

Abstract
Many examples of the emergence or re-emergence of infectious diseases involve the adaptation of zoonotic viruses to new amplification hosts or to humans themselves. These include several instances of simple mutational adaptations, often to hosts closely related to the natural reservoirs. However, based on theoretical grounds, arthropod-borne viruses, or arboviruses, may face several challenges for adaptation to new hosts. Here, we review recent findings regarding adaptive evolution of arboviruses and its impact on disease emergence. We focus on the zoonotic alphaviruses Venezuelan equine encephalitis and chikungunya viruses, which have undergone adaptive evolution that mediated recent outbreaks of disease, as well as the flaviviruses dengue and West Nile viruses, which have emerged via less dramatic adaptive mechanisms.
Keywords: adaptation, alphavirus, emergence, evolution, fitness, flavivirus, mosquito, vector
Several examples of viral disease emergence involving host switching, in some cases mediated by viral adaptation, have been studied in detail, including HIV [1], the SARS coronavirus [2,3] and feline parvoviruses transferring to dogs [4]. Most of these examples have involved viruses that infect a single host or a closely related group of hosts, such as primates or canids. However, there is also considerable interest in the emergence mechanisms of arthropod-borne viruses (arboviruses), most of which must infect and replicate in highly disparate amplifying vertebrate hosts, as well as mosquitoes, ticks or other hematophagous arthropods that transmit via infectious saliva. Many arboviruses, such as dengue (DENV) and chikungunya viruses (CHIKV), infect millions of individuals annually, with severe public health and economic consequences [5–7], and their ability to repeatedly emerge into urban, human–mosquito transmission cycles represents both a major public health challenge and a fascinating opportunity to study their adaptive landscapes.
Speculation has long centered on the hypothesis that arboviruses' requirement for infection of highly divergent hosts constrains their adaptive evolution because optimization for replication in one may reduce fitness for infection of the other [8]. This concept can be visualized as nonoverlapping adaptive landscapes in the vertebrate versus vector hosts, resulting in few fitness peaks that coincide in both host landscapes (Figure 1). The arboviruses must exist within the intersection of distinct vertebrate and vector landscapes, which theoretically requires them to traverse wider fitness valleys in order to find rare peaks that coincide in both host environments under different selective pressures. However, recent studies of arbovirus emergence history and mechanisms demonstrate that, despite these theoretical constraints on adaptive evolution, changes in host range and/or the efficiency of infection and replication in key amplification hosts or vectors can and do occur via simple point mutations. Examples include the emergence of Venezuelan equine encephalitis virus (VEEV) epidemics via single-point mutations that enhance equine amplification viremia or infection of mosquito bridge vectors [9], and CHIKV mutations that enhance infection of the invasive epidemic mosquito vector, Aedes albopictus [10,11], resulting in the dramatic geographic expansion of a major epidemic since 2004 [12].
Figure 1. Fitness landscapes for arboviruses.
(A) Theoretical, 3D landscape for infection of the vertebrate host; (B) theoretical, 3D landscape for infection of the vector; (C) superimposition of vertebrate and vector landscapes, demonstrating limited genotypes that are highly fit in both hosts (overlapping peaks). Peaks with partial overlap (arrows) could result in subtle shifts in the mutant swarm as an arbovirus alternates between vertebrate and vector infections. (D) Fitness landscape for CHIKV transmission by the epidemic mosquito vector, Aedes albopictus. The three envelope glycoprotein amino acids with major effects on A. albopictus infection are indicated, demonstrating sequential adaptive envelope glycoprotein substitutions in the IOL and epistatic limitations in the Asian lineage. The E2-L210Q mutation observed in India in 2009 is a second-step mutation that increases infectivity and dissemination. The Asian genotype needs one additional substitution in the E1 protein (T98A) compared with the IOL for the major fitness of the E1-A226V substitution to be manifested. This epistatic interaction apparently has prevented the Asian lineage from adapting to A. albopictus for the past six decades. Green lettering indicates ancestral residues; red lettering indicates derived residues associated with adaptation to A. albopictus.
CHIKV: Chikungunya virus; IOL: Indian Ocean lineage.
Adapted with permission from [108].
Despite the emergence examples cited above, there are far more examples of the lack of arboviral emergence for centuries despite apparent opportunities for the exploitation of alternative vectors or amplifying hosts. Other arboviruses such as West Nile virus (WNV) and DENV can apparently transfer into new geographic regions or habitats requiring the use of alternative vectors and/or hosts with little or no adaptation. Even CHIKV and VEEV, where emergence can involve single adaptive mutations in addition to population genetic and ecological mechanisms, rarely do so, despite what would appear to be nearly continuous opportunities, suggesting fundamental constraints on adaptive evolution that are poorly understood.
Here, we review recent findings on the adaptive evolution of arboviruses with an emphasis on the mechanisms implicated and constraints that may limit their frequency of adaptive emergence events. We focus on two RNA arbovirus groups that have received considerable attention: the genus Alphavirus in the family Togaviridae; and the genus Flavivirus in the family Flaviviridae.
Arboviruses
Arboviruses are maintained via arthropod vector transmission among vertebrates that serve as reservoir and/or amplification hosts [13]. Most arboviruses cycle horizontally with transmission during blood feeding, while a few are maintained by vertical transmission from adult arthropods to offspring or through venereal transmission during copulation. In enzootic cycles, vertebrates, including birds, primates and small mammals, serve as amplifying hosts by producing viremias (Figure 2). After ingestion from a viremic vertebrate host, the virus infects midgut epithelial cells and then disseminates to secondary sites of infection in the open body cavity (hemocoel) of the vector. Subsequent virus replication in salivary glands and deposition into saliva allows for transmission during subsequent feeding. Human arboviral disease usually results from spillover infections from enzootic cycles, and humans are often dead-end hosts. By contrast, a few arboviruses undergo urban transmission, with humans themselves acting as amplifying hosts via the generation of high-titered viremias.
Figure 2. Arbovirus transmission cycles showing ancestral enzootic cycles, epizootic cycles involving amplification by domesticated animals, such as horses, and endemic/epidemic cycles in urban habitats involving human amplification hosts.
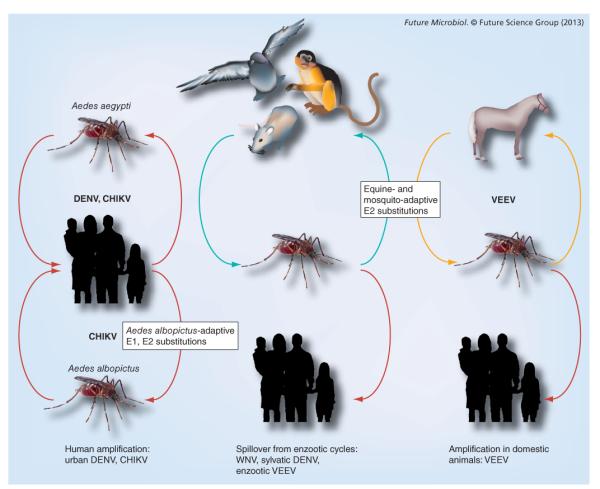
CHIKV: Chikungunya virus; DENV: Dengue virus; VEEV: Venezuelan equine encephalitis virus; WNV: West Nile virus.
Arboviruses typically exhibit relatively high host specificity for enzootic maintenance, with each virus using one or a few vertebrate and invertebrate species. Despite specializing in one or few host species, outbreaks of human or veterinary disease are sometimes associated with host-range changes where arboviruses adapt to new vectors or vertebrates, as discussed below.
Alphaviruses
Alphaviruses comprise a diverse group of 29 species that are nearly globally distributed and include three major categories: aquatic viruses, arthralgic viruses and encephalitic viruses [14]. All alphaviruses are mosquito-borne, except the aquatic viruses salmon pancreatic disease virus and southern elephant seal virus, which are either water-borne or vectored by ectoparasitic lice. The other alphaviruses are transmitted between mosquitoes and avian or mammalian hosts. The arthralgic alphaviruses are primarily found in the Old World, with the exception of Mayaro virus, which occurs in South America. Of the arthralgic alphaviruses, the most important human pathogen is CHIKV. Of the encephalitic alphaviruses, the most important human pathogens are VEEV and eastern equine encephalitis virus (EEEV). Finally, a `mosquito only' alphavirus, with vertebrate host infection incompetence demonstrated at the level of viral RNA replication, was recently described [15].
Alphaviruses are 70 nm in diameter, with nucleocapsids and enveloped outer shells that assemble into icosahedral structures with T-4 symmetry (Figure 3) [16]. The single-stranded, messenger sense, approximately 12-kb RNA genome is 5′-capped and 3′-polyadenylated. It encodes two open reading frames (ORFs) separated by an intergenic region and flanked by 5′- and 3′-untranslated regions (UTRs). Four nonstructural proteins (nsP1–4) are expressed as a polyprotein during cap-dependent translation of the 5′-ORF, and form the replicative complex that is responsible for viral genomic and subgenomic RNA replication. A subgenomic RNA encodes three main structural proteins (capsid, E2 and E1). A total of 240 copies of E2–E1 heterodimers form the outer shell of the alphavirus virion. E2 interacts with incompletely characterized cell surface receptor(s), while the E1 glycoprotein lies mostly below E2 and catalyzes a multistep fusion reaction within acidic endosomes to mediate entry. The viral genetic determinants associated with cross-species jumps and emergence of alphaviruses into new mosquito–human cycles have, to date, only been described for the E2 and E1 genes [9].
Figure 3. Virion morphology and genome organization for alphaviruses and flaviviruses.

CAP: Capsid; E: Envelope; prM: Premembrane.
Flaviviruses
Flaviviruses include a highly diverse group of arboviruses with a global distribution and a high human disease burden [17]. Among these are DENV and yellow fever virus, both of which cause extensive human disease, especially in resource-poor countries. Flaviviruses evolve in concert with their vectors, whereas the alphaviruses are more promiscuous in their vector usage [18]. Flaviviruses can be subdivided broadly into four groups: tick-borne; mosquito-borne (further subdivided into Culex- or Aedes-transmitted); no known vector; and mosquito only (not capable of infecting vertebrates). The tick-borne viruses are maintained mainly in the northern hemisphere, with tick-borne encephalitis virus stretching from Siberia to eastern Europe [19]. These viruses evolve less quickly than the mosquito-borne viruses due to the long life cycle of ticks, and therefore appear to be more constrained by their vectors than mosquito-borne viruses. Mosquito-borne flaviviruses are present on every continent except Antarctica, and their genetic relationship is highly correlated with geographic location; exceptions include DENV, which has been introduced across the tropics, yellow fever virus, which was introduced to South America with the slave trade, and, most recently, WNV, which was introduced into the Americas. These geographic and vector species constraints contrast with the alphaviruses, which are often transmitted by more than one genus of mosquito [18]. Although alphaviruses and flaviviruses occur in very similar niches, there is apparently a major disparity in the ability of flaviviruses to adapt to new vectors.
The flavivirus genome is encapsidated within an electron-dense core surrounded by a lipid bilayer, forming small spherical particles approximately 50 nm in diameter (Figure 3) [17,20]. The single-stranded, positive sense 11-kb RNA genome contains a single ORF that is flanked by UTRs ranging from approximately 100 nucleotides at the 5′-UTR to approximately 400–700 nucleotides at the 3′-UTR. Genomic RNA is translated to generate all viral proteins, including three structural proteins (capsid [C], pre-membrane/membrane [prM/M] and envelope [E]) and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5). The polyprotein is cleaved by host-encoded signal peptidases as well as a virus-encoded serine protease during and after translation to yield the ten viral proteins. The prM protein forms a scaffold for the viral E protein, which comprises the majority of the surface area on the mature virion and is responsible for receptor binding, and includes immunodominant epitopes, some of which appear to be under selective pressure [20,21]. Some positively selected codons are located within T- or B-cell epitopes, indicating they are probably involved in escaping host adaptive immunity, or in regions that enable host cell binding and entry, and could explain the fixation of DENV lineages in specific regions [20,21].
Historic evidence of arbovirus adaptive evolution
Venezuelan equine encephalitis virus
VEEV is so named because the disease it produces was first recognized in equids during the 1920s in Venezuela and Colombia [9]. Not until the 1950s was the connection made between equine and human disease. Subsequently, even during the absence of equine disease, sylvatic, enzootic and rodent–mosquito cycles of VEEV were discovered in Colombia, Mexico and Panama, including spillover cases in humans, some of which were fatal. Experimental equine infections combined with improved serological assays later determined that the VEEV strains isolated during equine epizootics, which fell into antigenic subtypes IAB and IC, were virulent for equids and generated viremia that was sufficient for amplification via mosquito vectors, while the remaining strains in the VEE complex (VEEV subtypes ID, IE and other species in the VEE complex of alphaviruses) were generally equine amplification incompetent [9]. Historical data indicate that the equine-amplified epizootics/epidemics occur every 10–20 years, with the last major outbreak in 1995 affecting approximately 100,000 persons in northern Venezuela and Colombia [9]. However, these epizootic/epidemic strains appear to persist only as long as susceptible equids are available (a minority survive infection and survivors become immune for life), and then disappear between outbreaks.
Because enzootic VEEV strains, which circulate continuously in forested or swamp habitats among rodents transmitted by mosquitoes in the subgenus Culex (Melanoconion), are antigenically distinct from the epizootic/epidemic strains, the origins of the latter remained an enigma for many years [9]. Initially, the hypothesis that the epizootic/epidemic IAB and IC strains evolve periodically and convergently from enzootic VEEV strains was supported through comparative genetic analyses, and ultimately by reverse genetic studies demonstrating that single amino acid substitutions in the E2 glycoprotein of subtype ID enzootic strains mediated adaptation for efficient replication, viremia induction and virulence in horses (Figure 2). Comparative infectivity studies also indicated that some epizootic/epidemic VEEV strains were also more efficient at infecting mosquito bridge vectors such as Aedes (Ochlerotatus) taeniorhynchus than enzootic strains [9]. Furthermore, recent epizootics on the Pacific coast of southern Mexico appear to have benefited from enhanced infection of this vector, mediated again by a single E2 amino acid substitution. Thus, adaptation to both equine amplification hosts and bridge vectors, both involving as little as single nucleotide substitutions, have major impacts on disease emergence.
Chikungunya virus
CHIKV, similar to VEEV, is maintained in two distinct ecological cycles. Sylvatic or enzootic CHIKV occurs throughout sub-Saharan Africa and employs forest-dwelling, primatophilic Aedes mosquitoes as vectors and nonhuman primate hosts, although other vertebrates may also participate in the cycle [9]. Spillovers from sylvatic cycles in west Africa are relatively common and occur predominantly during rainy seasons on the outskirts of villages, probably due to Aedes furcifer transmission [22]. Migration of viremic humans likely introduces CHIKV into cities, which can result in urban transmission via the anthropophilic Aedes aegypti and A. albopictus vectors. In contrast to Africa, CHIKV maintenance in Asia has only been convincingly associated with urban cycles [9].
CHIKV has been repeatedly introduced into Asia from Africa starting as early as the 18th century, with introductions from the 1920s to the 1950s, which led to the establishment of the endemic Asian CHIKV genotype, and in 2005 by the Indian Ocean lineage (IOL) [23,24]. The IOL evolved from the east-central-south-African (ECSA) enzootic genotype that emerged in 2004 in coastal Kenya and subsequently spread to the Indian Ocean islands, India, southeast Asia and Europe (Figure 4) [12,23,25]. A genetic adaptation of the IOL strains to the novel urban vector A. albopictus via an E1 substitution (E1-A226V) appears to be at least partly responsible for the evolutionary success of this emerging lineage [12,26], although this adaptation was not responsible for the initial emergence into the Indian Ocean archipelago, since early outbreak isolates lack the adaptive E1-226V mutation. E1-A226V mediates increased midgut infectivity, dissemination and transmission by A. albopictus, yet has little or no effect on infection of A. aegypti [10,26]. Epidemiological and phylogenetic studies show that E1-A226V has been selected convergently on at least four separate occasions from the ECSA CHIKV lineage in locations where A. aegypti is not known to occur, but where A. albopictus served as the principal epidemic vector [27–30]. Endemic Asian CHIKV strains, by contrast, which circulate in areas where both A. aegypti and A. albopictus occur, have not adapted to A. albopictus. Recent Asian outbreaks are instead attributed to introduced ECSA strains that are better adapted to A. albopictus.
Figure 4. Evolutionary history of Chikungunya virus emergence.
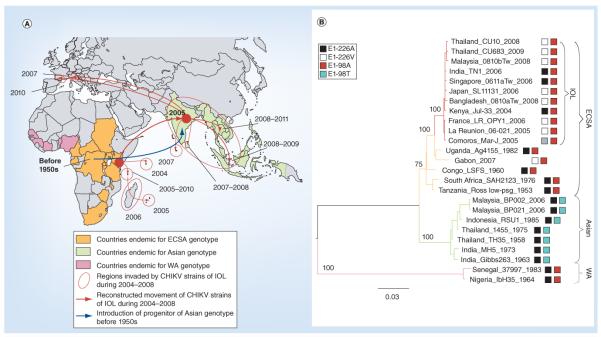
(A) Recent history of CHIKV outbreaks and (B) phylogenetic relationships of CHIKV genotypes. The phylogenetic tree for 26 representative CHIKV strains was constructed using the maximum likelihood method. Amino acid residues at positions E1-226 and E1-98 are indicated.
CHIKV: Chikungunya virus; ECSA: East-central-south-African; IOL: Indian Ocean lineage; WA: West Africa.
Adapted with permission from [24].
In contrast to adaptive VEEV emergence, the E1-A226V CHIKV substitution appears to have been only an initial event in a multistep process of CHIKV adaptation to A. albopictus. Several second-step adaptive mutations have recently been identified, which also increase CHIKV fitness in this mosquito [11]. One such mutation, E2-L210Q, was first described in IOL strains circulating in south India in 2009 [11,31]. This mutation provides a four- to five-fold increase in the ability of CHIKV to infect and disseminate in A. albopictus. However, this effect is significantly weaker than the effect of E1-A226V (a 50–100-fold fitness increase). Recently, novel second-step A. albopictus-adaptive mutations were found in IOL sublineages: E2-K252Q in strains that caused a 2008–2009 outbreak in Thailand, Malaysia and Singapore; and a double mutant E2-R198Q/E3-S18F, implicated in a 2008 outbreak in Sri Lanka [Tsetsarkin K, Weaver SC, Unpublished Data]. These findings indicate that the evolution of cross-species vector jumps for arboviruses can follow a multistep pattern similar to that postulated for single-host viruses, including SARS and pandemic influenza A [8,32,33]. The CHIKV adaptive cascade appears to have begun with a major jump on the fitness landscape (E1-A226V), followed by the ascent of several nearby fitness peaks by a series of optimizing, second-step adaptive mutations (Figure 1).
West Nile virus
WNV, first isolated in Uganda, traditionally caused epidemics in sub-Saharan Africa and the Middle East, beginning in the 1950s [34]. However, since the early 1990s, epidemics have been continuously reported in Europe. Genetically, WNV can be divided into at least four lineages [35]. Lineage I, which is almost globally distributed, has caused most of the detected disease. Historically, WNV probably spread from Africa to India and Australia in two separate incidents via trade, and then WNV adapted to new transmission cycles, usually using related Culex species as well as avian hosts. WNV has also been responsible for the best-studied introduction of a new virus into a new environment. In 1999, the first case of WNV in the western hemisphere was observed in New York City, followed by rapid spread across the USA and the establishment of endemicity. Since its introduction, several studies have demonstrated the ability of WNV to infect many different mosquito and vertebrate species [34]. However, WNV is generally maintained in an enzootic transmission cycle between corvid birds and Culex pipiens, Culex quinquefasciatus or Culex tarsalis mosquitoes.
Following its establishment in the USA, WNV has experienced limited adaptive genetic changes, resulting in a novel genotype (WN02) that swept across the USA and displaced the introduced 1999 strains. By 2005, it was thought that WNV had reached genetic homeostasis, as the WN02 genotype was found throughout the USA [36] and there was little or no consensus sequence difference among strains isolated at distant locations. Continued surveillance to 2012 has shown that this is not entirely true. Recent WNV studies indicate continued genetic change, with a new southwestern genotype spreading from the southwestern USA across the rest of the country [37].
The mechanism of WNV introduction into the USA has been the subject of intense speculation but remains unclear. Given the limited historical interhemispheric/intercontinental spread of flaviviruses, the successful introduction of WNV into India, Australia and the USA is surprising on the surface. No evidence has been presented for adaptive WNV evolution to North American birds, and mosquito-adaptive evolution remains controversial. Ebel et al. [38] and Moudy et al. [39] reported that the extrinsic incubation period following oral infection with the WN02 genotype, which apparently displaced the introduced, ancestral NY99 genotype following 2002, is shorter in C. pipiens and C. tarsalis mosquitoes, suggesting adaptation for more rapid transmission. However, Anderson et al. reported that C. tarsalis transmits both WNV genotypes with equal efficiency, suggesting that there has been limited adaptation to this important vector in western North America [40]. Experiments examining vector competence as well as vector feeding tropisms in additional populations of all major vectors are needed to further evaluate the role of mosquito-adaptive evolution in WNV establishment throughout the Americas.
Regardless of the extent of adaptive WNV evolution, few amino acid substitutions have been associated with the introduction into North America. One arose soon after the initial cases in 1999 (V159A in the envelope glycoprotein), which was rapidly fixed and partially defined the WN02 genotype (reviewed in [37]). No other mutations associated with WNV divergence and persistence in the Americas have been associated with phenotypic differences in natural hosts or vectors, suggesting that genetic drift may be the dominant mode of evolution.
The accumulated evidence that WNV has undergone limited adaptive evolution during 13 years of circulation in the Americas is surprising considering the dramatic adaptive changes reviewed above for alphaviruses. The rapid establishment and spread of WNV following its apparent 1999 introduction into New York suggests pre-existing fitness for North American vectors and avian hosts, which is consistent with the dearth of evidence of strong selection derived from WNV sequences [35]. This limited adaptive evolution could therefore reflect the worldwide distribution of the principal vectors in the C. pipiens complex, including C. quinquefasciatus, although there are significant differences in behavior and physiology among populations that are relatively isolated genetically, but also include hybrids [41]. Similar to many arboviruses, WNV uses a variety of passerine birds as amplification hosts, and its success in North America may reflect this flexible evolutionary strategy. However, the strong association of flavivirus lineages with particular vectors and vertebrate hosts, which contrasts with the more promiscuous evolutionary patterns depicted in alphavirus phylogenies, suggests that the former genus may also have intrinsic adaptive constraints compared with the latter. The availability of next-generation sequencing will allow a greater understanding of the viral diversity characteristic of these viruses, and may allow us to identify differences between flavivirus and alphaviruses that will answer some of these questions about adaptation to new environments.
Dengue virus
DENV, similar to CHIKV, has a sustained interhuman transmission cycle that is both ecologically and evolutionarily distinct from its zoonotic ancestors [7]. Unlike other arbo-viruses, DENV is restricted in its natural vertebrate host range, which most likely only includes primates (reviewed in [42]). A recent report from Latin America suggesting secondary transmission in a number of mammals (including bats, rodents and marsupials; reviewed in [42]) is in question because many similar studies in other DENV-endemic regions have not generated similar evidence of enzootic circulation. Although DENV has a long history of human contact dating back to the third century in China [43], human infections were first formally described in Philadelphia in 1789 [43], and for the next two centuries, DENV was only recognized as a pathogen of humans. The existence of the zoonotic transmission cycles was not documented until the 20th century, when DENV serotypes 1, 2 and 4 were found to be transmitted among nonhuman primates by arboreal Aedes spp. [43]. These cycles remain active in forests of southeast Asia (probably all serotypes) and in West Africa (DENV-2 only). Similarly, in its human transmission cycle, the four antigenically and genetically distinct serotypes (DENV-1–4) [21] are transmitted between humans and domestic and peridomestic Aedes mosquitoes, particularly A. aegypti and A. albopictus (Figure 5) [7]. While historical data suggest rolling epidemics that may have been unsustained due to human herd immunity, the establishment of trading routes in the 17th century, and more recently population movement facilitated by wars, social upheavals, jet travel and uncontrolled urbanization, have all led to an explosive increase in the geographic distribution of DENV. This has resulted in DENV hyperendemicity (coexistence of multiple serotypes) and rolling pandemics where a nearly half of the global population is at risk.
Figure 5. Emergence of urban dengue virus transmission cycles and lineages from sylvatic progenitors.
DENV: Dengue virus.
Emergence of human DENV transmission
While the origins of DENV have been debated for years, several lines of evidence provide support for a southeast Asian origin (reviewed in [42,44]). Subsequently, the extant, distinct human DENV serotypes have emerged independently and repeatedly in a series of divergence events that occurred after the establishment of urban populations capable of supporting the human transmission cycle (Figure 5) [43]. These emergences were facilitated by: vector switching from arboreal Aedes mosquitoes to peridomestic and domestic Aedes spp. mosquitoes; reservoir host switching from nonhuman primates to humans; and probably allopatric and ecological partitioning of ancestral sylvatic DENV strains in different species of nonhuman primates. The establishment of trading routes that facilitated the global spread of A. aegypti and waves of large-scale human movement allowed for serotype dispersal. Subsequently, antigenic divergence led to limited heterotypic cross-protection against challenge exhibited by the extensive genetic diversity of current DENV strains [45].
DENV hyperendemicity has led to rolling DENV pandemics, which may be facilitated by immune enhancement (augmented virus replication following heterologous infection), which hypothetically selects for higher virus replication and transmission efficiency due to limited cross-reactive immunity from previous, heterologous DENV infections [46]. Mosquitoes with limited oral susceptibility may also select for higher virus replication in humans via more efficient transmission with higher viremia [47]. Moreover, the fundamental basis of DENV genetic diversity can be attributed to its error-prone RNA-dependent RNA polymerase (RdRp), which lacks proofreading ability and produces approximately one mutation per round of genome replication [48]. As deep-sequencing technologies have become widely available [49,50], the interplay between immune involvement and viral inter- and intra-host evolution is becoming clearer, suggesting that immune responses (e.g., RNAi in mosquitoes and interferon in vertebrates) can drive viral evolution during DENV infection [51,52], and in general represent an important, understudied force shaping arbovirus evolution.
Evidence for constraints on adaptive arbovirus evolution in nature
The historic evidence that VEEV epizootics/epidemics emerge only every 10–20 years has been assumed to reflect the time required to replace susceptible equine amplification populations following their decimation due to mortality and the elimination of many survivors as potential amplifying hosts due to life-long immunity after infection [9]. However, the genetic link between enzootic subtype ID strains and epizootic/epidemic subtype IAB/C emergence left this explanation incompletely satisfactory. Subtype ID strains circulate from southern Florida (Everglades virus, subtype II in the VEEV complex, but defined genetically as a ID variant [9]) to Bolivia [53] in at least six major lineages, yet only one of these is known to generate epizootic/epidemic strains. The lack of outbreaks initiated in regions where other ID lineages circulate, yet which have been traversed by spreading epizootics/epidemics, suggests genetic constraints on the ability of most ID strains to adapt for equine amplification. However, the historic lack of sustained equine vaccination following VEEV outbreaks suggests that herd immunity cannot entirely explain the long intervals between outbreaks if epizootic mutants are constantly generated. Mosquito vector population sizes may also limit emergence, but another possible explanation for the limited frequency of outbreaks is that the vertebrate host-adaptive mutations are selected inefficiently in nature due to alternating host trade-offs or viral population genetic factors. Although direct experimental confirmation is lacking to confirm the hypothesis that equine-adaptive VEEV mutations may not be efficiently selected in nature, related studies shed light on this question. Inefficient adaptive evolution may stem from the need for VEEV and, by extension, other arboviruses, to maintain replication competence in both vertebrate and invertebrate hosts, where `generalist' genomes of high fitness for both hosts would be favored [54].
Despite the strong CHIKV fitness gains for A. albopictus transmission mediated by envelope glycoprotein amino acid substitutions resulting from single nucleotide mutations (see above) [10,11], these mutations occurred only after months to years of circulation in regions inhabited by this mosquito. This may in part be explained by relatively infrequent CHIKV–A. albopictus contact in areas where the vector primarily inhabits rural settings. In addition, the E1-A226V substitution never occurred in the Asian strain circulating since the 1950s, also suggesting that natural selection is not efficient, although a second epistatic mutation is apparently responsible for this constraint (see below) [24].
DENV-2 is believed to have undergone adaptive evolution after it emerged into the epidemic [55–57] and urban cycles, but no direct evidence of adaptive evolution of any of the DENV serotypes during urban emergence, or of DENV-1, -3 or -4 after emergence, has been obtained. Finally, as discussed above, little or no adaptive evolution has occurred in WNV since it was introduced into the Americas 13 years ago.
This accumulated evidence of inefficient positive selection of arboviruses in nature suggests a general evolutionary inefficiency, at least for members of two major genera, the alphaviruses and flaviviruses.
Hypothetical explanations for adaptive constraints on arboviruses
Since the majority of arboviruses are RNA viruses that lack polymerases with error correction, they exhibit error frequencies of approximately 10−4 per nucleotide copied [48]. These high mutation frequencies, coupled with large population sizes and fast replication, afford RNA viruses the ability to rapidly adapt to fluctuating environments. Despite this, sequence comparisons of strains of arboviruses isolated from nature show that their sequences are relatively stable and genetic studies indicate that they are subject to strong purifying selection [38,49,58,59]. This stability may stem from having to infect disparate host types that present conflicting replication and adaptation challenges, and that could constrain adaptation to either host alone by imposing a fitness cost where adaptations are antagonistic [54]. Only mutations that are beneficial or neutral in both hosts are maintained, resulting in the elimination of deleterious mutations by purifying selection.
As an extension to these observed genetic constraints on adaptation, phenotypic limitations (i.e., fitness trade-offs or antagonistic pleiotropy) should also be imposed on arboviruses, since phenotype is largely determined by genotype. A large number of experimental arbovirus evolution studies have focused on understanding mechanisms of fitness trade-offs in order to understand the unique ability of RNA arboviruses to simultaneously evolve in alternate hosts. These studies employed similar experimental designs: arboviruses were serially passaged in vertebrate or invertebrate cells, or alternately passaged between the two cell types to simulate natural cycling, and the fitness of progeny viruses was assessed relative to progenitors. Studies of this type reveal three general patterns of arbovirus evolution: fitness gains after serial passage in vertebrate or invertebrate cells (except in certain cases [60]) and losses in bypassed host cell types (DENV, EEEV, Sindbis, vesicular stomatitis [VSV] and Rift Valley fever viruses) [18,61–64], reduced fitness in novel cell types (VSV) [65] and fitness increases after alternating passage (DENV, EEEV, Sindbis and VSV) [18,54,61,62]. Together, these in vitro studies suggest that constraints on fitness differ in insect and vertebrate cells and can be virus-specific, but that arbovirus fitness in general is not limited by alternating between vertebrate and invertebrate hosts.
Although these studies challenge the fitness trade-off hypothesis, artificial selective pressures, such as adaptation for binding to heparan sulfate, which is not a natural receptor for most arboviruses, suggest limited relevance of in vitro models to natural in vivo arbovirus cycling. Furthermore, the results from some in vitro studies (St Louis encephalitis virus [SLEV]; e.g., [66]) did not translate into in vivo adaptations, suggesting that cell culture observations may not be relevant to natural arbovirus transmission, in that monocultures do not accurately represent the complexity of multicellular hosts. Moreover, some studies employing the same virus and similar experimental designs have also shown incongruent results, suggesting that differences in cell infection conditions, including cell type, temperature, multiplicities of infection, number and length of passage series, passage histories of virus strains, use of cell lines with defective innate immune responses and ways of measuring viral fitness, may affect outcomes. To circumvent these issues, in vivo evolution studies using arthropod vectors and vertebrate models of infection that better represent natural arbovirus transmissions have supplemented in vitro studies. Building on related studies from 1975, which showed that the alphavirus Ross River virus serially passaged in mice became more virulent while Ross River virus passaged alternately between mice and A. aegypti did not (reviewed in [18]), observations from in vivo studies largely support earlier in vitro conclusions. Artificially releasing an arbovirus from one host allows for specialization via increased fitness in the passaged host. VEEV passaged ten times in hamsters was five-times more fit than its progenitor, and mosquito-passaged VEEV was more infectious for mosquitoes [67]. In contrast to cell culture observations, alternately passaged VEEV experienced no detectable fitness gains in either host, supporting the idea that in vivo dual-host cycling selects for viruses that are adapted to both host types, but that fitness increases in both hosts are not a requisite for maintenance in alternating cycling [67]. Serial passage of WNV via inoculation into the C. pipiens thorax (artificially bypassing midgut infection) produced WNV that was more fit than its progenitor in C. pipiens, but that experienced no replication cost in chicks [68]. Using a more natural oral infection route that did not circumvent midgut infection, another WNV study found that chick-specialized virus showed fitness gains in chicks and C. pipiens, whereas mosquito-passaged virus experienced reduced fitness in chicks and little change in mosquitoes [69], again supporting cell culture data showing that artificially releasing an arbovirus from host alternation allows for rapid fitness gains in the passaged host. The fact that fitness losses are not always observed in bypassed hosts in vivo suggests that while arboviruses experience fitness trade-offs via host alternation, host-specific adaptation to sequential passage sometimes comes at little cost in the bypassed host, especially for WNV. Studies with another flavivirus, SLEV, show somewhat conflicting results. Neither mosquito-specialized SLEV passaged by intrathoracic inoculation nor chick-passaged virus experienced gains in host-specific fitness, suggesting that SLEV may already be highly adapted to both invertebrate and avian hosts [70]. The disparities in patterns of in vivo adaptation between SLEV, WNV and VEEV may reflect virus-specific evolutionary traits (e.g., differences in host utilization, genome organization, rates of recombination, composition and breadth of mutant swarm, mechanisms of transmission and seasonality) or, alternatively, could result from differences in experimental designs. While intrathoracic inoculations ensured the infection of Culex mosquitoes in SLEV and some WNV studies [68,70], infection, replication and dissemination from the mosquito midgut may present additional selective constraints on the virus that were experimentally circumvented. To ensure high-titer blood meals for serial mosquito cycling, the VEEV study pooled multiple infected mosquitoes, which also potentially confounded results by representing viruses from many vector infections instead of a single mosquito [67]. Nevertheless, despite the problems of in vivo arbovirus evolution studies, the use of vectors and relevant vertebrate hosts in lieu of cells better simulates the complex environmental pressures faced by arboviruses.
Population genetic arboviral bottlenecks
Mosquito vectors present anatomical barriers to productive arbovirus transmission, sometimes blocking midgut infection and other times preventing escape from the midgut and dissemination into the hemocoel in order to reach the salivary glands for transmission. The four stages of mosquito infection presenting anatomical barriers include: midgut infection; midgut escape; salivary gland infection; and transmission to the vertebrate host (Figure 6). Experimental infection studies using VEEV [71], WNV [72] and VEEV replicon particles [71] expressing fluorescent proteins, which are capable of only a single round of infection, showed that few midgut epithelial cells become infected, suggesting that only certain `portal' cells are susceptible, even at high ingested doses. Interestingly, this was only the case for an epidemic VEEV strain; for enzootic VEEV, the enzootic vector midgut is uniformly susceptible, suggesting that a long-term vector–virus association may lead to higher midgut infectivity [73].
Figure 6. Route of infection and potential bottlenecks during arbovirus infection of a mosquito.
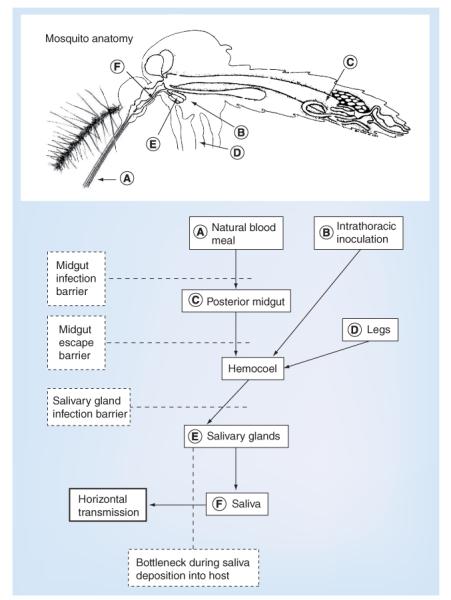
The route of virus dissemination from an infectious blood meal is shown by solid boxes, and the anatomical placement of each stage is designated by the letters (A–F) on the mosquito. The potential bottlenecks associated with physical barriers that the virus encounters during dissemination are shown in dashed boxes.
Adapted with permission from [113].
Genetic bottlenecks corresponding to anatomical barriers may also affect mosquito infection and transmission dynamics. Bottlenecks, so-called because they reduce genetic diversity and distance (the number of mutations by which each RNA differs from consensus) of a virus population compared with the input (but do not necessarily change the consensus), can profoundly affect arbovirus infection dynamics. This is because genetically diverse RNA virus populations exhibit greater phenotypic plasticity than homogenous populations, since they are more likely to possess variant genomes with adaptive mutations. However, despite barriers that create bottlenecks, genetic variation in whole mosquitoes after experimental WNV passage is much greater than that seen in birds [74], suggesting that arboviruses can circumvent genetic bottlenecks due to anatomical barriers by regenerating diversity during replication in downstream organs. Recent studies have focused on characterizing how the mutant swarm changes during the four stages of mosquito infection (Figure 7). WNV populations in C. quinquefasciatus midguts, hemolymph and saliva have comparable levels of genetic diversity, and although some individual variants compartmentalize to specific organs, as has been seen in poliovirus in mice [75], anatomical barriers do not impose significant bottlenecks on WNV populations [76]. In support of WNV findings, a similar experiment with VEEV revealed that, despite a bottleneck at midgut escape, measured as a significant reduction in genetic diversity on the days following escape of the virus into the hemocoel, the number of marked variants remained constant over time [77]. CHIKV studies parallel WNV and VEEV findings; although population diversity in the midgut and salivary gland was reduced compared with the blood meal input or midgut, respectively, diversity was recovered downstream of each barrier [Coffey LL, Unpublished Data]. It is unclear how diversity relates to the amount of virus transmitted by feeding mosquitoes; while A. taeniorhynchus inoculate an average of 11 PFU of VEEV into mice [78] and transmitted doses of WNV [76,79] and CHIKV [Coffey LL, Unpublished Data] range from 102 to 104 PFU, no studies have examined how variant composition and diversity relate to transmitted doses. Results from these three studies showing maintenance of diversity but changes in composition of the arbovirus mutant spectrum contrast with findings from another WNV study. WNV in C. pipiens showed a decrease in numbers of RNAs with mutations following midgut infection and transmission, which correlated with time since infection of the mosquito, suggesting that WNV swarms in that species are subject to temporal sweeps that decrease intrahost diversity [80].
Figure 7. Arbovirus population dynamics in vector infection.
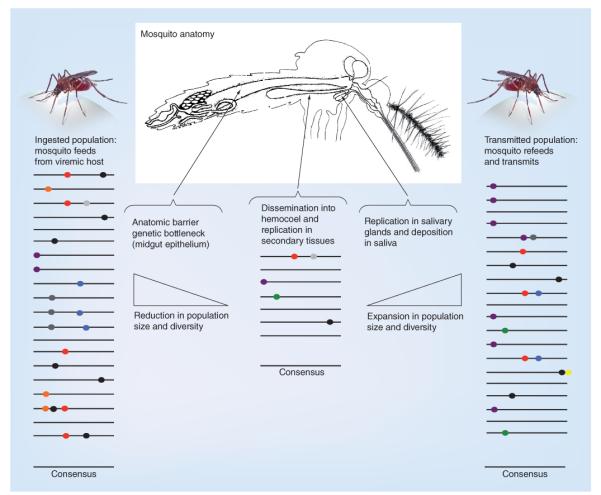
Lines represent individual arbovirus genomes; colored dots show mutations. The average sequence (consensus) in each population is shown, and is often unchanged during infection. Studies with West Nile virus, Venezuelan equine encephalitis virus and Chikungunya virus show that the composition of the mutant spectrum changes as viruses traverse anatomical barriers and experience genetic bottlenecks (diminishing slope showing decreased population size), including during infection and escape from the midgut epithelium, but that replication in secondary tissues regenerates genetic diversity (increasing slope) at levels comparable to populations ingested from the vertebrate host. In some cases, compartmentalization of individual variants (e.g., purple transmitted mutation) occurs in selected tissues, and some mutations (e.g., green) arise de novo in secondary tissues. In general, these studies show that arboviruses can circumvent anatomical barriers that produce genetic bottlenecks in mosquitoes, such that the size of the mutant spectrum that disseminates and is transmitted by vectors is not significantly reduced.
These results show that despite anatomical barriers to transmission, arboviruses typically circumvent reductions in genetic diversity and distance during mosquito infection, and that for WNV, C. pipiens and C. quinquefasciatus exert different pressures on population dynamics, despite being closely related. Together, these studies suggest that arboviruses retain high mutation frequencies in order to rapidly replenish diversity lost traversing anatomical barriers. Indeed, a CHIKV strain unable to generate wild-type-like genetic diversity showed reduced dissemination in vivo [81].
The mutant swarm of viral RNAs may in part determine arbovirus adaptability. Evolutionary theory predicts that diverse populations of viral RNAs are more likely than homogeneous populations to possess phenotypic plasticity and adaptability because, by chance, they contain more variant genomes with potentially adaptive mutations. Error rates of RdRp affect mutation frequencies in viral RNA populations and, in large part, control genetic diversity. Studies with fidelity variants of the vertebrate-only RNA poliovirus showed that reduced genetic diversity negatively affects virus dissemination and pathogenesis in mice [82]. By similar reasoning, arbovirus populations with less diversity might also be less fit, where fewer variant genomes would afford less phenotypic plasticity in both host types. In vitro studies using CHIKV [83] and WNV [84] and in vivo WNV experiments [74] support this concept. CHIKV populations alternately passaged between vertebrate and invertebrate cells possessed less genetic diversity than populations passaged serially on either cell type and were less capable of adapting to new cells or escaping neutralization. WNV after serial or alternating mosquito–avian passages showed increases in intrahost diversity that coincided with fitness increases (although alternately in vivo-passaged WNV, in contrast to in vitro-passaged CHIKV, showed the greatest diversity [74]), suggesting that minority genomes can augment fitness. In further support of this mechanism, when 24 different strains of WNV were mixed to create a population that was 1.5-or four-times more diverse than populations comprising mixes of eight and four strains, respectively, the more diverse populations out-competed the reference population better than the less diverse populations in mosquitoes (but not in chickens) [85]. WNV diversity in serial chicken-passaged lineages was also consistently lower than in mosquito-passaged lineages, suggesting that purifying selection is relaxed during mosquito infection (although a caveat of these studies was the use of intrathoracic inoculation of mosquitoes that circumvents midgut infection) [86].
The observed differences in diversity between serial versus alternately passaged arboviruses suggests that selection during alternating passages is focused on maintaining replication competence in both hosts. Greater genetic diversity (as observed in serial CHIKV passages and in mosquito-passaged WNV) may not always be achievable after alternation because minority mutations that would drift the mutant spectrum away from overlapping invertebrate and vertebrate adaptive peaks would be removed by purifying selection with each alternating passage. The expected outcome would be evolutionary stasis, which may, in part, explain the relatively slow evolutionary rates exhibited by arboviruses [8]. Thus, cell culture studies suggest that alternating host cycles restrict the expansion of genetic diversity compared with single-host serial passages, but still increase fitness at the cost of less adaptability. These in vitro and in vivo results indicate an evolutionary trade-off between maximizing fitness for alternating host infections and maximizing adaptability, where the most adaptable populations are those that have enhanced population diversity, but are not necessarily the best generalists.
To experimentally modulate genetic diversity instead of just characterizing it after passage, an arbovirus fidelity variant was isolated via selection of a mutagen-resistant variant with a single amino acid change in the nsP4 RdRp gene that increases replication fidelity by approximately 30%, but still replicates at wild-type levels [81]. Compared with its wild-type parent, this high-fidelity CHIKV variant infects and disseminates less effectively in A. aegypti and produces shorter viremias and lower organ titers in neonatal mice. These results suggest that, as for poliovirus, increased arbovirus replication fidelity imposes a fitness cost in both mosquitoes and vertebrates. This supports the idea that `sloppy' replication lends itself to arbovirus adaptability and may in part explain why arboviruses do not evolve higher-fidelity polymerases.
Epistatic constraints on CHIKV adaptation
As discussed above, CHIKV strains from the IOL have repeatedly adapted to A. albopictus by acquiring a single alanine-to-valine substitution in the E1 glycoprotein (E1-A226V) [10,26,29,30]. Interestingly, the same substitution has never been detected in any Asian-genotype CHIKV strain. Laboratory studies showed that the infectivity of several Asian-genotype CHIKV strains for A. albopictus is not significantly different from the ECSA genotype, including IOL strains that lack the E1-A226V change [24]. This indicates that although A. albopictus is highly abundant in southeast Asia, where the Asian genotype has persisted for more than 60 years [23,87], CHIKV nevertheless has failed to efficiently adapt to A. albopictus, and has probably used only A. aegypti for transmission and maintenance in the region. However, the invasion and establishment of A. albopictus-adapted CHIKV strains of the IOL into southeast Asia in recent years has demonstrated that A. albopictus can be a highly efficient vector, enabling a dramatic CHIKV expansion [29,88–91]. Thus, the inability of the Asian CHIKV lineage to occupy a human–A. albopictus niche, possibly due to differences in the distributions of the two vectors in southeast Asia versus the Indian Ocean islands and the Indian subcontinent, may have enabled the invasion and establishment of the IOL in southeast Asia, which may eventually promote local extinction of the Asian genotype. Furthermore, A. aegypti may enable the spread of IOL in Asia, which are equally infectious for this species compared with the Asian lineage, especially in areas where it predominates over A. albopictus.
The failure of the Asian lineage to exploit the E1-A226V substitution is explained by negative epistatic interactions with a threonine at position E1-98, which is invariant in Asian-lineage CHIKV strains. All strains of the ECSA genotype, including IOL, have an alanine at E1-98, which is neutral in its effect on the fitness of the E1-A226V substitution. Therefore, epistatic interactions between E1-226V and E1-98T residues are responsible for the increase in length of the fitness plateau (since two mutations are required instead of one) that populations of Asian-lineage CHIKV strains would have to traverse in order to reach the A. albopictus-adaptive peak. We call it a `plateau' instead of a `valley' because no negative effect on CHIKV fitness was detected in the alternative mosquito vector (A. aegypti) or in vertebrate hosts (newborn mice as a model for human infection). Since strains of the IOL are not affected by this epistatic interaction, they traverse this plateau more readily to occupy an A. albopictus-adaptive peak (via the E1-A226V mutation). Thus, the existence of different adaptive landscapes for Asian-genotype and Indian Ocean CHIKV lineages could explain the ongoing displacement of Asian-genotype strains by the IOL [24].
Constraints on adaptive WNV evolution
Similar to other RNA viruses, WNV exists as a swarm of mutants [74] that likely influences infection of both vertebrate and invertebrate hosts. Mosquitoes sampled from nature possess more WNV mutants compared with avian hosts. This result was confirmed by experiments that released the virus from its dual-host replication pattern through serial passage in chickens, mosquitoes, or alternately between chickens and mosquitoes, all of which were passaged 20-times each. However, similar to the naturally sampled WNV described above, 40 passages of WNV released from dual host cycling generated a greater increase in sequence diversity in mosquitoes than in chickens [92]. The level of intrahost diversity was very low in chickens and the pattern of mutations indicated strong purifying selection. However, the mosquito-only passages (intrathoracic infections that bypassed oral infection) exhibited a high degree of change from the consensus sequence. Interestingly, these results appear to contradict the standard dual-host trade-off hypothesis, which envisions that alternating replication in vertebrates and invertebrates requires fitness compromises in both hosts in order to maintain the transmission cycle. However, the chicken-only WNV passages yielded equal fitness in both chicken and C. pipiens mosquitoes, but not in a related species of mosquito [68,69,74]. The mosquito-only passages showed fitness increases in C. pipiens but not in the related C. quinquefasciatus mosquitoes, but did show severe fitness losses in chickens. Thus, at least for WNV, there appear to be several constraints involved in infecting both hosts. These results suggest that WNV adaptation to mosquitoes is species-specific, even in the congeneric mosquitoes C. pipiens and C. quinquefasciatus [92].
As discussed above, WNV may have constraints on the generation of high-fitness-adaptive mutants. The mutant swarm appears to be critical to the successful infection of both mosquito and avian hosts. In particular, the high diversity that is a consequence of mosquito infection by WNV appears to suppress high-fitness variants that can arise in chickens [84,93]. This apparent ability of the mutant swarm to affect the production of high-fitness variants has implications for the further evolution of WNV in the Americas. The experimental evidence suggests that WNV may be evolving towards a less lethal phenotype in birds, but evidence from natural samples will be required to test this hypothesis.
DENV intrahost genetic variation
Similar to other RNA viruses, DENV genetic diversity can be attributed to its error-prone RdRp. Prior to the advent of deep sequencing, the presence of DENV intrahost genetic diversity had been confirmed by sequencing large numbers of clonal amplicons derived from unpassaged, natural isolates (reviewed in [94]). Surprisingly, the extent of diversity included genomes with stop codons in their E genes, as well as mixed genotypes and putative recombinants. Some of those defective RNA genomes (E gene stop codon variants) were observed in human [50,95] and mosquito [95] samples, suggesting a mechanism of long-term transmission maintenance through complementation. This would require the preservation of relatively high multiplicities of infection throughout all stages of the transmission cycle, which would require experimental validation in mosquitoes, where transmission bottlenecks are likely to occur, based on studies of other mosquito-borne flaviviruses [80] or alphaviruses [71]. Currently, the amount of DENV inoculated by mosquitoes while probing or feeding on a live host is not known, although a recent report suggested an average salivary secretion titer of 50 PFU [96]. This estimate is much lower than that reported for WNV (102–104 PFU) [76,97]. Some studies suggest that the dose transmitted may affect vertebrate viremia. Chicks that receive higher doses of WNV from mosquitoes develop higher early viremias, and chicks infected by multiple mosquitoes produce viremias up to 50-times higher than chicks infected by a single mosquito [97]. These results indicate that doses delivered by vectors correlate with viremia levels in hosts, where higher viremias are sometimes associated with more severe disease. However, studies estimating doses secreted by vectors are biased, in that mosquitoes that salivate in vitro eject higher titers than they inoculate into vertebrates, and infection by needle, as is conventional for most pathogenesis studies, may influence vertebrate pathogenesis [78]. Higher viremias in vertebrates render them infectious to naive mosquitoes for longer periods. The minimum viremia levels necessary for infection of vectors vary according to virus and vector species; generally, primary vector species are identified by high susceptibility to infection at low ingested titers.
Sylvatic DENV epidemics & human contact
DENV emergence and the role of adaptation to new hosts and vectors are important issues for arbovirology and have enormous public health implications, especially considering the potential for eradicating the human transmission cycle with the effective vaccines now under development. Humans become infected with sylvatic DENV [98,99], probably very regularly, as evidenced by serologic and surveillance studies in southeast Asia and west Africa [42]. Spillover from sylvatic cycles could potentially generate large outbreaks [100]. However, the limited detection of sylvatic DENV is partially attributed to misdiagnosis of human DENV infections or confusion with other etiological agents that share similar clinical signs and symptoms, or nondetection due to subclinical presentation. Another possible explanation for the limited spillover potential is the requirement for adaptation to peridomestic vectors and/or human hosts. The latter hypothesis was tested experimentally using surrogate models of human infection, in vitro model systems and in mosquitoes, described below.
Vertebrate model studies of adaptive constraints on DENV evolution
Mechanisms of human DENV emergence due to host-range expansion of sylvatic strains by adaptation to the use of humans as reservoirs (via increased magnitude of replication) were evaluated in two surrogate models of human infection: monocyte-derived dendritic cells and severe combined immune deficiency mice xenografted with human hepatoma cells [57]. Select DENV-2 strains representing all four genotypes, including Asian and African sylvatic strains, as well as Asian, African and American human strains of various pathogenic potentials (classical dengue fever to severe dengue disease), were evaluated. In both models, there was significant variation in mean replication titers of DENV-2 strains. However, there was no overall difference in replication between sylvatic and endemic strains. Interestingly, sylvatic strains replicated to lower titers than human Asian strains, but did not differ consistently from the endemic American strains, suggesting that the American strains have maintained or regained their ancestral phenotype. These observations suggest that the historical emergence of DENV from the ancestral sylvatic transmission cycle into human cycles may not have required adaptation in order to replicate in humans as reservoir hosts, which implies that the probability of re-emergence into human transmission is high.
Mosquito model studies of adaptive constraints on DENV evolution
A number of studies have addressed the question of whether human DENV emergence was mediated by adaptation to the peridomestic mosquito vectors A. aegypti and A. albopictus. Moncayo et al. supported this hypothesis [101], but more recent studies that included an expanded repertoire of DENV strains suggest that sylvatic and human DENV-2 strains are equally infectious for both mosquito species, indicating that the emergence of sylvatic DENV into human transmission did not require adaptation to these vectors [Hanley K & Vasilakis N, Unpublished Data], echoing observations from the vertebrate model studies described above. Given the increased ecologic pressures and widespread conversion of native forests into oil palm plantations and other agricultural settings in sylvatic DENV foci, the question of whether human strains could reinvade the forest cycle becomes epidemiologically relevant. Diallo and colleagues examined whether human DENV strains have lost fitness for transmission by sylvatic vectors by comparing the vector competence of sylvatic and various peridomestic populations of Senegalese mosquitoes using both human and sylvatic DENV-2 strains (reviewed in [42]). The results of this study refuted the hypothesis that any adaptation of human strains to domestic vectors was species specific, and supported the hypothesis that human strains have the potential to become established in a forest cycle.
In vitro studies of adaptive constraints on DENV evolution
The observed DENV intrahost genetic diversity suggests that variants play a dynamic role in viral fitness, replication and their ability to successfully adapt to new environments, a mechanism attributed to the trade-off hypothesis mentioned above. The extent of DENV diversity was examined with similar methodologies as those used previously [8,61,67]. Early studies based on partial genomic consensus sequences indicated that adaptation in mosquito cells, unlike adaptation in vertebrates or alternating cycles, has a minimal effect on DENV evolution. These results contrasted with those for WNV [84,102], where genetic diversity was shown to correspond to substantial phenotypic diversity. This discrepancy could be attributed to the inherent limitation of consensus (Sanger) sequencing, where each nucleotide at a given position of the viral genome only reflects the majority and does not represent the true mutant swarm. Subsequently, a comprehensive study simulating the DENV transmission cycle or adaptive specialization in vertebrate or mosquito cell lines examined the trade-off hypothesis by employing clonal (plaque-purified, with a defined sequence) or passaged, uncloned (where the determined sequence represents the consensus of the population) DENV [62]. While an inherent limitation of this study was the utilization of consensus sequencing to monitor the evolution of viral populations, which could mask the presence of minority mutant populations, the data supported the hypothesis that releasing DENV from host alternation facilitates adaptation. However, there was limited support for the hypothesis that such alternation necessitates a fitness trade-off. An interesting observation of this study was that alternately passaged clonal DENV exhibited fitness gains, an observation attributed to the acquisition of both host cell-specific and amphi-cell-specific adaptations, or to recovery from fitness losses arising from bottlenecking due to biological cloning [62]. An equally important observation indicated that DENV adapted exclusively in the vertebrate cell line or alternating between vertebrate and mosquito host cells led to the emergence of a qualitatively and quantitatively distinct mutation spectrum from exclusive passage in the invertebrate cell line, which signifies the role of the depth and breadth of intrahost genetic diversity in shaping the adaptive phenotype [84].
Constraints imposed by epistatic interactions
The process of cross-species jumps and adaptive evolution can be viewed as the movement of viral populations across adaptive landscapes. Epistatic interactions (interdependent effects of different genetic loci on viral fitness) could create multiple peaks in the fitness landscape, which could constrain the evolution of given species by forcing a population to occupy a local peak. The transition of a viral population to new peaks of possibly higher fitness in recipient species would therefore be constrained by movement across adaptive valleys or plateaus of lower fitness [8]. Although the existence of multipeaked landscapes in nature is still debatable, several studies have confirmed their role in the constraints of bacterial and viral evolution in laboratory settings [103–105]. Interestingly, the adaptive constraints of Asian-genotype CHIKV to A. albopictus mosquitoes, discussed above, can also be explained in terms of multipeaked landscapes.
Future perspective
There appear to be fundamental differences in the evolutionary patterns and mechanisms of emergence among alphaviruses and flaviviruses. The alphaviruses, perhaps due to intrinsic properties that are not fully understood, can sometimes undergo dramatic adaptive evolution, while flaviviruses are associated with emergence through geographic expansion and urbanization, with little or no evidence that adaptation plays a role in the initial establishment of endemicity. There is evidence (which is stronger for alphaviruses than for flaviviruses) that the requirement for alternating infection of vertebrate and invertebrate hosts is a constraining force on adaptive evolution. In addition, for CHIKV, epistatic interactions that vary among viral lineages can have dramatic effects of epidemic emergence. Finally, for both alphaviruses and flaviviruses, marked changes in viral population sizes, including significant bottlenecks that accompany midgut infection and viral dissemination into the hemocoel, may limit the efficiency of natural selection. Improvements in study design to sample populations immediately after a barrier has been circumvented (instead of long after, where founder genomes may be masked by new variants arising subsequently to the bottleneck) will better define how bottlenecks shape populations. Studies of this type will be helpful in determining whether these bottlenecks often lead to viral extinction, as predicted by Muller's ratchet [77], and if so, how the arboviruses compensate in order to persist in nature.
The above conclusions suggest several risks for future arboviral emergence. First, the ability of both alphaviruses and flaviviruses to urbanize and cause explosive epidemics through transmission by A. aegypti and A. albopictus, as exemplified by DENV and CHIKV, as well as yellow fever virus, is a major concern [106]. Several other viruses with known exposure to tropical urban populations and the ability to infect these potential urban vectors, including VEEV and Mayaro virus, as well as the flavivirus Zika virus, are of particular concern. Mayaro virus is of special interest because it regularly infects people in major urban areas of South America, but is grossly under-reported due to misdiagnosis as DENV. Undoubtedly, there are also as-yet undiscovered arboviruses with emergence potential, and recent advances in deep sequencing provide new opportunities to identify them contingent on continued surveillance activities, especially in the tropics, where viral diversity is highest. The continuing global expansion of A. albopictus, which is a competent laboratory vector of many arboviruses, will increase this risk, as well as climate change, which is predicted to expand the distribution of A. aegypti further outside the tropics. A more complete understanding of the molecular interactions associated with the adaptive emergence of VEEV and CHIKV will be especially useful in improving our ability to predict the likelihood of adaptive urbanization of Mayaro virus. Dramatic advances in determining the structures of both alphavirus and flavivirus particles and envelope proteins [16,20,107] have increased opportunities for advances in this area. However, the identification of key cellular receptors for many alphaviruses and flaviviruses remains an obstacle to progress, especially receptors in vector midguts, which are the key portals of entry leading to transmission.
Executive summary.
Arboviruses
-
■
Most arboviruses are zoonotic agents that can cause disease in domesticated animals and/or humans via direct spillover from enzootic cycles, sometimes after introduction into a new geographic region, amplification in domesticated animals to increase circulation and human exposure or the initiation of human–mosquito–human transmission cycles in peridomestic habitats.
-
■
The latter two emergence mechanisms sometimes, but not always, involve adaptive evolution in order to enhance transmission by mosquito vectors or more efficient amplification by vertebrate hosts.
Historic evidence of arbovirus adaptive evolution
-
■
The alphaviruses Venezuelan equine encephalitis and chikungunya viruses have undergone dramatic emergence via equine- and mosquito-adaptive amino acid substitutions involving single-point mutations in their envelope glycoprotein genes.
Evidence for constraints on adaptive arbovirus evolution in nature
-
■
Despite these examples of dramatic adaptive emergence mediated by single mutations, and the high mutation frequencies exhibited by most if not all arboviruses, these emergence events occur only occasionally, suggesting fundamental constraints on the selection of adaptive mutations.
Hypothetical explanations for adaptive constraints on arboviruses
-
■
Evidence from retrospective studies of adaptive emergence, as well as prospective studies of arbovirus adaptation, suggest three major constraint mechanisms: the requirement for alternating infection and replication in vertebrate and invertebrate hosts imposes constraints on adaptation due to fitness trade-offs in the two hosts or differing fitness landscapes for the infection of each; epistatic interactions that can differ dramatically between closely related arboviral lineages sometimes limit the penetrance of adaptive mutations in different geographic regions; and the arbovirus transmission cycle imposes population bottlenecks that constrain adaptive evolution by limiting the efficiency of selection.
Acknowledgements
The authors would like to thank S Rossi for expert graphic design of Figure 5.
The research described was supported by NIH grants R01-AI071192, R01-AI069145 and R01-AI48807.
No writing assistance was utilized in the production of this manuscript.
Footnotes
Financial & competing interests disclosure
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
Papers of special note have been highlighted as:
■ of interest
■■ of considerable interest
- 1.Sharp PM, Hahn BH. Origins of HIV and the AIDS pandemic. Cold Spring Harb. Perspect. Med. 2011;1(1):a006841. doi: 10.1101/cshperspect.a006841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balboni A, Battilani M, Prosperi S. The SARS-like coronaviruses. the role of bats and evolutionary relationships with SARS coronavirus. New Microbiol. 2012;35(1):1–16. [PubMed] [Google Scholar]
- 3.Bolles M, Donaldson E, Baric R. SARS-CoV and emergent coronaviruses: viral determinants of interspecies transmission. Curr. Opin. Virol. 2011;1(6):624–634. doi: 10.1016/j.coviro.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoelzer K, Parrish CR. The emergence of parvoviruses of carnivores. Vet. Res. 2010;41(6):39. doi: 10.1051/vetres/2010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weaver SC, Osorio JE, Livengood JA, Chen R, Stinchcomb DT. Chikungunya virus and prospects for a vaccine. Expert Rev. Vaccines. 2012;11(9):1087–1101. doi: 10.1586/erv.12.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simon F, Javelle E, Oliver M, Leparc-Goffart I, Marimoutou C. Chikungunya virus infection. Curr. Infect. Dis. Rep. 2011;13(3):218–228. doi: 10.1007/s11908-011-0180-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gubler DJ. The economic burden of dengue. Am. J. Trop. Med. Hyg. 2012;86(5):743–744. doi: 10.4269/ajtmh.2012.12-0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holmes EC. The Evolution and Emergence of RNA Viruses. Oxford University Press; NY, USA: 2009. [Google Scholar]
- 9.Weaver SC, Winegar R, Manger ID, Forrester NL. Alphaviruses: population genetics and determinants of emergence. Antiviral Res. 2012;94(3):242–257. doi: 10.1016/j.antiviral.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsetsarkin KA, Vanlandingham DL, Mcgee CE, Higgs S. A single mutation in chikungunya virus affects vector specificity and epidemic potential. PLoS Pathog. 2007;3(12):e201. doi: 10.1371/journal.ppat.0030201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsetsarkin KA, Weaver SC. Sequential adaptive mutations enhance efficient vector switching by chikungunya virus and its epidemic emergence. PLoS Pathog. 2011;7(12):e1002412. doi: 10.1371/journal.ppat.1002412. [DOI] [PMC free article] [PubMed] [Google Scholar]; ■ Describes sequential Aedes albopictus-adaptive mutations and their role in midgut epithelial cell infection during epidemic transmission of chikungunya virus (CHIKV) in the Indian Ocean and India from 2005 to 2009.
- 12.Tsetsarkin KA, Chen R, Sherman MB, Weaver SC. Chikungunya virus: evolution and genetic determinants of emergence. Curr. Opin. Virol. 2011;1(4):310–317. doi: 10.1016/j.coviro.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barrett ADT, Weaver SC. Medical Microbiology. 18th Edition Elsevier; UK: 2012. Arboviruses: alphaviruses, flaviviruses and bunyaviruses; pp. 520–536. [Google Scholar]
- 14.Powers AM, Huang HV, Roehrig JT, Strauss EG, Weaver SC. Togaviridae. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ, editors. Virus Taxonomy, Ninth Report of the International Committee on Taxonomy of Viruses. Elsevier; UK: 2011. pp. 1103–1110. [Google Scholar]
- 15.Nasar F, Palacios G, Gorchakov RV, et al. Eilat virus, a unique alphavirus with host range restricted to insects by RNA replication. Proc. Natl Acad. Sci. USA. 2012;109(36):14622–14627. doi: 10.1073/pnas.1204787109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuhn RJ. Togaviridae: the viruses and their replication. In: Knipe DM, Howley PM, editors. Fields' Virology. 5th Edition Lippincott, Williams and Wilkins; NY, USA: 2007. pp. 1001–1022. [Google Scholar]
- 17.Simmonds P, Becher P, Collett MS, et al. Flaviviridae. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ, editors. Virus Taxonomy, Ninth Report of the International Committee on Taxonomy of Viruses. UK; Elsevier: 2011. pp. 1003–1020. [Google Scholar]
- 18.Hanley KA, Weaver SC. Arbovirus evolution. In: Domingo E, Parrish CR, Holland JJ, editors. Origin and Evolution of Viruses. Elsevier; UK: 2008. pp. 351–392. [Google Scholar]
- 19.Heinze DM, Gould EA, Forrester NL. Revisiting the clinal concept of evolution and dispersal for the tick-borne flaviviruses by using phylogenetic and biogeographic analyses. J. Virol. 2012;86(16):8663–8671. doi: 10.1128/JVI.01013-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lindenbach BD, Thiel H-J, Rice CM. Flaviviridae: the viruses and their replication. In: Knipe DM, Howley PM, editors. Fields' Virology. 5th Edition Lippincott, Williams and Wilkins; NY, USA: 2007. pp. 1101–1152. [Google Scholar]
- 21.Chen R, Vasilakis N. Dengue: quo tu et quo vadis? Viruses. 2011;3(9):1562–1608. doi: 10.3390/v3091562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Diallo D, Sall AA, Buenemann M, et al. Landscape ecology of sylvatic chikungunya virus and mosquito vectors in southeastern Senegal. PLoS Negl. Trop. Dis. 2012;6(6):e1649. doi: 10.1371/journal.pntd.0001649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Volk SM, Chen R, Tsetsarkin KA, et al. Genome-scale phylogenetic analyses of chikungunya virus reveal independent emergences of recent epidemics and various evolutionary rates. J. Virol. 2010;84(13):6497–6504. doi: 10.1128/JVI.01603-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsetsarkin KA, Chen R, Leal G, et al. Chikungunya virus emergence is constrained in Asia by lineage-specific adaptive landscapes. Proc. Natl Acad. Sci. USA. 2011;108(19):7872–7877. doi: 10.1073/pnas.1018344108. [DOI] [PMC free article] [PubMed] [Google Scholar]; ■■ Describes epistatic constraints on envelope glycoprotein amino acid substitutions that constrained the emergence of A. albopictus-adaptive CHIKV in Asia, lasting for more than 50 years.
- 25.Schwartz O, Albert ML. Biology and pathogenesis of chikungunya virus. Nat. Rev. Microbiol. 2010;8(7):491–500. doi: 10.1038/nrmicro2368. [DOI] [PubMed] [Google Scholar]
- 26.Vazeille M, Moutailler S, Coudrier D, et al. Two chikungunya isolates from the outbreak of La Reunion (Indian Ocean) exhibit different patterns of infection in the mosquito, Aedes albopictus. PLoS One. 2007;2(11):e1168. doi: 10.1371/journal.pone.0001168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schuffenecker I, Iteman I, Michault A, et al. Genome microevolution of chikungunya viruses causing the Indian Ocean outbreak. PLoS Med. 2006;3(7):e263. doi: 10.1371/journal.pmed.0030263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumar NP, Joseph R, Kamaraj T, Jambulingam P. A226V mutation in virus during the 2007 chikungunya outbreak in Kerala, India. J. Gen. Virol. 2008;89(Pt 8):1945–1948. doi: 10.1099/vir.0.83628-0. [DOI] [PubMed] [Google Scholar]
- 29.Hapuarachchi HC, Bandara KB, Sumanadasa SD, et al. Re-emergence of chikungunya virus in south-east Asia. virological evidence from Sri Lanka and Singapore. J. Gen. Virol. 2010;91(Pt 4):1067–1076. doi: 10.1099/vir.0.015743-0. [DOI] [PubMed] [Google Scholar]
- 30.de Lamballerie X, Leroy E, Charrel RN, Ttsetsarkin K, Higgs S, Gould EA. Chikungunya virus adapts to tiger mosquito via evolutionary convergence: a sign of things to come? Virol. J. 2008;5:33. doi: 10.1186/1743-422X-5-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Niyas KP, Abraham R, Unnikrishnan RN, et al. Molecular characterization of chikungunya virus isolates from clinical samples and adult Aedes albopictus mosquitoes emerged from larvae from Kerala, South India. Virol. J. 2010;7:189. doi: 10.1186/1743-422X-7-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Domingo E. Mechanisms of viral emergence. Vet. Res. 2010;41(6):38. doi: 10.1051/vetres/2010010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nijhuis M, Van Maarseveen NM, Boucher CA. Antiviral resistance and impact on viral replication capacity: evolution of viruses under antiviral pressure occurs in three phases. Handb. Exp. Pharmacol. 2009;(189):299–320. doi: 10.1007/978-3-540-79086-0_11. [DOI] [PubMed] [Google Scholar]
- 34.Kramer LD, Styer LM, Ebel GD. A global perspective on the epidemiology of West Nile virus. Annu. Rev. Entomol. 2008;53:61–81. doi: 10.1146/annurev.ento.53.103106.093258. [DOI] [PubMed] [Google Scholar]
- 35.May FJ, Davis CT, Tesh RB, Barrett AD. Phylogeography of West Nile virus: from the cradle of evolution in Africa to Eurasia, Australia, and the Americas. J. Virol. 2011;85(6):2964–2974. doi: 10.1128/JVI.01963-10. [DOI] [PMC free article] [PubMed] [Google Scholar]; ■ The origin of the introduction of West Nile virus into the USA was reanalyzed using the latest methods, revealing that the virus was more likely introduced from Africa than from Israel. Instead, both the Israeli and US viruses share a common African ancestor, which is likely the source of both outbreaks.
- 36.Davis CT, Li L, May FJ, et al. Genetic stasis of dominant West Nile virus genotype, Houston, Texas. Emerg. Infect. Dis. 2007;13(4):601–604. doi: 10.3201/eid1304.061473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mcmullen AR, May FJ, Li L, et al. Evolution of new genotype of West Nile virus in North America. Emerg. Infect. Dis. 2011;17(5):785–793. doi: 10.3201/eid1705.101707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ebel GD, Carricaburu J, Young D, Bernard KA, Kramer LD. Genetic and phenotypic variation of West Nile virus in New York, 2000–2003. Am. J. Trop. Med. Hyg. 2004;71(4):493–500. [PubMed] [Google Scholar]
- 39.Moudy RM, Meola MA, Morin LL, Ebel GD, Kramer LD. A newly emergent genotype of West Nile virus is transmitted earlier and more efficiently by Culex mosquitoes. Am. J. Trop. Med. Hyg. 2007;77(2):365–370. [PubMed] [Google Scholar]
- 40.Anderson JF, Main AJ, Cheng G, Ferrandino FJ, Fikrig E. Horizontal and vertical transmission of West Nile virus genotype NY99 by Culex salinarius and genotypes NY99 and WN02 by Culex tarsalis. Am. J. Trop. Med. Hyg. 2012;86(1):134–139. doi: 10.4269/ajtmh.2012.11-0473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fonseca DM, Keyghobadi N, Malcolm CA, et al. Emerging vectors in the Culex pipiens complex. Science. 2004;303(5663):1535–1538. doi: 10.1126/science.1094247. [DOI] [PubMed] [Google Scholar]
- 42.Vasilakis N, Cardosa J, Hanley KA, Holmes EC, Weaver SC. Fever from the forest: prospects for the continued emergence of sylvatic dengue virus and its impact on public health. Nat. Rev. Microbiol. 2011;9(7):532–541. doi: 10.1038/nrmicro2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vasilakis N, Weaver SC. The history and evolution of human dengue emergence. Adv. Virus Res. 2008;72:1–76. doi: 10.1016/S0065-3527(08)00401-6. [DOI] [PubMed] [Google Scholar]
- 44.Vasilakis N, Hanley KA, Weaver SC. Dengue virus emergence from its sylvatic cycle. In: Hanley KA, Weaver SC, editors. Frontiers in Dengue Virus Research. Horizon Press; UK: 2010. pp. 183–217. [Google Scholar]
- 45.Vasilakis N, Durbin AP, Da Rosa AP, Munoz-Jordan JL, Tesh RB, Weaver SC. Antigenic relationships between sylvatic and endemic dengue viruses. Am. J. Trop. Med. Hyg. 2008;79(1):128–132. [PubMed] [Google Scholar]
- 46.Rothman AL. Immunity to dengue virus: a tale of original antigenic sin and tropical cytokine storms. Nat. Rev. Immunol. 2011;11(8):532–543. doi: 10.1038/nri3014. [DOI] [PubMed] [Google Scholar]
- 47.Rico-Hesse R. Dengue virus markers of virulence and pathogenicity. Future Virol. 2009;4(6):581–589. doi: 10.2217/fvl.09.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Duffy S, Shackelton LA, Holmes EC. Rates of evolutionary change in viruses: patterns and determinants. Nat. Rev. Genet. 2008;9(4):267–276. doi: 10.1038/nrg2323. [DOI] [PubMed] [Google Scholar]
- 49.Parameswaran P, Charlebois P, Tellez Y, et al. Genome-wide patterns of intrahuman dengue virus diversity reveal associations with viral phylogenetic clade and interhost diversity. J. Virol. 2012;86(16):8546–8558. doi: 10.1128/JVI.00736-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thai KT, Henn MR, Zody MC, et al. High-resolution analysis of intrahost genetic diversity in dengue virus serotype 1 infection identifies mixed infections. J. Virol. 2012;86(2):835–843. doi: 10.1128/JVI.05985-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Blair CD. Mosquito RNAi is the major innate immune pathway controlling arbovirus infection and transmission. Future Microbiol. 2011;6(3):265–277. doi: 10.2217/fmb.11.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morrison J, Aguirre S, Fernandez-Sesma A. Innate immunity evasion by dengue virus. Viruses. 2012;4(3):397–413. doi: 10.3390/v4030397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aguilar PV, Adams AP, Suarez V, et al. Genetic characterization of Venezuelan equine encephalitis virus from Bolivia, Ecuador and Peru. identification of a new subtype ID lineage. PLoS Negl. Trop. Dis. 2009;3(9):e514. doi: 10.1371/journal.pntd.0000514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Turner PE, Morales NM, Alto BW, Remold SK. Role of evolved host breadth in the initial emergence of an RNA virus. Evolution. 2010;64(11):3273–3286. doi: 10.1111/j.1558-5646.2010.01051.x. [DOI] [PubMed] [Google Scholar]
- 55.Cologna R, Armstrong PM, Rico-Hesse R. Selection for virulent dengue viruses occurs in humans and mosquitoes. J. Virol. 2005;79(2):853–859. doi: 10.1128/JVI.79.2.853-859.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]; ■■ Describes the adaptation of Asian-genotype dengue virus (DENV)2 strains for more efficient human viremia and Aedes aegypti infection, which allowed it to displace the American genotype in Latin America.
- 56.Vasilakis N, Fokam EB, Hanson CT, et al. Genetic and phenotypic characterization of sylvatic dengue virus type 2 strains. Virology. 2008;377(2):296–307. doi: 10.1016/j.virol.2008.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]; ■■ Compares the genotypes and phenotypes of sylvatic versus human DENV with in vivo and ex vivo surrogate models of human infection. Suggests that DENV emergence in humans encounters little or no adaptive barrier.
- 57.Vasilakis N, Shell EJ, Fokam EB, et al. Potential of ancestral sylvatic dengue-2 viruses to re-emerge. Virology. 2007;358(2):402–412. doi: 10.1016/j.virol.2006.08.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Davis CT, Ebel GD, Lanciotti RS, et al. Phylogenetic analysis of North American West Nile virus isolates, 2001–2004: evidence for the emergence of a dominant genotype. Virology. 2005;342(2):252–265. doi: 10.1016/j.virol.2005.07.022. [DOI] [PubMed] [Google Scholar]
- 59.Jenkins GM, Holmes EC. The extent of codon usage bias in human RNA viruses and its evolutionary origin. Virus Res. 2003;92(1):1–7. doi: 10.1016/s0168-1702(02)00309-x. [DOI] [PubMed] [Google Scholar]
- 60.Cooper LA, Scott TW. Differential evolution of eastern equine encephalitis virus populations in response to host cell type. Genetics. 2001;157(4):1403–1412. doi: 10.1093/genetics/157.4.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Greene IP, Wang E, Deardorff ER, Milleron R, Domingo E, Weaver SC. Effect of alternating passage on adaptation of Sindbis virus to vertebrate and invertebrate cells. J. Virol. 2005;79(22):14253–14260. doi: 10.1128/JVI.79.22.14253-14260.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vasilakis N, Deardorff ER, Kenney JL, Rossi SL, Hanley KA, Weaver SC. Mosquitoes put the brake on arbovirus evolution: experimental evolution reveals slower mutation accumulation in mosquito than vertebrate cells. PLoS Pathog. 2009;5(6):e1000467. doi: 10.1371/journal.ppat.1000467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moutailler S, Roche B. Thiberge JM, Caro V, Rougeon F, Failloux AB. Host alternation is necessary to maintain the genome stability of rift valley fever virus. PLoS Negl. Trop. Dis. 2011;5(5):e1156. doi: 10.1371/journal.pntd.0001156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Presloid JB, Ebendick-Corpus BE, Zarate S, Novella IS. Antagonistic pleiotropy involving promoter sequences in a virus. J. Mol. Biol. 2008;382(2):342–352. doi: 10.1016/j.jmb.2008.06.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Novella IS, Quer J, Domingo E, Holland JJ. Exponential fitness gains of RNA virus populations are limited by bottleneck effects. J. Virol. 1999;73(2):1668–1671. doi: 10.1128/jvi.73.2.1668-1671.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ciota AT, Lovelace AO, Ngo KA, et al. Cell-specific adaptation of two flaviviruses following serial passage in mosquito cell culture. Virology. 2007;357(2):165–174. doi: 10.1016/j.virol.2006.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Coffey LL, Vasilakis N, Brault AC, Powers AM, Tripet F, Weaver SC. Arbovirus evolution in vivo is constrained by host alternation. Proc. Natl Acad. Sci. USA. 2008;105(19):6970–6975. doi: 10.1073/pnas.0712130105. [DOI] [PMC free article] [PubMed] [Google Scholar]; ■■ Provides the first in vivo experimental evidence that the adaptative evolution of arboviruses is constrained by obligate host alternation and predicts that arboviral emergence via host range changes may be less frequent than that of single-host animal RNA viruses.
- 68.Ciota AT, Lovelace AO, Jia Y, Davis LJ, Young DS, Kramer LD. Characterization of mosquito-adapted West Nile virus. J. Gen. Virol. 2008;89(Pt 7):1633–1642. doi: 10.1099/vir.0.2008/000893-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Deardorff ER, Fitzpatrick KA, Jerzak GV, Shi PY, Kramer LD, Ebel GD. West Nile virus experimental evolution in vivo and the trade-off hypothesis. PLoS Pathog. 2011;7(11):e1002335. doi: 10.1371/journal.ppat.1002335. [DOI] [PMC free article] [PubMed] [Google Scholar]; ■■ Provides the first in vivo experimental evidence that flavivirus infection of birds involves strong purifying selection, but that infection of mosquito vectors results in increased genetic diversity due to weak purifying selection.
- 70.Ciota AT, Jia Y, Payne AF, et al. Experimental passage of St. Louis encephalitis virus in vivo in mosquitoes and chickens reveals evolutionarily significant virus characteristics. PLoS One. 2009;4(11):e7876. doi: 10.1371/journal.pone.0007876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Smith DR, Adams AP, Kenney JL, Wang E, Weaver SC. Venezuelan equine encephalitis virus in the mosquito vector Aedes taeniorhynchus: infection initiated by a small number of susceptible epithelial cells and a population bottleneck. Virology. 2008;372(1):176–186. doi: 10.1016/j.virol.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Scholle F, Girard YA, Zhao Q, Higgs S, Mason PW. Trans-packaged West Nile virus-like particles: infectious properties in vitro and in infected mosquito vectors. J. Virol. 2004;78(21):11605–11614. doi: 10.1128/JVI.78.21.11605-11614.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kenney JL, Adams AP, Gorchakov R, Leal G, Weaver SC. Genetic and anatomic determinants of enzootic Venezuelan equine encephalitis virus infection of Culex (Melanoconion) taeniopus. PLoS Negl. Trop. Dis. 2012;6(4):e1606. doi: 10.1371/journal.pntd.0001606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jerzak GV, Brown I, Shi PY, Kramer LD, Ebel GD. Genetic diversity and purifying selection in West Nile virus populations are maintained during host switching. Virology. 2008;374(2):256–260. doi: 10.1016/j.virol.2008.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kuss SK, Etheredge CA, Pfeiffer JK. Multiple host barriers restrict poliovirus trafficking in mice. PLoS Pathog. 4(6):e1000082–2008. doi: 10.1371/journal.ppat.1000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brackney DE, Pesko KN, Brown IK, Deardorff ER, Kawatachi J, Ebel GD. West Nile virus genetic diversity is maintained during transmission by Culex pipiens quinquefasciatus mosquitoes. PLoS One. 2011;6(9):e24466. doi: 10.1371/journal.pone.0024466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Forrester NL, Guerbois M, Seymour RL, Spratt H, Weaver SC. Vector-borne transmission imposes a severe bottleneck on an RNA virus population. PLoS Pathog. 2012;8(9):e1002897. doi: 10.1371/journal.ppat.1002897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Smith DR, Aguilar PV, Coffey LL, Gromowski GD, Wang E, Weaver SC. Venezuelan equine encephalitis virus transmission and effect on pathogenesis. Emerg. Infect. Dis. 2006;12(8):1190–1196. doi: 10.3201/eid1208.050841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Styer LM, Kent KA, Albright RG, Bennett CJ, Kramer LD, Bernard KA. Mosquitoes inoculate high doses of West Nile virus as they probe and feed on live hosts. PLoS Pathog. 2007;3(9):1262–1270. doi: 10.1371/journal.ppat.0030132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ciota AT, Ehrbar DJ, Van Slyke GA, et al. Quantification of intrahost bottlenecks of West Nile virus in Culex pipiens mosquitoes using an artificial mutant swarm. Infect. Genet. Evol. 2012;12(3):557–564. doi: 10.1016/j.meegid.2012.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Coffey LL, Beeharry Y, Borderia AV, Blanc H, Vignuzzi M. Arbovirus high fidelity variant loses fitness in mosquitoes and mice. Proc. Natl Acad. Sci. USA. 2011;108(38):16038–16043. doi: 10.1073/pnas.1111650108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vignuzzi M. Viruses: foe, freeloader or friend? Curr. Opin. Microbiol. 2012;15(4):486–489. doi: 10.1016/j.mib.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 83.Coffey LL, Vignuzzi M. Host alternation of chikungunya virus increases fitness while restricting population diversity and adaptability to novel selective pressures. J. Virol. 2011;85(2):1025–1035. doi: 10.1128/JVI.01918-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ciota AT, Ehrbar DJ, Van Slyke GA, Willsey GG, Kramer LD. Cooperative interactions in the West Nile virus mutant swarm. BMC Evol. Biol. 2012;12:58. doi: 10.1186/1471-2148-12-58. [DOI] [PMC free article] [PubMed] [Google Scholar]; ■■ Using cloned sequences, the fitness levels of West Nile virus swarms were determined during in vitro infections. This is the first paper to evaluate the effect of flavivirus diversity on arboviral fitness and how the mutant virus swarm affects overall viral fitness.
- 85.Fitzpatrick KA, Deardorff ER, Pesko K, et al. Population variation of West Nile virus confers a host-specific fitness benefit in mosquitoes. Virology. 2010;404(1):89–95. doi: 10.1016/j.virol.2010.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jerzak GV, Bernard K, Kramer LD, Shi PY, Ebel GD. The West Nile virus mutant spectrum is host-dependant and a determinant of mortality in mice. Virology. 2007;360(2):469–476. doi: 10.1016/j.virol.2006.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Powers AM, Brault AC, Tesh RB, Weaver SC. Re-emergence of chikungunya and O'nyong-nyong viruses: evidence for distinct geographical lineages and distant evolutionary relationships. J. Gen. Virol. 2000;81(Pt 2):471–479. doi: 10.1099/0022-1317-81-2-471. [DOI] [PubMed] [Google Scholar]
- 88.Ng LC, Tan LK, Tan CH, et al. Entomologic and virologic investigation of chikungunya, Singapore. Emerg. Infect. Dis. 2009;15(8):1243–1249. doi: 10.3201/eid1508.081486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rianthavorn P, Prianantathavorn K, Wuttirattanakowit N, Theamboonlers A, Poovorawan Y. An outbreak of chikungunya in southern Thailand from 2008 to 2009 caused by African strains with A226V mutation. Intl J. Infect. Dis. 2010;14(Suppl. 3):e161–e165. doi: 10.1016/j.ijid.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 90.Sam IC, Chan YF, Chan SY, et al. Chikungunya virus of Asian and central/east African genotypes in Malaysia. J. Clin. Virol. 2009;46(2):180–183. doi: 10.1016/j.jcv.2009.07.016. [DOI] [PubMed] [Google Scholar]
- 91.Auksornkitti V, Pongsiri P, Theamboonlers A, et al. Whole-genome characterisation of chikungunya virus from Aedes albopictus collected in Thailand. Ann. Trop. Med. Parasitol. 2010;104(3):265–269. doi: 10.1179/136485910X12647085215778. [DOI] [PubMed] [Google Scholar]
- 92.Ciota AT, Kramer LD. Insights into arbovirus evolution and adaptation from experimental studies. Viruses. 2010;2(12):2594–2617. doi: 10.3390/v2122594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ebel GD, Fitzpatrick KA, Lim PY, et al. Nonconsensus West Nile virus genomes arising during mosquito infection suppress pathogenesis and modulate virus fitness in vivo. J. Virol. 2011;85(23):12605–12613. doi: 10.1128/JVI.05637-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Weaver SC, Vasilakis N. Molecular evolution of dengue viruses: contributions of phylogenetics to understanding the history and epidemiology of the pre-eminent arboviral disease. Infect. Genet. Evol. 2009;9(4):523–540. doi: 10.1016/j.meegid.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Aaskov J, Buzacott K, Thu HM, Lowry K, Holmes EC. Long-term transmission of defective RNA viruses in humans and Aedes mosquitoes. Science. 2006;311(5758):236–238. doi: 10.1126/science.1115030. [DOI] [PubMed] [Google Scholar]; ■ Describes DENV from humans and mosquitoes in Myanmar that contain stop-codon envelope gene mutations, reflecting defective RNA genomes. Sustained transmission of such variants was apparently mediated by complementation via coinfection of host cells with wild-type and defective DENV.
- 96.Cox J, Mota J, Sukupolvi-Petty S, Diamond MS, Rico-Hesse R. Mosquito bite delivery of dengue virus enhances immunogenicity and pathogenesis in humanized mice. J. Virol. 2012;86(14):7637–7649. doi: 10.1128/JVI.00534-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Styer LM, Bernard KA, Kramer LD. Enhanced early West Nile virus infection in young chickens infected by mosquito bite: effect of viral dose. Am. J. Trop. Med. Hyg. 2006;75(2):337–345. [PubMed] [Google Scholar]
- 98.Cardosa J, Ooi MH, Tio PH, et al. Dengue virus serotype 2 from a sylvatic lineage isolated from a patient with dengue hemorrhagic fever. PLoS Negl. Trop. Dis. 2009;3(4):e423. doi: 10.1371/journal.pntd.0000423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Franco L, Palacios G, Martinez JA, et al. First report of sylvatic DENV-2-associated dengue hemorrhagic fever in West Africa. PLoS Negl. Trop. Dis. 2011;5(8):e1251. doi: 10.1371/journal.pntd.0001251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Vasilakis N, Tesh RB, Weaver SC. Sylvatic dengue virus type 2 activity in humans, Nigeria, 1966. Emerg. Infect. Dis. 2008;14(3):502–504. doi: 10.3201/eid1403.070843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Moncayo AC, Fernandez Z, Ortiz D, et al. Dengue emergence and adaptation to peridomestic mosquitoes. Emerg. Infect. Dis. 2004;10(10):1790–1796. doi: 10.3201/eid1010.030846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ciota AT, Ngo KA, Lovelace AO, et al. Role of the mutant spectrum in adaptation and replication of West Nile virus. J. Gen. Virol. 2007;88(Pt 3):865–874. doi: 10.1099/vir.0.82606-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Weinreich DM, Delaney NF, Depristo MA, Hartl DL. Darwinian evolution can follow only very few mutational paths to fitter proteins. Science. 2006;312(5770):111–114. doi: 10.1126/science.1123539. [DOI] [PubMed] [Google Scholar]
- 104.Da Silva J, Coetzer M, Nedellec R, Pastore C, Mosier DE. Fitness epistasis and constraints on adaptation in a human immunodeficiency virus type 1 protein region. Genetics. 2010;185(1):293–303. doi: 10.1534/genetics.109.112458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dawid A, Kiviet DJ, Kogenaru M, De Vos M, Tans SJ. Multiple peaks and reciprocal sign epistasis in an empirically determined genotype-phenotype landscape. Chaos. 2010;20(2):026105. doi: 10.1063/1.3453602. [DOI] [PubMed] [Google Scholar]
- 106.Weaver SC, Reisen WK. Present and future arboviral threats. Antiviral Res. 2009;85(2):328–345. doi: 10.1016/j.antiviral.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Voss JE, Vaney MC, Duquerroy S, et al. Glycoprotein organization of chikungunya virus particles revealed by x-ray crystallography. Nature. 2010;468(7324):709–712. doi: 10.1038/nature09555. [DOI] [PubMed] [Google Scholar]; ■■ The atomic structure of the E3–D2–E1 glycoprotein complex of CHIKV was solved using x-ray crystallography; this is the first such structure for an alphavirus. This information provides a structural basis for understanding the biological properties of alphaviruses.
- 108.Scott TW, Weaver SC, Mallampalli V. Evolution of mosquito-borne viruses. In: Morse SS, editor. Evolutionary Biology of Viruses. Raven Press; NY, USA: 1994. pp. 293–324. [Google Scholar]
- 109.Zhang R, Hryc CF, Cong Y, et al. 4.4 Å cryo-EM structure of an enveloped alphavirus Venezuelan equine encephalitis virus. EMBO J. 2011;30(18):3854–3863. doi: 10.1038/emboj.2011.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lok SM, Kostyuchenko V, Nybakken GE, et al. Binding of a neutralizing antibody to dengue virus alters the arrangement of surface glycoproteins. Nat. Struct. Biol. 2008;15(3):312–317. doi: 10.1038/nsmb.1382. [DOI] [PubMed] [Google Scholar]
- 111.Zhang W, Chipman PR, Corver J, et al. Visualization of membrane protein domains by cryo-electron microscopy of dengue virus. Nat. Struct. Biol. 2003;10(11):907–912. doi: 10.1038/nsb990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yu IM, Zhang W, Holdaway HA, et al. Structure of the immature dengue virus at low pH primes proteolytic maturation. Science. 2008;319(5871):1834–1837. doi: 10.1126/science.1153264. [DOI] [PubMed] [Google Scholar]
- 113.Weaver SC, Lorenz LH, Scott TW. Distribution of western equine encephalomyelitis virus in the alimentary tract of Culex tarsalis (Diptera: Culicidae) following natural and artificial blood meals. J. Med. Entomol. 1993;30(2):391–397. doi: 10.1093/jmedent/30.2.391. [DOI] [PubMed] [Google Scholar]