

Abstract
Siderophores are small-molecule iron chelators produced by bacteria and other microorganisms for survival under iron limiting conditions, such as found in a mammalian host. Siderophore biosynthesis is essential for the virulence of many important Gram-negative pathogens including Acinetobacter baumannii, Klebsiella pneumoniae, Pseudomonas aeruginosa, and Escherichia coli. We performed high-throughput screening of against BasE, which is involved in siderophore biosynthesis in A. baumannii and identified 6-phenyl-1-(pyridin-4-ylmethyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid 15. Herein we report the synthesis, biochemical, and microbiological evaluation of a systematic series of analogues of the HTS hit 15. Analogue 67 is the most potent analogue with a KD of 2 nM against BasE. Structural characterization of the inhibitors with BasE reveal they bind in a unique orientation in the active site occupying all three substrate binding sites, and thus can be considered multisubstrate inhibitors. These results provide a foundation for future studies aimed at both increasing enzyme potency and antibacterial activity.
Introduction
The increase of antibacterial resistance coupled with the lack of new antibiotics is cause for great concern.1, 2 This is highlighted by the Gram-negative bacteria Acinetobacter baumannii, an opportunistic organism that has emerged over the last couple of decades as one of the most insidious pathogens.3 A. baumannii now accounts for more than 10% of hospital-acquired infections and is the leading cause of wound infections in soldiers in Iraq and Afghanistan.4, 5 Additionally, up to 30% of A. baumannii isolates in intensive care units are resistant to almost all known antibiotics including the β-lactams, fluoroquinolones, aminoglycosides, and tetracyclines.5 According to the MYSTIC susceptibility data from 15 North American medical centers, acinetobacter sensitivity is now below 60% for ceftazidime, cefepime, piperacillin/tazobactam, meropenem, imipenem, aztreonam, and gentamicin.6 Comparative genomic studies of A. baumannii identified an unprecedented 86-kb cluster of 45 resistance genes in one particular strain.7, 8 The prevalence of the multidrug resistance (MDR) phenotype among Gram-negative pathogens including A. baumannii has led infectious disease physicians to reintroduce the colistins and polymyxins.9 These related cationic lipopeptides were first introduced in the 1950’s, but their use had been largely curtailed by the 1980’s as a result of their considerable nephrotoxicity.10 A. baumannii clinical isolates have been reported that are no longer susceptible to these antibiotics of last resort.10 It is astonishing to think that we may soon enter an era when antibiotic therapy is unavailable for previously treatable infections.
All bacteria with the exception of Borrelia burgdorferei11 require micromolar levels of iron (Fe2+/Fe3+) for growth since iron serves as a cofactor in numerous biochemical processes.12 However, the concentrations of iron in serum and human body fluids is approximately 10−24 M. The extraordinarily low concentration of free iron provides innate immunity to bacterial infections and is a result of the insolubility of iron (III) under aerobic conditions and the sequestration of the remaining free iron by the iron-binding proteins such as transferrin and lactoferrin. Many pathogens like A. baumannii overcome this iron limitation via the synthesis of siderophores, which are small molecule high-affinity iron-chelators secreted by bacteria and reimported from the external milieu after successfully chelating non-heme host iron (Figure 1).12–15 The critical role that siderophores play in virulence has been demonstrated in A. baumannii,16 as well as in numerous other significant Gram-negative pathogens including Klebsiella pneumoniae,17 Pseudomonas aeruginosa,18 and Escherichia coli19 Siderophores are also critical for the virulence of many Gram-positive bacteria including Bacillus anthracis,20 Staphylococcus aureus,21 and the acid fast Mycobacterium tuberculosis.22 Consequently, inhibition of siderophore biosynthesis represents a promising new strategy for antibacterial drug development; an approach that is further bolstered by the observation that many bactericidal antibiotics ultimately operate through disruption of bacterial iron homeostasis and generation of reactive oxygen species (ROS).23, 24
Figure 1.

Structure of representative aryl-capped siderophores
A. baumannii produces acinetobactin, a mixed ligand siderophore containing a catechol and imidazole for iron coordination.25, 26 The biosynthesis of acinetobactin is initiated by BasE that activates and loads 2,3-dihydroxybenzoic acid (DHB) onto a nonribosomal peptide synthetase (NRPS) pathway comprised of four other proteins (BasF, BasD, BasA, and BasB).27, 28 This assembly line of proteins condenses DHB, L-threonine, and N-hydroxyhistamine to afford preacinetobactin 11, which spontaneously rearranges to acinetobactin 1 (Figure 2).29, 30 BasE represents an ideal target since it does not possess a mammalian homologue, the protein has been biochemically and structurally characterized,31 and the functionally related aminoacyl t-RNA synthetases are validated antibiotic targets with mupirocin the first in class inhibitor.32 Homologues from other organisms as shown in Table 1 suggest inhibitors may also be useful to combat several significant bacterial pathogens.
Figure 2. Biosynthesis of Acinetobactin.
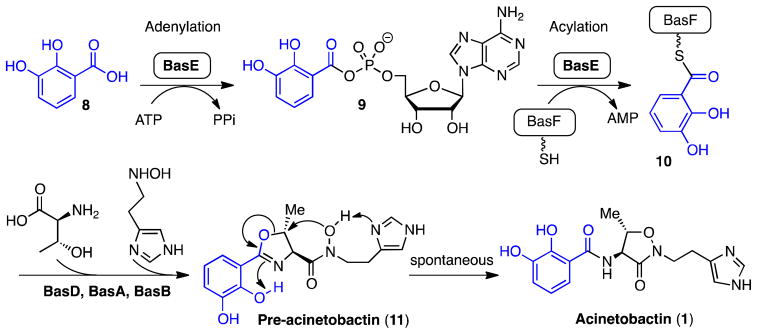
BasE binds 2,3-dihydroxybenzoic acid 8 and ATP then catalyzes their condensation to form an intermediate acyl-adenylate 9 that remains tightly bound. In a second half reaction, BasE catalyzes the transfer of the acyl group (blue) onto a nucleophilic sulfur atom of the aryl carrier domain of BasF to provide the acylated complex 10 with the release of AMP. Further steps are catalyzed by BasD, BasA and BasB, incorporating threonine and N-hydroxyhistamine to yield pre-acinetobactin 11. This molecule undergoes a facile rearrangement to the isoxazolidinone isomer of acinetobactin 1 that is likely promoted via an internal hydrogen bond with the oxazoline nitrogen atom in conjunction with the proximal imidazole moiety that serves to deprotonate the N-hydroxyamide functional group.
Table 1.
Aryl-capped siderophores producing pathogens and corresponding AAAEs.
| Organism | Siderophore | AAAE | AAAE Substratea | |
|---|---|---|---|---|
| Gram-negative | A. baumannii | acinetobactin | BasE | DHB |
| E. coli | enterobactin | EntE | DHB | |
| K. pneumoniae | yersiniabactin | YbtE | DHB | |
| enterobactin | EntE | SAL | ||
| P. aeruginosa | pyochelin | PchD | DHB | |
| Y. pestis | yersiniabactin | YbtE | SAL | |
| Y. pseudotuberculosis | yersiniabactin | YbtE | SAL | |
| V. cholerae | vibriobactin | VibE | DHB | |
| V. vulnificus | vulnibactin | VibE1 | DHB | |
| VibE2 | SAL | |||
|
| ||||
| Gram-positive | B. subtilis | bacillibactin | DhbE | DHB |
| B. anthracis | petrobactin | AsbC | 3,4-DHB | |
|
| ||||
| Acid-fast | M. tuberculosis | mycobactin | MbtA | SAL |
SAL, salicylic acid; DHB, 2,3-dihydroxybenzoic acid; 3–4-DHB, 3,4-dihydroxybenzoic acid
BasE is an aryl acid adenylating enzyme (AAAE) and catalyzes the condensation of DHB 8 with ATP to form an acyl-adenylate intermediate 9, whereby the carboxy group is activated as a mixed anhydride. Following liberation of pyrophosphate, BasE binds the phosphopantetheinylated aryl carrier protein (ArCP) domain of BasF to form a ternary complex and then catalyzes the transfer of the activated DHB onto the terminal thiol of the pantetheine moiety of BasF to provide thioester 10 (Figure 2).33 AAAEs are members of the ANL superfamily of enzymes that contain a large N-terminal domain and a smaller C-terminal domain with the active site located at the domain interface.34 AAAEs thus carry out two reactions, an adenylation and thioesterification, at the same active site. In the adenylation reaction, a catalytic lysine from the C-terminal subdomain coordinates the carboxylate substrate and directs nucleophilic attack on the α-phosphate of ATP. The enzyme then undergoes a ~140° rigid body rotation about a hinge residue (Lys437 in BasE), which allows the phosphopantetheine arm of the carrier domain to insert into the active-site for thioester formation.34
5′-O-[N-(Salicyl)sulfamoyl]adenosine (Sal-AMS, 12) and the related 5′-O-[N-(2,3-dihydroxybenzoyl)sulfamoyl]adenosine (2,3-DHB-AMS, 13) are the first confirmed AAAE inhibitors of siderophore biosynthesis with potent nanomolar apparent Ki values against a range of AAAEs including BasE, YbtE, and MbtA from A. baumannii, Yersinia sp., and M. tuberculosis, respectively (Figure 3).31, 35–37 Sal-AMS and 2,3-DHB-AMS mimic the acyladenylate intermediate 9 (Figure 2) through replacement of the labile acylphosphate moiety with a stable acylsulfamate isostere. Sal-AMS displays impressive activity against the acid-fast Gram-positive M. tuberculosis with a minimum inhibitory concentration (MIC) of 0.39 μM under iron-deficient conditions. However, the antibacterial potency of Sal-AMS is more than 100 times weaker against Gram-negative Y. pestis and Y. pseudotuberculosis under the same conditions, despite possessing potent nanomolar enzyme inhibition of YbtE, the respective AAAE from these organisms.35, 38 Moreover, Sal-AMS and 2,3-DHB-AMS display no activity against other Gram-negative organisms including A. baumannii, K. pneumoniae, E. coli, and P. aeruginosa (unpublished results, Brian Beck, Laura Celia, ATCC). The reason for such a striking difference could be that the highly polar (ClogP ~ −2) and negatively charged nucleoside derivatives may prevent cellular uptake, although many other mechanisms of intrinsic resistance may be involved.
Figure 3.
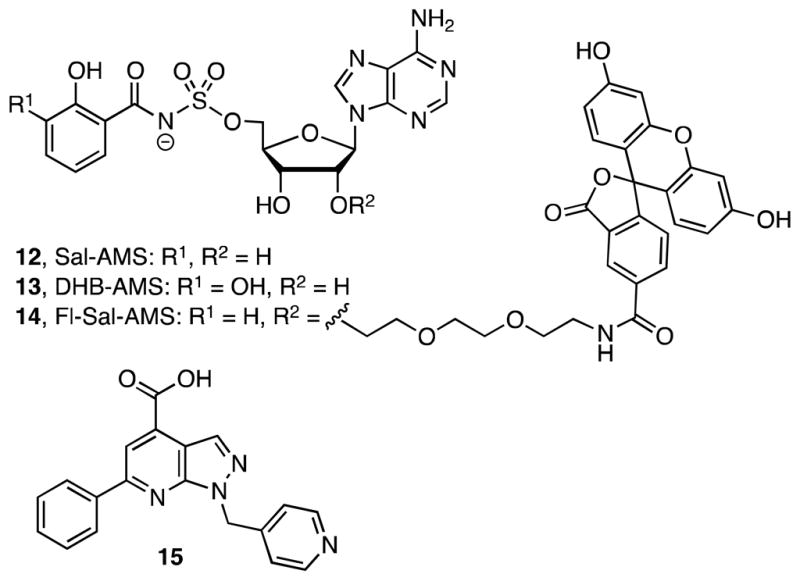
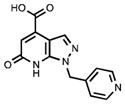
Inhibitor and probe structures. 5′-O-[N-(salicyl)sulfamoyl]adenosine (Sal-AMS, 12), 5′-O-[N-(2,3-dihydroxybenzoyl)sulfamoyl]adenosine (2,3-DHB-AMS, 13), fluorescent probe 14, and HTS hit 6-phenyl-1-(pyridin-4-ylmethyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid (15).
We have recently reported the discovery of a new class of potent non-nucleoside AAAE inhibitors through high-throughput screening (HTS) using a fluorescence polarization (FP) displacement assay with a fluorescent analogue of Sal-AMS (Fl-Sal-AMS, 14) as a ligand.39 The most potent hit discovered, pyrazolopyridine 15 (Figure 3) binds BasE with a submicromolar dissociation constant as determined independently by isothermal titration calorimetry and our FP assay.39 Further kinetic characterization of 15 reveals it exhibits competitive inhibition with respect to both substrates 2,3-DHB and ATP.31 Herein we report the design, synthesis, biochemical, and biological evaluation of a systematic series of analogues of 15 that comprehensively explores the SAR of this promising scaffold. Structural information is also reported for the complexes of BasE with two of the most potent analogues of 15 as well as in vitro data demonstrating that deletion of basE in A. baumannii impairs growth under irondeficient conditions.
Results
Chemistry
The structure of 6-phenyl-1-(pyridin-4-ylmethyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid (15) discovered from high-throughput screening39 can be divided into four domains for SAR purposes as depicted in Figure 4. This compound is part of a large compound library supplied by Enamine (Ukraine) and there is no literature available regarding its synthesis or any other properties.
Figure 4.
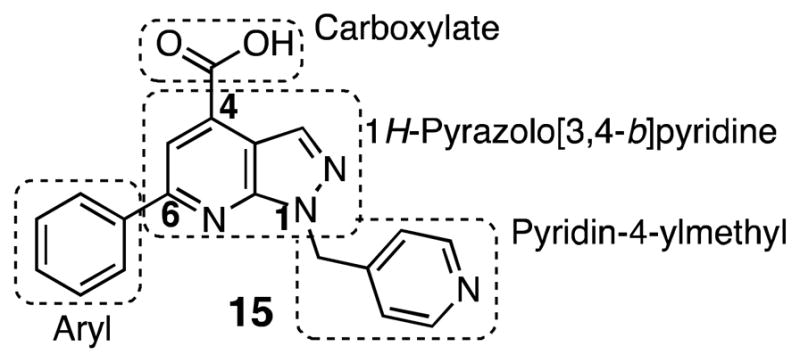
Dissection of pyrazolopyridine hit 15 into 4 domains for SAR analysis.
For the synthesis of 15 and analogues, the key intermediate ethyl 6-oxo-1-(pyridin-4-ylmethyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylate 109 was initially prepared, as well as the p-methoxyphenyl (PMP) analogue 108 (Scheme 1). The methodology devised by Hohn40 was used to synthesize the 1-aryl-1H-pyrazol-5-amines 104 and 105, through the multicomponent condensation of acrylonitrile 101, hydrazine, and the appropriate aryl aldehydes to afford the intermediate imines 102 and 103. These were not isolated but directly converted to the corresponding pyrazoles via base-promoted cyclization and subsequent redox isomerization. Pyridone annulation was accomplished in a two-step process involving condensation of 104 and 105 with diethyl oxalacetate to provide 106 and 107, which were then cyclized to the respective 1H-pyrazolo[3,4-b]pyridones 108 and 109 by refluxing in glacial acetic acid using the procedure described by Dorn and Müller.41
Scheme 1a.

aReaction conditions: (a) H2NNH2·H2O, EtOH, rt, 24 h; (b) ArCHO, 0 °C to rt, 2 h; (c) n-BuONa, n-BuOH, 120 °C, 3 h (76%, 3 steps, from 101); (d) EtO2C(C=O)CH2CO2Et, benzene, 65 °C, 20 h, 48–63%; (e) glacial AcOH, reflux, 3 h, 78–81%.
Elaboration of intermediates 108–109 to the final C-6 aryl pyrazolopyridines requires activation of the tautomerizable pyridone carbonyl as a halide or pseudohalide followed by Suzuki coupling and ester hydrolysis. We initially began with PMP analogue 108 since this provides p-hydroxybenzyl and p-methoxybenzyl analogues 31–32, and selective deprotection of the PMP moiety would allow access to a wide variety of analogues at N-1 by direct alkylation of the resultant free amino group. Chlorination (POCl3) of 108 was not successful (not shown), but the triflate 110 was readily obtained using triflic anhydride and 2,6-di-t-butyl-4-methylpyridine as base (Scheme 2). Suzuki coupling of 110 with phenylboronic acid under standard Suzuki conditions furnished the intermediate 111. Cleavage of the PMP group proceeded optimally in neat TFA at 70 °C to afford compound 112. Alkylation of 112 with 4-(bromomethyl)pyridine under a variety of conditions was then attempted. Although the desired product 15 was obtained, the reaction was not regioselective and alkylation occurred at both N-1 and N-2 of the pyrazole, leading to poor yields of the desired product (not shown). The intermediates 111 and 112 were readily hydrolyzed with NaOH in THF–H2O at room temperature providing compounds 31 and 36. The p-methoxy group of 111 was hydrolyzed using boron trifluoride·dimethyl sulfate complex, yielding the p-hydroxybenzyl analogue 32. Buchwald-Hartwig coupling of triflate 110 with aniline followed by hydrolysis of the ethyl ester furnished the phenylamino analogue 100.
Scheme 2a.

aReaction conditions: (a) 2,6-di-t-butyl-4-methylpyridine, Tf2O, CH2Cl2, −78 to 0 °C, 4 h, 68%; (b) Pd(PPh3)4, Cs2CO3, PhB(OH)2, dioxane, 100 °C, 5 h, 96%; (c) TFA, 70 °C, 24 h, 85%; (d) aq. NaOH, THF, rt, 4 h, 84% (average); (e) (CH3)2S·BF3, 0 °C to rt, 18 h, 17%; (f) Pd(OAc)2, BINAP, Cs2CO3, PhNH2, dioxane, 100 °C, 22 h, 90%.
To explore the SAR of the 6-aryl domain of the lead compound 15, we first attempted to prepare the triflate of 109 using the procedure described for PMP analogue 108. However triflation was not successful in this case, probably due to the presence of the pyridine moiety. We then explored the conditions developed by Kang and co-workers42 for the PyBroP-mediated activation of tautomerizable heterocycles. Treatment of 109 with PyBroP at room temperature for 2 hours resulted in the formation of the activated intermediate 113 as monitored by mass spectrometry that was not isolated, but directed coupled to phenylboronic acid under standard Suzuki conditions to afford the desired product in a respectable 74% yield (Scheme 3). Hydrolysis of the ethyl ester and HPLC purification provided lead compound 15 in 81% yield that possessed identical 1H NMR, 13C NMR, HRMS data to the compound obtained from commercial vendor Enamine. The success of the Kang protocol for activation of 109 highlights the chemoselectivity of PyBroP–mediated activation for highly functionalized heterocycles.
Scheme 3a,b.

aReaction conditions: (a) PyBroP, Et3N, dioxane, rt, 2 h; (b) ArB(OH)2 or ArB(OH)2-pinacolate ester, PdCl2dppf, Na2CO3, 5:1 dioxane–H2O, 100 °C, 4 h, 64% (average, 2 steps, from 109); (c) aq. NaOH, THF, rt, 4 h, 80% (average); (d) R–C=C–B(OH)2 or respective pinacolate ester, PdCl2(PPh3)2, Na2CO3, 5:1 dioxane–H2O, 100 °C, 4 h, 76–84% (2 steps, from 109); (e) BOP, DBU, HNR1R2, dioxane, 70 °C, 5 h, 22–79%. bThe structures of 37–59, 61–62 and 66–90 are shown in Tables 5 and 6.
We used parallel synthesis to prepare a systematic series of fifty-four (hetero)aryl-substituted analogues with substitution at C-6 (Scheme 3). Parallel synthesis was performed on a 0.33 millimole scale employing 100 mg of pyrazolopyridone 109 in 8 mL sealed vials using stock solution of all reagents. Extraction of the products from the reaction mixtures was achieved with dichloromethane using phase-separation cartridges. The esters were purified by flash chromatography with an average yield of 64% (range 7–97%), an excellent result considering that a standard purification method was utilized for all compounds. In some cases, when halogenated (hetero)arylboronic acids where used, more than one product was obtained due to a second Suzuki coupling (compounds 73, 74 and 85, tables 5 and 6). Whenever possible the two products were isolated. Hydrolysis of the esters was performed with NaOH in THF–H2O with good yields affording compounds 15, 37–59, 61–62 and 66–90 (structures in Tables 5 and 6), which were purified by HPLC and lyophilized. The PyBroP-activated intermediate 113 was also used to successfully introduce alkene groups using similar conditions to provide analogues 91 and 92 after ester hydrolysis (Scheme 3).
Table 5.
SAR of C-6 phenyl group.
| |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | KD (nM) | |
| BasE | MbtA | ||||
| 15 | H | H | H | 36.1 ± 4.9 | (3.74 ± 0.41)•103 |
| 37 | Me | H | H | (2.15 ± 0.11)•103 | >100•103 |
| 38 | H | Me | H | 158 ± 24 | (7.40 ± 0.90)•103 |
| 39 | H | H | Me | 212 ± 22 | (1.44 ± 0.15)•103 |
| 40 | OH | H | H | 255 ± 36 | (6.50 ± 0.90)•103 |
| 41 | H | OH | H | 124 ± 17 | (7.67 ± 1.15)•103 |
| 42 | H | H | OH | 8.85 ± 3.16 | (3.70 ± 0.50)•103 |
| 43 | H | H | CH2OH | 247 ± 33 | (3.70 ± 0.60)•103 |
| 44 | Cl | H | H | (2.48 ± 0.19)•103 | >100•103 |
| 45 | H | Cl | H | ≤ 2.0 | (4.60 ± 0.50)•103 |
| 46 | H | H | Cl | 153 ± 21 | 953 ± 90 |
| 47 | H | H | Br | 20.6 ± 7.0 | 560 ± 28 |
| 48 | H | H | F | 28 ± 4.7 | (1.89 ± 0.21)•103 |
| 49 | F | H | F | 42.6 ± 8.3 | (8.00 ± 0.90)•103 |
| 50 | H | H | CF3 | 191 ± 27 | 887 ± 96 |
| 51 | H | H | NO2 | 3.91 ± 2.27 | 202 ± 38 |
| 52 | H | H | NH2 | 78.2 ± 13.2 | (4.08 ± 0.63)•103 |
| 53 | H | H | N(Me)2 | 628 ± 98 | (1.70 ± 0.30)•103 |
| 54 | H | H | NH(C=O)Me | 747 ± 65 | (4.20 ± 0.80)•103 |
| 55 | H | H | SO2Me | 13.0 ± 6.1 | 130 ± 23 |
| 56 | H | H | CN | 22.7 ± 6.2 | 380 ± 46 |
| 57 | H | H | C(=O)NH2 | 288 ± 45 | (1.00 ± 0.30)•103 |
| 58 | H | H | COOH | 776 ± 119 | (6.10 ± 0.80)•103 |
| 59 | H | OMe | COOH | (1.70 ± 0.15)•103 | (49.9 ± 3.7)•103 |
| 60 | H | OH | COOH | 169 ± 13.4 | 495 ± 34 |
| 61 | H | H | SMe | 23.5 ± 5.5 | 178 ± 15 |
| 62 | H | H | OMe | 78.2 ± 13.2 | (3.23 ± 0.38)•103 |
| 63 | H | H | O(CH2)7CH3 | 191 ± 13 | >50•103 |
| 64 | H | H | O(CH2)11CH3 | (28.4 ± 3.9)•103 | >50•103 |
| 65 | H | H | O(CH2)15CH3 | - | - |
| 66 | H | H | OPh | 32.5 ± 6.3 | 187 ± 28 |
| 67 | H | H | OCH2Ph | 2.14 ± 1.46 | (5.40 ± 0.90)•103 |
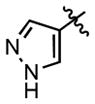
| 68 | H | H | OCH2(p-PhOMe) | 44.5 ± 7.7 | (5.80 ± 0.90)•103 |
| 69 | H | H | C(=O)Me | 104 ± 11 | 134 ± 22 |
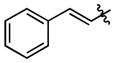
| 70 | H | H | C(=O)Ph | 19.1 ± 4.9 | 19.1 ± 2.4 |
| 71 | H | Ph | H | 164 ± 20 | (3.60 ± 0.38)•103 |
| 72 | H | H | Ph | 125 ± 9 | (3.50 ± 0.30)•103 |
| 73 | H | H | p-PhBr | 181 ± 39 | (19.7 ± 0.9)•103 |
| 74 | o-PhCl | H | H | (19.7 ± 0.9)•103 | >50•103 |
Table 6.
SAR at C-6: substitution with heterocyles, alkynes, alkenes, and amines.
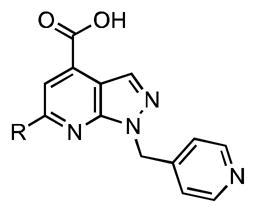
| |||
|---|---|---|---|
| Entry | R | KD (nM) | |
| BasE | MbtA | ||
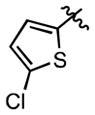
| 15 | Ph | 36.1 ± 4.9 | (3.74 ± 0.41)•103 |
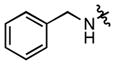
| 75 |
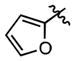
|
342 ± 53 | (19.5 ± 2.2)•103 |
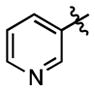
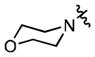
| 76 |
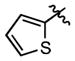
|
443 ± 74 | (6.68 ± 0.50)•103 |
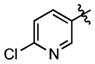
| 77 |
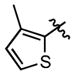
|
340 ± 32 | (19.2 ± 1.7)•103 |
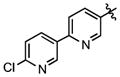
| 78 |
|
86.9 ± 18.7 | (2.54 ± 0.41)•103 |
| 79 |
|
7.60 ± 2.49 | (2.60 ± 0.60)•103 |
| 80 |
|
36.3 ± 7.5 | 318 ± 56 |
| 81 |
|
62.4 ± 8.5 | 701 ± 163 |
| 82 |
|
39.2 ± 7.5 | 264 ± 30 |
| 83 |
|
132 ± 16 | (16.8 ± 1.7)•103 |
| 84 |
|
121 ± 19 | 901 ± 109 |
| 85 |
|
(1.17 ± 0.14)•103 | 380 ± 35 |
| 86 |
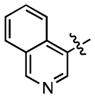
|
(7.73 ± 0.43)•103 | > 100•103 |
| 87 |
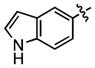
|
61.8 ± 10.4 | (2.90 ± 0.20)•103 |
| 88 |
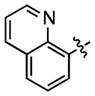
|
776 ± 52 | (22.5 ± 2.1)•103 |
| 89 |
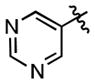
|
583 ± 44 | (13.6 ± 0.7)•103 |
| 90 |
|
(2.30 ± 0.20)•103 | (27.9 ± 1.6)•103 |
| 91 |
|
(1.90 ± 0.20)•103 | > 100•103 |
| 92 |
|
280 ± 43 | (5.10 ± 0.70)•103 |
| 93 |
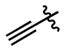
|
(2.04 ± 0.20)•103 | (18.5 ± 2.0)•103 |
| 94 |
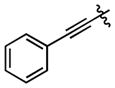
|
421 ± 61 | (3.00 ± 0.60)•103 |
| 95 |
|
(2.32 ± 0.29)•103 | (6.02 ± 0.72)•103 |
| 96 |
|
(1.45 ± 0.16)•103 | (29.1 ± 2.0)•103 |
| 97 |
|
(2.46 ± 0.24)•103 | (97.7 ± 9.8)•103 |
| 98 |
|
> 100•103 | > 100•103 |
BOP-mediated coupling of the pyrazolopyridone 109 with three different amines was performed successfully using the procedure developed by Wan et al.,43 further increasing the scope of this key intermediate and the range of substituents in the pyrazolopyridine at C-6 (Scheme 3). Analogues 95–97 were thus obtained following ester hydrolysis. Simple hydrolysis of the pyrazolopyridone intermediate 109 afforded analogue 98, which lacks a 6-aryl moiety (Scheme 3).
A further series of C-6 substituted analogues was prepared as shown in Scheme 4. Analogue 60 incorporating salicylic acid at C-6 was prepared from 59-Ethyl ester through simultaneous cleavage of the methyl ether and ethyl esters by treatment with boron trifluoride–dimethyl sulfate complex. We were also interested in evaluating the impact of increased lipophilicity of the lead compound 15, hence a short series of analogues was synthesized bearing a 4-alkoxyphenyl group with 8, 12 and 16-carbon linear alkyl chains (compounds 63–65). This strategy was successful in our previous SAR studies of Sal-AMS (12), maintaining or even increasing the respective inhibitory potency against MbtA, the AAAE from M. tuberculosis.44 These compounds were synthesized through alkylation of 42-Ethyl ester with the corresponding alkyl bromides (Scheme 4). The resulting esters were then hydrolyzed with NaOH to afford compounds 63–65. We also prepared two alkyne analogues by PyBroP activation of 109 and subsequent palladium-catalyzed Sonogashira with triethylsilylacetylene and phenylacetylene under copper-free conditions employing PdCl2(CH3CN)2 and Buchwald’s 2-(dicyclohexylphosphino)biphenyl ligand to afford 114 and 115 (Scheme 4).45 Notably, standard Sonogashira conditions did not afford any of the desired product. Deprotection of the terminal TES group in 114 with TBAF furnished 116. Saponification of 115 and 116 with NaOH in aqueous THF provided 94 and 93, respectively.
Scheme 4a.

aReaction conditions: (a) (CH3)2S·BF3, DCM, 0 °C to rt, 3.5 h, 37%; (b) R-Br, Na2CO3, dioxane–H2O, 80 °C, 12 h; (c) NaOH, MeOH, rt, 2 h, 40–70 % (2 steps, from 42-Ethyl ester); (d) i) PyBrOP, Et3N, dioxane, rt, 2 h; ii) R-C≡C-H, PdCl2(CH3CN)2, 2-(dicyclohexylphosphino)biphenyl, Cs2CO3, dioxane–H2O, 85 °C, 6 h, 77–87%; (e) TBAF, THF, 0 °C, 3 h, 35%; (f) NaOH, MeOH, rt, 2 h, 41–65%.
A small series of analogues was prepared as shown in Scheme 5 to study the modification at C-4 of the lead compound 15. LiAlH4 reduction of 18 provided hydroxy analogue 19. A series of amides 22–25 and 27–28 was synthesized by conversion of 15 to the corresponding acid chloride employing oxalyl chloride followed by aminolysis. Functional group interconversion of carboxylic acid 15 to amine 21 was achieved by Curtius rearrangement of the respective acyl azide.
Scheme 5a.

aReaction conditions: (a) LiAlH4, THF, 0 °C to rt, 2 h, 48%; (b) i) (COCl)2, CH2Cl2, DMF (1 equiv), 0 °C, 1 h; ii) HNR1R2, DMAP, rt, 1 h, 39% (average); (c) i) (COCl)2, CH2Cl2–THF (3:1), rt, 4 h; ii) NaN3, acetone–H2O (1:1); iii) TFA, benzene, reflux, 16 h; iv) K2CO3, MeOH, rt, 8 h, 31%.
To explore the importance of the N-2 and N-7 atoms of the pyrazolo[3,4-b]pyridine scaffold we prepared analogues 16 and 17 from 6-bromo-1H-indazole-4-carboxylate 117 and 6-bromo-1H-indole-4-carboxylate 118 (Scheme 6). Alkylation of indazole 117 with 4-(bromomethyl)pyridine hydrobromide employing Cs2CO3 afforded a mixture of regioisomers 119 and 121 in 31 and 19% yield, respectively favoring the desired N-1 alkylated product. Indole 120 was prepared analogously from 118 in 77% yield. All three compounds were subjected to Suzuki coupling with phenylboronic acid and the methyl esters were hydrolyzed to provide the final compounds 16, 17 and 99.
Scheme 6a.

aReaction conditions: (a) Cs2CO3, 4-(bromomethyl)pyridine hydrobromide, DMF, rt, 3 h, 50– 77%; (b) PdCl2dppf, Cs2CO3, PhB(OH)2, dioxane, 100 °C, 5 h, 63–77% (c) aq. NaOH, THF, rt, 4 h, 20–77%.
Direct binding studies
The binding of all analogues to BasE was measured using a fluorescence polarization (FP) assay that measures displacement of the active-site directed fluorescent probe Fl-Sal-AMS 14 (see Figure 3) from BasE, which results in an increase in polarization.39 Fitting of the resultant displacement curve following the analysis described by Wagner and co-workers enables determination of the ligand dissociation constant.46 We had previously demonstrated that our direct-binding FP assay agrees closely with results obtained by isothermal titration calorimetry and a functional steady-state kinetic assay that measures acylation of the native aryl-carrier protein domain of BasF.31 Although this manuscript is primarily focused on A. baumannii and inhibition of BasE, we also evaluated all compounds against the homologue MbtA from M. tuberculosis as a means to assess inhibitor specificity toward other AAAEs. MbtA was selected since it is a representative AAAE that utilizes salicylic acid (SAL) instead of 2,3-dihydroxybenzoic acid (DHB) as the native aryl acid substrate.
Direct binding experiments were performed in a 96-well plate format in a 100 μL volume containing 20 nM Fl-Sal-AMS 14 and 200 nM BasE or 50 nM MbtA. Fitting of the experimental data in the form of measured anisotropy (AOBS) versus test compound concentration (LST) to equations 1 and 2 (see Experimental Section) provides the equilibrium dissociation constant (KD) for each compound. The KD of the lead compound 15, previously determined as 78 nM against BasE from a purchased sample39 was re-determined here as 36 nM for BasE and 3.7 μM for MbtA for a newly synthesized and purified sample.
The importance of the 1H-pyrazolo[3,4-b]pyridine scaffold was evaluated with indazole 16 and indole 17 analogues wherein the N-2 and N-7 atoms are replaced with CH isosteres (Table 2). Deletion of N-7 in indazole 16 is well tolerated resulting in a 2-fold loss of potency toward BasE. By contrast, removal of N-2 in indole 17 results in a drastic 1400-fold loss of affinity toward BasE. A similar trend for 15–17 was observed for MbtA; however, the relative magnitudes difference in binding affinities were different. These results demonstrate that the pyrazole N-2 nitrogen atom is essential while the N-7 nitrogen atom is dispensable for potent activity.
Table 2.
SAR of the core heterocycle.
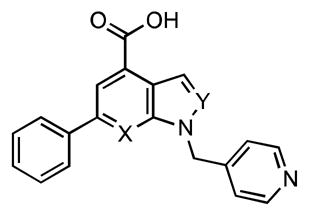
| ||||
|---|---|---|---|---|
| Compound | X | Y | KD (nM) | |
| BasE | MbtA | |||
| 15 | N | N | 36.1 ± 4.9 | (3.74 ± 0.41)•103 |
| 16 | C | N | 84.8 ± 10.0 | (27.6 ± 4.2)•103 |
| 17 | C | C | (52.2 ± 4.8)•103 | (161 ± 7)•103 |
Next, the role of the carboxylic acid at C-4 in 15 was assessed with a series of twelve analogues (Table 3). All modifications to the carboxylic acid lead to a decrease in binding affinity. Deletion of the carboxy group in 20 results in a complete loss of affinity while ethyl ester 18 is 60-fold less potent. Notably, the neutral carboxamide 22 and hydroxymethyl 19 are the best tolerated of all modification producing a modest 1.8- and 2.8-fold loss in affinities for BasE. In the amide series (compounds 23–28), substitution of the amide provides a minor-to-modest decrease in potency toward BasE when compared to the carboxamide 22, varying from 2.2- to 13.6-fold. However, no clear trend in regards to steric or electrostatic interactions is observed. Finally, replacement of the carboxy group with an amino group in 21 is surprisingly well tolerated for BasE resulting in an approximately 6-fold loss in potency. Collectively, these results suggest that a hydrogen-bond donor at C-4 is required (18 and 20 vs. 19 and 21), the electrostatic interaction of the negatively charged carboxylate is not important (15 vs. 22), and that small alkyl substituents are reasonably tolerated (25–27). Similar trends were observed with MbtA except that substituted amides were not accepted.
Table 3.
SAR at C-4.
| |||
|---|---|---|---|
| Compound | R | KD (nM) | |
| BasE | MbtA | ||
| 15 |
|
36.1 ± 4.9 | (3.74 ± 0.41)•103 |
| 18 |
|
(2.16 ± 0.30)•103 | >100•103 |
| 19 |
|
102 ± 8 | (16.8 ± 4.0)•103 |
| 20 |
|
>100•103 | >100•103 |
| 21 |
|
200 ± 25 | (36.4 ± 7.2)•103 |
| 22 |
|
66.8 ± 7.3 | (8.57 ± 0.79)•103 |
| 23 |
|
420 ± 54 | >50•103 |
| 24 |
|
908 ± 69 | >50•103 |
| 25 |
|
249 ± 27 | >50•103 |
| 26 |
|
164 ± 13 | >100•103 |
| 27 |
|
150 ± 19 | >100•103 |
| 28 |
|
334 ± 38 | >50•103 |
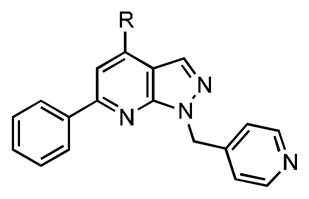
All modifications performed at N-1 lead to either complete loss in affinity or drastic loss of potency against both BasE and MbtA (Table 4). Even a minimal change, such as transposition of the pyridine ring nitrogen from the para to the meta position causes complete loss of affinity against both enzymes. The only compound in this series that maintains modest activity (a 15-fold decrease in affinity toward BasE and no activity against MbtA) is p-hydroxyphenylmethyl 32, which possesses a hydrogen-bond acceptor moiety at an equivalent position as the 4-pyridyl substituent in 15.
Table 4.
SAR at N-1.
| |||
|---|---|---|---|
| Compound | R | KD (nM) | |
| BasE | MbtA | ||
| 15 |
|
36.1 ± 4.9 | (3.74 ± 0.41).103 |
| 29 |
|
>100•103 | >100•103 |
| 30 |
|
>100•103 | >100•103 |
| 31 |
|
>100•103 | >100•103 |
| 32 |
|
556 ± 66 | >100•103 |
| 33 |
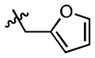
|
>100•103 | >100•103 |
| 34 |
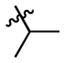
|
>500•103 | >500•103 |
| 35 |
|
>500•103 | >500•103 |
| 36 |
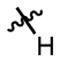
|
>100•103 | >100•103 |
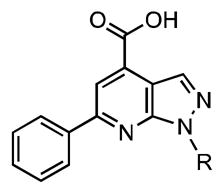
Extensive SAR studies were then carried out at C-6 of 15, as it soon became evident that this was the most amenable to modification. Accordingly, sixty-two analogues were synthesized and the respective results are shown in table 5, for phenyl derivatives, and table 6, for other analogues including heterocycles, amines, alkenes and alkynes. The results obtained in this series are quite promising, as there are sixteen compounds that improved or equaled the potency of the lead compound 15, notably compounds 42, 45, 51, 67 and 79, with KD’s under 10 nM toward BasE. The actual KD values for these compounds may be even lower since these are already at the lower detection limit of the fluorescence polarization assay.
Thirty-eight analogues (37–74) were synthesized in the C-6 phenyl series (Table 5). Unless explicitly stated the SAR refers to BasE and the differences in activity are relative to lead compound 15. A methyl, hydroxy and chloro scan of the ortho-, meta-, and para-positions was performed (compounds 37–42, 44–46) to assess the ability to tolerate substitution at each position. All substitutions at the ortho position result in a decrease in affinity ranging from 7- to nearly 70-fold. Both o-methyl 37 and o-chloro 44 possess a drastic loss in affinity (60- to 70-fold), whereas the o-hydroxy 40 is only 7-fold less potent, perhaps due to a possible intramolecular H-bond formed with the adjacent pyridine nitrogen, which could stabilize the structure in a favorable conformation. Substitution at both the meta- and para-positions is better tolerated. While most compounds result in a modest decrease in affinity ranging from 3- to 6- fold, m-chloro 45 and p-hydroxy 42 are more than 4-fold more potent than 15. The observed SAR with respect to MbtA has a nearly identical trend except that p-methyl 39 is the most potent toward MbtA with a 2.6-fold increase in affinity.
Based on the ability to tolerate substitution at the para-position, we prepared a systematic series of seventeen analogues with a range of functional groups (43, 47–62). Among this initial series p-nitro 51 is the only analogue with substantially improved potency resulting in a 9-fold increase in affinity. Several other analogues including p-fluoro 48, p-bromo 47, p-methylthio 61, p-cyano 56, and p-methylsulfonyl 55 possess a modest increase in affinity ranging from 1.3- to 2.8 fold. However, the majority of analogues are less potent resulting in a modest decrease in affinity from 2.2-fold for p-amino 52 and p-methoxy 62 to over 20-fold for p-acetylamino 54 and p-carboxylate 58. The combination of a para-carboxylate and a meta-hydroxy in salicylate 60 is only 4.7-fold less potent, showing that the additional meta-hydroxy is able to partially restore binding affinity. Overall, no clear trends emerged in regards to steric or electronic effects. The SAR of this series with respect to MbtA does not parallel that observed for BasE with p-methylsulfonyl 55 displaying the highest affinity (29-fold greater than 15) and p-carboxy 58 the lowest affinity (2-fold lower than 15) among the para-substituted analogues.
We next explored bulky hydrophobic groups (63–74) to define a steric boundary of the active-site (Table 5). Long chain alkoxy groups (compounds 63–65) reduce binding affinity and result in poor solubility (compound 65 was insoluble in the assay conditions). Since the bulky phenyl group in p-phenoxy 66 does not adversely affect affinity, we decided to further explore the SAR with other groups and synthesized p-biphenyl 72 that is 3.5-less potent and p-benzoyl 70, which is 2-fold more potent. Introduction of a CH2 spacer in p-phenoxy 66 provided p-benzyloxy 67 that possesses an impressive 17-fold increase in affinity. Further addition of a p-methoxy group to 67 affords 4-methoxybenzyloxy 68 that is a 21-fold less potent than 67. The overall SAR trends for this series of compounds with respect to MbtA do not correlate very closely with BasE. p-Benzoyl 70 is the most potent analogue of MbtA with a KD of 19 nM, which is an astonishing 197-fold more potent than the lead compound 15. Among the series of compounds in Table 5, p-benzyloxy 67 and p-benzoyl 70 emerged as the most attractive due to their high affinities toward BasE. Additionally, 70 was deemed particularly interesting as a result of its balanced activity against both MbtA and BasE.
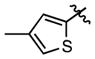
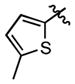
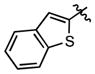
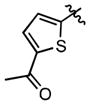
We also synthesized a series of 24 analogues 75–98 containing a wide variety of heterocycles, alkenes, alkynes and amines at C-6 (Table 6). The 5-membered furan and thiophene heterocycles were initially examined with analogues 75–76 and these are nearly 10-fold less potent than 15. A methyl scan in the thiophene ring with compounds 77–79 showed that substitution at positions 3 and 4 (77–78) is not well tolerated resulting in a respective 9- and 2.4-fold decrease in affinity; however, a 5-fold increase in affinity is conspicuously observed at position 5 (compound 79). Given the enhanced affinity of 5-methyl substituted thiophen-2-yl group, we also evaluated benzothiophene 80, 5-(acetyl)thiophene 81, and 5-(chloro)thiophene 82, but none of these enhance affinity. All of the nitrogen-containing heterocyclic analogues including pyridines 83–84, bipyridine 85, isoquinoline 86, indole 87, quinoline 88, pyrimidine 89 and pyrazole 90 result in a loss of potency ranging from 1.7-fold for indole 87 to 215-fold for isoquinoline 86. Similarly, alkene and alkyne analogues 91–94 bind with lower affinities ranging from ~10-fold for phenylethenyl 92 and phenylethynyl 94 to ~55-fold for unsubstituted analogues ethenyl 91 and ethynyl 93. Amino analogues including benzylamine 95, morpholine 96 and 3-hydroxypropylamine 97 are 40-fold to 68-fold less potent. Analogue 98 lacking an aryl moiety at C-6 is completely inactive (> 2800-fold less potent). Collectively, the results from this series of analogues demonstrate that a phenyl or isosteric heterocycle is required at C-6 (91, 93, 95–98) and nonpolar heterocyles are optimally tolerated (79, 80, 82 vs. 89 and 90). Overall, no improvement in affinity was achieved for 75–98 with BasE. The SAR of this series for MbtA is markedly different and several compounds were identified that are more potent including benzothiophene 80, 5-(acetyl)thiophene 81, 5-(chloro)thiophene 82, 6-chloropyridine 84, and bipyridine 85, which possess 12-, 5-, 14-, 4-, and 10-fold higher affinities relative to 15.
To complete our cursory SAR studies of 15, we also evaluated two compounds prepared during the course of our studies that involve double modifications (Table 7). Compound 99 containing an indazole core, but with the (4-pyridyl)methyl group at position 2 instead of 3, retains some potency toward MbtA (15.5-fold loss) and a pronounced 1800-fold loss in affinity toward BasE. Compound 100 with two unfavorable modifications (replacement of the 1-(4-pyridyl)methyl group and introduction of an amino group at position 6) displays no affinity toward either BasE or MbtA.
Table 7.
Miscellaneous SAR.
| Compound | KD (nM) | |
|---|---|---|
| BasE | MbtA | |
99
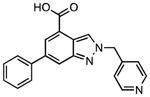
|
(66.4 ± 6.3)•103 | (58.0 ± 4.1)•103 |
100
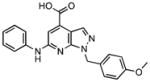
|
> 100•103 | > 100•103 |
Structural Characterization of Inhibitors with BasE
The active site of BasE, like all AAAE enzymes, contains three subsites that are used to bind the nucleotide, the aromatic acid, and the pantetheine chain of the incoming carrier protein that is used in the thioesterification reaction.34 The binding pocket for the nucleotide base is bordered on one side by a conserved aromatic residue and on the other by main chain interactions. The aromatic acid binds in a well-defined pocket of the AAAE.47 Finally, the pantetheine moiety of the cofactor enters the active site through a long tunnel formed between the larger N- and smaller C-terminal domain of the adenylating enzyme. This tunnel forms in the related acyl-CoA synthetases through rotation of the C-terminal domain upon completion of the initial adenylation reaction to form a conformation that is competent for thioester-formation.31
We have previously reported on the crystal structure of BasE bound to DHB-AMS 13, a derivative of 13 bearing an aliphatic chain on the C-2 position of the adenine, and the parent compound 15.31 The structure of BasE bound to 15 shows that the pyridine approximates the binding mode of the DHB substrate. The nitrogen of the pyridine moiety hydrogen bonds to the side chain of Asn242, mimicking the binding interaction of the 2- and 3-hydroxy groups of DHB in the structure of BasE bound to 13.31 Unexpectedly, the binding of 15 did not utilize the carboxylate to mimic the adenylate phosphate nor occupy the adenine binding pocket. The phenyl moiety was placed into the pantetheine tunnel. The binding of the phenyl group in this tunnel provides room for the larger inhibitors that were observed to result in higher affinity for BasE. We therefore determined the crystal structure of BasE bound to 67 and 70, to examine how the larger aromatic substituent would fill the pantetheine tunnel.
Crystals of BasE complexes with compounds 67 and 70 diffract well, and the structures were determined by difference Fourier methods. Data collection and refinement statistics are presented in Table 8. As with the previous structures of BasE31 and other ANL adenylating enzymes,48, 49 the conformationaly dynamic C-terminal domain is disordered and is not included in the final models.
Table 8.
Crystallographic Diffraction and Refinement data.
| BasE • 67 | BasE • 70 | |
|---|---|---|
| PDB Accession Code | 3U16 | 3U17 |
| Resolution | 40.0 – 2.1 Å | 50.0 – 2.1 Å |
| Space Group | P212121 | P212121 |
| Unit Cell | a=66.1 Å | a=65.5 Å |
| b=144.8 Å | b=143.3 Å | |
| c=148.7 Å | c=148.8 Å | |
| Rmergea | 10.2 % (54.2%) | 7.0 % (34.2 %) |
| Completenessa | 98.0 % (88.4%) | 90.2 % (51.9 %) |
| I/sa | 11.8 | 10.1 |
| # Observations | 365612 | 280779 |
| # Reflections | 82980 | 74749 |
| Rcryst (Overall/Highest Resolution Shell) a | 18.5 % (26.8 %) | 19.0 % (26.7 %) |
| Rfree (Overall/Highest Resolution Shell) a | 21.9 % (31.9 %) | 22.1 % (31.0 %) |
| Wilson B-factor | 32.1 Å2 | 28.9 Å2 |
| Average B-Factor, Proteinb | A=36.6 Å2 | A=41.3 Å2 |
| B=36.8 Å2 | B=41.2 Å2 | |
| Average B-Factor, Ligand, Solvent, Ions (Å2) | 34.4, 39.9, 46.6 | 34.9, 41.5, 55.5 |
| Number of Water Molecules, Ions | 511 H2O, 6 Ca2+ ions | 379 H2O, 1 Ca2+ ion |
| RMS Deviation Bond Lengths, Angles | 0.007 Å, 1.06° | 0.007 Å, 1.05° |
Values for the highest resolution shell are given in parentheses.
The two ligands bind in the active site of BasE in a manner identical to that of 15 reported previously.31 The pyridin-4-yl-methyl group enters into the DHB binding pocket, forming a hydrogen bond with the side chain of Asn242. This interaction is important for binding, as seen in compounds 29 through 36 (Table 4) where only compound 32 retained some binding affinity. SAR of the core heterocycle (Table 2) demonstrates the importance of N-2 of the pyrazole ring. The N-2 of the pyrazole ring accepts a hydrogen bond from the main chain amine of Gly338. Loss of this interaction results in a reduction of affinity of three orders of magnitude. This interaction is unique to the HTS ligands as the amide of Gly338 does not interact with the DHB-AMS analog of the adenylate 13. In contrast, the N-7 nitrogen of the pyrazolopyridine makes no interactions with the BasE active site residues and therefore substitution with a carbon has relatively little impact on binding. The carboxylate groups of 67 and 70 interact with the side chain of Arg435. The interaction of compound 67 with Arg435 is different from that of compounds 15 and 70. However we note that the electron density for this residue, which is only 2 residues from the hinge that separates the N-terminal domain from the disordered C-terminal domain, is disordered in one chain in the asymmetric unit for each protein model, therefore it is likely that Arg435 adopts multiple conformations. Nevertheless, the density is of sufficient quality to be modeled in the second chain in the asymmetric unit for each complex. In the structure of BasE bound to 70, the side chain of Arg435 appears to make a bivalent interaction with the carboxylate. In the model for BasE bound to 67, the side chain of Arg435 interacts with just a single oxygen from the carboxylate. Furthermore, in the previously reported complex with 15, the side chain of Arg435 does not interact directly with the carboxylate of this compound, rather this is mediated through a water molecule. This fact further strengthens our hypothesis of multiple conformations adopted by this residue, and could also justify the higher affinity to BasE observed for compounds 67 and 70 (direct interaction between Arg435 and the carboxylate) when compared to 15. SAR with compounds altered in this carboxylate (18 through 28, Table 3) illustrates a complicated relationship of this group to binding affinity. Replacement of the carboxylate with an alcohol or a carboxamide are reasonably well tolerated in compounds 19 and 22, resulting in only a 3- and 2-fold increase in KD. This shows that a strict ionic interaction is not required with the side chain of Arg435.
The longer hydrophobic moieties present in 67 and 70 continue into the pantetheine tunnel (Figure 5). The phenyl ring shared by 15, 67, and 70 stacks against the side chain of His241. The benzyloxy group of 67 and the benzoyl ring of 70 adopt different conformations. The ring in 67 is positioned closer to the pocket formed by Pro266, Val286, and Ala 289 whereas the ring of 70 is positioned near the top of the groove near residues Leu109 and Pro238. The binding of the phenyl group into this hydrophobic pocket helps to explain the affect of changes at the ortho-position in compounds 37 and 44. The ring is 4.1–4.3 A from the side chain of Phe243 on one side. The ortho-carbon on the other side points toward the location of the disordered C-terminal domain and we cannot determine if it could be accommodated here. The phenyl ring and the pyrazolopyridine core are nearly coplanar, with inter-planar torsion angles ranging from 3° to 15° in the three inhibitor molecules, and the o-hydroxy analog 40 may be tolerated because the hydroxyl could hydrogen bond with the adjacent N-7 nitrogen from the pyrazolopyridine core.
Figure 5.

Structural characterization of inhibitor binding. Ribbon diagrams are shown for the BasE enzyme bound to A. Inhibitor 67 and B. Inhibitor 70. Superimposed on both panels is the nucleotide DHB-AMS 13 (yellow) from PDB 3O82, demonstrating the interaction between Asp420 and the ribose hydroxyls and how the pyridyl group of the inhibitors mimics DHB moiety. Arg435, which is weakly ordered in both chains, interacts with the inhibitor carboxylate. Residues that form the hydrophobic binding pocket are shown in side chain representation.
Interestingly, whereas 70 serves as a potent inhibitor for both BasE and MbtA, 67 serves as a nanomolar inhibitor for BasE, but is 1000-fold weaker with MbtA (Table 5). Examination of the binding pockets of the two inhibitor complex and a sequence alignment of BasE and MbtA shows that the residues labeled in Figure 5 are conserved between BasE and MbtA, with the exception of Pro238 and Ala289, which are replaced by alanine and leucine, respectively, in MbtA. The replacement of Ala289 with the bulkier leucine is likely the reason why 67 is unable to bind in the same manner as observed in the BasE crystal structure, resulting in the observed micromolar binding constant. We note, however, that the disordered C-terminal domain does form a portion of the pantetheine tunnel in which the aromatic groups bind. Therefore, together with the differences in residues between BasE and MbtA, the C-terminus may also contribute to binding of the inhibitors and be responsible for the different binding affinities observed.
Antibacterial activity
All of the final compounds 15–100 were evaluated for antibacterial activity against A. baumannii ATCC 19606 under iron–deficient (1 μM FeCl3 and 200 μM dipyridyl as chelating agent) and iron-replete conditions (200 μM FeCl3) by broth microdilution in M9 minimal media supplemented with casamino acids (see Experimental Section). However, none of the compounds exhibited antibacterial activity against this bacterium. Several ester intermediates of the most potent inhibitors were also tested to assess whether the higher lipophilicity of these could lead to improved uptake by the bacterium, assuming that a bacterial esterase would hydrolyze the esters. Again, no activity was observed for the esters tested. The resistance of A. baumannii to many antibiotics is caused by numerous mechanisms including multidrug efflux pumps and permeability defects due to loss of porins.50 This could explain the resistance to 15 and its analogues, but further studies are required to assess this hypothesis. A small selection of compounds that exhibited potent activity toward MbtA, namely 15, 18, 66, 70, and 82 and their ethyl esters were also tested against M. tuberculosis H37Rv, under iron-deficient and iron-replete conditions (Table 9). While some of these compounds displayed very modest activity, the activity under iron replete conditions suggests these compounds may operate by a secondary mechanism of action since siderophore synthesis in M. tuberculosis is dispensable under rich-conditions.22
Table 9.
MIC99 determined against M. tuberculosis H37Rv (μM).
| Compound | MIC (μM) | MIC (μM) |
|---|---|---|
| Iron-deficient (GAST/−Fe) | Iron-replete (GAST/+Fe) | |
| 15 | >125 | >125 |
| 18 | >125 | 125 |
| 66 | 25 | 25 |
| 66-ethyl ester | 50 | 100 |
| 70 | >125 | >125 |
| 70-ethyl ester | >125 | >125 |
| 82 | 50 | 50 |
| 82-ethyl ester | 25 | 25 |
Disruption of BasE in A. baumannii
Earlier studies had rigorously demonstrated the importance of acinetobactin production for growth under iron limiting conditions by insertional inactivation of BasD, the cyclase-condensation didomain NRPS involved in acinetobactin biosynthesis.27, 28 Based on our inability to obtain antibacterial activity against A. baumannii ATCC 19606, we hypothesized that BasE may be functionally redundant, although no other AAAE’s are present in the genome. In order to unequivocally demonstrate the role of BasE in acinetobactin biosynthesis, we deleted basE by homologous recombination replacing it with a kanamycin resistance gene on the chromosome. The deletion was confirmed by PCR (data not shown). Under iron deficient conditions,27 the mutant was severely impaired for growth (Figure 6A). However, under iron-replete conditions,27 there was little observable difference between the growth rates between the wild-type and mutant knockout strain, except for a slight increase in lag-time for entry in exponential growth (Figure 6B). The modest growth observed for the basE mutant in Figure 6A under iron deficient conditions is caused by residual bacterial iron stores present in the initial inoculum. If the strain is pre-conditioned under iron-deficient conditions and then inoculated into iron deficient media, it is unable to grow. Reintroduction of basE on a plasmid was able to partially complement the deletion phenotype (Figure 6A). The inability to fully complement the basE knockout strain may be caused by a polar effect on the downstream pathway. The gene immediately downstream of basE, basF may be transcriptionally coupled to its upstream neighbor as there are only 18 bp between the two genes and there is no easily recognizable Shine-Dalgarno sequence in this region. Attempts to remove the kanamycin resistance gene and create a clean deletion of basE were unsuccessful. In summary, these results in conjunction with prior genetic studies on acinetobactin synthesis suggest BasE is nonreduntant and required for growth of A. baumannii under iron-restricted conditions with inorganic iron as the sole source of iron.
Figure 6.

In vitro growth curves of wild type A. baumannii ATCC 19606 (●), ΔbasE::kan mutant (■), and the ΔbasE::kan mutant complemented with plasmid pCDD140 (▲). The absorbance at 600 nm is plotted versus incubation time. All strains were inoculated directly into the indicated media at an A600 of 0.0003 and were not pre-conditioned under iron limitation. A. Growth under iron-deficient conditions containing 1 μM FeCl3 and 200 μM dipyridyl in M9 minimal media. All measurements were performed in triplicate and error bars represent the standard deviation. B. Growth under iron-rich conditions containing 200 μM FeCl3 in M9 minimal media.
Discussion
The importance of iron for bacterial pathogenesis has led to an increasing interest in targeting iron acquisition pathways for antibacterial development.51–53 The most ubiquitous strategy employed by bacteria to obtain iron is the synthesis of siderophores.12 Many bacteria also possess a heme uptake pathway, but this is only important to support bacteremia or bloodstream infections.54 However, inhibition of siderophore biosynthesis is unlikely to provide broad spectrum antibiotics due to large number of structurally different siderophores produced by bacteria.55 Given the alarming rise of antibacterial resistance and the extreme challenges of developing new classes of broad-spectrum agents, the synthesis of narrow spectrum antibiotics is becoming more attractive, particularly for serious infections like A. baumannii for which there are few other treatment options.56
Collins and co-workers have shown that bactericidal antibiotics generate hydroxyl radicals through the Fenton reaction caused by release of Fe2+ from bacterial iron-sulfur proteins.23, 24 We hypothesize that siderophores may protect bacteria from ROS by chelating free iron. A recent study demonstrated that enterobactin (the prototypical aryl-capped siderophore from E. coli) protected this bacteria from oxidative stress.57 Thus, inhibition of siderophore biosynthesis may have the additional benefit of enhancing the bactericidal activity of existing antibiotics.
Quadri and co-workers were the first to report an inhibitor of siderophore biosynthesis with the synthesis of Sal-AMS, the prototypical AAAE inhibitor.35–37 Unfortunately, Sal-AMS and related nucleoside analogues have only limited activity against Gram-negative pathogens. We believe this is a result of their highly polar nature and formal negative charge that likely prevents uptake across the negatively charged outer lipopolysaccharide-rich membrane of Gram-negative organisms. The confirmed role of aryl-capped siderophores for virulence in Gram-negative infection including acinetobactin, enterobactin, and yersiniabactin, coupled with lack of activity of Sal-AMS toward A. baumannii, K. pneumoniae, P. aeruginosa, and E. coli motivated us to search for alternate scaffolds as AAAE inhibitors.
We identified pyrazolopyridine 15 using a high-throughput fluorescence polarization assay with BasE from A. baumannii and performed detailed SAR studies.39 The N-2 nitrogen atom of the pyrazolopyridine scaffold 15 is essential while the N-7 nitrogen is not required for potent activity. Analysis of the co-crystal structures of 15, 67, and 70 show a hydrogen bond between N-2 of the pyrazolopyridine and the amide NH of Gly338 whereas no interaction is observed with N-7. The pyridylmethyl substituent at N-1 is optimal and binds in the DHB pocket with the pyridine substituent occupying a position nearly identical to the native DHB ligand forming a hydrogen bond to Asn242. The importance of this hydrogen bond was assessed by isosteric replacement of the pyridine N with a CH, which resulted in a greater than 2800-fold decrease in binding affinity that corresponds to a staggering loss of more than 4.8 kcal/mol in binding energy. The carboxylate substituent at C-4 is preferred, but can be replaced by neutral isosteres such as a carboxamido or hydroxymethyl with only a modest 2–3-fold attenuation in potency. Presumably these analogues can maintain the interaction with Arg435 observed with 15, 67, and 70. The phenyl substituent at C-6 is the most tolerant to modification and resulted in the identification of p-hydroxyphenyl 42, p-nitro 51, p-methylsulfonylphenyl 55, p-benzyloxyphenyl 67 and p-benzophenone 70, which are up to 18-fold more potent than 15. Analysis of the co-crystal structures of 67 and 70 reveals that the larger para-substituents are accommodated in the pantetheine tunnel. Thus, the pyrazolopyridine analogues are considered multisubstrate inhibitors since they occupy the binding sites of all three BasE substrates (DHB, ATP, and pantetheine cofactor of BasF).
MbtA was also studied as a representative salicylate adenylating enzyme, which are found in M. tuberculosis, Yersinia sp. and K. pneumoniae. The lead compound 15 and its analogues are generally more active against BasE than MbtA. Benzophenone 70 is the most potent inhibitor of MbtA with a KD of 19 nM, a value approximately 200-fold lower than the lead compound 15. Moreover, 70 is equipotent against both MbtA and BasE demonstrating the feasibility of identifying an inhibitor with balanced activity despite differences in the active-site architecture between these enzymes.
Surprisingly, in spite of the low nanomolar dissociation constants of some of the BasE inhibitors in the biochemical assay, they failed to inhibit growth of A. baumannii in vitro. A selection of compounds was also tested against M. tuberculosis that encodes for MbtA, a homologue of BasE. Modest bacteriostatic activity was observed with MICs varying between 25 and 100 μM. However the MICs were identical under iron-deficient and iron-replete conditions indicating that inhibition of MbtA is not fully responsible for the observed activity. Further work will be necessary in order to improve and verify the ability of this series of compounds to penetrate A. baumannii and reach their enzyme target BasE as discussed above.
The structure for acinetobactin was described in 1994 as 11 (Figure 2).25 In 2008 Walsh and Sattely revised the structure of acinetobactin to 1 based on their astute observation of the structural dissimilarities of pseudomonine and acinetobactin despite a common organization of their respective biosynthetic gene clusters.29, 30 The iron-binding properties of acinetobactin have not been evaluated, but it is expected to possess a substantially lower affinity for Fe3+ than pre-acinetobactin, which contains a oxazoline and hydroxamate functions. In acinetobactin these functional groups rapidly rearrange (t1/2 ~ 1 hour) to provide the isoxazolidinone in 1 (Figure 2). The revised structure 1 has been confirmed via total synthesis.26
The importance of acinetobactin for iron acquisition was first studied in A. baumannii strain ATCC 19606.27, 28, 58 Insertional inactivation of basD, which encodes for a didomain protein responsible for the condensation of the DHB and L-Thr building blocks in acinetobactin biosynthesis, results in a strain incapable of producing acinetobactin.28 The basD knockout is impaired in the ability to replicate under iron deficient conditions,27 in human A549 alveolar epithelial cells,59 and in a mouse sepsis model.60 While these studies clearly demonstrate the importance of basD for virulence, we wished to confirm that basE also phenocopies the basD mutant since these isogenic mutants will potentially produce different siderophore intermediates that may partially rescue loss of acinetobactin. In this study we examined the importance of basE for virulence of A. baumannii strain ATCC 19606. Deletion of basE results in a strain unable to replicate under iron deficient conditions, but that grows at the same rate as the wildtype strain under iron-replete conditions. These results confirm BasE as a valid target and suggest the inability of the pyrazolopyridine inhibitors to exhibit whole-cell activity toward A. baumannii is due to other factors such as limited accumulation and/or lack of vulnerability of BasE to inhibition by small-molecules. Vulnerability or amount that a target must be inhibited is another important consideration that cannot be assessed by a simple knockout strain.
The initial studies of iron acquisition in A. baumannii focused on strain ATCC 19606 and demonstrated acinetobactin is the only siderophore produced by this organism.25 Genome sequencing of the related strain A. baumannii ATCC 17978, reveals it encodes for an additional siderophore pathway of an uncharacterized aryl-capped siderophore.16 As a result acinetobactin is dispensable in ATCC 17978.16 Analysis of the second siderophore gene cluster reveals it encodes for two 2,3-dihydroxybenzoate-AMP ligases (A1S_2573 and A1S_2574), which we expect can also be inhibited by our pyrazolopyridine BasE inhibitors.59, 61, 62 Comparative genomics studies of six fully sequenced A. baumannii strains and PCR analysis of 50 clinical isolates were recently described providing the most detailed picture yet reported of iron acquisition systems in this pathogen.62 The acinetobactin gene cluster is highly conserved among clinical isolates. Another prevalent gene cluster was identified, which encodes for a putative hydroxamate siderophore. Genes encoding for ferrous uptake and heme uptake are also observed in virtually all strains. The importance of these multiple iron acquisition systems for virulence remains to be evaluated, but suggests this pathogen is capable of using alternate iron sources for survival under different environmental conditions.
Conclusion
A comprehensive analysis of the structure–activity relationships of the HTS hit 15 was performed that examined the importance of the pyrazolopyridine heterocycle, the 4-pyridylmethyl substituent at N-1, the carboxylic acid at C-4, and the phenyl group at C-6 for binding to BasE and MbtA. BasE from A. baumannii was the primary focus of the work and the initial SAR studies defined the crucial interactions necessary to maintain potency and also identified sites amenable to modification. The pyrazolopyridine heterocycle ideally positions the N-1, C-4, and C-6 substituents into the DHB, ATP, and pantetheine binding pockets. The N-2 nitrogen of the pyrazolopyridine forms a key hydrogen bond with the amide backbone of Gly338, but N-7 is dispensable for potent activity. The pyridylmethyl substituent at N-1 is crucial illustrated by the almost 5 kcal/mol loss in binding affinity by simple deletion of the nitrogen atom in the pyridine. The carboxylic acid at C-4 is not required and can be replaced with alternate hydrogen-bond acceptor moieties including hydroxymethyl and carboxamido with only a modest attenuation in binding affinity demonstrating the ionic interaction observed between the carboxylic acid and Arg435 in the co-crystal structures of BasE with three different pyrazolopyridine ligands is not critical. The C-6 phenyl group is most tolerant to substitution and hydrophobic (hetero)aryl substituents are preferred. p-Benzyloxyphenyl 67 was identified as the most potent analogue toward BasE with a KD of 2 nM. The entire compound series was also evaluated against MbtA, a representative AAAE that activates salicylic acid and similar SAR trends were observed. HTS hit 15 is considerably less potent toward MbtA with KD of only 3.7 μM. However, benzophenone 70 was found to have balanced activity against both BasE and MbtA with a KD of 19 nM, which represents a nearly 200-fold increase in potency toward MbtA. The SAR and structural characterization of ligands with BasE described herein provide a foundation for future studies to improve upon the antibacterial activity and exploit the unique multisubstrate modality of inhibition.
Experimental Section
Chemistry: General Methods and Materials
All commercial reagents (Sigma-Aldrich, Acros, Alfa-Aesar) were used as provided. Boronic acids and boronic acid pinacolate esters were purchased from Aldrich, Boron Molecular (Research Triangle, NC), and Frontier Scientific (Logan, UT). Compounds 20, 26, 29–30 and 33–35 were purchased from Enamine (Ukraine). Compounds 117 and 118 were obtained from Sinova (Bethesda, MD). Compounds 104,63 114,45 and 115 45 were prepared according to the respective literature procedure. Purity (≥95%) of all final compounds was confirmed by reverse-phase HPLC using the indicated method (see Supporting Information). An anhydrous solvent dispensing system (JC Meyer, Laguna Beach, CA) using 2 packed columns of neutral alumina was used for drying THF, DMF and CH2Cl2 and the solvents were dispensed under argon. All reactions were performed under an inert atmosphere of dry Ar or N2 in oven-dried (150 °C) glassware. Flash chromatography was performed with an ISCO Combiflash Companion® purification system with prepacked silica gel cartridges supplied by Luknova, with the indicated solvent system. Preparative HPLC was performed on a Varian Microsorb MV 100-8 C18 column (41.4 × 250 mm, 8 μm particle size) operating at 40 mL/min with detection at 254 nm in the conditions described in the Supporting Information. 1H and 13C NMR spectra were recorded on either Varian 600 MHz or Bruker Avance 400 MHz spectrometers. Proton chemical shifts are reported in ppm from an internal standard of residual chloroform (7.26 ppm), dimethylsulfoxide (2.50 ppm) or methanol (3.31 ppm), and carbon chemical shifts are reported using an internal standard of residual chloroform (77.1 ppm), dimethylsulfoxide (39.5 ppm) or methanol (49.0 ppm). Proton chemical data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet, br = broad, ovlp = overlapping), coupling constant, and integration. High-resolution mass spectra were obtained on an Agilent TOF II TOF/MS instrument equipped with an ESI interface.
Compounds from Scheme 1
Diethyl 2-(5-amino-1-(4-methoxybenzyl)-1H-pyrazol-4-yl)-2-hydroxysuccinate (106)
A solution of diethyl oxalacetate (3.46 g, 18.4 mmol, 1.01 equiv) and 10463 (3.7 g, 18.2 mmol, 1.0 equiv) in benzene (40 mL) was heated at 65 °C for 20 h. The mixture was concentrated and purification by flash chromatography (3:2 hexanes/EtOAc) afforded the title compound (4.5 g, 63%) as a yellow oil: Rf 0.65 (EtOAc); 1H NMR (600 MHz, CDCl3) δ 1.23–1.27 (m, 6H), 2.92 (d, J = 16.8 Hz, 1H), 3.32 (d, J = 16.8 Hz, 1H), 3.78 (s, 3H), 4.08 (br s, 2H, NH2), 4.16 (q, J = 7.2 Hz, 2H), 4.24 (q, J = 7.2 Hz, 2H), 5.06 (s, 2H), 6.85 (d, J = 8.4 Hz, 2H), 7.10 (d, J = 8.4 Hz, 2H), 7.23 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 14.17, 14.23, 43.9, 51.5, 55.4, 61.2, 62,4, 73.3, 102.7, 114.5, 128.1, 128.5, 135.8, 143.3, 159.4, 171.3, 173.5; HRMS (ESI+) calcd for C19H26N3O6 [M + H]+ 392.1816, found 392.1825 (error 2.3 ppm).
Ethyl 1-(4-methoxybenzyl)-6-oxo-6,7-dihydro-1H-pyrazolo[3,4-b]pyridine-4-carboxylate (108)
A suspension of 106 (5.2 g, 13.2 mmol) in glacial acetic acid (40 mL) was refluxed for 3 h. The mixture was concentrated to approximately half of the volume, then isopropanol (100 mL) was added and the solution was cooled to room temperature. The product crystallized as a white solid (3.5 g, 81%): mp 235–236 °C; Rf 0.65 (1:1 EtOAc/hexane); 1H NMR (600 MHz, DMSO-d6) δ 1.37 (t, J = 7.2 Hz, 3H), 3.70 (s, 3H), 4.40 (q, J = 7.2 Hz, 2H), 5.46 (s, 2H), 6.86 (d, J = 8.4 Hz, 2H), 6.95 (br s, 1H), 7.16 (d, J = 8.4 Hz, 2H), 8.11 (s, 1H), 12.05 (br s, D2O-exchangeable), 1H); 13C NMR (150 MHz, DMSO-d6) δ 14.0, 49.5, 55.1, 61.7, 113.9, 128.7, 129.1, 133.0, 134.0, 158.7, 163.5, 164.2 (unable to observe 3 carbons-estimated at 99, 135, and 147 ppm due to quadrupolar coupling with nitrogen); HRMS (ESI+) calcd for C19H26N3O6 [M + H]+ 328.1292, found 328.1303 (error 3.4 ppm).
1-(Pyridin-4-ylmethyl)-1H-pyrazol-5-amine (105)
To a solution of acrylonitrile (4.42 g, 83 mmol, 1.05 equiv) at 0 °C in absolute EtOH (80 mL), hydrazine hydrate (3.93 g, 79 mmol, 1.0 equiv) was added dropwise with vigorous stirring over 10 min. The ice bath was removed and the reaction was stirred for 24 h at rt. The reaction mixture was cooled to 0 °C and 4-pyridinecarboxaldehyde (8.8 g, 82 mmol, 1.04 equiv) was slowly added and stirring continued for 2 h at rt. The mixture was concentrated, the residue was dissolved in dry n-butanol (30 mL) and a 16% sodium n-butoxide solution in n-butanol (100 mL, 167 mmol, 2.1 equiv) was added. The resulting solution was refluxed for 1 h, cooled to rt, and concentrated. The residue was partitioned between H2O (150 mL) and EtOAc (3 × 150 mL). The combined organic layers were dried (MgSO4) and concentrated to afford the title compound (10.6 g, 76%) as a pale brown solid: Rf 0.19 (9:1 EtOAc/MeOH); 1H NMR (600 MHz, CDCl3) δ 3.43 (br s, 2H, NH2), 5.20 (s, 2H), 5.63 (d, J = 1.8 Hz, 1 H), 7.00 (d, J = 6.0 Hz, 2H), 7.35 (d, J = 1.8 Hz, 1H), 8.54 (d, J = 6.0 Hz, 2H); 13C NMR (150 MHz, CDCl3) δ 50.3, 92.8, 121.7, 139.5, 144.6, 146.0, 150.4; HRMS (ESI+) calcd for C9H11N4 [M + H]+ 175.0978, found 175.0976 (error 1.1 ppm).
Diethyl 2-(5-amino-1-(pyridin-4-ylmethyl)-1H-pyrazol-4-yl)-2-hydroxysuccinate (107)
A solution of diethyl oxalacetate (10.6 g, 56.1 mmol, 1.2 equiv) and 105 (8.1 g, 46.7 mmol, 1.0 equiv) in benzene (100 mL) was heated at 65 °C for 15 h. The mixture was concentrated and the residue was recrystallized from EtOH/Et2O to afford the title compound (10.6 g 48%) as a pale yellow solid: mp 118–120 °C; Rf 0.61 (7:3 EtOAc/MeOH); 1H NMR (600 MHz, CDCl3) δ 1.27 (ovlp t, J = 7.2 Hz, 3H), 1.28 (ovlp t, J = 7.2 Hz, 3H), 2.95 (d, J = 16.8 Hz, 1H), 3.35 (d, J = 16.8 Hz, 1H), 4.16 (br s, 2H, NH2), 4.19 (q, J = 7.2 Hz, 2H), 4.26 (q, J = 7.2 Hz, 2H), 5.13 (s, 2H), 7.01 (d, J = 6.0 Hz, 2H), 7.29 (s, 1H), 8.55 (d, J = 6.0 Hz, 2H); 13C NMR (150 MHz, CDCl3) δ 14.20, 14.25, 43.9, 50.4, 61.3, 62.5, 73.3, 103.2, 121.8, 136.8, 143.6, 145.5, 150.4, 171.3, 173.4; HRMS (ESI+) calcd for C17H23N4O5 [M + H]+ 363.1663, found 363.1668 (error 1.4 ppm).
Ethyl 6-oxo-1-(pyridin-4-ylmethyl)-6,7-dihydro-1H-pyrazolo[3,4-b]pyridine-4-carboxylate (109)
A suspension of 107 (9.7 g, 26.9 mmol) in glacial acetic acid (60 mL) was refluxed for 4 h. The mixture was concentrated and the residue was triturated with isopropanol (100 mL). The white solid that formed was filtered, washed with isopropanol/ether and dried under vacuum to afford the title compound (6.29 g, 78%) as an off-white solid: Rf 0.25 (9:1 EtOAc/MeOH); 1H NMR (600 MHz, DMSO-d6) δ 1.39 (t, J = 7.2 Hz, 3H), 4.42 (q, J = 7.2 Hz, 2H), 5.61 (s, 2H), 7.00 (br s, 1H), 7.07 (d, J = 6.0 Hz, 2H), 8.20 (s, 1H), 8.49 (d, J = 6.0 Hz, 2H), 12.10 (br s, 1H); 13C NMR (150 MHz, DMSO-d6) δ 14.0, 49.0, 61.7, 106.4, 109.0, 121.8, 133.6, 134.1, 146.1, 149.8, 163.8, 164.1 (missing 1 C); HRMS (ESI+) calcd for C15H15N4O3 [M + H]+ 299.1139, found 299.1128 (error 3.7 ppm).
Compounds from Scheme 2
Ethyl 1-(4-methoxybenzyl)-6-(trifluoromethylsulfonyloxy)-1H-pyrazolo[3,4-b]pyridine-4-carboxylate (110)
To a solution of 108 (3.38 g, 10.3 mmol, 1.0 equiv) and 2,6-di-tert-butyl-4-methylpyridine (3.18 g, 15.5 mmol, 1.5 equiv) in CH2Cl2 (50 mL) at −78 °C, was added dropwise a solution of trifluoromethanesulfonic anhydride (2.98 mL, 17.7 mmol, 1.7 equiv) in CH2Cl2 (10 mL). The reaction mixture was stirred at 0 °C for 4 h. The solvent volume was reduced to one-third in vacuo and diluted with EtOAc (40 mL). The resulting solution was washed consecutively with saturated aqueous NaHCO3 (3 × 20 mL), H2O (20 mL), 1 M aqueous HCl (3 × 20 mL), H2O (20 mL) and saturated aqueous NaCl (20 mL). The organic layer was dried (MgSO4) and concentrated. Purification by flash chromatography (4:1 hexane/EtOAc) afforded the title compound (3.2 g, 68%) as a white solid: Rf 0.43 (4:1 hexane/EtOAc); 1H NMR (600 MHz, CDCl3) δ 1.48 (t, J = 7.2 Hz, 3H), 3.76 (s, 3H), 4.52 (q, J = 7.2 Hz, 2H), 5.59 (s, 2H), 6.84 (d, J = 8.4 Hz, 2H), 7.37 (d, J = 8.4 Hz, 2H), 7.56 (s, 1H), 8.46 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 14.3, 51.3, 55.3, 62.7, 109.1, 113.7, 114.2, 118.8 (q, 1JC-F = 319 Hz, CF3), 128.0, 130.0, 133.8, 136.3, 148.3, 154.5, 159.7, 163.4; HRMS (ESI+) calcd for C18H17F3N3O6S [M + H]+ 460.0785, found 460.0762 (error 5.0 ppm).
Ethyl 1-(4-methoxybenzyl)-6-phenyl-1H-pyrazolo[3,4-b]pyridine-4-carboxylate (111)
A mixture of 110 (0.50 g, 1.08 mmol, 1.0 equiv), Pd(PPh3)4 (62.5 mg, 0.054 mmol, 0.05 equiv), Cs2CO3 (0.704 g, 2.16 mmol, 2.0 equiv), PhB(OH)2 (0.198 g, 1.62 mmol, 1.5 equiv) and dioxane (20 mL) was stirred at 100 °C for 5 h. The reaction mixture was cooled to rt, filtered through a plug of Celite and concentrated. Purification by flash chromatography (7:3 hexanes/EtOAc) afforded the title compound (0.39 g, 96%) as a white solid: Rf 0.49 (7:3 hexanes/EtOAc); 1H NMR (600 MHz, CDCl3) δ 1.50 (t, J = 7.2 Hz, 3H), 3.76 (s, 3H), 4.53 (q, J = 7.2 Hz, 2H), 5.74 (s, 2H), 6.84 (d, J = 9.0 Hz, 2H), 7.40 (d, J = 9.0 Hz, 2H), 7.49 (t, J = 7.2 Hz, 1H), 7.55 (t, J = 7.2 Hz, 2H), 8.22 (d, J = 7.2 Hz, 2H), 8.25 (s, 1H), 8.39 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 14.5, 50.6, 55.3, 62.0, 112.1, 114.1, 115.4, 127.7, 129.0, 129.3, 129.8, 129.9, 132.0, 133.2, 138.6, 151.6, 156.9, 159.4, 165.4; HRMS (ESI+) calcd for C23H22N3O3 [M + H]+ 388.1656, found 388.1666 (error 2.6 ppm).
1-(4-Methoxybenzyl)-6-phenyl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid (31)
To a solution of 111 (25 mg, 0.065 mmol) in THF (1 mL) was added 1 N aqueous NaOH (2 mL). The resulting solution was stirred at rt for 3 h. The solvent was partially evaporated, the reaction mixture diluted with H2O (10 mL) and the pH adjusted to 4–5 with 1 N aqueous HCl. The resulting suspension was extracted with EtOAc (3 × 15 mL), the combined organic layers were washed with saturated aqueous NaCl, dried (MgSO4), and concentrated to afford the title compound (23 mg, 98%) as a white solid: 1H NMR (600 MHz, DMSO–d6) δ 3.69 (s, 3H), 5.71 (s, 2H), 6.88 (d, J = 9.0 Hz, 2H), 7.31 (d, J = 9.0 Hz, 2H), 7.53 (t, J = 7.2 Hz, 1H), 7.58 (t, J = 7.2 Hz, 2H), 8.21 (s, 1H), 8.27 (d, J = 7.2 Hz, 2H), 8.37 (s, 1H); 13C NMR (150 MHz, DMSO–d6) δ 49.8, 55.0, 111.9, 113.9, 114.7, 127.3, 129.06, 129.18, 129.25, 130.0, 132.5, 133.3, 137.8, 150.9, 155.9, 158.7, 166.0; HRMS (ESI+) calcd for C21H16N3O3 [M − H]− 358.1197, found 358.1200 (error 0.8 ppm).
1-(4-Hydroxybenzyl)-6-phenyl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid (32)
To a solution of 111 (20 mg, 0.052 mmol, 1.0 equiv) in CH2Cl2 (2 mL) was added dropwise neat boron trifluoride–dimethylsulfide complex (55 μL, 0.052 mmol, 1.0 equiv) at 0 °C then the reaction was stirred at rt for 18 h. The reaction mixture was slowly poured over ice cold 0.5 M aqueous HCl (10 mL) and extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with saturated aqueous NaHCO3 (10 mL), H2O (10 mL), saturated aqueous NaCl (10 mL), dried (MgSO4) and concentrated. The residue was dissolved in THF (1.0 mL) then 1 N aqueous NaOH (1.0 mL) was added and the resulting solution was stirred at rt for 2 h. The mixture was concentrated and the residue was purified by preparative reverse-phase HPLC (solvent A: 10 mM NH4·HCO3, pH 7.5, solvent B: MeCN) using a linear gradient of 20%B to 100%B over 20 min (see General Methods and Materials for further details) to afford the title compound (3.0 mg, 17%) as a white solid: RT 9.6 min; 1H NMR (600 MHz, CD3OD) δ 5.68 (s, 2H), 6.71 (d, J = 8.4 Hz, 2H), 7.25 (d, J = 8.4 Hz, 2H), 7.49 (t, J = 7.2 Hz, 1H), 7.54 (t, J = 7.2 Hz, 2H), 8.23 (ovlp d, J = 7.2 Hz, 2H), 8.24 (ovlp s, 1 H), 8.40 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 51.4, 113.9, 116.2, 116.3, 128.6, 129.4, 130.0, 130.5, 130.8, 134.3. 134.5, 140.1, 152.7, 158.2, 158.6, 165.2; HRMS (ESI−) calcd for C20H14N3O3 [M − H]− 344.1041, found 344.1038 (error 0.9 ppm).
Ethyl 6-phenyl-1H-pyrazolo[3,4-b]pyridine-4-carboxylate (112)
A mixture of 111 (340 mg, 0.88 mmol) and TFA (4 mL) was heated at 70 °C for 24 h. Evaporation under vacuum followed by purification of the residue by flash chromatography (7:3 hexanes/EtOAc) afforded the title compound (199 mg, 85%) as a white solid: Rf 0.51 (3:2 hexanes/EtOAc); 1H NMR (600 MHz, CDCl3) δ 1.53 (t, J = 7.2 Hz, 3H), 4.57 (q, J = 7.2 Hz, 2H), 7.51 (t, J = 7.2 Hz, 1H), 7.56 (t, J = 7.2 Hz, 2H), 8.16 (d, J = 7.2 Hz, 2H), 8.28 (s, 1H), 8.52 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 14.5, 62.2, 111.8, 116.2, 127.9, 129.2, 130.1, 132.4, 135.1, 138.6, 153.4, 158.0, 165.3; HRMS (ESI+) calcd for C15H14N3O2 [M + H]+ 268.1081, found 268.1056 (error 9.3 ppm).
6-Phenyl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid (36)
To a solution of 112 (30 mg, 0.11 mmol) in THF (1.0 mL) was added 1 N aqueous NaOH (0.6 mL). The resultant solution was stirred at rt for 5 h. The mixture was concentrated and the residue was purified by preparative reverse-phase HPLC (solvent A: 10 mM NH4·HCO3, pH 7.5, solvent B: MeCN) using a linear gradient of 10%B to 40%B over 20 min (see General Methods and Materials for further details) to afford the title compound (23 mg, 86%) as a white solid: RT 18.9 min; 1H NMR (600 MHz, CD3OD) δ 7.45 (t, J = 7.2 Hz, 1H), 7.51 (t, J = 7.2 Hz, 2H), 8.13 (s, 1 H), 8.15 (d, J = 7.2 Hz, 2H), 8.47 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 113.8, 115.8, 128.6, 129.8, 130.5, 135.9, 140.7, 142.2, 150.3, 159.9, 172.5; HRMS (ESI−) calcd for C13H8N3O2 [M − H]−, 238.0622; found 238.0617 (error 2.1 ppm).
1-(4-Methoxybenzyl)-6-(phenylamino)-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid (100)
A mixture of 110 (100 mg, 0.22 mmol, 1.0 equiv), Pd(OAc)2 (5 mg, 0.022 mmol, 0.1 equiv), BINAP (20.4 mg, 0.033 mmol, 0.15 equiv), Cs2CO3 (107 mg, 0.33 mmol, 1.5 equiv), aniline (30 μL, 0.33 mmol, 1.5 equiv) and dioxane (1.5 mL) in a pressure vessel was stirred at 100 °C for 22 h. The reaction mixture was cooled to rt, diluted with EtOAc (10 mL), filtered through a plug of Celite and concentrated. Purification by flash chromatography (4:1 hexanes/EtOAc) afforded ethyl 1-(4-methoxybenzyl)-6-(phenylamino)-1H-pyrazolo[3,4-b]pyridine-4-carboxylate (83 mg, 90%) as a light yellow solid: Rf 0.53 (3:2 hexanes/EtOAc); 1H NMR (600 MHz, CDCl3) δ 1.45 (t, J = 7.2 Hz, 3H), 3.76 (s, 3H), 4.46 (q, J = 7.2 Hz, 2H), 5.55 (s, 2H), 6.84 (d, J = 8.4 Hz, 2H), 6.87 (br s, 1H, NH), 7.11 (t, J = 7.8 Hz, 1H), 7.22 (s, 1H), 7.33 (d, J = 8.4 Hz, 2H), 7.38 (t, J = 7.8 Hz, 2H), 7.63 (d, J = 7.8 Hz, 2H), 8.18 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 14.5, 50.7, 55.5, 62.1, 107.5, 108.6, 114.2, 120.1, 123.4, 129.4, 129.6, 129.7, 133.2, 133.6, 140.1, 150.9, 155.1, 159.4, 165.4.
To a solution of ethyl 1-(4-methoxybenzyl)-6-(phenylamino)-1H-pyrazolo[3,4-b]pyridine-4-carboxylate prepared above (30 mg, 0.074 mmol) in THF (1.0 mL) was added 1 N aqueous NaOH (2 mL). The resulting solution was stirred at rt for 3 h. The mixture was concentrated and H2O (10 mL) was added to the residue. The pH was adjusted to 4–5 with 1 N aqueous HCl and the solution extracted with EtOAc (3 × 15 mL). The combined organic layers were dried (MgSO4) and concentrated. Purification by flash chromatography (3:7 EtOAc/MeOH) afforded the title compound (19 mg, 69%) as a yellow solid: 1H NMR (600 MHz, CDCl3) δ 3.72 (s, 3H), 5.47 (s, 2H), 6.83 (d, J = 9.0 Hz, 2H), 6.97 (t, J = 7.8 Hz, 1H), 7.16 (s, 1H), 7.26 (d, J = 9.0 Hz, 2H), 7.29 (t, J = 7.8 Hz, 2H), 7.79 (d, J = 7.8 Hz, 2H), 8.23 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 51.1, 55.7, 108.9, 110.2, 114.9, 120.1, 122.8, 129.7, 130.1, 131.0, 135.0, 141.3, 142.5, 151.9, 157.5, 160.7, 173.2; HRMS (ESI−) calcd for C21H17N4O3 [M − H]− 373.1306, found 373.1282 (error 6.4 ppm).
Compounds from Scheme 3
See Supporting Information for the experimental details and data for 37–41, 43–62, and 66–92, which were prepared analogously to 15 and 42, whose experimentals are included below as representative examples.
General Procedure for Suzuki Coupling of 109
To a solution of 109 (100 mg, 0.335 mmol, 1.0 equiv) and triethylamine (141 μL, 1.00 mmol, 3.0 equiv) in dioxane (2.5 mL) in an 8 mL heavy-walled screw-cap round-bottom vial was added PyBroP (187 mg, 0.402 mmol, 1.2 equiv). The reaction mixture was shaken on a Glas-Col heater/shaker (Glas-Col LLC, Terre Haute, IN) at rt for 2 h. The desired boronic acid (0.67 mmol, 2.0 equiv), PdCl2dppf (12 mg, 0.05 equiv) and Na2CO3 (0.7 mL of a 0.254 g/mL aqueous solution, 5.0 equiv) were added, the vial was sealed, and heated at 100 °C for 4 h with shaking. The reaction mixture was cooled to rt and the solvent removed on a Genevac instrument at 45 °C. The residue was resuspended in H2O (2 mL) and extracted with CH2Cl2 (3 mL) using a 6 mL Biotage Isolute phase separator cartridge (Biotage, Charlottesville, Virginia). The organic layer was concentrated and the residue purified by flash chromatography (hexanes/EtOAc gradient) on a Combiflash Companion system, using a 4 g pre-packed silica column, to afford the desired product.
General Procedure for Ester Hydrolysis
To a solution of the ester (typically 50 mg, 1 equiv) in THF (1 mL) in an 8 mL vial was added 1 N aqueous NaOH (5.0 mL, 5 equiv). The resulting cloudy solution was stirred at rt for 4 h. The solvent was removed under vacuum at 45 °C on a Genevac instrument. The residue was resuspended in H2O (2 mL) and neutralized with 12 N HCl (0.29 mL, 3.5 equiv). Methanol or DMSO were added when necessary to fully dissolve the products, which were purified by preparative reverse-phase HPLC using the indicated methods. Lyophilisation of the fractions containing the product afforded the title compounds.
Ethyl 6-phenyl-1-(pyridin-4-ylmethyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylate (18)
The title compound was prepared using the general procedure for Suzuki coupling of 109 (0.40 g, 1.34 mmol, 1.0 equiv) using phenylboronic acid (327 mg, 2.68 mmol, 2.0 equiv). Purification by flash chromatography afforded the title compound (326 mg, 74%) as a white solid: Rf 0.39 (EtOAc); 1H NMR (600 MHz, CDCl3) δ 1.52 (t, J = 7.2 Hz, 3H), 4.55 (q, J = 7.2 Hz, 2H), 5.82 (s, 2H), 7.22 (d, J = 6.0 Hz, 2H), 7.49 (t, J = 7.2 Hz, 1H), 7.53 (t, J = 7.2 Hz, 2H), 8.17 (d, J = 7.2 Hz, 2H), 8.29 (s, 1H), 8.46 (s, 1H), 8.55 (d, J = 6.0 Hz, 2H); 13C NMR (150 MHz, CDCl3) δ 14.5, 49.8, 62.2, 112.1, 115.8, 122.6, 127.6, 129.0, 130.1, 132.4, 134.0, 138.3, 145.8, 150.3, 151.9, 157.4, 165.2; HRMS (ESI+) calcd for C21H19N4O2 [M + H]+ 359.1503; found 359.1527 (error 6.7 ppm).
6-Phenyl-1-(pyridin-4-ylmethyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid (15)
Compound 18 (120 mg, 0.324 mmol, 1.0 equiv) was converted to the title compound using the general procedure for ester hydrolysis. Purification by preparative reverse-phase HPLC using method 2 followed by lyophilisation of the pooled product fractions afforded the title compound (87 mg, 81%) as a white solid: RT 11.0 min; 1H NMR (600 MHz, CD3OD) δ 5.86 (s, 2H), 7.30 (d, J = 6.0 Hz, 2H), 7.45 (t, J = 7.2 Hz, 1H), 7.50 (t, J = 7.2 Hz, 2H), 8.18 (d, J = 7.2 Hz, 2H), 8.19 (s, 1H), 8.46 (d, J = 6.0 Hz, 2H), 8.53 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 50.3, 114.7, 116.1, 124.1, 128.6, 129.8, 130.6, 135.9, 140.4, 143.1, 149.3, 150.3, 153.2, 158.9, 172.4; HRMS (ESI−) calcd for C19H13N4O2 [M − H]− 329.1044, found 329.1040 (error 1.2 ppm).
6-(4-Hydroxyphenyl)-1-(pyridin-4-ylmethyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid (42)
Ethyl 6-(4-hydroxyphenyl)-1-(pyridin-4-ylmethyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylate (42-Ethyl ester) was prepared using the general procedure for Suzuki coupling of 109 with 4-hydroxyphenylboronic acid (92 mg, 0.67 mmol, 2 equiv). Purification by flash chromatography afforded 42-Ethyl ester (78 mg, 62%) as a white solid: Rf 0.22 (EtOAc); 1H NMR (600 MHz, DMSO-d6) δ 1.44 (t, J = 7.2 Hz, 3H), 4.49 (q, J = 7.2 Hz, 2H), 5.83 (s, 2H), 6.92 (d, J = 8.4 Hz, 2H), 7.19 (d, J = 5.4 Hz, 2H), 8.10 (d, J = 8.4 Hz, 2H), 8.16 (s, 1H), 8.41 (s, 1H), 8.50 (d, J = 5.4 Hz, 2H), 9.98 (s, 1H, OH); 13C NMR (150 MHz, DMSO-d6) δ 14.1, 48.9, 61.8, 110.6, 114.1, 115.9, 122.2, 128.3, 129.0, 131.9, 133.2, 146.1, 149.9, 151.4, 156.4, 159.6, 164.6.
42-Ethyl ester (50 mg, 0.133 mmol, 1.0 equiv) prepared above was converted to the title compound using the general procedure for ester hydrolysis. Purification by preparative reverse-phase HPLC using method 2 afforded the title compound (43 mg, 93%) as a white solid: RT 8.0 min; 1H NMR (600 MHz, CD3OD) δ 5.83 (s, 2H), 6.89 (d, J = 8.4 Hz, 2H), 7.29 (d, J = 5.4 Hz, 2H), 8.05 (d, J = 8.4 Hz, 2H), 8.11 (s, 1H), 8.46 (d, J = 5.4 Hz, 2H), 8.48 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 50.2, 113.9, 115.5, 116.7, 124.1, 130.1, 131.5, 142.8, 149.4, 150.3, 153.2, 159.1, 159.4, 160.8, 172.6; HRMS (ESI−) calcd for C19H13N4O3 [M − H]− 345.0993, found 345.0999 (error 1.7 ppm).
General procedure for Amination of 109
To a solution of 109 (100 mg, 0.335 mmol, 1.0 equiv) and BOP (193 mg, 0.437 mmol, 1.3 equiv) in dioxane (3 mL) in an 8 mL vial was added DBU (76 μL, 0.504 mmol, 1.5 equiv) and the resulting mixture was stirred for 15 min at rt. The desired amine (1.01 mmol, 3 equiv) was added, the vial was sealed, and the reaction mixture was heated at 70 °C with stirring for 5 h. The reaction mixture was concentrated, then the residue was resuspended in H2O (10 mL) and extracted with EtOAc (3 × 15 mL). The combined organic layers were dried (MgSO4) and concentrated. Purification by flash chromatography (hexane/EtOAc gradient) on a Combiflash Companion system, using a 4 g pre-packed silica column, afforded the desired product.
6-(Benzylamino)-1-(pyridin-4-ylmethyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid (95)
Ethyl 6-(benzylamino)-1-(pyridin-4-ylmethyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylate was prepared using the general procedure for amination of 109 with benzylamine (110 μL, 1.01 mmol, 3 equiv). Purification by flash chromatography afforded the ethyl ester of the title compound (28 mg, 22%) as a white solid: Rf 0.42 (EtOAc); 1H NMR (600 MHz, CDCl3) δ 1.44 (t, J = 7.2 Hz, 3H), 4.43 (q, J = 7.2 Hz, 2H), 4.66 (d, J = 6.0 Hz, 2H), 5.50 (ovlp t, J = 6.0 Hz, 1H, NH), 5.52 (ovlp s, 2H), 7.02 (s, 1H), 7.07 (d, J = 5.4 Hz, 2H), 7.26 (t, J = 7.2 Hz, 1H), 7.29 (t, J = 7.2 Hz, 2H), 7.33 (d, J = 7.2 Hz, 2H), 8.15 (s, 1H), 8.46 (d, J = 5.4 Hz, 2H); 13C NMR (150 MHz, CDCl3) δ 14.4, 45.9, 49.4, 61.9, 105.1, 108.3, 122.7, 127.5, 127.7, 128.8, 132.9, 134.2, 139.0, 146.3, 150.0, 151.7, 158.0, 165.4.
Ethyl 6-(benzylamino)-1-(pyridin-4-ylmethyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylate prepared above (20 mg, 0.052 mmol, 1.0 equiv) was converted to the title compound using the general procedure for ester hydrolysis. Purification by preparative reverse-phase HPLC using method 4 afforded the title compound as a white solid (17 mg, 91%): RT 10.0 min; 1H NMR (600 MHz, CD3OD) δ 4.60 (s, 2H), 5.51 (s, 2H), 6.92 (s, 1H), 7.10 (d, J = 6.0 Hz, 2H), 7.17 (t, J = 7.2 Hz, 1H), 7.21 (t, J = 7.2 Hz, 2H), 7.31 (d, J = 7.2 Hz, 2H), 8.13 (s, 1H), 8.34 (d, J = 6.0 Hz, 2H); 13C NMR (150 MHz, DMSO-d6) δ 44.2, 48.5, 104.9, 122.3, 122.5, 127.4, 127.7, 128.0, 128.3, 139.8, 146.4, 149.6, 149.8, 151.2, 158.3, 166.3; HRMS (ESI−) calcd for C20H16N5O2 [M − H]− 358.1309, found 358.1316 (error 2.0 ppm).
6-Morpholino-1-(pyridin-4-ylmethyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid (96)
Ethyl 6-morpholino-1-(pyridin-4-ylmethyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylate was prepared using the general procedure for amination of 109 with morpholine (88 μL, 1.01 mmol, 3 equiv). Purification by flash chromatography afforded the ethyl ester of the title compound (98 mg, 79%) as a pale yellow solid: Rf 0.37 (EtOAc); 1H NMR (600 MHz, CDCl3) δ 1.48 (t, J = 7.2 Hz, 3H), 3.68 (t, J = 4.8 Hz, 4H), 3.83 (t, J = 4.8 Hz, 4H), 4.49 (q, J = 7.2 Hz, 2H), 5.56 (s, 2H), 7.12 (d, J = 6.0 Hz, 2H), 7.27 (s, 1H), 8.18 (s, 1H), 8.52 (d, J = 6.0 Hz, 2H); 13C NMR (150 MHz, CDCl3) δ 14.5, 45.8, 49.4, 62.1, 66.8, 105.5, 106.3, 122.6, 133.3, 134.2, 146.4, 150.1, 151.5, 158.9, 165.6.
Ethyl 6-morpholino-1-(pyridin-4-ylmethyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylate prepared above (98 mg, 0.267 mmol, 1.0 equiv) was converted to the title compound using the general procedure for ester hydrolysis. Purification by preparative reverse-phase HPLC using method 4 afforded the title compound (45 mg, 97%) as a pale yellow solid: RT 6.5 min; 1H NMR (600 MHz, DMSO–d6) δ 3.62 (t, J = 4.8 Hz, 4H), 3.70 (t, J = 4.8 Hz, 4H), 5.56 (s, 2H), 7.14 (d, J = 6.0 Hz, 2H), 7.26 (s, 1H), 8.10 (s, 1H), 8.48 (d, J = 6.0 Hz, 2H); 13C NMR (150 MHz, CD3OD) δ 45.2, 48.6, 65.9, 105.1, 105.7, 122.4, 133.2, 133.9, 146.4, 149.8, 150.9, 158.6, 166.3; HRMS (ESI−) calcd for C17H16N5O3 [M − H]− 338.1259, found 338.1262 (error 0.9 ppm).
6-(3-Hydroxypropylamino)-1-(pyridin-4-ylmethyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid (97)
Ethyl 6-(3-hydroxypropylamino)-1-(pyridin-4-ylmethyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylate was prepared using the general procedure for amination of 109 with 3-hydroxypropylamine (77 μL, 1.01 mmol, 3 equiv). The crude product obtained after extraction was converted to the title compound using the general procedure for ester hydrolysis. Purification by preparative reverse-phase HPLC using method 4 afforded the title compound (22 mg, 18% over 2 steps) as a white solid: RT 8.6 min; 1H NMR (600 MHz, DMSOd6) δ 1.70 (pent, J = 6.6 Hz, 2H), 3.37 (t, J = 6.6 Hz, 2H), 3.48 (t, J = 6.6 Hz, 2H), 5.50 (s, 2H), 7.01 (s, 1H), 7.15 (d, J = 4.8 Hz, 2H), 7.98 (s, 1H), 8.47 (d, J = 4.8 Hz, 2H); 13C NMR (150 MHz, DMSO-d6) δ 31.9, 38.0, 48.7, 58.7, 99.5, 104.6, 122.6, 132.5, 133.3, 146.8, 149.8, 151.8, 158.8, 166.4; HRMS (ESI−) calcd for C16H16N5O3 [M − H]− 326.1259, found 326.1260 (error 0.3 ppm).
6-Oxo-1-(pyridin-4-ylmethyl)-6,7-dihydro-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid (98)
Compound 109 (40 mg, 0.134 mmol, 1.00 equiv) was converted to the title compound using the general procedure for ester hydrolysis. Purification by preparative HPLC using method 5 afforded the title compound (26 mg, 72%) as a white solid: RT = 8.0 min; 1H NMR (600 MHz, CD3OD) δ 5.59 (s, 2H), 6.86 (s, 1H), 7.17 (d, J = 6.0 Hz, 2H), 8.21 (s, 1H), 8.44 (d, J = 6.0 Hz, 2H); 13C NMR (150 MHz, CD3OD) δ 50.0, 108.4, 111.5, 123.8, 136.4, 144.9, 149.5, 150.2, 150.9, 169.5, 172.8; HRMS (ESI−) calcd for C13H9N4O3 [M − H]− 269.0680, found 269.0682 (error 0.7 ppm).
Compounds from Scheme 4
See Supporting Information for the experimental details and data for 60 and 64–65, which were prepared analogously to 43, whose experimentals are included below as a representative example.
General Procedure for Alkylation of 42-Ethyl ester and Hydrolysis
To a solution of 42-Ethyl ester (112.3 mg, 0.30 mmol, 1.0 equiv) and Na2CO3 (63.6 mg, 0.6 mmol, 2.0 equiv) in 1:1 dioxane–H2O (6 mL) was added the respective alkylbromide (1.2 mmol, 4.0 equiv). The mixture was heated at 80 °C for 12 h, then cooled to rt and the crude mixture was extracted with EtOAc (3 × 15 mL). The combined organic layers were washed with saturated aqueous NaCl (20 mL), dried (MgSO4), and concentrated. The crude residue was purified by flash chromatography (4:1 EtOAc–hexane) to afford the ethyl esters of the title compounds. Hydrolysis of the ethyl esters was accomplished by dissolving the intermediate ethyl esters (0.30 mmol, 1.0 equiv) in THF (3 mL) followed by the addition of 1 N aqueous NaOH (3 mL, 3 mmol, 10 equiv). The resulting solution was stirred at 60 °C for 12 h, then cooled to rt. The pH was adjusted to ~6 by the addition of 1 N aqueous HCl and the crude mixture was extracted with 10:1 EtOAc–MeOH (5 × 15 mL). The combined organic extracts were washed with saturated aqueous NaCl (20 mL), dried (MgSO4), and concentrated. The products were dissolved in EtOAc with a small amount of MeOH to aid in dissolution then triturated with hexane, whereupon the products precipitated as analytically pure white solids in yields ranging from 40–70%.
6-[4-(Octyloxy)phenyl]-1-(pyridin-4-ylmethyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid (63)
The title compound was prepared using the general procedure for alkylation of 42-Ethyl ester and hydrolysis employing octylbromide (210 μL, 1.2 mmol) to afford the title compound (38 mg, 28% over two steps) as a white solid: Rf 0.22 (1:4 MeOH/EtOAc); 1H NMR (600 MHz, DMSO–d6) δ 0.86 (t, J = 7.2 Hz, 3H), 1.27–1.32 (m, 8H), 1.41–1.42 (m, 2H), 1.72–1.75 (m, 2H), 4.04 (t, J = 7.2 Hz, 2H), 5.84 (s, 2H), 7.07 (d, J = 8.2 Hz, 2H), 7.20 (d, J = 3.2 Hz, 2H), 8.17–8.19 (m, 3H), 8.41 (s, 1H), 8.50 (d, J = 3.2 Hz, 2H); 13C NMR (150 MHz, DMSO–d6) δ 14.0, 22.1, 25.5, 28.62, 28.65, 28.7, 31.2, 48.9, 67.6, 111.6, 114.3, 114.8, 122.3, 128.8, 130.0, 133.5, 141.5, 146.2, 149.9, 151.4, 156.0, 160.3, 170.4; HRMS (ESI−) calcd for C27H29N4O3 [M − H]− 457.2245, found 457.2220 (error 5.5 ppm).
6-Ethynyl-1-(pyridin-4-ylmethyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid (93)
A 1.0 M solution of TBAF in THF (0.25 mL, 1.0 equiv) was added to a solution of 114 45 (105 mg, 0.25 mmol, 1.0 equiv) in THF (10 mL) at 0 °C and stirred for 3 h. The reaction mixture was diluted with H2O (10 mL) and extracted with CH2Cl2 (3 × 15 mL). The combined organic layers were dried (MgSO4), and concentrated. Purification by flash chromatography (hexane–EtOAc gradient) afforded 116 (27 mg, 35%) as a white solid: Rf 0.54 (EtOAc); 1H NMR (600 MHz, CDCl3) δ 1.48 (t, J = 7.2 Hz, 3H), 3.32 (s, 1H), 4.52 (q, J = 7.2 Hz, 2H), 5.75 (s, 2H), 7.13 (d, J = 4.8 Hz, 2H), 7.94 (s, 1H), 8.48 (s, 1H), 8.53 (d, J = 4.8 Hz, 2H); 13C NMR (150 MHz, CDCl3) δ 14.4, 49.8, 62.4, 72.3, 82.7, 113.0, 122.0, 122.6, 132.1, 134.1, 141.5, 145.4, 150.3, 151.3, 164.4.
Compound 116 prepared above (20 mg, 0.065 mmol, 1.0 equiv) was converted to the title compound using the general procedure for ester hydrolysis. Purification by preparative reverse-phase HPLC using method 4 afforded the title compound (7.4 mg, 41%) as an off-white solid: RT 8.3 min; 1H NMR (400 MHz, DMSO-d6) δ 4.64 (s, 1H), 5.78 (s, 2H), 7.12 (d, J = 6.0 Hz, 2H), 7.74 (s, 1H), 8.50 (d, J = 6.0 Hz, 2H), 8.51 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 49.0, 82.4, 82.8, 99.5, 112.9, 121.0, 122.0, 133.7, 140.7, 145.9, 149.9, 150.8, 165.3; HRMS (ESI−) calcd for C15H9N4O2 [M − H]− 277.0731, found 277.0736 (error 1.8 ppm).
6-(Phenylethynyl)-1-(pyridin-4-ylmethyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid (94)
Compound 115 45 (60 mg, 0.157 mmol, 1.0 equiv) was converted to the title compound using the general procedure for ester hydrolysis. Purification by preparative reverse-phase HPLC using method 2 afforded the title compound (36 mg, 65%) as a white solid: RT 11.8 min; 1H NMR (600 MHz, DMSO-d6) δ 5.77 (s, 2H), 7.10 (d, J = 5.4 Hz, 2H), 7.45–7.50 (m, 3H), 7.68 (dd, J = 7,8, 2.4 Hz, 2H), 7.77 (s, 1H), 8.49 (d, J = 5.4 Hz, 2H), 8.54 (s, 1H); 13C NMR (150 MHz, DMSO-d6) δ 48.9, 89.5, 89.6, 113.7, 121.1, 121.2, 121.9, 128.9, 129.7, 131.9, 134.7, 141.1, 146.4, 149.8, 151.1, 165.8 (missing one aryl carbon); HRMS (ESI−) calcd for C21H13N4O2 [M − H]− 353.1044, found 353.1035 (error 2.5 ppm).
Compounds from Scheme 5
See Supporting Information for the experimental details and data for 23–25, 27–28, which were prepared analogously to 22, whose experimental is included below as a representative example.
4-Hydroxymethyl-6-phenyl-1-(pyridin-4-ylmethyl)-1H-pyrazolo[3,4-b]pyridine (19)
A solution of 18 (358 mg, 1.0 mmol, 1.0 equiv) in THF (2 mL) was slowly added to a suspension of LiAlH4 (57 mg, 1.5 mmol, 1.5 equiv) in THF (8.0 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 10 min and gradually warmed up to 22 °C over 1 h. After stirring for 2 h at 22 °C, the reaction was quenched with MeOH (5 mL) and diluted with CH2Cl2 (15 mL). The organic layer was washed consecutively with saturated aqueous NH4Cl (10 mL), H2O (10 mL), saturated aqueous NaCl (10 mL), then dried (Na2SO4) and concentrated. Purification by flash chromatography (10:1 to 5:1 CH2Cl2–MeOH) afforded the title compound 19 (143 mg, 45%) as a white solid: Rf 0.36 (EtOAc); 1H NMR (600 MHz, CDCl3) δ 5.09 (s, 2H), 5.75 (s, 2H), 7.15 (d, J = 6.0 Hz, 2H), 7.43 (t, J = 7.2 Hz, 1H), 7.47 (t, J = 7.2 Hz, 2H), 7.65 (s, 1H), 8.08 (d, J = 7.2 Hz, 2H), 8.16 (s, 1H), 8.42 (d, J = 6.0 Hz, 2H); 13C NMR (150 MHz, CDCl3) δ 49.5, 62.7, 111.7, 112.6, 122.8, 127.6, 128.9, 129.7, 132.3, 139.2, 146.2, 146.6, 149.8, 151.3, 157.4; HRMS (ESI+) calcd for C19H17N4O [M]+ 317.1397, found 317.1395 (error 0.6 ppm).
6-Phenyl-1-(pyridin-4-ylmethyl)-1H-pyrazolo[3,4-b]pyridin-4-amine (21)
Compound 15 (50 mg, 0.168 mmol, 1.0 equiv) was dissolved in 3:1 CH2Cl2–THF (4 mL), and oxalyl chloride (17.6 μL, 0.205 mmol, 1.2 equiv) was slowly added at rt. The reaction mixture was stirred for 4 h then concentrated in vacuo. The residue was dissolved in acetone (5 mL) and added to a solution of NaN3 (47 mg, 0.73 mmol, 4.3 equiv) in H2O (5 mL). The solution was immediately extracted with EtOAc (2 × 20 mL). The combined organic layers were dried (MgSO4) and the solvent removed under vacuum. The residue was dissolved in benzene (5 mL), then TFA (19 μL, 0.25 mmol, 1.5 equiv) was added, and the solution was refluxed for 16 h. The solution was concentrated and the residue redissolved in MeOH (5 mL). Solid K2CO3 (51 mg, 0.37 mmol, 2.2 equiv) was added and the reaction mixture was stirred vigorously for 8 h at rt. The mixture was concentrated and the residue partitioned between H2O (10 mL) and EtOAc (3 × 15 mL). The combined organic layers were dried (MgSO4) and the solvent removed under vacuum. Purification by flash chromatography (hexanes/EtOAc gradient) afforded the title compound (16 mg, 31%) as a white solid: Rf 0.36 (EtOAc); 1H NMR (600 MHz, CDCl3) δ 4.80 (br s, 2H, NH2), 5.70 (s, 2H), 6.78 (s, 1H), 7.21 (d, J = 4.8 Hz, 2H), 7.41 (t, J = 7.2 Hz, 1H), 7.46 (t, J = 7.2 Hz, 2H), 7.97 (s, 1H), 8.02 (d, J = 7.2 Hz, 2H), 8.54 (br s, 2H); 13C NMR (150 MHz, CDCl3) δ 49.6, 98.5, 104.7, 122.8, 127.5, 128.7, 129.3, 130.2, 139.9, 146.5, 148.2, 150.1, 152.9, 158.6; HRMS (ESI−) calcd for C18H14N5O [M − H]− 300.1255, found 300.1255 (error 0 ppm).
General procedure for the synthesis of 6-phenyl-1-(pyridin-4-ylmethyl)-1H-pyrazolo[ 3,4-b]pyridine-4-carboxamide analogues
To a solution of 15 (15 mg, 0.045 mmol, 1.0 equiv) and DMF (3.5 μL, 0.045 mmol, 1.0 equiv) in dry CH2Cl2 (2 mL) at 0 °C was added oxalyl chloride (7.7 μL, 0.090 mmol, 2.0 equiv). The reaction was stirred for 1 hour, then concentrated in vacuo. A solution of the desired amine (2–10 equiv) and DMAP (1.8 mg, 0.015 mmol, 0.3 equiv) in CH2Cl2 (2.0 mL) were added and the reaction was stirred 1 h at rt. The mixture was concentrated and the residue resuspended in H2O (2 mL). Methanol or DMSO were added if necessary to fully dissolve the products. Purification by preparative HPLC using method 2, followed by lyophilization of the pooled product fractions afforded the title compounds.
6-Phenyl-1-(pyridin-4-ylmethyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxamide (22)
The title compound was prepared using the general procedure for the synthesis of 4-carboxamide analogues from 15, using a 14.8 M ammonium hydroxide solution (34 μL, 0.50 mmol, 10 equiv). Purification by preparative reverse-phase HPLC using method 2 afforded the title compound (4.9 mg, 30%) as a white solid: RT 12.2 min; 1H NMR (600 MHz, CD3OD) δ 5.88 (s, 2H), 7.32 (d, J = 5.4 Hz, 2H), 7.48 (t, J = 7.2 Hz, 1H), 7.52 (t, J = 7.2 Hz, 2H), 8.17 (s, 1H), 8.23 (d, J = 7.2 Hz, 2H), 8.45 (s, 1H), 8.47 (d, J = 5.4 Hz, 2H); 13C NMR (150 MHz, CD3OD) δ 50.4, 113.2, 114.3, 124.1, 128.7, 130.0, 131.1, 134.7, 138.3, 139.7, 149.0, 150.4, 153.0, 159.1, 169.9; HRMS (ESI+) calcd for C19H16N5O [M + H]+ 330.1349, found 330.1353 (error 1.2 ppm).
Compounds from Scheme 6
Methyl 6-bromo-1-(pyridin-4-ylmethyl)-1H-indazole-4-carboxylate (119) and Methyl 6-bromo-2-(pyridin-4-ylmethyl)-2H-indazole-4-carboxylate (121)
To a solution of methyl 6-bromo-1H-indazole-4-carboxylate 117 (300 mg, 1.18 mmol, 1.0 equiv) in DMF (10 mL) was added Cs2CO3 (2.3 g, 7.1 mmol, 6.0 equiv) and the resulting suspension was stirred at rt for 30 min. 4-(Bromomethyl)pyridine hydrobromide (0.89 g, 3.53 mmol, 3.0 equiv) was added and stirring continued at rt for 3 h. The reaction mixture was diluted with EtOAc (20 mL), filtered through a bed of Celite, and concentrated. Purification by flash chromatography (hexanes/EtOAc gradient) afforded the two title compounds as white solids:
Data for 119 (128 mg, 31%): Rf 0.37 (EtOAc); 1H NMR (600 MHz, CDCl3) δ 4.01 (s, 3H), 5.58 (s, 2H), 6.98 (d, J = 6.0 Hz, 2H), 7.65 (s, 1H), 8.01 (s, 1H), 8.52–8.54 (m, 3H); 13C NMR (150 MHz, CDCl3) δ 51.9, 52.7, 116.3, 120.4, 121.7, 122.0, 124.4, 127.9, 135.3, 140.9, 145.1, 150.5, 165.4; HRMS (ESI+) calcd for C15H13BrN3O2 [M + H]+ 348.0186, found 348.0176 (error 2.8 ppm).
Data for 121 (76 mg, 19%): Rf 0.19 (EtOAc); 1H NMR (600 MHz, CDCl3) δ 3.95 (s, 3H), 5.60 (s, 2H), 7.07 (d, J = 6.0 Hz, 2H), 7.97 (s, 1H), 8.09 (s, 1H), 8.47 (s, 1H), 8.57 (d, J = 6.0 Hz, 2H); 13C NMR (150 MHz, CDCl3) δ 52.5, 56.5, 119.2, 119.3, 122.1, 123.9, 125.5, 126.0, 130.0, 144.3, 150.1, 150.6, 165.5; HRMS (ESI+) calcd for C15H13BrN3O2 [M + H]+ 348.0186, found 348.0175 (error 3.2 ppm).
6-Phenyl-1-(pyridin-4-ylmethyl)-1H-indazole-4-carboxylic acid (16)
Methyl 6-phenyl-1-(pyridin-4-ylmethyl)-1H-indazole-4-carboxylate was prepared using the general procedure for Suzuki coupling from 119 (50 mg, 0.144 mmol, 1.0 equiv). Purification by flash chromatography (hexanes/EtOAc gradient) afforded the methyl ester of the title compound (28 mg, 57%) as a white solid: Rf 0.40 (EtOAc); 1H NMR (600 MHz, CDCl3) δ 4.06 (s, 3H), 5.69 (s, 2H), 7.03 (d, J = 6.0 Hz, 2H), 7.40 (t, J = 7.2 Hz, 1H), 7.47 (t, J = 7.2 Hz, 2H), 7.61 (d, J = 7.2 Hz, 2H), 7.63 (s, 1H), 8.22 (s, 1H), 8.54 (d, J = 6.0 Hz, 2H), 8.60 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 51.9, 52.5, 111.6, 121.7, 122.2, 123,7, 125.0, 127.7, 128.2, 129.2, 135.1, 140.1, 140.3, 141.0, 145.6, 150.5, 166.6.
Methyl 6-phenyl-1-(pyridin-4-ylmethyl)-1H-indazole-4-carboxylate (23 mg, 0.067 mmol, 1.0 equiv) prepared above was converted to the title compound using the general procedure for ester hydrolysis. Purification by preparative reverse-phase HPLC using method 3 afforded the title compound (16 mg, 72%) as a white solid: RT 9.5 min; 1H NMR (400 MHz, DMSO-d6) δ 5.87 (s, 2H), 7.13 (d, J = 6.0 Hz, 2H), 7.42 (t, J = 7.2 Hz, 1H), 7.51 (t, J = 7.2 Hz, 2H), 7.78 (d, J = 7.2 Hz, 2H), 8.08 (s, 1H), 8.29 (s, 1H), 8.48–8.50 (m, 3H); 13C NMR (100 MHz, DMSO-d6) δ 50.6, 111.4, 121.6, 122.0 (2 C), 123.1, 127.3, 127.9, 129.0, 134.4, 138.4, 139.5, 141.1, 146.4, 149.9, 167.2; HRMS (ESI−) calcd for C20H14N3O2 [M − H]− 328.1092, found 328.1089 (error 0.9 ppm).
6-Phenyl-2-(pyridin-4-ylmethyl)-2H-indazole-4-carboxylic acid (99)
Methyl 6-phenyl-2-(pyridin-4-ylmethyl)-2H-indazole-4-carboxylate was prepared using the general procedure for Suzuki coupling from 121 (50 mg, 0.144 mmol, 1.0 equiv). Purification by flash chromatography (hexanes/EtOAc gradient) afforded the methyl ester of the title compound (38 mg, 77%) as a white solid: Rf 0.22 (EtOAc); 1H NMR (600 MHz, CDCl3) δ 3.99 (s, 3H), 5.65 (s, 2H), 7.12 (d, J = 6.0 Hz, 2H), 7.39 (t, J = 7.2 Hz, 1H), 7.48 (t, J = 7.2 Hz, 2H), 7.69 (d, J = 7.2 Hz, 2H), 8.13 (s, 1H), 8.24 (s, 1H), 8.51 (s, 1H), 8.59 (d, J = 6.0 Hz, 2H); 13C NMR (150 MHz, CDCl3) δ 52.3, 56.5, 119.8, 120.8, 122.1, 122.9, 125.6, 127.4, 127.5, 127.9, 129.1, 139.1, 140.1, 144.7, 150.3, 150.5, 166.6.
Methyl 6-phenyl-2-(pyridin-4-ylmethyl)-2H-indazole-4-carboxylate (34 mg, 0.099 mmol, 1.0 equiv) prepared above was converted to the title compound using the general procedure for ester hydrolysis. Purification by preparative reverse-phase HPLC using method 3 afforded the title compound (25 mg, 77%) as a white solid: RT 9.0 min; 1H NMR (400 MHz, DMSO-d6) δ 5.82 (s, 2H), 7.24 (d, J = 6.0 Hz, 2H), 7.40 (t, J = 7.2 Hz, 1H), 7.50 (t, J = 7.2 Hz, 2H), 7.77 (d, J = 7.2 Hz, 2H), 8.08 (s, 1H), 8.15 (s, 1H), 8.54 (d, J = 6.0 Hz, 2H), 8.87 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 55.1, 119.0, 119.6, 122.5, 124.3, 125.3, 126.3, 127.0, 127.7, 129.1, 137.4, 139.8, 145.6, 149.6, 149.9, 167.0; HRMS (ESI−) calcd for C20H14N3O2 [M − H]− 328.1092, found 328.1084 (error 2.4 ppm).
Methyl 6-bromo-1-(pyridin-4-ylmethyl)-1H-indole-4-carboxylate (120)
To a solution of methyl 6-bromo-1H-indole-4-carboxylate 118 (50 mg, 0.20 mmol, 1.0 equiv) in DMF (3 mL) was added Cs2CO3 (391 mg, 1.2 mmol, 6.0 equiv) and the resulting suspension was stirred at rt for 30 min. 4-(Bromomethyl)pyridine hydrobromide (152 mg, 0.6 mmol, 3.0 equiv) was added and stirring continued at rt for 2 h. The reaction mixture was diluted with EtOAc (10 mL), filtered through a bed of Celite and concentrated. Purification by flash chromatography (hexanes/EtOAc gradient) afforded the title compound (53 mg, 77%) as a white solid: Rf 0.30 (EtOAc); 1H NMR (600 MHz, CDCl3) δ 3.98 (s, 3H), 5.32 (s, 2H), 6.89 (d, J = 5.4 Hz, 2H), 7.20 (d, J = 3.0 Hz, 1H), 7.23 (d, J = 3.0 Hz, 1H), 7.50 (s, 1H), 8.01 (s, 1H), 8.53 (d, J = 5.4 Hz, 2H); 13C NMR (150 MHz, CDCl3) δ 49.3, 52.2, 104.1, 114.8, 116.9, 121.2, 123.3, 125.5, 127.4, 131.1, 137.8, 145.7, 150.6, 166.7; HRMS (ESI+) calcd for C16H14BrN2O2 [M + H]+ 347.0213, found 347.0222 (error 2.6 ppm).
6-Phenyl-1-(pyridin-4-ylmethyl)-1H-indole-4-carboxylic acid (17)
Methyl 6-phenyl-1-(pyridin-4-ylmethyl)-1H-indole-4-carboxylate was prepared using the general procedure for Suzuki coupling from 120 (50 mg, 0.145 mmol, 1.0 equiv). Purification by flash chromatography (hexanes/EtOAc gradient) afforded the methyl ester of the title compound (59 mg, 63%) as a white solid: Rf 0.30 (EtOAc); 1H NMR (600 MHz, CDCl3) δ 4.02 (s, 3H), 5.42 (s, 2H), 6.95 (d, J = 6.0 Hz, 2H), 7.25 (d, J = 3.0 Hz, 1H), 7.29 (d, J = 3.0 Hz, 1H), 7.33 (t, J = 7.2 Hz, 1H), 7.43 (t, J = 7.2 Hz, 2H), 7.55 (s, 1H), 7.58 (d, J = 7.2 Hz, 2H), 8.21 (s, 1H), 8.53 (d, J = 6.0 Hz, 2H); 13C NMR (150 MHz, CDCl3) δ 49.3, 52.0, 103.9, 112.5, 121.3, 122.5, 123.4, 127.3, 127.5, 127.7, 129.0, 131.0, 135.4, 137.7, 141.2, 146.3, 150.5, 167.9.
Methyl 6-phenyl-1-(pyridin-4-ylmethyl)-1H-indole-4-carboxylate (50 mg, 0.146 mmol, 1.0 equiv) prepared above was converted to the title compound using the general procedure for ester hydrolysis. Purification by preparative reverse-phase HPLC using method 7 afforded the title compound (9.4 mg, 20%) as a white solid: RT 14.5 min; 1H NMR (600 MHz, DMSO-d6) δ 5.54 (s, 2H), 7.07 (d, J = 6.0 Hz, 2H), 7.19 (d, J = 3.0 Hz, 1H), 7.27 (d, J = 3.0 Hz, 1H), 7.33 (t, J = 7.2 Hz, 1H), 7.39–7.43 (m, 3H), 7.64 (d, J = 7.2 Hz, 2H), 7.90 (s, 1H), 8.44 (d, J = 6.0 Hz, 2H); 13C NMR (150 MHz, DMSO-d6) δ 48.1, 104.5, 108.3, 120.7, 121.9, 126.6, 126.9, 128.0, 129.0, 129.6, 133.0, 137.4, 141.9, 148.0, 149.9, 157.0, 170.8; HRMS (ESI−) calcd for C21H15N2O2 [M − H]− 327.1139, found 327.1145 (error 1.8 ppm).
Enzyme Binding Studies
BasE and MbtA were expressed in E. coli and purified as described.38, 39 Determination of equilibrium dissociation constants KD for each compound was performed using the fluorescence polarization assay with Fl-Sal-AMS 14 as ligand in black flat bottom 96-well plates (Costar #3915) as described.39 A three-fold serial dilution of each compound (10 μL) was added to 90 μL of a solution of 14 (20 nM final concentration) and 200 nM BasE or 50 nM MbtA, in 30 mM Tris-HCl pH 7.5, 1 mM MgCl2 and 0.0025% Igepal CA-630. The fluorescence anisotropy was measured after a 30 min incubation at 25 °C. The KD’s of each compound tested were determined by fitting the displacement curves (AOBS, the experimentally measured anisotropy vs. LT, the test compound concentration) to Equations 1 and 246 using Mathematica 7 (Wolfram Research Inc.). In these equations, Q is the ratio of the fluorescence intensity of the probe in the bound and free states (1.07 for BasE and 1.02 for MbtA39), FSB is the fraction of bound 14, AB (0.035) and AF (0.220 for BasE and 0.308 for MbtA39), represent the anisotropies of bound and free probe 14, KD1 is the equilibrium dissociation constant of 14 (84.3 nM for BasE and 9.26 nM for MbtA39), LST is the concentration of 14, RT is the receptor protein concentration and KD2 is the test compound’s equilibrium dissociation constant.46
| (1) |
| (2) |
with
Acinetobacter baumannii MIC Assay
A single LB agar plate was streaked with 19606 WT and grown overnight at 30° C. The plate was flooded with 2 mL of medium 1 (M9 minimal media, 0.2% casamino acids, 200 μM dipyridyl) and transferred to a 15 mL centrifuge tube. Cells were spun down at room temperature and washed twice with 10 mL medium 1 and then resuspended in 2 mL medium 1. The A600 was measured and cells were diluted in medium 1 to an A600 of 1.7 – 2.0 and then further diluted to 0.0003 in either medium 1 supplemented with 1 μM FeCl3 or Medium 2 (M9 minimal media, 0.2% casamino acids) supplemented with 200 μM FeCl3 and 200 μL of each solution was dispensed in triplicate into a 96 well plate containing 2 μL of each inhibitor in DMSO for a final concentration of 100 μM. Tetracycline was used as a positive control (100 ng/mL) and 1% DMSO was used as a negative control. The plates were incubated at 30° C and the A600 was measured at 15 hours.
Mycobacterium tuberculosis H37RV MIC Assay
Minimum inhibitory concentrations (MICs) were experimentally determined as previously described.36 MICs were determined in quadruplicate in iron-deficient GAST and GAST supplemented with 200 μM FeCl3 according to the broth microdilution method using compounds from DMSO stock solutions or with control wells treated with an equivalent amount of DMSO. Isoniazid was used as a positive control while DMSO was employed as a negative control. All measurements reported herein used an initial cell density of 104–105 cells/assay and growth was monitored at 10–14 days, with the untreated and DMSO-treated control cultures reaching an OD620 0.2–0.3. Plates were incubated at 37 °C (100 μL/well) and growth was recorded by measurement of optical density at 620 nm.
Protein Crystallography
The BasE enzyme from A. baumannii strain AB900 for crystallographic studies was produced and crystallized as described.31 BasE was co-crystallized with 67 and 70; crystals were isomorphic with the prior BasE structures. Data were collected at SSRL beamline 11-1. Initial phases were provided by difference Fourier methods using the previous structure of BasE bound to 15 as an initial refinement model; all non-protein atoms were removed prior to refinement. The models were refined to completion as described previously.31
Insertional deletion of BasE
Flanking regions of basE from A. baumannii ATCC 19606 were amplified by PCR using the primers 5′ F, 5′ R (CACCGAGCTCGGATCCACTGGATGTGGTGAGAAGC, CCCGGGACTAGTGACATTCTAAATATTCAATTTAATTTAATG) and 3′ F, 3′ R (CACCCGGGCTAGCTAAATATTGAGCAGCATATGG, CCTGCAGGATCCATGTGCTCTGAAGGACACG). The kanamycin resistance gene (kanR) was amplified from pCR2.1 TOPO (Invitrogen) using the primers Kan F, Kan R (CACCACTAGTTAACCGGAATTGCCAGCTGGG, GCTAGCTCAGAAGAACTCGTCAAG). The three PCR fragments were cloned into pENTR/TEV/D-TOPO (Invitrogen) for ease of propagation creating pCDD129, pCDD130 and pCDD131 for the 5′, KanR, and 3′ fragments respectively. The knockout fragments were cloned sequentially into pUC18 by first ligating the 5′ fragment after digesting pCDD129 and pUC18 with SacI and SmaI creating pCDD132. The 3′ fragment was added next by ligating SmaI, SbfI digested pCDD131 and pCDD132, creating pCDD133. KanR was removed from pCDD130 by digestion with SpeI, NheI and ligated into similarly cut pCDD133 creating pCDD134. The knock out cassette was removed from pCDD134 by digestion with BamHI and cloned into pKC1139 a Streptomyces/E. coli shuttle vector to create pCDD135, which will act as a suicide vector in A. baumannii. The knock out plasmid was transformed into ATCC 19606 using electroporation as previously described.64, 65 Clones resistant to kanamycin and sensitive to apramycin were confirmed by PCR to have the correct genome insertion using the primer pairs 5′ confirm (CACCACGAGGTATTTTGTGCTGGG), KanR, and KanF, 3′ confirm (CACCACGAGGTATTTTGTGCTGGG), creating 19606 ΔbasE.
Complementation of 19606 ΔbasE
The E. coli, A. baumannii shuttle vector pWH1266 (ATCC, Manassas VA) was used to complement 19606 ΔbasE. The primers basE comp F (CACCGGATCCTTGTTAATCATTTCCAATTTTG), and basE comp R (GCATGCTTAAGATGTTGTAGATGTATTTAAAATGC) were used to PCR amplify basE and its promoter region from ATCC19606. Primers Apr F (CACCAAGCTTTAAGGTTCATGTGCAGCTCCATC) and Apr R (GGATCCTCAGCCAATCGACTGGCG) were used to amplify the apramycin resistance (aprR) gene from pKC1139. The basE fragment was digested with BamHI and SphI, and AprR was digested with BamHI and HindIII. Both fragments were ligated into pWH1266 digested with HindIII and SphI creating pCDD140. The final plasmid was transformed into 19606 ΔbasE using the method described above.
Growth of A. baumannii strains
LB agar plates were streaked with 19606 WT, 19606 ΔbasE, or 19606 ΔbasE pCDD140 and grown overnight at 30° C. Each plate was flooded with 2 mL of medium 1 (M9 minimal media, 0.2% casamino acids, 200 μM dipyridyl) and transferred to a 15 mL centrifuge tube. Cells were spun down at rt and washed twice with 10 mL medium 1 and then resuspended in 2 mL medium 1. The A600 was measured and cells were diluted in medium 1 to an A600 of 1.7–2.0 and then further diluted to 0.0003 in either medium 1 supplemented with 1 μM FeCl3 or Medium 2 (M9 minimal media, 0.2% casamino acids) supplemented with 200 μM FeCl3 and placed into a 96 well plate in six 200 μL replicates. Positive controls for iron limiting and rich conditions consisted of WT supplemented with 100 ng/mL tetracycline. The plates were incubated at 30° C and the A600 was measured at 2, 4, 6, 7, 8, 9, 10, 12, 13 and 22 h.
Supplementary Material
Acknowledgments
This research was supported by grants from the National Institutes of Health (AI070219 to C.C.A. and GM-068440 to A.M.G.) and the Intramural Research Program of the NIAID in the National Institutes of Health (to Clifton E. Barry 3rd). We thank Dr. Michael Walters (Institute for Therapeutics Discovery and Development, University of Minnesota) for assistance with the parallel synthesis and Prof. David H. Sherman (Life Sciences Institute, University of Michigan) for plasmids pUC18 and pKC1139.
Abbreviations
- AAAE
aryl acid adenylating enzyme
- ArCP
aryl carrier protein
- BOP
(benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate
- DHB
2,3-dihydroxybenzoic acid
- DHB-AMS
5′-O-[N-(2,3-dihydroxybenzoyl)sulfamoyl]adenosine
- Fl-Sal-AMS
22-O-{2-[2-(2-{[(fluorescein-5-yl)carbonyl]amino}ethoxy)ethoxy]ethoxy}-52-O-[N-(salicyl)sulfamoyl]adenosine
- FP
fluorescence polarization
- HPLC
high-performance liquid chromatography
- HTS
high-throughput screening
- MDR
multidrug resistant
- MIC
minimum inhibitory concentration
- NRPS
nonribosomal peptide synthetase
- PMP
p-methoxyphenyl
- PyBroP
bromotripyrrolidinophosphonium hexafluorophosphate
- ROS
reactive oxygen species
- SAL
salicylic acid
- Sal-AMS
5′-O-[N-(salicyl)sulfamoyl]adenosine
- SAR
structure–activity relationship
- TES
triethylsilyl
- TFA
trifluoroacetic acid
Footnotes
Supporting Information Available: The synthetic procedures and characterization data for compounds 23–25, 27–28, 37–62, and 64–92 as well as representative 1H and 13C NMR spectra for 15, 16, 17, 18, 19, 21, 22, 37, 60, 63, 91, 94, and 98 can be found in the Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Projan SJ, Bradford PA. Late stage antibacterial drugs in the clinical pipeline. Curr Opin Microbiol. 2007;10:441–446. doi: 10.1016/j.mib.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 2.Wright GD. Antibiotics: a new hope. Chem Biol. 2012;19:3–10. doi: 10.1016/j.chembiol.2011.10.019. [DOI] [PubMed] [Google Scholar]
- 3.Peleg AY, Seifert H, Paterson DL. Acinetobacter baumannii: emergence of a successful pathogen. Clin Microbiol Rev. 2008;21:538–582. doi: 10.1128/CMR.00058-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calhoun JH, Murray CK, Manring MM. Multidrug-resistant organisms in military wounds from Iraq and Afghanistan. Clin Orthop Relat Res. 2008;466:1356–1362. doi: 10.1007/s11999-008-0212-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lockhart SR, Abramson MA, Beekmann SE, Gallagher G, Riedel S, Diekema DJ, Quinn JP, Doern GV. Antimicrobial resistance among Gram-negative bacilli causing infections in intensive care unit patients in the United States between 1993 and 2004. J Clin Microbiol. 2007;45:3352–3359. doi: 10.1128/JCM.01284-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rhomberg PR, Jones RN. Summary trends for the Meropenem Yearly Susceptibility Test Information Collection Program: a 10-year experience in the United States (1999–2008) Diagn Microbiol Infect Dis. 2009;65:414–426. doi: 10.1016/j.diagmicrobio.2009.08.020. [DOI] [PubMed] [Google Scholar]
- 7.Vallenet D, Nordmann P, Barbe V, Poirel L, Mangenot S, Bataille E, Dossat C, Gas S, Kreimeyer A, Lenoble P, Oztas S, Poulain J, Segurens B, Robert C, Abergel C, Claverie JM, Raoult D, Medigue C, Weissenbach J, Cruveiller S. Comparative analysis of Acinetobacters: three genomes for three lifestyles. PLoS ONE. 2008;3:e1805. doi: 10.1371/journal.pone.0001805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adams MD, Goglin K, Molyneaux N, Hujer KM, Lavender H, Jamison JJ, MacDonald IJ, Martin KM, Russo T, Campagnari AA, Hujer AM, Bonomo RA, Gill SR. Comparative genome sequence analysis of multidrug-resistant Acinetobacter baumannii. J Bacteriol. 2008;190:8053–8064. doi: 10.1128/JB.00834-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Falagas ME, Kasiakou SK. Colistin: the revival of polymyxins for the management of multidrug-resistant gram-negative bacterial infections. Clin Infect Dis. 2005;40:1333–1341. doi: 10.1086/429323. [DOI] [PubMed] [Google Scholar]
- 10.Cai Y, Chai D, Wang R, Liang B, Bai N. Colistin resistance of Acinetobacter baumannii: clinical reports, mechanisms and antimicrobial strategies. J Antimicrob Chemother. 2012;67:1607–1615. doi: 10.1093/jac/dks084. [DOI] [PubMed] [Google Scholar]
- 11.Posey JE, Gherardini FC. Lack of a role for iron in the lyme disease pathogen. Science. 2000;288:1651–1653. doi: 10.1126/science.288.5471.1651. [DOI] [PubMed] [Google Scholar]
- 12.Miethke M, Marahiel MA. Siderophore-based iron acquisition and pathogen control. Microbiol Mol Biol Rev. 2007;71:413–451. doi: 10.1128/MMBR.00012-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crosa JH, Walsh CT. Genetics and assembly line enzymology of siderophore biosynthesis in bacteria. Microbiol Mol Biol Rev. 2002;66:223–249. doi: 10.1128/MMBR.66.2.223-249.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sandy M, Butler A. Microbial iron acquisition: marine and terrestrial siderophores. Chem Rev. 2009;109:4580–4595. doi: 10.1021/cr9002787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hider RC, Kong X. Chemistry and biology of siderophores. Nat Prod Rep. 2010;27:637–657. doi: 10.1039/b906679a. [DOI] [PubMed] [Google Scholar]
- 16.Zimbler DL, Penwell WF, Gaddy JA, Menke SM, Tomaras AP, Connerly PL, Actis LA. Iron acquisition functions expressed by the human pathogen Acinetobacter baumannii. Biometals. 2009;22:23–32. doi: 10.1007/s10534-008-9202-3. [DOI] [PubMed] [Google Scholar]
- 17.Lawlor MS, O’Connor C, Miller VL. Yersiniabactin is a virulence factor for Klebsiella pneumoniae during pulmonary infection. Infect Immun. 2007;75:1463–1472. doi: 10.1128/IAI.00372-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takase H, Nitanai H, Hoshino K, Otani T. Impact of siderophore production on Pseudomonas aeruginosa infections in immunosuppressed mice. Infect Immun. 2000;68:1834–1839. doi: 10.1128/iai.68.4.1834-1839.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fischbach MA, Lin H, Zhou L, Yu Y, Abergel RJ, Liu DR, Raymond KN, Wanner BL, Strong RK, Walsh CT, Aderem A, Smith KD. The pathogen-associated iroA gene cluster mediates bacterial evasion of lipocalin 2. Proc Natl Acad Sci USA. 2006;103:16502–16507. doi: 10.1073/pnas.0604636103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cendrowski S, MacArthur W, Hanna P. Bacillus anthracis requires siderophore biosynthesis for growth in macrophages and mouse virulence. Mol Microbiol. 2004;51:407–417. doi: 10.1046/j.1365-2958.2003.03861.x. [DOI] [PubMed] [Google Scholar]
- 21.Dale SE, Doherty-Kirby A, Lajoie G, Heinrichs DE. Role of siderophore biosynthesis in virulence of Staphylococcus aureus: identification and characterization of genes involved in production of a siderophore. Infect Immun. 2004;72:29–37. doi: 10.1128/IAI.72.1.29-37.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Voss JJ, Rutter K, Schroeder BG, Su H, Zhu Y, Barry CE., 3rd The salicylate-derived mycobactin siderophores of Mycobacterium tuberculosis are essential for growth in macrophages. Proc Natl Acad Sci USA. 2000;97:1252–1257. doi: 10.1073/pnas.97.3.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. A common mechanism of cellular death induced by bactericidal antibiotics. Cell. 2007;130:797–810. doi: 10.1016/j.cell.2007.06.049. [DOI] [PubMed] [Google Scholar]
- 24.Dwyer DJ, Kohanski MA, Collins JJ. Role of reactive oxygen species in antibiotic action and resistance. Curr Opin Microbiol. 2009;12:482–489. doi: 10.1016/j.mib.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamamoto S, Okujo N, Sakakibara Y. Isolation and structure elucidation of acinetobactin, a novel siderophore from Acinetobacter baumannii. Arch Microbiol. 1994;162:249–254. doi: 10.1007/BF00301846. [DOI] [PubMed] [Google Scholar]
- 26.Takeuchi Y, Ozaki S, Satoh M, Mimura K, Hara S, Abe H, Nishioka H, Harayama T. Synthesis of acinetobactin. Chem Pharm Bull (Tokyo) 2010;58:1552– 1553. doi: 10.1248/cpb.58.1552. [DOI] [PubMed] [Google Scholar]
- 27.Dorsey CW, Tomaras AP, Connerly PL, Tolmasky ME, Crosa JH, Actis LA. The siderophore-mediated iron acquisition systems of Acinetobacter baumannii ATCC 19606 and Vibrio anguillarum 775 are structurally and functionally related. Microbiology. 2004;150:3657–3667. doi: 10.1099/mic.0.27371-0. [DOI] [PubMed] [Google Scholar]
- 28.Mihara K, Tanabe T, Yamakawa Y, Funahashi T, Nakao H, Narimatsu S, Yamamoto S. Identification and transcriptional organization of a gene cluster involved in biosynthesis and transport of acinetobactin, a siderophore produced by Acinetobacter baumannii ATCC 19606T. Microbiology. 2004;150:2587–2597. doi: 10.1099/mic.0.27141-0. [DOI] [PubMed] [Google Scholar]
- 29.Sattely ES, Walsh CT. A latent oxazoline electrophile for N-O-C bond formation in pseudomonine biosynthesis. J Am Chem Soc. 2008;130:12282–12284. doi: 10.1021/ja804499r. [DOI] [PubMed] [Google Scholar]
- 30.Wuest WM, Sattely ES, Walsh CT. Three siderophores from one bacterial enzymatic assembly line. J Am Chem Soc. 2009;131:5056–5057. doi: 10.1021/ja900815w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Drake EJ, Duckworth BP, Neres J, Aldrich CC, Gulick AM. Biochemical and structural characterization of bisubstrate inhibitors of BasE, the self-standing nonribosomal peptide synthetase adenylate-forming enzyme of acinetobactin synthesis. Biochemistry. 2010;49:9292–9305. doi: 10.1021/bi101226n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hurdle JG, O’Neill AJ, Chopra I. Prospects for aminoacyl-tRNA synthetase inhibitors as new antimicrobial agents. Antimicrob Agents Chemother. 2005;49:4821– 4833. doi: 10.1128/AAC.49.12.4821-4833.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sikora AL, Wilson DJ, Aldrich CC, Blanchard JS. Kinetic and inhibition studies of dihydroxybenzoate-AMP ligase from Escherichia coli. Biochemistry. 2010;49:3648–3657. doi: 10.1021/bi100350c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gulick AM. Conformational dynamics in the Acyl-CoA synthetases, adenylation domains of non-ribosomal peptide synthetases, and firefly luciferase. ACS Chem Biol. 2009;4:811–827. doi: 10.1021/cb900156h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ferreras JA, Ryu JS, Di Lello F, Tan DS, Quadri LE. Small-molecule inhibition of siderophore biosynthesis in Mycobacterium tuberculosis and Yersinia pestis. Nat Chem Biol. 2005;1:29–32. doi: 10.1038/nchembio706. [DOI] [PubMed] [Google Scholar]
- 36.Somu RV, Boshoff H, Qiao CH, Bennett EM, Barry CE, Aldrich CC. Rationally designed nucleoside antibiotics that inhibit siderophore biosynthesis of Mycobacterium tuberculosis. J Med Chem. 2006;49:31–34. doi: 10.1021/jm051060o. [DOI] [PubMed] [Google Scholar]
- 37.Miethke M, Bisseret P, Beckering CL, Vignard D, Eustache J, Marahiel MA. Inhibition of aryl acid adenylation domains involved in bacterial siderophore synthesis. FEBS J. 2006;273:409–419. doi: 10.1111/j.1742-4658.2005.05077.x. [DOI] [PubMed] [Google Scholar]
- 38.Somu RV, Wilson DJ, Bennett EM, Boshoff HI, Celia L, Beck BJ, Barry CE, Aldrich CC. Antitubercular nucleosides that inhibit siderophore biosynthesis: SAR of the glycosyl domain. J Med Chem. 2006;49:7623–7635. doi: 10.1021/jm061068d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neres J, Wilson DJ, Celia L, Beck BJ, Aldrich CC. Aryl acid adenylating enzymes involved in siderophore biosynthesis: Fluorescence polarization assay, ligand specificity, and discovery of non-nucleoside inhibitors via high-throughput screening. Biochemistry. 2008;47:11735–11749. doi: 10.1021/bi801625b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hohn H. Ein neues Verfahren zur Darstellung von 5-Amino-pyrazolen. Z Chem. 1970;10:386–388. [Google Scholar]
- 41.Dorn H, Müller T. Zur direkten (C-4)-Substitution von Amino-pyrazolen mit β-Keto-carbonyl-Verbindungen — 1-Benzyl-6, 7-dihydro-4-ethoxycarbonyl-6-oxo-pyrazolo[3,4-b]pyridin. Z Chem. 1980;20:95. [Google Scholar]
- 42.Kang FA, Sui ZH, Murray WV. Pd-catalyzed direct arylation of tautomerizable heterocycles with aryl boronic acids via C-OH bond activation using phosphonium salts. J Am Chem Soc. 2008;130:11300–11302. doi: 10.1021/ja804804p. [DOI] [PubMed] [Google Scholar]
- 43.Wan ZK, Wacharasindhu S, Levins CG, Lin M, Tabei K, Mansour TS. The scope and mechanism of phosphonium-mediated S(N)Ar reactions in heterocyclic amides and ureas. J Org Chem. 2007;72:10194–10210. doi: 10.1021/jo7020373. [DOI] [PubMed] [Google Scholar]
- 44.Gupte A, Boshoff HI, Wilson DJ, Neres J, Labello NP, Somu RV, Xing C, Barry CE, Aldrich CC. Inhibition of siderophore biosynthesis by 2-triazole substituted analogues of 5′-O-[N-(salicyl)sulfamoyl]adenosine: antibacterial nucleosides effective against Mycobacterium tuberculosis. J Med Chem. 2008;51:7495–7507. doi: 10.1021/jm8008037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shi C, Aldrich CC. Efficient Pd-catalyzed coupling of tautomerizable heterocycles with terminal alkynes via C-OH bond activation using PyBroP. Org Lett. 2010;12:2286–2289. doi: 10.1021/ol100657n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roehrl MHA, Wang JY, Wagner G. A general framework for development and data analysis of competitive high-throughput screens for small-molecule inhibitors of protein - Protein interactions by fluorescence polarization. Biochemistry. 2004;43:16056–16066. doi: 10.1021/bi048233g. [DOI] [PubMed] [Google Scholar]
- 47.Stachelhaus T, Mootz HD, Marahiel MA. The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chem Biol. 1999;6:493–505. doi: 10.1016/S1074-5521(99)80082-9. [DOI] [PubMed] [Google Scholar]
- 48.Auld DS, Lovell S, Thorne N, Lea WA, Maloney DJ, Shen M, Rai G, Battaile KP, Thomas CJ, Simeonov A, Hanzlik RP, Inglese J. Molecular basis for the high-affinity binding and stabilization of firefly luciferase by PTC124. Proc Natl Acad Sci USA. 2010;107:4878–4883. doi: 10.1073/pnas.0909141107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee TV, Johnson LJ, Johnson RD, Koulman A, Lane GA, Lott JS, Arcus VL. Structure of a eukaryotic nonribosomal peptide synthetase adenylation domain that activates a large hydroxamate amino acid in siderophore biosynthesis. J Biol Chem. 2010;285:2415–2427. doi: 10.1074/jbc.M109.071324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gordon NC, Wareham DW. Multidrug-resistant Acinetobacter baumannii: mechanisms of virulence and resistance. Int J Antimicrob Agents. 2010;35:219–226. doi: 10.1016/j.ijantimicag.2009.10.024. [DOI] [PubMed] [Google Scholar]
- 51.Quadri LE. Strategic paradigm shifts in the antimicrobial drug discovery process of the 21st century. Infect Disord Drug Targets. 2007;7:230–237. doi: 10.2174/187152607782110040. [DOI] [PubMed] [Google Scholar]
- 52.Frederick RE, Mayfield JA, DuBois JL. Iron trafficking as an antimicrobial target. Biometals. 2009;22:583–593. doi: 10.1007/s10534-009-9236-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ballouche M, Cornelis P, Baysse C. Iron metabolism: a promising target for antibacterial strategies. Recent Pat Antiinfect Drug Discov. 2009;4:190–205. doi: 10.2174/157489109789318514. [DOI] [PubMed] [Google Scholar]
- 54.Cescau S, Cwerman H, Letoffe S, Delepelaire P, Wandersman C, Biville F. Heme acquisition by hemophores. Biometals. 2007;20:603–613. doi: 10.1007/s10534-006-9050-y. [DOI] [PubMed] [Google Scholar]
- 55.Budzikiewicz H. Microbial Siderophores. Vol. 92. Spinger-Verlag; Wien: 2010. p. 75. [DOI] [PubMed] [Google Scholar]
- 56.Fischbach MA, Walsh CT. Antibiotics for emerging pathogens. Science. 2009;325:1089–1093. doi: 10.1126/science.1176667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Adler C, Corbalan NS, Seyedsayamdost MR, Pomares MF, de Cristobal RE, Clardy J, Kolter R, Vincent PA. Catecholate siderophores protect bacteria from pyochelin toxicity. PLoS One. 2012;7:e46754. doi: 10.1371/journal.pone.0046754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dorsey CW, Tolmasky ME, Crosa JH, Actis LA. Genetic organization of an Acinetobacter baumannii chromosomal region harbouring genes related to siderophore biosynthesis and transport. Microbiology. 2003;149:1227–1238. doi: 10.1099/mic.0.26204-0. [DOI] [PubMed] [Google Scholar]
- 59.Penwell WF, Arivett BA, Actis LA. The Acinetobacter baumannii entA gene located outside the acinetobactin cluster is critical for siderophore production, iron acquisition and virulence. PLoS One. 2012;7:e36493. doi: 10.1371/journal.pone.0036493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gaddy JA, Arivett BA, McConnell MJ, Lopez-Rojas R, Pachon J, Actis LA. Role of acinetobactin-mediated iron acquisition functions in the interaction of Acinetobacter baumannii strain ATCC 19606T with human lung epithelial cells, Galleria mellonella caterpillars, and mice. Infect Immun. 2012;80:1015–1024. doi: 10.1128/IAI.06279-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Eijkelkamp BA, Hassan KA, Paulsen IT, Brown MH. Investigation of the human pathogen Acinetobacter baumannii under iron limiting conditions. BMC Genomics. 2011;12:126. doi: 10.1186/1471-2164-12-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Antunes LC, Imperi F, Towner KJ, Visca P. Genome-assisted identification of putative iron-utilization genes in Acinetobacter baumannii and their distribution among a genotypically diverse collection of clinical isolates. Res Microbiol. 2011;162:279–284. doi: 10.1016/j.resmic.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 63.Misra RN, Rawlins DB, Xiao HY, Shan WF, Bursuker I, Kellar KA, Mulheron JG, Sack JS, Tokarski JS, Kimball SD, Webster KR. 1H-pyrazolo[3,4-b]pyridine inhibitors of cyclin-dependent kinases. Bioorg Med Chem Lett. 2003;13:1133–1136. doi: 10.1016/s0960-894x(03)00034-9. [DOI] [PubMed] [Google Scholar]
- 64.Higgins PG, Poirel L, Lehmann M, Nordmann P, Seifert H. OXA-143, a novel carbapenem-hydrolyzing class D beta-lactamase in Acinetobacter baumannii. Antimicrob Agents Chemother. 2009;53:5035–5038. doi: 10.1128/AAC.00856-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Choi KH, Kumar A, Schweizer HP. A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. J Microbiol Methods. 2006;64:391–397. doi: 10.1016/j.mimet.2005.06.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.