and DMEM supplemented with 10% FBS/2 mM methionine/2 mM cysteine was added with or without TNF and MG132. Total cell lysates as well as cytosolic/nuclear extracts were essentially obtained as described above. For immunoprecipitation nuclei were lysed in RIPA buffer, and cytosolic fractions were diluted in RIPA at a ratio of 1/10. RelA was pulled down from lysates with RelA-specific antibody coupled to protein-A Sepharose. Radioactive labeled RelA in precipitates was visualized by SDS-PAGE and autoradiography.
Quantitative real time-polymerase chain reaction (qPCR)
qPCR was performed as described [19]. For a complete list of primer sequences used see the Online Supplement.
Results
RelA phospho-serine 205, 276 and 281 mutants are constitutively ubiquitinated
We have shown earlier that inhibition of RelA phosphorylation in the RHD at serines 205, 276 and 281 negatively influences NF-κB transcriptional activity [19]. In our efforts to identify the mechanism responsible for this regulation, we have found that transcriptionally impaired RelA phospho-serine to alanine (SA) mutants exhibit increased and prolonged nuclear localization when compared to wild-type (wt) RelA [23]. Recent reports describe a key role for ubiquitination of nuclear RelA in downregulating NF-κB transcriptional activity [11, 12, 24]; therefore, we wanted to investigate if ubiquitination could contribute to the negative regulation of RelA S205A, S276A and S281A activity. To examine the ubiquitination status of RelA we ectopically expressed wt and mutant proteins together with 6× histidine-ubiquitin and detected ubiquitinated RelA in Ni-NTA pull-downs. Under these conditions ubiquitination of RelA wt was barely detectable, while RelA S205A, S276A and S281A were ubiquitinated (Fig. 1a). In contrast ubiquitination levels of RelA S311A, another RHD phospho-serine mutant whose transcriptional activity was in our hands unaffected by the mutation [19], was found to be comparable to RelA wt. Similar results were obtained when expressing RelA proteins in combination with HA-ubiquitin (Fig S1). Next we detected ubiquitination of endogenous RelA wt and S276A. For this study we employed RelA-deficient mouse embryonic fibroblasts (RelA−/− 3T3) [25] retrovirally reconstituted with RelA wt or S276A. After pull-down of RelA from cell lysates under denaturing conditions, ubiquitinated RelA was detected with an anti-ubiquitin antibody. As shown in Fig. 1b, comparable to HEK293T, resting 3T3 cells had only low amounts of ubiquitinated RelA wt, but substantial amounts of ubiquitinated RelA S276A. We have previously established that RelA S205A, S276A and S281A are predominantly localized to the nuclear compartment of 3T3 cells [23] and therefore thought that the nuclear localization of these mutants compared to RelA wt could trigger RelA ubiquitination. This raises the question of whether nuclear translocation of RelA wt after tumor necrosis factor (TNF) stimulation could induce RelA wt ubiquitination. However, the ubiquitination levels of RelA wt as well as S276A remained unchanged after TNF stimulation for 30 and 120 min (Fig. 1b), indicating that transient nuclear localization as seen after TNF stimulation is not sufficient to trigger RelA ubiquitination. Therefore, we reason that ubiquitination of RelA is specifically induced when its cytosolic redistribution is defective.
Fig. 1.
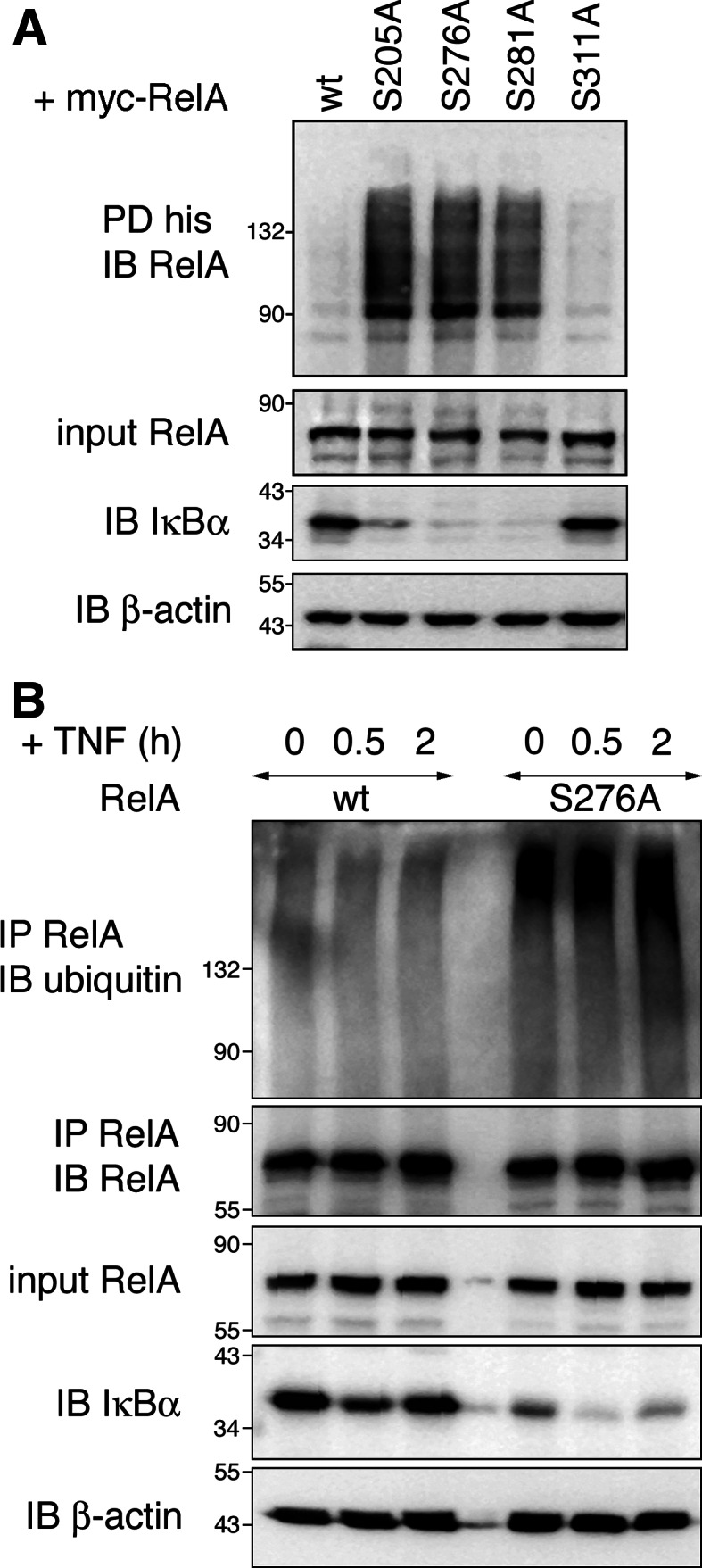
Mutation of NF-κB RelA serines 205, 276 and 281 leads to increased RelA ubiquitination. a HEK293T cells were transfected with myc-RelA wt, S205A, S276A, S281A or S311A together with his-ubiquitin. His-ubiquitin-coupled proteins were pulled down under denaturing conditions with nickel-conjugated beads. Cell lysates and precipitates were probed with RelA, IκBα and β-actin-specific antibodies. b RelA−/− 3T3 were retrovirally reconstituted with either RelA wt or S276A mutant and stimulated with TNF for 0, 0.5 and 2 h. After lysis endogenous RelA was pulled down with anti-RelA antibody. Precipitates were examined for the presence of ubiquitinated RelA with anti-ubiquitin antibody. RelA, IκBα and β-actin protein levels were detected in input cell lysates. IB immunoblot, IP immunoprecipitation, PD pull-down
Ubiquitination of hypo-phosphorylated RelA does not enhance RelA protein degradation
It was suggested that ubiquitination of RelA limits its transcriptional activity by enhancing its degradation [8, 11, 12]; therefore, we wished to test whether RelA SA mutants, due to their increased ubiquitination, would be less stable than RelA wt. First we over-expressed RelA wt and each mutant, and examined RelA protein levels compared to β-actin in whole cell extracts in the presence of the protein synthesis inhibitor cycloheximide (CHX) for up to 16 h in HEK293T cells. RelA wt as well as SA mutant levels remained stable at all time points tested (Fig. 2a). Next we repeated the experiment in RelA−/− 3T3 reconstituted with RelA wt or mutants stimulated with lipopolysaccharide (LPS) prior to CHX for 30 min to induce NF-κB nuclear translocation and activity. Whereas the presence of CHX inhibited IκBα resynthesis and thus confirmed the translational block, we did not observe reduced RelA SA protein stability when compared to RelA wt (Fig. 2b). Stimulation of fibroblasts with TNF in conjunction with protein synthesis inhibition leads to apoptotic cell death [26, 27]. Therefore, we determined the effect of TNF induction on RelA protein stability in 3T3 by [35S]-pulse chase experiments. Similar to LPS, pre-stimulation with TNF for 30 min did not significantly affect the half-life of RelA S276A compared to RelA wt (Fig. 2c). We have shown so far that the increased ubiquitination does not affect turnover of total cellular RelA SA proteins. Because ubiquitination might specifically target nuclear RelA, we next investigated RelA wt and RelA S276A stabilities in nuclear fractions of LPS-stimulated 3T3. As shown in Fig. 2d relative amounts of both RelA wt and S276A were gradually increased in the nucleus after CHX addition. At the same time RelA levels in corresponding cytosolic extracts were decreased. This pattern reflects the accumulation of RelA in the nuclear compartment after consecutive LPS/CHX treatment due to blockage of IκBα resynthesis [28]. In summary, extensively ubiquitinated RelA SA proteins are as stable as RelA wt.
Fig. 2.

Protein stability of RelA is not influenced by its ubiquitination status. a HEK293T cells were transiently transfected with RelA wt, S205A, S276A or S281A, and treated with cycloheximide (CHX) for indicated time points. Expression of RelA and β-actin was monitored. b RelA−/− 3T3 retrovirally reconstituted with RelA wt or mutants were stimulated for 30 min with LPS and treated with CHX for indicated time points. Immunoblots were probed with RelA-, IκBα- and β-actin-specific antibodies. 0*, w/o addition of CHX, was harvested with 16-h time points. c RelA−/− 3T3 reconstituted with RelA wt or S276A were stimulated with TNF for 30 min in the presence of [35S]-methionine and chased for 0, 0.5, 1, 2 and 4 h in presence of TNF. Immunoprecipitated RelA protein was detected by autoradiography, and RelA protein levels from four experiments were quantified (lower panel = representative autoradiograph). d RelA−/− 3T3 reconstituted with RelA wt or S276A were stimulated for 30 min with LPS and CHX for indicated time points. Cell lysates were separated into cytosolic and nuclear fractions. The presence of RelA was detected with RelA-specific antibody. Separation of cytosol and nuclei was ensured by exclusive detection of GAPDH in cytosolic and lamin B in nuclear fractions. Relative cytosolic and nuclear RelA from two experiments were quantified by normalization to GAPDH and lamin B protein levels, respectively. IB immunoblot
Hypo-phosphorylated RelA is monoubiquitinated on multiple lysine residues
The functional consequence of ubiquitination on the fate of a target protein is highly dependent on the molecular nature of ubiquitin polymers attached to a substrate. Our data thus far indicate that ubiquitination of RelA SA proteins does not trigger RelA degradation, possibly implying that RelA SA mutants might not be targeted by K48-linked polyubiquitination. We therefore studied RelA ubiquitination by pull-down using a K48R ubiquitin mutant deficient in K48-anchored branch formation. Whereas no ubiquitinated RelA wt could be detected in any of the pull-downs, RelA SA proteins were readily precipitated with both wt and K48R ubiquitin (Fig. 3a). This result implies that RelA is not targeted by K48-linked polyubiquitination and hence is in accordance with a non-degradative effect of ubiquitin on RelA. Out of the remaining six lysines of ubiquitin used for chain formation K29 and K63 were most prominently shown to regulate protein activities independently of proteolysis [29]. Therefore, we tested next whether K29R and K63R ubiquitins could be conjugated to RelA. Deficiency of neither K29 nor K63 had any influence on RelA SA ubiquitination (Fig. 3a). Finally, we employed an ubiquitin mutant where all seven lysines were substituted by arginines (K7xR), thus rendering it unable to form ubiquitin chains. Interestingly, ubiquitination of RelA S276A was still evident with K7xR ubiquitin (Fig. 3b), suggesting RelA to be monoubiquitinated. One limitation of our experimental setup could be that the endogenous ubiquitin present in cells could form mixed chains with the introduced K7xR ubiquitin mutant resulting in similar ubiquitin conjugation patterns as ubiquitin wt. Therefore, we investigated the ubiquitination status of two proteins known to be polyubiquitinated, IκBα after proteasomal inhibition [30] and RelA wt with co-expression of the ubiquitin ligase PDLIM2 [12], in the presence of ubiquitin wt and K7xR. In contrast to RelA S276A ubiquitination, both MG132-mediated IκBα and PDLIM2-triggered RelA wt ubiquitination were clearly reduced with the ubiquitin mutant (Fig S2), thus showing the validity of our approach. To further confirm the observed data, we analyzed RelA ubiquitination by pulling down RelA from HEK293T cell extracts overexpressing either RelA wt or mutants and detecting ubiquitinated RelA in precipitates with two different anti-ubiquitin antibodies specific for mono- and polyubiquitin (P4D1) and polyubiquitin only (FK1) [31]. Before executing the assay we tested both antibodies positively for their functionality by detection of polyubiquitinated IκBα with FK1 as well as P4D1 and exclusive detection of monoubiquitinated Eps15 with P4D1 (Fig S3). In line with data from the pull-downs, ubiquitinated RelA was only detectable with P4D1 antibody, but not with the polyubiquitin-specific antibody FK1 (Fig. 3c). The same result was obtained for endogenous RelA in 3T3 cells (Fig. 3d). Since ubiquitinated RelA is detected as multiple bands or higher molecular weight smears, we assume that RelA is monoubiquitinated on more than one lysine residue. This is also reflected by the fact that no single or double mutation of any of the 18 lysines encoded in human RelA S276A resulted in inhibition of RelA ubiquitination (Fig. 3e). In summary, our data support that RelA is multiply monoubiquitinated, a modification that does not lead to proteasomal degradation.
Fig. 3.

NF-κB RelA phospho-mutants are monoubiquitinated on multiple lysine residues. a His-ubiquitin without lysines conventionally used for chain formation is still efficiently conjugated to RelA. HEK293T expressing his-ubiquitin wt, K29R, K48R, K63R or K29/48/63R (K3xR) together with myc-RelA proteins were lysed, and coupled proteins were pulled down with nickel-conjugated beads. Precipitates were probed with RelA-specific antibody. b Monoubiquitin is readily conjugated to RelA S276A. HEK293T cells were transfected with RelA S276A and ubiquitin wt, K3xR or K7xR. His-tagged proteins were precipitated from whole cell extracts using Ni-agarose beads, and RelA was detected in immunoblots. c RelA is immunoreactive with P4D1 antibody that recognizes mono- and polyubiquitin, but not with FK1 antibody specific for polyubiquitin. HEK293T cells were transfected with his-ubiquitin and myc-RelA wt, S205A, S276A or S281A. Lysates were precipitated with myc-affinity agarose followed by detection of RelA in immunoblot with anti-myc, anti-ubiquitin P4D1 and anti-polyubiquitin FK1 antibodies. d Ubiquitin attached to endogenous RelA S276A in 3T3 is not detectable with a polyubiquitin-specific antibody. RelA−/− 3T3 reconstituted with either RelA wt or S276A were left untreated or treated with TNF for 30 min. Extracts were subject to immunoprecipitation with anti-RelA antibody, and ubiquitinated RelA was detected in precipitates with either P4D1 or FK1 anti-ubiquitin antibodies. Equal precipitation of RelA was ensured by staining of precipitates with RelA. e All RelA S276A lysine-to-arginine mutants are ubiquitinated. HEK293T were transfected with his-ubiquitin and indicated myc-RelA KR mutants. Assay was carried out as described above. IB immunoblot, IP immunoprecipitation, PD pull-down
Nuclear localization and absence of IκBα are a prerequisite for RelA ubiquitination
Next we determined the circumstances under which RelA ubiquitination occurs. On one hand, RelA de-phosphorylation could directly regulate RelA ubiquitination; however, we considered this scenario as unlikely, because all mutants are differentially phosphorylated [19] while exhibiting the same ubiquitination pattern. On the other hand the prolonged nuclear localization of SA mutants due to lack of IκBα [23] could trigger ubiquitination in order to regulate NF-κB-dependent transcription in the absence of IκBα. Since RelA mutants shared both extended nuclear localization and ubiquitination, and nuclear localization had been suggested earlier as a possible regulator of NF-κB ubiquitination [11, 12], we further tested this possibility. The subcellular localization of RelA mutants has so far been investigated only in 3T3 stimulated with TNF [23]; therefore, we first determined RelA localization in HEK293T where most ubiquitination assays were performed. We observed that extensively ubiquitinated RelA proteins S205A, S276A and S281A were predominantly nuclear, whereas non-ubiquitinated RelA wt and S311A were largely cytosolic (Fig S4a). The nuclear retention of RelA SA mutants was correlated with lower IκBα protein levels in RelA S205A, S276A and S281A cells compared to RelA wt and S311A cells (Fig. 1). To investigate a possible function for IκBα in RelA ubiquitination we tested if co-expression of IκBα in RelA SA mutant cells would have any impact on RelA ubiquitination. Interestingly re-introduction of IκBα completely abolished RelA ubiquitination (Fig. 4a). Since introduction of IκBα into RelA SA cells results in increased IκBα-RelA binding as well as cytosolic relocation of the NF-κB/IκB complex [23], the decrease in RelA ubiquitination as a result of IκBα expression could be attributed to both. To test if IκBα would inhibit RelA ubiquitination when cytosolic relocation is blocked, we introduced IκBα into RelA S276A cells in the presence of the nuclear export inhibitor leptomycin B (LMB). Addition of LMB largely inhibited IκBα-mediated cytosolic translocation of RelA S276A (Fig S4b), but did not re-establish RelA ubiquitination (Fig. 4b). Next we treated RelA wt-expressing cells with LMB for up to 4 h. This treatment led to sustained nuclear accumulation of RelA wt (Fig S4c), but was not sufficient to trigger RelA ubiquitination (Fig. 4c). In contrast, RelA S276A was ubiquitinated at all time points in the absence of exogenous IκBα. Together these results reveal that IκBα blocks RelA ubiquitination independently of RelA subcellular localization, but does not answer whether nuclear localization is a prerequisite for RelA ubiquitination. To this end we constructed a RelA S276A nuclear localization signal mutant (RelA S276A mNLS) and confirmed by immunofluorescence that nuclear translocation of this mutant was blocked (Fig S4d). When comparing the ubiquitination status of RelA S276A and S276A mNLS, we found RelA S276A was substantially ubiquitinated, whereas ubiquitination of the NLS mutant was only marginal (Fig. 4d). Hence, nuclear translocation is required for RelA to be efficiently ubiquitinated. Altogether our data indicate that hypo-phosphorylated RelA is ubiquitinated in the nucleus when not bound to IκBα. We showed earlier that RelA SA mutation did not alter NF-κB complex formation and p50 heterodimerization [19]. However, due to the overrepresentation of exogenous RelA in our experimental setup, it appears likely that RelA homodimer formation is favored in these cells over RelA-p50 heterodimer formation. To investigate if p50 association has an impact on RelA ubiquitination, we co-expressed p50 in equimolar concentration to RelA and tested for RelA ubiquitination levels. As shown in Fig. S5a, RelA ubiquitination remained constant in the presence of p50, indicating that the dimer composition plays no role in RelA ubiquitination.
Fig. 4.

Extended nuclear localization and absence of IκBα are required for RelA ubiquitination. a IκBα inhibits RelA ubiquitination. HEK293T cells were transfected with his-ubiquitin, myc-RelA, and either empty vector (−) or HA-IκBα. Ubiquitination assay was performed under denaturing conditions. b Nuclear RelA S276A is not ubiquitinated in the presence of IκBα. HEK293T were transfected with myc-RelA S276A in the absence or presence of HA-IκBα. To maintain RelA nuclear with co-expression of IκBα LMB was added for 2 h. Assay was performed as in a. c Nuclear accumulation of RelA wt in the presence of IκBα does not lead to an increase of RelA ubiquitination. HEK293T cells transfected with myc-RelA and his-ubiquitin were treated with leptomycin B (LMB) for indicated time points. Cells were harvested in guanidine hydrochloride-containing buffer, and his pull-down was carried out. d Reduced nuclear translocation of RelA S276A results in diminished ubiquitination. HEK293T cells were transfected with his-ubiquitin and either empty vector control (−), RelA wt, RelA S276A or RelA S276A NLS mutant (mNLS). Ubiquitination of RelA was determined by his-ubiquitin pull-down and RelA antibody staining. IB immunoblot, PD pull-down
Proteasomal blockage in IκBα−/− cells leads to nuclear translocation of RelA
Ubiquitination of RelA was recently demonstrated to play a key role in regulating the NF-κB response in IκBα−/− cells [11]. Whereas in our hands RelA protein stability was not affected by ubiquitination (Fig. 2), it was suggested that termination of NF-κB-dependent transcription in IκBα−/− cells is achieved by ubiquitin-dependent degradation of RelA. Therefore, we decided to monitor RelA stability and subcellular localization in IκBα−/− cells. According to published protocols [11] we stimulated cells with TNF for 15 min, followed by wash out and incubation for 6 h with proteasomal inhibitors. After confirming ubiquitination of RelA (Fig. 5a) as previously seen also by Saccani et al. [11], we examined RelA protein levels in nuclear and corresponding cytosolic fractions of metabolically labeled cells under the same treatment paradigm with and without MG132. RelA protein levels decreased in both cytosolic and nuclear fractions over a time course of 6 h after TNF stimulation independently of MG132 (Fig. 5b), indicating that a certain amount of total cellular RelA protein is degraded in a proteasome-independent manner. However, the relative rate of RelA decay in both compartments differed depending on MG132 addition. Without MG132 we observed a more pronounced decrease in nuclear RelA levels compared to MG132-treated cells. This was however compensated by a smaller decline in cytosolic RelA in cells not treated with MG132 (Fig. 5b). These data indicate a shift of cytosolic to nuclear RelA after proteasomal inhibition. To test this hypothesis we detected RelA in TNF-stimulated IκBα−/− cells by indirect immunofluorescence after 1, 3 and 6 h MG132 treatment. In spite of IκBα absence, nuclear RelA was completely cleared over the time course of 6 h medium after a pulse of 15 min TNF. Addition of MG132, however, led to nuclear retention of RelA with simultaneous loss of cytoplasmic immunoreactivity (Fig. 5c). Taken together our data imply that the increase of nuclear RelA after proteasomal blockage is a result of nuclear retention and is not due to increased RelA protein stability.
Fig. 5.

Proteasomal inhibition results in nuclear accumulation rather than stabilization of RelA in IκBα−/− cells. a Treatment of IκBα knockout cells with β-lactone and MG132 leads to an increase in RelA ubiquitination. IκBα−/− 3T3 were stimulated with a pulse of TNF for 15 min, then either left untreated or incubated with β-lactone or MG132 for 6 h. Ubiquitinated RelA was pulled down with RelA antibody and detected with anti-ubiquitin antibody. b Addition of MG132 causes a shift in cytosolic to nuclear RelA, but does not stabilize RelA protein. IκBα−/− fibroblasts were pulse-labeled with [35S]-methionine and during chase treated as in a. Cells were lysed to obtain cytosolic and nuclear extracts. Radiolabeled RelA was detected in autoradiography. The graph shows results obtained from three experiments. Percent levels of RelA after 6 h with or without MG132 treatment were compared to 100% RelA at time point 0 (represents RelA levels observed after 15 min TNF). Lower panel shows representative autoradiograph (non-relevant lanes were digitally removed). c MG132 treatment prolongs RelA nuclear localization. After IκBα−/− 3T3 were treated with TNF for 15 min and then either left untreated or stimulated with MG132 for 1, 3 and 6 h, cells were processed for immunofluorescence with RelA antibody. Nuclei were stained with DAPI. Bar 10 μm. The graph shows ratios of cytosolic and nuclear fluorescence obtained by automatic quantification of RelA compared to DAPI staining (n = 195–244 cells, derived from 3 experiments). Values N/C > 1 indicate predominantly nuclear RelA. IB immunoblot, IP immunoprecipitation
Inhibition of the proteasome leads to monoubiquitination of nuclear RelA
After establishing that inhibition of the proteasome in IκBα−/− cells leads to ubiquitination and nuclear translocation of RelA, we wanted to investigate whether proteasomal inhibition leads to monoubiquitination as we had shown earlier for RelA SA mutants. Accordingly we analyzed RelA wt ubiquitination in the presence of proteasome inhibitors. RelA wt was only minimally ubiquitinated, while ubiquitination of a RelA SA triple mutant was abundant (Fig. 6a). Treatment of cells with proteasome inhibitors MG132 or MG262 for 3 h increased RelA wt ubiquitination to a level comparable to RelA 3xSA. This augmentation was not altered by TNF treatment. In contrast, proteasomal inhibition did not further enhance RelA 3xSA ubiquitination. Next we determined RelA wt and 3xSA subcellular localizations in the presence or absence of MG132. RelA 3xSA was located in the nucleus regardless of MG132, which is consistent with proteasomal inhibition independent ubiquitination of RelA 3xSA (Fig. 6b). RelA wt, in contrast, was mainly cytosolic in the absence of MG132, but translocated to the nucleus after addition of the proteasome inhibitor. Next we probed the molecular structure of RelA ubiquitination induced by proteasomal inhibition by ectopically expressing RelA wt together with wt-, K29R-, K48R-, K63R- or K3xR-ubiquitin in the presence of MG132 and assessing RelA ubiquitination by pull-down assay. Like for RelA S276A we observed that all ubiquitin variants were efficiently conjugated to RelA (Fig. 6c). In addition, we determined if RelA wt would be ubiquitinated in the presence of K7xR-ubiquitin when the proteasome is blocked. As shown in Fig. 6d, RelA wt was equally conjugated to wt and K7xR-ubiquitin. Finally, we tested the nature of endogenous ubiquitinated RelA obtained by proteasomal inhibition with ubiquitin conjugation-specific antibodies. Similarly to RelA S276A (Fig. 3d), ubiquitinated RelA was not detectable with the poly-ubiquitin-specific FK1 antibody, but was highly reactive with the P4D1 antibody recognizing also monoubiquitin (Fig. 6e). Consequently, we propose that proteasomal inhibition-induced nuclear translocation of RelA leads to RelA monoubiquitination.
Fig. 6.

Proteasomal inhibition results in nuclear accumulation and monoubiquitination of RelA. a Blockage of the proteasome leads to increased RelA wt ubiquitination levels. HEK293T cells were transfected with myc-RelA wt or mutant S205/276/281A (RelA 3xSA) together with his-ubiquitin. Where indicated cells were treated for 3 h with 50 μM MG132 or 1 μM MG262 to block proteasomal protein degradation. Ubiquitinated RelA was detected in his-ubiquitin pull-down fractions. Expression of myc-RelA, endogenous IκBα and β-actin in cell lysates was monitored by immunoblotting. b Addition of MG132 leads to nuclear retention of RelA wt. HEK293T cells transfected with myc-RelA wt or 3xSA were left untreated or exposed to MG132 for 3 h. RelA proteins were detected in immunostaining with myc-specific antibody. Nuclei were visualized with DAPI staining. Size bar 10 μm. c MG132 does not trigger conventional RelA K29, K48 or K63 ubiquitination. HEK293T cells were transfected with myc-RelA wt together with his-ubiquitin variants. Cells were treated with MG132 for 3 h and processed to determine RelA ubiquitination. d K7xR ubiquitin is efficiently conjugated to RelA after proteasomal inhibition. HEK293T cells were transfected with myc-RelA, his-ubiquitin wt, K3xR and K7xR. After treatment with MG132 for 3 h cells were lysed, and ubiquitinated RelA was pulled down with Ni-agarose. e Endogenous ubiquitinated RelA detected after proteasomal inhibition of 3T3 cells is not reactive with a polyubiquitin-specific antibody. IκBα−/− 3T3 cells were treated with TNF for 30 min followed by MG132 for 3 h and then harvested to immuno-precipitate endogenous RelA with RelA-specific antibody. Ubiquitinated RelA species were detected in immunoblotting with anti-ubiquitin P4D1, but not with anti-ubiquitin FK1 antibody. IB immunoblot, PD pull-down
Fusion of a single ubiquitin to RelA wt alters NF-κB transcriptional activity in a similar way as S276A mutation
Thus far, we have demonstrated that RelA nuclear translocation caused either by hypo-phosphorylation or proteasomal inhibition results in RelA monoubiquitination. However, we did not address whether monoubiquitination changes RelA transcriptional activity. The best approach to determine the physiological consequences of RelA ubiquitination would be the transcriptional characterization of a constitutively ubiquitinated or non-ubiquitinated RelA mutant protein, which requires the identification of RelA ubiquitination sites. However, we had failed to identify ubiquitin acceptor sites responsible for RelA S276A ubiquitination by a mutagenesis approach targeting all 18 RelA lysine residues by single or double KR substitutions (Fig. 3e). In another effort to identify RelA ubiquitin acceptor sites after proteasomal inhibition, we applied tandem mass spectrometry (MS) for determining ubiquitinated lysine residues. After tryptic digestion of ubiquitinated RelA pulled down from MG132-treated HEK293T cells, we identified three ubiquitinated RelA lysine residues, including K62, K123 and K315 (Table 1; Fig S6). This MS result provides direct evidence that RelA is ubiquitinated at more than one position after proteasomal inhibition; however, a RelA mutant lacking these three residues still underwent extensive ubiquitination (data not shown). A very recently published article by Li et al. [32] also addressed the location of RelA ubiquitination sites. While MS led to the identification of seven ubiquitinated RelA lysines, among them also K62, K123 and K315, it was necessary to eliminate all 18 lysines contained in RelA to abolish RelA ubiquitination. Since such an extensive alteration of RelA leads to a non-functional protein [32], it cannot be used for further studies.
Table 1.
Identification of ubiquitinated peptides of RelA protein by nanoLC-MS/MS analysis
| Sequence | Modification | XCorr | Δ Score | Intensity | Charge | m/z [Da] | MH+ [Da] | ΔM [ppm] |
|---|---|---|---|---|---|---|---|---|
| 57-THPTIkInGYTGPGTVR-73 | K62 (GlyGly), N64 (Deamidated) | 3.33 | 0.47 | 7.70E+04 | 3 | 643.0068 | 1,927.0047 | 0.78 |
| 123-kRDLEQAISQR-133 | K123 (GlyGly) | 4.23 | 0.07 | 8.47E+04 | 3 | 486.5989 | 1,457.7823 | 0.19 |
| 315-kSPFSGPTDPRPPPR-329 | K315 (GlyGly) | 3.02 | 0.26 | 4.35E+04 | 3 | 583.9725 | 1,749.9029 | −0.18 |
Therefore, as an alternative approach to mimic constitutive monoubiquitination of RelA, we fused a single ubiquitin moiety to the amino terminus of RelA wt (Ub-RelA) (Fig. 7a). This method was previously established to reveal the role of monoubiquitination in regulating p53 and FOXO4 transcriptional activities [33, 34]. We characterized the fusion protein by testing its ability to interact with p50 and IκBα by co-immunoprecipitation and found that the Ub-RelA fusion associated efficiently with both proteins (Fig S5b and data not shown). To ascertain that the effects on RelA transcriptional activity were not a result of the amino-terminal tagging of RelA, we used as a control a portion of the LacZ gene (pLacZ) with similar biochemical features to ubiquitin (Fig. 7a). After ensuring comparable protein expression levels of RelA wt, RelA S276A, Ub-RelA and pLacZ-RelA stably introduced into RelA−/− 3T3 (Fig. 7b), we studied their transcriptional activity in TNF-stimulated cells by monitoring mRNA levels of four NF-κB-dependent genes. As shown in Fig. 7c, RelA wt and pLacZ-RelA efficiently induced transcription of all genes tested. In contrast RelA S276A and Ub-RelA showed gene-specific transcriptional impairment. While both severely reduced transcription from IL-6 and ICAM-I genes, there was no effect on IP-10 and only minor effects on MHC-I mRNA expression (Fig. 7c). The similarity in transcriptional potencies between RelA S276A and Ub-RelA indicates that the increased monoubiquitination of RelA SA mutants could indeed account for the decrease in RelA transcriptional activity. Supportive for this conclusion is also the subcellular localization of Ub-RelA at different time points after TNF stimulation. As shown earlier for RelA S276A [23], Ub-RelA translocated to the nucleus 30 min after TNF addition, but did not relocate to the cytoplasm after 2 and 4 h of TNF stimulation (Fig. 7d). The control fusion protein pLacZ-RelA, on the other hand, readily translocated back to the cytoplasm 2 h after TNF was added.
Fig. 7.

Conjugation of monoubiquitin to RelA wt leads to a similar alteration of NF-κB-dependent transcription as RelA S276A mutation. a Schematic representation of RelA constructs used in this study. b Equal expression levels of RelA proteins in 3T3. RelA−/− 3T3 were retrovirally reconstituted with indicated constructs. Expression levels of RelA proteins were monitored by immunoblot with RelA-specific antibody. c Ubiquitin conjugation to RelA wt leads to a similar change in NF-κB transcriptional activity as seen for RelA S276A mutation. RelA−/− 3T3 expressing empty vector (control), RelA wt, RelA S276A, Ub-RelA and pLacZ-RelA were stimulated with TNF for 0.5, 1.5, 3 and 6 h; total RNA was isolated and mRNA levels of IL-6, ICAM-I, IP-10 and MHC-I were analyzed by qPCR. Fold induction of experimental groups was calculated relative to the expression level of unstimulated cells, which was set to 1. Error bars represent mean + SEM of triplicates derived from three independent experiments. d Attachment of ubiquitin but not pLacZ results in prolonged nuclear localization of RelA. RelA−/− 3T3 stably expressing Ub-RelA and pLacZ-RelA were stimulated with TNF for the indicated time points and processed for immunostaining with RelA antibody. Nuclei were stained with DAPI. Size bar 10 μm. e RelA S276A and Ub-RelA bind less efficiently to CBP than RelA wt. Lysates from RelA−/− 3T3 expressing empty vector (control), RelA wt, RelA S276A or Ub-RelA were subjected to immunoprecipitation with CBP antibody. Presence of RelA in precipitates was tested with RelA-specific antibody. IB immunoblot, IP immunoprecipitation
Hypo-phosphorylation of RelA at S276 was shown to inhibit NF-κB activity by abrogating the interaction with the transcriptional co-activator CBP [35]. Since hypo-phosphorylated RelA resembled the transcriptional profile of monoubiquitinated RelA, we wanted to examine if monoubiquitin attachment to RelA is affecting CBP binding. Indeed, we found that both RelA S276A and to a larger extent Ub-RelA exhibited a decreased affinity for CBP when compared to RelA wt (Fig. 7e). Thus, impaired recruitment of CBP to the transcriptional machinery could be responsible for the decreased expression of NF-κB target genes by RelA S276A and Ub-RelA. Taken together, our data indicate that monoubiquitination plays an important role in regulating RelA transcriptional activity in a gene-specific manner by influencing the interaction between RelA and CBP.
Discussion
The strength and duration of NF-κB activity are controlled at multiple levels. Whereas binding of its inhibitor IκB constitutes the main mechanism for silencing of NF-κB, post-translational modification of the DNA-binding subunits by phosphorylation, acetylation and ubiquitination has been described to fine-tune the activity of nuclear NF-κB [36]. While previous reports have linked RelA ubiquitination to nuclear degradation, here we provide evidence that nuclear sequestered RelA is targeted by multiple monoubiquitination, resulting in RelA transcriptional silencing without affecting its cellular half-life.
Here we show that inhibition of phosphorylation at three serine residues within the RHD as well as inhibition of the proteasome lead to monoubiquitination of RelA. The unexpected finding that RelA also undergoes monoubiquitination is in contrast to previously reported RelA polyubiquitination, which was mainly triggered by addition of proteasome inhibitors and the ubiquitin ligases SOCS1/COMMD1 and PDLIM2 [8, 10–12, 32]. By MS analysis it has been determined that polyubiquitin chains linked via K29, K33, K48 and K63 can be attached to RelA after proteasomal blockage [32], indicating that inhibition of the proteasome also induces degradation-independent RelA modification [37]. On the other hand, the nature of ubiquitin additions conjugated by co-expression of SOCS1 and PDLIM2 ubiquitin ligase components has not been addressed, but it has been described to result in destabilization of RelA [8, 10, 12, 38, 39], pointing to the polyubiquitination RelA.
The observation that RelA is polyubiquitinated is often based on the detection of a high molecular weight ubiquitinated RelA species in immunoblotting. This alone, however, is not a conclusive evidence for RelA polyubiquitination, since also multiple monoubiquitin attachments would lead to the same ubiquitination pattern. The fact that mutation of all lysines in RelA is necessary to prevent its ubiquitination after addition of MG132 [32] provides basic evidence that enough monoubiquitin moieties could be conjugated to RelA lysine residues to shift a substantial portion of ubiquitinated RelA past a molecular weight of 100 kDa.
In our study we detected mainly monoubiquitin additions to RelA. A slight decrease in ubiquitinated RelA species with addition of KR ubiquitin variants (Figs. 3a, b; 6c, d), however, indicates that RelA is not exclusively modified with monoubiquitin. The amount of polyubiquitinated RelA however lies below the detection range of the polyubiquitin-specific FK1 antibody, which could easily detect polyubiquitinated IκBα (Figs. 3c, d; 6e; S3). In conclusion, contemplating all available published data on RelA ubiquitination, it becomes apparent that RelA is not exclusively targeted by one type of ubiquitin modification, but is likely conjugated to the full spectrum of possible ubiquitin variants on multiple ubiquitin acceptor residues.
Two paradigms led to RelA ubiquitination. First, we found that ubiquitination of RelA was markedly enhanced by inhibiting phosphorylation at S205, S276 and S281. While in most cases phosphorylation induces ubiquitination, as reported for p53 [40] and c-myc [41], phosphorylation can also inhibit ubiquitination, as seen in c-Jun [42] or ATF-2 [43]. Both circumstances have been shown for RelA. Phosphorylation at S468 by the IKK complex induces its ubiquitination by enabling the association with the COMMD1/SOCS-1 E3 ligase complex [38, 39], whereas, confirming our result, phosphorylation at S276 by the kinase Pim-1 inhibits RelA ubiquitination [44]. Whether inhibition of RelA phosphorylation at S205, S276 and S281 directly contributes to RelA ubiquitination by, e.g., permitting the binding of an ubiquitin ligase, remains to be determined. However, we propose a different mechanism where phosphorylation deficiency at these discrete sites only indirectly induces RelA ubiquitination. Inhibition of RelA phosphorylation at S205, S276 and S281 reduces NF-κB transcriptional activity, resulting in decreased cellular IκBα levels [19, 23]. This is reflective of IκBα−/− cells, where prolonged nuclear presence of NF-κB leads to RelA ubiquitination [11]. Therefore, we believe that RelA S205A, S276A and S281A display an elevated ubiquitination level because of their extended nuclear localization caused by insufficient IκBα induction. Data presented in our work support this conclusion. First, inhibition of phosphorylation at S311, which neither results in decreased cellular IκBα levels nor nuclear accumulation of NF-κB [23], did not enhance RelA ubiquitination. Second, reintroduction of IκBα into RelA mutant cells completely abolished RelA ubiquitination, suggesting that RelA cannot be ubiquitinated when bound by IκBα. In line with this, we find that nuclear accumulation of RelA caused by LMB, a specific inhibitor of Crm1-dependent nuclear export [45], was not sufficient to induce RelA ubiquitination (Fig. 4c). Apart from nuclear retention of RelA, LMB treatment also leads to nuclear sequestration of IκBα resulting in RelA-IκBα complex formation and transcriptional silencing [46–48]. Interestingly and similar to RelA-IκBα interactions, ubiquitination of NF-κB p50 is negatively regulated by its binding to the IκB family member Bcl-3 [49]. Third, inhibition of nuclear translocation of RelA S276A diminished its ubiquitination (Fig. 4d), underlining the importance of nuclear transfer for RelA to be ubiquitinated.
The second inducer of RelA ubiquitination was proteasomal inhibition. The most prevalent and immediate interpretation for this phenomenon is that proteasomal inhibition blocks RelA degradation, resulting in accumulation of ubiquitinated RelA species [8, 11, 12]. However, this explanation is not entirely satisfactory. In contrast to short-lived transcription factors like p53, c-myc or c-Jun, whose activities are primarily regulated by degradation and resynthesis [14, 16, 50], RelA protein was reported to be comparably stable [28, 51, 52]. This coheres with the fact that NF-κB activation does not require de novo protein synthesis [53, 54] and its termination is mainly regulated by an auto-regulatory feedback loop involving proteins produced in response to NF-κB activation [55, 56]. In line with a proteasome-independent regulatory function of RelA ubiquitination, we found in this study that ubiquitinated RelA phospho-mutants were stable under all examined conditions. Although it cannot be entirely ruled out that RelA is degraded by the proteasome under certain conditions, we show evidence herein that the impact of proteasomal inhibition on RelA ubiquitination is the result of nuclear retention rather than stabilization of RelA. This is based on the observation that nuclear localization, as evidenced by RelA phospho-mutants, was sufficient to induce ubiquitination and ubiquitination was not further increased by treatment with MG132 (Fig. 6a). In addition we observed in IκBα−/− cells that increase in nuclear RelA after MG132 treatment was paralleled by a decrease in cytosolic RelA (Fig. 5b, c), supporting the hypothesis that proteasomal inhibition leads to nuclear translocation and retention of RelA. The mechanism for RelA nuclear translocation induced by proteasomal inhibition warrants further investigation. On the one hand, proteasomal inhibition was shown to inhibit NF-κB activity by blocking IκBα degradation [30, 57], but on the other hand it has also been shown to result in NF-κB induction due to IKK activation [58–60]. In the latter case IκBα is degraded independently of the proteasome, a mechanism that has been shown to regulate inducible as well as constitutive NF-κB activation [61, 62]. The lack of IκBα then consequently leads to nuclear retention and enhanced DNA binding of NF-κB [58, 60].
Finally, we provide evidence that monoubiquitination negatively regulates RelA transcriptional activity in a gene-specific manner. Fusion of a single ubiquitin to RelA was sufficient to cause prolonged nuclear retention after TNF stimulation while at the same time inhibiting its transcriptional activity on some genes, but not on others (Fig. 7c). The transcriptional profile was similar to the RelA S276A mutant, which, comparable to the ubiquitin-RelA fusion, was also monoubiquitinated (Fig. 3) and predominantly nuclear localized [23]. While an increase in NF-κB-dependent transcription has been described after proteasomal inhibition in IκBα−/− cells [11], it is not clear to what extent this effect is RelA-dependent. We observed substantial induction of NF-κB-dependent genes in TNF-stimulated RelA−/− 3T3 after MG132 treatment (Fig S7). It is therefore likely that proteasomal inhibition can increase transcription at least of some NF-κB-dependent genes without the involvement of RelA.
How RelA multiple monoubiquitination influences NF-κB activity remains largely elusive, but our data indicate that ubiquitinated RelA loses its ability to bind to CBP (Fig. 7e). This could be either the result of lower binding affinity due to changes in protein properties by ubiquitin attachment or due to an induced spatial separation of RelA from CBP. For example, ubiquitination could translocate RelA to transcriptionally selective nuclear compartments such as PML bodies [12] or lead to nucleolar sequestration of RelA shown to be preceded by ubiquitination after proteasomal inhibition [24]. Another regulatory mechanism could involve competition of ubiquitin and acetyl groups for RelA lysine residues. Interestingly, acetylation of K123, an ubiquitin-acceptor site identified in this study and by others [32], promotes IκBα-mediated nuclear export [7]. Thus, it is possible that ubiquitination at this site leads to nuclear retention of RelA by inhibiting acetyl-lysine-mediated IκBα interaction.
In summary, we present RelA monoubiquitination as a novel regulatory modification mediating gene-specific silencing of NF-κB activity. In future studies it will be important to identify proteins involved in the RelA monoubiquitination process. The identification and characterization of these new mediators in the NF-κB pathway will not only improve our knowledge and understanding of mechanisms involved in inflammation and associated diseases, but will also provide new targets for therapy and drug discovery.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
We are grateful to the following persons for providing materials used in this study: Drs. Amer A. Beg (RelA−/− 3T3), Dirk Bohmann (pMT107), Rainer De Martin (pKSII/ECI-6), Ivan Dikic (pcDNA3-HA-ubiquitin) and Alexander Hoffmann (IκBα−/− 3T3). We also thank Dr. Wei Chen for performing the MS analysis. This work was supported by a National Institutes of Health grant [HL077308 to J.A.] and American Heart Association Scientist Development grant [10SDG2600298 to K.H.].
References
- 1.Hoffmann A, Natoli G, Ghosh G. Transcriptional regulation via the NF-κB signaling module. Oncogene. 2006;25:6706–6716. doi: 10.1038/sj.onc.1209933. [DOI] [PubMed] [Google Scholar]
- 2.Ghosh S, Hayden MS. New regulators of NF-κB in inflammation. Nat Rev Immunol. 2008;8:837–848. doi: 10.1038/nri2423. [DOI] [PubMed] [Google Scholar]
- 3.Anrather J, Csizmadia V, Soares MP, Winkler H. Regulation of NF-κB RelA phosphorylation and transcriptional activity by p21ras and protein kinase Cζ in primary endothelial cells. J Biol Chem. 1999;274:13594–13603. doi: 10.1074/jbc.274.19.13594. [DOI] [PubMed] [Google Scholar]
- 4.Naumann M, Scheidereit C. Activation of NF-κB in vivo is regulated by multiple phosphorylations. EMBO J. 1994;13:4597–4607. doi: 10.1002/j.1460-2075.1994.tb06781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhong HH, Suyang H, Erdjumentbromage H, Tempst P, Ghosh S. The transcriptional activity of NF-κB is regulated by the IκB-associated PKAc subunit through a cyclic AMP-independent mechanism. Cell. 1997;89:413–424. doi: 10.1016/S0092-8674(00)80222-6. [DOI] [PubMed] [Google Scholar]
- 6.Chen L, Shinde U, Ortolan TG, Madura K. Ubiquitin-associated (UBA) domains in Rad23 bind ubiquitin and promote inhibition of multi-ubiquitin chain assembly. EMBO Rep. 2001;2:933–938. doi: 10.1093/embo-reports/kve203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kiernan R, Bres V, Ng RW, Coudart MP, El Messaoudi S, Sardet C, Jin DY, Emiliani S, Benkirane M. Post-activation turn-off of NF-κB-dependent transcription is regulated by acetylation of p65. J Biol Chem. 2003;278:2758–2766. doi: 10.1074/jbc.M209572200. [DOI] [PubMed] [Google Scholar]
- 8.Ryo A, Suizu F, Yoshida Y, Perrem K, Liou YC, Wulf G, Rottapel R, Yamaoka S, Lu KP. Regulation of NF-κB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol Cell. 2003;12:1413–1426. doi: 10.1016/S1097-2765(03)00490-8. [DOI] [PubMed] [Google Scholar]
- 9.Yang XD, Huang B, Li M, Lamb A, Kelleher NL, Chen LF. Negative regulation of NF-κB action by Set9-mediated lysine methylation of the RelA subunit. EMBO J. 2009;28:1055–1066. doi: 10.1038/emboj.2009.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maine GN, Mao X, Komarck CM, Burstein E. COMMD1 promotes the ubiquitination of NF-κB subunits through a cullin-containing ubiquitin ligase. EMBO J. 2007;26:436–447. doi: 10.1038/sj.emboj.7601489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saccani S, Marazzi I, Beg AA, Natoli G. Degradation of promoter-bound p65/RelA is essential for the prompt termination of the nuclear factor κB response. J Exp Med. 2004;200:107–113. doi: 10.1084/jem.20040196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanaka T, Grusby MJ, Kaisho T. PDLIM2-mediated termination of transcription factor NF-κB activation by intranuclear sequestration and degradation of the p65 subunit. Nat Immunol. 2007;8:584–591. doi: 10.1038/ni1464. [DOI] [PubMed] [Google Scholar]
- 13.Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000;19:94–102. doi: 10.1093/emboj/19.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Treier M, Staszewski LM, Bohmann D. Ubiquitin-dependent c-Jun degradation in vivo is mediated by the delta domain. Cell. 1994;78:787–798. doi: 10.1016/S0092-8674(94)90502-9. [DOI] [PubMed] [Google Scholar]
- 15.Gross-Mesilaty S, Reinstein E, Bercovich B, Tobias KE, Schwartz AL, Kahana C, Ciechanover A. Basal and human papillomavirus E6 oncoprotein-induced degradation of Myc proteins by the ubiquitin pathway. Proc Natl Acad Sci USA. 1998;95:8058–8063. doi: 10.1073/pnas.95.14.8058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chowdary DR, Dermody JJ, Jha KK, Ozer HL. Accumulation of p53 in a mutant cell line defective in the ubiquitin pathway. Mol Cell Biol. 1994;14:1997–2003. doi: 10.1128/mcb.14.3.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang TT, Kudo N, Yoshida M, Miyamoto S. A nuclear export signal in the N-terminal regulatory domain of IκBα controls cytoplasmic localization of inactive NF-κB/IκBα complexes. Proc Natl Acad Sci USA. 2000;97:1014–1019. doi: 10.1073/pnas.97.3.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson C, Van Antwerp D, Hope TJ. An N-terminal nuclear export signal is required for the nucleocytoplasmic shuttling of IκBα. EMBO J. 1999;18:6682–6693. doi: 10.1093/emboj/18.23.6682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anrather J, Racchumi G, Iadecola C. Cis-acting element-specific transcriptional activity of differentially phosphorylated nuclear factor-κB. J Biol Chem. 2005;280:244–252. doi: 10.1074/jbc.M409344200. [DOI] [PubMed] [Google Scholar]
- 20.Beg AA, Ruben SM, Scheinman RI, Haskill S, Rosen CA, Baldwin AS., Jr IκB interacts with the nuclear localization sequences of the subunits of NF-κB: a mechanism for cytoplasmic retention. Genes Dev. 1992;6:1899–1913. doi: 10.1101/gad.6.10.1899. [DOI] [PubMed] [Google Scholar]
- 21.Stack JH, Whitney M, Rodems SM, Pollok BA. A ubiquitin-based tagging system for controlled modulation of protein stability. Nat Biotechnol. 2000;18:1298–1302. doi: 10.1038/82422. [DOI] [PubMed] [Google Scholar]
- 22.Zhang S, Van Pelt CK, Henion JD. Automated chip-based nanoelectrospray-mass spectrometry for rapid identification of proteins separated by two-dimensional gel electrophoresis. Electrophoresis. 2003;24:3620–3632. doi: 10.1002/elps.200305585. [DOI] [PubMed] [Google Scholar]
- 23.Hochrainer K, Racchumi G, Anrather J. Hypo-phosphorylation leads to nuclear retention of NF-κB p65 due to impaired IκBα gene synthesis. FEBS Lett. 2007;581:5493–5499. doi: 10.1016/j.febslet.2007.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thoms HC, Loveridge CJ, Simpson J, Clipson A, Reinhardt K, Dunlop MG, Stark LA. Nucleolar targeting of RelA (p65) is regulated by COMMD1-dependent ubiquitination. Cancer Res. 2010;70:139–149. doi: 10.1158/0008-5472.CAN-09-1397. [DOI] [PubMed] [Google Scholar]
- 25.Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature. 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- 26.Sanchez-Alcazar JA, Ruiz-Cabello J, Hernandez-Munoz I, Pobre PS, de la Torre P, Siles-Rivas E, Garcia I, Kaplan O, Munoz-Yague MT, Solis-Herruzo JA. Tumor necrosis factor-α increases ATP content in metabolically inhibited L929 cells preceding cell death. J Biol Chem. 1997;272:30167–30177. doi: 10.1074/jbc.272.48.30167. [DOI] [PubMed] [Google Scholar]
- 27.Woods KM, Chapes SK. Three distinct cell phenotypes of induced-TNF cytotoxicity and their relationship to apoptosis. J Leukoc Biol. 1993;53:37–44. doi: 10.1002/jlb.53.1.37. [DOI] [PubMed] [Google Scholar]
- 28.Rice NR, Ernst MK. In vivo control of NF-κB activation by IκBα. EMBO J. 1993;12:4685–4695. doi: 10.1002/j.1460-2075.1993.tb06157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ikeda F, Dikic I. Atypical ubiquitin chains: new molecular signals. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep. 2008;9:536–542. doi: 10.1038/embor.2008.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Traenckner EB, Wilk S, Baeuerle PA. A proteasome inhibitor prevents activation of NF-κB and stabilizes a newly phosphorylated form of IκBα that is still bound to NF-κB. EMBO J. 1994;13:5433–5441. doi: 10.1002/j.1460-2075.1994.tb06878.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haglund K, Sigismund S, Polo S, Szymkiewicz I, Di Fiore PP, Dikic I. Multiple monoubiquitination of RTKs is sufficient for their endocytosis and degradation. Nat Cell Biol. 2003;5:461–466. doi: 10.1038/ncb983. [DOI] [PubMed] [Google Scholar]
- 32.Li H, Wittwer T, Weber A, Schneider H, Moreno R, Maine GN, Kracht M, Schmitz ML, Burstein E. Regulation of NF-κB activity by competition between RelA acetylation and ubiquitination. Oncogene. 2011 doi: 10.1038/onc.2011.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li M, Brooks CL, Wu-Baer F, Chen D, Baer R, Gu W. Mono- versus polyubiquitination: differential control of p53 fate by Mdm2. Science. 2003;302:1972–1975. doi: 10.1126/science.1091362. [DOI] [PubMed] [Google Scholar]
- 34.van der Horst A, de Vries-Smits AM, Brenkman AB, van Triest MH, van den Broek N, Colland F, Maurice MM, Burgering BM. FOXO4 transcriptional activity is regulated by monoubiquitination and USP7/HAUSP. Nat Cell Biol. 2006;8:1064–1073. doi: 10.1038/ncb1469. [DOI] [PubMed] [Google Scholar]
- 35.Zhong H, Voll RE, Ghosh S. Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol Cell. 1998;1:661–671. doi: 10.1016/S1097-2765(00)80066-0. [DOI] [PubMed] [Google Scholar]
- 36.Perkins ND. Post-translational modifications regulating the activity and function of the nuclear factor κB pathway. Oncogene. 2006;25:6717–6730. doi: 10.1038/sj.onc.1209937. [DOI] [PubMed] [Google Scholar]
- 37.Chen ZJ, Sun LJ. Nonproteolytic functions of ubiquitin in cell signaling. Mol Cell. 2009;33:275–286. doi: 10.1016/j.molcel.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 38.Geng H, Wittwer T, Dittrich-Breiholz O, Kracht M, Schmitz ML. Phosphorylation of NF-κB p65 at Ser468 controls its COMMD1-dependent ubiquitination and target gene-specific proteasomal elimination. EMBO Rep. 2009;10:381–386. doi: 10.1038/embor.2009.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mao X, Gluck N, Li D, Maine GN, Li H, Zaidi IW, Repaka A, Mayo MW, Burstein E. GCN5 is a required cofactor for a ubiquitin ligase that targets NF-κB/RelA. Genes Dev. 2009;23:849–861. doi: 10.1101/gad.1748409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chernov MV, Bean LJ, Lerner N, Stark GR. Regulation of ubiquitination and degradation of p53 in unstressed cells through C-terminal phosphorylation. J Biol Chem. 2001;276:31819–31824. doi: 10.1074/jbc.M103170200. [DOI] [PubMed] [Google Scholar]
- 41.Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, Ishida N, Okumura F, Nakayama K, Nakayama KI. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 2004;23:2116–2125. doi: 10.1038/sj.emboj.7600217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Musti AM, Treier M, Bohmann D. Reduced ubiquitin-dependent degradation of c-Jun after phosphorylation by MAP kinases. Science. 1997;275:400–402. doi: 10.1126/science.275.5298.400. [DOI] [PubMed] [Google Scholar]
- 43.Fuchs SY, Tappin I, Ronai Z. Stability of the ATF2 transcription factor is regulated by phosphorylation and dephosphorylation. J Biol Chem. 2000;275:12560–12564. doi: 10.1074/jbc.275.17.12560. [DOI] [PubMed] [Google Scholar]
- 44.Nihira K, Ando Y, Yamaguchi T, Kagami Y, Miki Y, Yoshida K. Pim-1 controls NF-κB signalling by stabilizing RelA/p65. Cell Death Differ. 2009;17:689–698. doi: 10.1038/cdd.2009.174. [DOI] [PubMed] [Google Scholar]
- 45.Wolff B, Sanglier JJ, Wang Y. Leptomycin B is an inhibitor of nuclear export: inhibition of nucleo-cytoplasmic translocation of the human immunodeficiency virus type 1 (HIV-1) Rev protein and Rev-dependent mRNA. Chem Biol. 1997;4:139–147. doi: 10.1016/S1074-5521(97)90257-X. [DOI] [PubMed] [Google Scholar]
- 46.Birbach A, Gold P, Binder BR, Hofer E, de Martin R, Schmid JA. Signaling molecules of the NF-κB pathway shuttle constitutively between cytoplasm and nucleus. J Biol Chem. 2002;277:10842–10851. doi: 10.1074/jbc.M112475200. [DOI] [PubMed] [Google Scholar]
- 47.Rodriguez MS, Thompson J, Hay RT, Dargemont C. Nuclear retention of IκBα protects it from signal-induced degradation and inhibits nuclear factor κB transcriptional activation. J Biol Chem. 1999;274:9108–9115. doi: 10.1074/jbc.274.13.9108. [DOI] [PubMed] [Google Scholar]
- 48.Tam WF, Lee LH, Davis L, Sen R. Cytoplasmic sequestration of rel proteins by IκBα requires CRM1-dependent nuclear export. Mol Cell Biol. 2000;20:2269–2284. doi: 10.1128/MCB.20.6.2269-2284.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carmody RJ, Ruan Q, Palmer S, Hilliard B, Chen YH. Negative regulation of toll-like receptor signaling by NF-κB p50 ubiquitination blockade. Science. 2007;317:675–678. doi: 10.1126/science.1142953. [DOI] [PubMed] [Google Scholar]
- 50.Salghetti SE, Kim SY, Tansey WP. Destruction of Myc by ubiquitin-mediated proteolysis: cancer-associated and transforming mutations stabilize Myc. EMBO J. 1999;18:717–726. doi: 10.1093/emboj/18.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krappmann D, Scheidereit C. Regulation of NF-κB activity by IκBα and IκBβ stability. Immunobiology. 1997;198:3–13. doi: 10.1016/S0171-2985(97)80022-8. [DOI] [PubMed] [Google Scholar]
- 52.Scott ML, Fujita T, Liou HC, Nolan GP, Baltimore D. The p65 subunit of NF-κB regulates IκB by two distinct mechanisms. Genes Dev. 1993;7:1266–1276. doi: 10.1101/gad.7.7a.1266. [DOI] [PubMed] [Google Scholar]
- 53.Hohmann HP, Remy R, Scheidereit C, van Loon AP. Maintenance of NF-κB activity is dependent on protein synthesis and the continuous presence of external stimuli. Mol Cell Biol. 1991;11:259–266. doi: 10.1128/mcb.11.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sen R, Baltimore D. Inducibility of κ immunoglobulin enhancer-binding protein NF-κB by a posttranslational mechanism. Cell. 1986;47:921–928. doi: 10.1016/0092-8674(86)90807-X. [DOI] [PubMed] [Google Scholar]
- 55.Sun SC, Ganchi PA, Beraud C, Ballard DW, Greene WC. Autoregulation of the NF-κB transactivator RelA (p65) by multiple cytoplasmic inhibitors containing ankyrin motifs. Proc Natl Acad Sci USA. 1994;91:1346–1350. doi: 10.1073/pnas.91.4.1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wertz IE, O’Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, Ma A, Koonin EV, Dixit VM. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature. 2004;430:694–699. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- 57.Chen Z, Hagler J, Palombella VJ, Melandri F, Scherer D, Ballard D, Maniatis T. Signal-induced site-specific phosphorylation targets IκBα to the ubiquitin-proteasome pathway. Genes Dev. 1995;9:1586–1597. doi: 10.1101/gad.9.13.1586. [DOI] [PubMed] [Google Scholar]
- 58.Dolcet X, Llobet D, Encinas M, Pallares J, Cabero A, Schoenenberger JA, Comella JX, Matias-Guiu X. Proteasome inhibitors induce death but activate NF-κB on endometrial carcinoma cell lines and primary culture explants. J Biol Chem. 2006;281:22118–22130. doi: 10.1074/jbc.M601350200. [DOI] [PubMed] [Google Scholar]
- 59.Li C, Chen S, Yue P, Deng X, Lonial S, Khuri FR, Sun SY. Proteasome inhibitor PS-341 (bortezomib) induces calpain-dependent IκBα degradation. J Biol Chem. 2010;285:16096–16104. doi: 10.1074/jbc.M109.072694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nemeth ZH, Wong HR, Odoms K, Deitch EA, Szabo C, Vizi ES, Hasko G. Proteasome inhibitors induce inhibitory κB (IκB) kinase activation, IκBα degradation, and nuclear factor κB activation in HT-29 cells. Mol Pharmacol. 2004;65:342–349. doi: 10.1124/mol.65.2.342. [DOI] [PubMed] [Google Scholar]
- 61.Han Y, Weinman S, Boldogh I, Walker RK, Brasier AR. Tumor necrosis factor-α-inducible IκBα proteolysis mediated by cytosolic m-calpain. A mechanism parallel to the ubiquitin-proteasome pathway for nuclear factor-κB activation. J Biol Chem. 1999;274:787–794. doi: 10.1074/jbc.274.2.787. [DOI] [PubMed] [Google Scholar]
- 62.Miyamoto S, Chiao PJ, Verma IM. Enhanced IκBα degradation is responsible for constitutive NF-κB activity in mature murine B-cell lines. Mol Cell Biol. 1994;14:3276–3282. doi: 10.1128/mcb.14.5.3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.