

Abstract
Cadmium (Cd) is a widely dispersed environmental agent that causes oxidative toxicity through mechanisms that are sensitive to thioredoxin-1 (Trx1). Trx1 is a cytoplasmic protein that translocates to nuclei during oxidative stress. Recent research shows that interaction of Trx1 with actin plays a critical role in cell survival and that increased nuclear Trx-1 potentiates proinflammatory signaling and death in cell and mouse models. These observations indicate that oxidative toxicity caused by low-dose Cd could involve disruption of actin-Trx1 interaction, nuclear Trx1 translocation, and potentiation of proinflammatory cell death mechanisms. In this study, we investigated the role of nuclei-localized Trx1 in Cd-induced inflammation and cytotoxicity using in vitro and in vivo models. The results show that Cd stimulated nuclear translocation of Trx1 and p65 of NF-κB. Elevation of Trx1 in nuclei in in vitro cells and kidney of transgenic mice potentiated Cd-stimulated NF-κB activation and cell death. Cd-stimulated Trx1 nuclear translocation and NF-κB activation were inhibited by cytochalasin D, an inhibitor of actin polymerization, suggesting that actin regulates Trx1 nuclear translocation and NF-κB activation by Cd. A nuclear-targeted dominant negative form of Trx1 blocked Cd-stimulated NF-κB activation and decreased cell death. Addition of zinc, known to antagonize Cd toxicity by increasing metallothionein, had no effect on Cd-stimulated nuclear translocation of Trx1 and NF-κB activation. Taken together, the results show that nuclear translocation and accumulation of redox-active Trx1 in nuclei play an important role in Cd-induced inflammation and cell death.
Key Words: actin, compartmental redox regulation, environmental toxicant, NF-κB, nuclear redox state, thiol/disulfide.
Concern about environmental cadmium (Cd) has increased due to the recognition that Cd is present in food (daily adult intake of Cd; 30 µg/day) and tobacco products (U.S. Department of Health and Human Services, 2011) and accumulates in humans because Cd is not effectively excreted (Waalkes, 2003). Metallothionein (MT), a protein central to zinc homeostasis, provides an important protective system against Cd toxicity (Klaassen et al., 1999). Cd promotes nuclear translocation and activation of the metal response element-binding transcription factor 1, which in turn stimulates MT gene expression (Smirnova et al., 2000). MT genes are readily induced by various physiologic and pathologic stimuli and protect against toxicity by sequestering Cd in protein-bound forms (Klaassen et al., 1999). Increased MT levels are associated with Cd exposure/accumulation and toxicity such as cell death (Gurel et al., 2007).
Cd has multiple roles in cell pathophysiology, including activation of proinflammatory signaling, cell death signaling, estrogen-receptor signaling, and carcinogenesis. For instance, Cd in low concentration triggered proliferation in mice lung cells and caused severe inflammation prior to pathologic outcomes (Kundu et al., 2009). Cd activates human estrogen receptors (Stoica et al., 2000) through an interaction with the hormone-binding domain of the receptor. Although Cd results in stimulation of MAP kinase phosphorylation and DNA synthesis associated with cell proliferation (Zang et al., 2009), it can also cause tissue damage by stimulating apoptotic and necrotic cell death (Templeton and Liu, 2010). In addition, Cd is classified as a human carcinogen, affecting renal, lung, liver, prostate, hematopoietic, and other systems (Nordberg et al., 1992; U.S. Department of Health and Human Services, 2011). However, mechanistic studies indicate that some carcinogenic mechanisms of Cd may not be oxidative in nature (Qu et al., 2005). Thus, despite continuing research advances to understand molecular mechanisms for Cd-induced cell toxicity, details remain to be fully elucidated.
Thioredoxin-1 (Trx1) and mitochondrial thioredoxin-2 (Trx2) are major cellular oxidoreductases regulating cellular redox homeostasis (Holmgren, 1985) and are sensitive to oxidation by Cd (Hansen et al., 2006). Each system has an associated Trx reductase supported by NADPH as reductant and functions in defense against oxidative stress through peroxiredoxins, enzymes that reduce H2O2 and other peroxides. Trx1 is localized mostly in cytoplasm and translocates into nuclei upon stress signals, for example, H2O2, NO, UV, and viral infection (Arai et al., 2006; Go et al., 2011; Schroeder et al., 2007).
Trx1 in different subcellular locations functions differently in controlling oxidative stress signaling. For instance, during cell stress induced by nutrient deprivation, proinflammatory signals, oxidants, or reactive electrophiles, the nuclear Trx1 is more resistant to oxidation than cytoplasmic Trx1 or mitochondrial Trx2 (Go and Jones, 2010; Go et al., 2007). Accordingly, translocation of Trx1 into nuclei appears to be a critical factor to regulate nuclear redox state. In nuclei, Trx1 stimulates activation of numerous transcription factors, including NF-κB (Hirota et al., 1999; Toledano and Leonard, 1991), through regulation of redox sensitive cysteine(s) in the DNA-binding region. In the nuclei, excessive oxidant production oxidizes a critical cysteine residue in the DNA binding region of NF-κB and inhibits DNA binding (Toledano and Leonard, 1991). Increased nuclear Trx1 enhances NF-κB activity (Go et al., 2011; Hirota et al., 1999), suggesting that the nuclear activation by Trx1 counters an endogenous H2O2-dependent transcriptional termination mechanism.
Modulation of NF-κB signaling by nuclear Trx1 raises the possibility that low-dose Cd could cause a proinflammatory response with hyper-responsive immune signaling due to excessive nuclear Trx1. Indeed, our previous study using a transgenic mouse model (nuclear localization signal [NLS]-Trx1 Tg), in which nuclear Trx1 was increased by expression of a fusion protein containing human Trx1 with NLS, showed that nuclear Trx1 controls NF-κB activity in vivo. NLS-Trx1 Tg mice with H1N1 influenza infection had greater inflammatory responses, including elevated NF-κB activity, IL-6 and TNF-α induction, and increased death (Go et al., 2011).
The aim of this study was to test the hypothesis that a low concentration of Cd stimulates nuclear translocation of Trx1 and contributes to toxicity by increasing proinflammatory signaling. Studies were performed using molecular and cellular methods in HeLa cells, a cell line previously well characterized with regard to sensitivity to Cd toxicity, and in NLS-Trx1 Tg mice, a mouse line that shows enhanced sensitivity to proinflammatory signaling. Results show stimulation of nuclear Trx1 translocation by low-dose Cd and increased NF-κB activation. Accumulated nuclear Trx1 potentiated cell death. Cd-stimulated nuclear translocation of Trx1 and NF-κB activation was blocked by inhibition of actin polymerization.
MATERIALS AND METHODS
Cell culture, transfection, plasmids, luciferase assay, and Cd treatment.
HeLa and NRK-52 cells were obtained from American Tissue Culture Collection (Rockville, MD) and were cultured in Dulbecco’s modified eagle medium with 10% fetal bovine serum (FBS) supplemented with antibiotics (penicillin/streptomycin). Cells were maintained in a humidified incubator at 5% CO2 at 37°C. HeLa cells transfected with the plasmids using Lipofectamine (Invitrogen) were maintained in a growth medium and transfected cells were treated with Cd (0.5, 1.0µM as Cd Cl2) in a 2% FBS media. The plasmids used in this study are as follows: Vector control (VC, pCMV mammalian expression vector, Clontech Laboratory, Mountain View, CA), NLS-Trx1 wild type (WT, myc epitope tag; Go et al., 2011), NLS-Trx1-dominant negative mutant (DN, Cys 35 mutation to Ser 35), nuclear exporting signal (NES)-Trx1 (hemagglutinin (HA) epitope tag at the N-terminus and NES at the C-terminus), mitochondrial Trx2 (Go et al., 2010), pNF-κB-Luc (Clontech), and lacZ (Invitrogen). For NF-κB activity measurement, cells were cotransfected with pcDNA3.1, NLS-wt Trx1, or NLS-dn Trx1, NES-Trx1, Trx2 and with pNF-κB-Luc plus pcDNA3.1 containing lacZ gene (Invitrogen). Two days after transfection, cells exposed to Cd were examined for luciferase activity. Measured luciferase activity was normalized for transfection efficiency by β-galactosidase activity. To initiate luciferase activity, cell lysates (20 µl) were added to 100 µl of reaction buffer (Promega, Madison, WI), and luminescence was recorded at 30°C using a luminometer. β-Galactosidase activity was quantified by monitoring cleavage of o-nitrophenyl-β-d-galactopyranoside. All plasmids were prepared using an endotoxin-free maxiprep kit following the manufacturer’s instruction (Qiagen Inc, Valencia, CA). CdCl2, Zn acetate, and cytochalasin D were purchased from Sigma-Aldrich.
Glutathione (GSH) and cysteine (Cys) redox measurement.
HeLa cells after Cd treatment or with no treatment were analyzed for GSH, glutathione disulfide (GSSG), Cys, and cystine (CySS) by high-performance liquid chromatography (HPLC) with fluorescence detection (Jones and Liang, 2009). Values were used to calculate the steady-state redox potential for cellular GSH/GSSG (EhGSSG) and extracellular Cys/CySS (EhCySS) using the measured concentrations, the Nernst equation, and respective EoGSSG (−240 mV, pH 7.0) and EoCySS (−250 mV, pH 7.4) (Jones and Liang, 2009).
Determination of gene expression levels by quantitative real-time PCR.
Total mRNA was isolated from cells treated with none or Cd (0.5, 1.0µM) for 18h using RNeasy mini kit (Qiagen) following the manufacturer’s protocol, and reverse transcription was performed to generate cDNAs (Clontech). For quantitative real-time (qRT) PCR, amplification was performed in triplicate on an iCycler IQ Multicolor RT-PCR Detection System (Bio-Rad Laboratories, Hercules, CA) for 30 cycles as follows: 95°C for 30 s, 58°C for 30 s, and 72°C for 1min. Quantification and melting curves were analyzed with iCycler software. Primers for Bax and MT were designed using a program provided by Integrated DNA Technologies (Coralville, IA). Details of PCR primer sequences for Bax and MT used in the analyses of extracted RNA for qRT-PCR are as follows: human Bax, forward: 5′-atgttttctgacggcaacttc-3′, reverse: 5′-atc agt tcc ggc acct tg-3′; mouse Bax, forward: 5′-gtg agc ggc tgc ttg tct-3′, reverse: 5′-ggt ccc gaa gta gga gag ga-3′; human MT1, forward: 5′-GCA CCT CCT GCA AGA AAA GCT-3′, reverse: 5′-GCA GCT GCA CTT CTC TGA TGC-3′; mouse MT1, forward: 5′-ATG GAC CCC AAC TGC TCC TGC TCC ACC-3′, reverse: 5′-GGC ACA GCA CGT GCA CTT GTC CGC-3′; mouse MT2, forward: 5′-ATG GAC CCC AAC TGC TCC TGT GCC-3′, reverse: 5′-GCT GCA CTT GTC GGA AGC CTC TTT-3′.
Cd uptake assay.
HeLa cells cultured in 96-well and 6-well plates were treated with Cd (0, 0.5µM) to measure the amounts of Cd left in the media or in cells, respectively. Four hours after Cd treatment, 10 µl of medium was analyzed for Cd using the Measure-iT assay kit following procedures provided by the manufacturer (Molecular Probes, Eugene, OR). Differences in Cd levels in medium were quantified according to fluorescence levels of Cd standards in triplicates. To test for increase in intracellular Cd, HeLa cells treated with Cd or none were analyzed using change in fluorescence (excitation/emission; 490/520nm) due to binding of Cd to Leadmium Green dye in cells preloaded with Leadmium Green AM according to manufacturer’s instructions (Molecular Probes). For each assay, a group of cells treated with cytochalasin D (Cyt D) (0.1µM) 30min prior to Cd exposure were also analyzed for extracellular and intracellular Cd detection as described above.
Subcellular fractionation, Western blotting, and electrophoretic mobility shift assay.
To examine nuclear translocation of Trx1 and p65 NF-κB, and DNA-binding activity of NF-κB by Cd, subcellular fractionation of cells (1–2 × 107) and kidneys (45–55mg) exposed to Cd were isolated using a nuclear and cytoplasmic extraction kit (Thermo Scientific, Rockford, IL) following the procedures provided by the manufacturer. Isolated fractions were then confirmed by Western blotting probed with antibodies (ab) against β-actin and lamin for cytoplasm and nuclei, respectively. Trx1 and p65 NF-κB levels in these fractions were determined by Western blotting probed with antibody against Trx1 (AbFrontier, Seoul, Korea) and p65 (Cell Signaling Technology, Boston, MA). Expression levels for NLS-wt Trx1 (myc epitope tag) and NLS-dn Trx1 (myc epitope tag) were probed with ab specific to Myc (Cell Signaling Technology), whereas expression levels for NES-Trx1 and Trx2 were probed with ab specific to HA and V5, respectively. For Western blot analysis, Alexa Fluor 680-conjugated antirabbit or antimouse secondary antibody (Invitrogen) was used and a band corresponding to each protein was visualized using an Odyssey scanner and Odyssey 2.1 software (Li-Cor, Lincoln, NE). Additionally, DNA-binding activity of NF-κB was examined by electrophoretic mobility shift assay (EMSA). Nuclear extracts prepared from cells exposed to Cd were analyzed using an EMSA kit (Affymetrics, Clara, CA) by incubating a biotin-labeled or -unlabeled probe containing an NF-κB DNA-binding consensus sequence (5′-AGTTGAGGGGACTTTCCCAGGC-3′) with a nuclear extract (5 µg) without added reductant for 30min at 15°C. The samples were analyzed on a 6% nondenaturing polyacrylamide gel electrophoresis and electroblotted for 30min. Signals were detected by chemiluminescence imaging according to the manufacturer’s protocol. DNA-binding activity of NF-κB was also examined in nuclear extracts of mouse kidney by EMSA following the procedures as described above.
Subcellular localization of Trx by immunocytochemistry and fluorescence microscopy.
Two days after transfection, cells were washed, fixed, and stained with anti-Myc (NLS-Trx1), HA (NES-Trx1), and V5 (Trx2) ab followed by Cy3-conjugated goat antirabbit ab (Jackson Immuno Research, West Grove, PA) to visualize subcellular localization of ectopic Trx. Alexa Fluor 488 phalloidin was used to stain actin. Nuclear expressions of NLS-wt Trx1 and NLS-dn Trx1 were confirmed individually using anti-myc ab. Fluorescein isothiocyanate (FITC, Invitrogen, Molecular Probes) conjugated and Cy3-conjugated goat antirabbit ab (Jackson Immuno Research) were used for wt Trx1 and dn Trx1, respectively. Immunofluorescence was visualized using an Olympus X-70 fluorescence microscope system.
Cell survival/death by WST-1 assay.
Cell survival and death were quantified by measuring absorbance of the dye product from the nonradioactive quantitative reagent WST-1 (Roche, Basel, Switzerland) and also examined by visualizing images using an Olympus X-70 fluorescence microscope system under bright field.
NLS-Trx1 transgenic mouse and Cd exposure.
All animal experiments and husbandry for the studies presented were conducted under the review and approval of the Emory University Institutional Animal Care and Use Committee (IACUC, approval ID: DAR-2000040-062113). Mice were maintained in the Emory University Division of Animal Resources Facility. C57BL/6 (WT) mice were purchased from Charles River Laboratories and used to maintain the transgenic mouse colony, with WT mice in experiments being littermates of transgenic mice (Tg) expressing the human Trx1 (hTrx1) gene. The hTrx1 was modified to contain c-Myc epitope tag at the N-terminus and NLS at the C-terminus (designated NLS-Trx1 Tg) as described (Go et al., 2011). The presence of the NLS-Trx1 transgene was confirmed by PCR analysis using mouse genomic DNA prepared from tail biopsy as a template and the following oligonucleotide primers: forward primer, 5′-ATGGCATCAATGCAGAAGCTGATCT-3′; and reverse primer, 5′-GCCGCTGGATCTTCTACCTTTCTCT-3′ (Integrated DNA Technologies). WT and NLS-Trx1 transgenic mice (8–10 weeks, six mice per group) were used for ip injection of Cd (10mg/kg) or saline (Bartosiewicz et al., 2001) and euthanized at 6h post injection to collect kidney samples. This treatment has been used previously to characterize effects of Cd on gene expression (Garrett et al., 2011); results show significant effects on stress genes (MT1/MT2, heat shock proteins, c-jun, and jun-b) and xenobiotic metabolizing enzymes (cyp2F2, methyltransferase, and acetyltransferase). To address whether this high acute dose was cytotoxic to kidney, we examined plasma and urinary creatinine levels and renal GSH/GSSG redox state. Results showed no significant differences from control mice, suggesting that this dose does not have a severe effect on kidney function during this exposure period (Supplementary data). Additional histological examination of kidney showed no observable difference in morphology or size of glomeruli present in outer cortex region of kidney by Cd compared with those in control group (data not shown).
RESULTS
Nuclear Translocation of Trx-1 and p65 NF-κB by Low-Dose Cd
Based upon previous research showing Cd-induced activation of apoptotic cell death (Oh et al., 2004), we examined whether low-dose Cd increased the proapoptotic molecule, Bax. Results show that mRNA levels of Bax determined by RT-PCR were enhanced by Cd exposure (0µM Cd 0, 5.0±0.2 pmol; 0.5µM Cd, 6.0±0.2 pmol; 1.0µM Cd, 6.7±0.4 pmol; Fig. 1A). MTs are a family of stress responsive proteins that play an important role in the detoxification of Cd (Lu et al., 2001) and are useful as a biomarker of Cd exposure. MT1 expression was measured by RT-PCR and showed substantial elevation of mRNA levels for MT1 by Cd (0.5µM, 24.4±2.2 pmol; 1.0µM, 58.3±8.5 pmol; Fig 1A). Under these conditions, Cd had no detectable effect on cellular GSH or cysteine redox couples (Table 1). Together, the increase in mRNA for Bax and MT1 shows that HeLa cells respond to Cd at doses that do not perturb the major GSH antioxidant system.
FIG. 1.
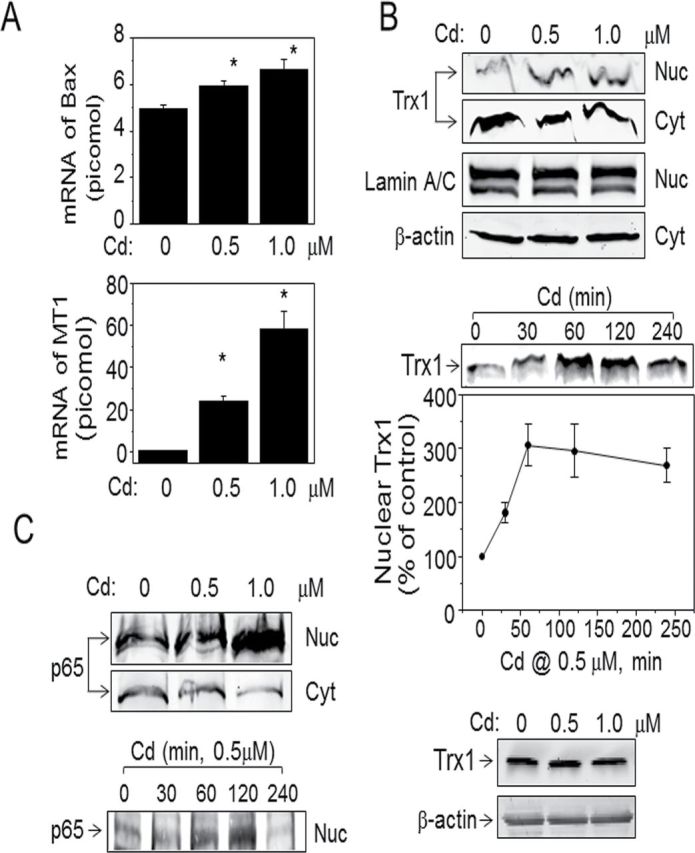
Nuclear translocation of Trx1 and p65 NF-κB by low-dose Cd. Cells treated with Cd as indicated concentration and time were analyzed to examine mRNA levels of Bax and MT1 by qRT-PCR (A). Subcellular localization of Trx1 (B) and p65 NF-κB (C) and the amount of Trx1 protein extracted from whole cells (D) were examined by Western blotting. Experiment was repeated three times. Results are shown as mean ± SE; n = 3. *p < 0.05 vs. control group (Cd 0). Subcellular fractions including cytoplasm and nuclei were analyzed to determine nuclear translocation of Trx1 and p65 NF-κB by Western blotting. Fractionated samples were confirmed by Western blotting probed with lamin and actin as markers to verify purity of nuclear and cytoplasmic fractions.
TABLE 1.
Redox States for Cellular Ehgssg and Extracellular Ehcyss Were Not Affected by Low Levels of Cd Treatment in HeLa Cells )
| Cellular | Extracellular | |||||
|---|---|---|---|---|---|---|
| Cd (µM) | GSH (mM) | GSSG (µM) | EhGSSG (mV) | Cys (µM) | CySS (µM) | EhCySS (mV) |
| 0 | 27±3 | 78±3 | −269±2 | 4.7±1.3 | 8.5±2.6 | −112±3 |
| 0.5 | 30±3 | 70±8 | −274±1 | 4.1±0.3 | 12.8±0.5 | −104±1 |
| 1.0 | 30±2 | 81±3 | −271±2 | 3.7±0.5 | 9.5±3.9 | −106±2 |
Notes. Cells exposed to Cd by indicated amounts were analyzed by HPLC to measure GSH/GSSG and Cys/CySS. Redox states (redox potential, mV) were calculated using the Nernst equation (Jones and Liang, 2009).
Thioredoxins complement antioxidant functions of GSH, providing a parallel and nonredundant system for cellular defense. Trx1 controls cellular redox state in response to a variety of stress signals with distinct functions in different subcellular compartments. Previous studies showed that Trx1 translocates to nuclei from cytoplasm by stress signals (Arai et al., 2006; Go et al., 2011; Schroeder et al., 2007); so, we evaluated whether a Cd-induced stress signal affects Trx1 localization. Results show that Cd (0.5, 1.0µM) increased Trx1 in nuclei (Fig. 1B, top). This increase was associated with a small decrease in cytoplasm, suggesting that Cd at low concentrations stimulates nuclear increase in Trx1 without depletion of cytoplasmic Trx1. β-Actin and lamin A/C of the same samples were examined to verify purity of cytoplasmic and nuclear fractions, respectively (Fig. 1B). Time-course studies (0–4h, 0.5µM) showed that increased Trx1 was observed at 30min (180.9±19.6) after addition of Cd and maximal between 1h (306.8±38.7) and 2h (295.5±48.9) (Fig. 1B, middle). There was no observable change in the amount of Trx1 protein by Cd (Fig. 1B, bottom).
In nuclei, Trx1 stimulates activity of transcription factors, for example, NF-κB, AP-1, p53, and glucocorticoid receptor, by reducing critical cysteine residues required for DNA binding as described above. Previous studies demonstrated that Cd alters activities of oxidative stress–sensitive transcription factors, including NF-κB, a prominent factor regulating cell death/survival balance. Therefore, we examined dose and time-dependent effects of Cd on the p65 subunit of NF-κB. Results showed that 0.5µM Cd stimulated nuclear translocation of p65 maximally by 2h after Cd exposure (Fig. 1C).
Cd-induced cellular toxicity and responses are associated with cellular accumulation of Cd. To evaluate Cd uptake by HeLa cells, we examined the amount of Cd left in medium (Fig. 2A) and accumulation in cells (Fig. 2B) after 4h Cd treatment. Cd (0.5µM) added to the culture medium with HeLa cells was significantly reduced 4h after treatment (Cd 0h, 0.47±0.08µM; Cd 4h, 0.10±0.02µM; Fig 2A), whereas Cd in medium alone showed no change in concentration (Cd 0h, 0.51±0.05µM; Cd 4h, 0.49±0.04µM; Fig 2A). These results suggest that HeLa cells take up about 80% of Cd 4h after exposure. Additionally, we examined Cd level in cells using an intracellular indicator (Fig. 2B). Although Cd levels were not calibrated using this method, cellular fluorescence was increased 1.4-fold with 0.5µM Cd compared with control (no Cd treatment). To determine whether disruption of actin polymerization affected Cd uptake, cells treated with Cyt D prior to Cd exposure (0.5µM, 4h) were also examined. The results showed no significant effect by Cyt D (0.1µM) on Cd uptake by cells as measured in both medium and cells (Figs. 2A and 2B).
FIG. 2.
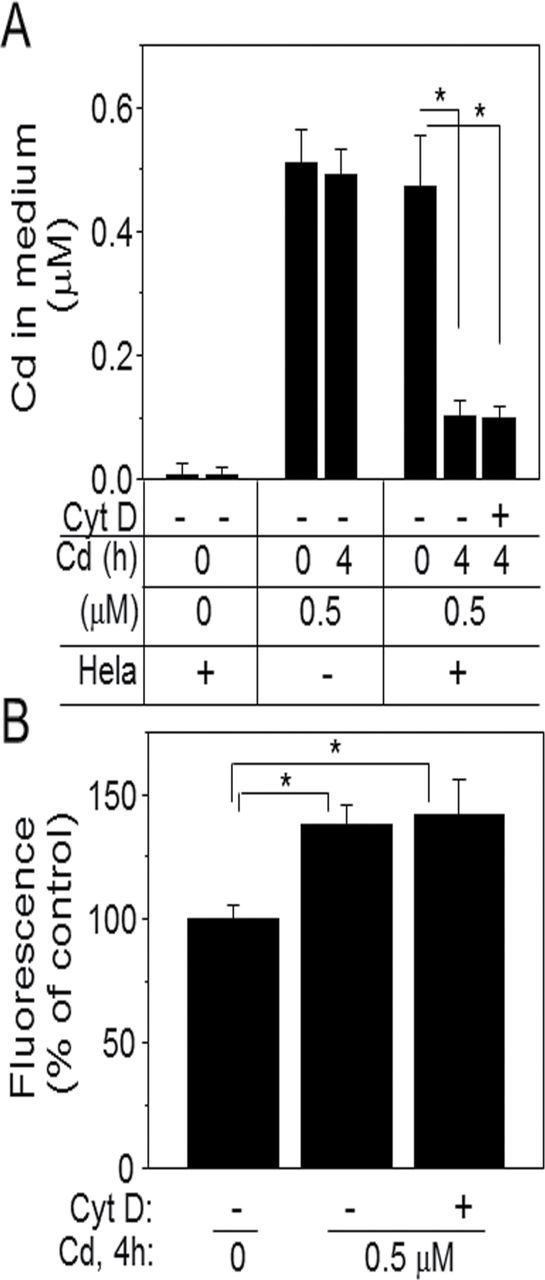
Cd uptake by HeLa cells. Cells cultured in 96-well or 6-well plates treated with Cd as indicated amounts and time were analyzed for Cd measurement by fluorescence plate reader. The amount of Cd left in medium was quantified (A) and increased Cd levels in cells (B) were shown as % of control. In some studies, cells were pretreated with Cyt D for 30min before Cd exposure for 4h to test Cyt D effect on Cd uptake. Data in bar chart represent means ± SE of eight measurements for (A) and three measurements for (B) using three different experiments. *p < 0.05 vs. control group.
Increased Expression of Nuclei-Targeted Trx1 Sensitizes Cells to Death by Low-Dose Cd
To evaluate the effect of increased nuclear Trx1 on cell toxicity, we investigated Cd-induced cell stress/death in cells with increased nuclear Trx1 obtained using transient transfection. Cells expressing Trx1 targeted to nuclei (NLS-Trx1) were examined for Cd-induced cell death/survival. Compared with cells increasing Trx expression in cytoplasm (NES-Trx1) or mitochondria (Trx2), Trx1 elevation in nuclei (NLS-Trx1) potentiated Cd-induced cell death as examined by microscopy (Fig. 3A) and quantified by the WST-1 cell death assay (% of live cells indicated in Figure 3B: none, 100%; vector control, 85.5±9.6; NLS-Trx1, 58.5±7.0; NES-Trx1, 80.9±9.0; Trx2, 88.4±9.1). Transient overexpression of Trx1 in different subcellular locations was confirmed by immunofluorescence examination (see distribution of Cy3-conjugated orange color fluorescence as indication of Trx1 expression in different subcellular compartments; epitope tags are as follows: NLS-Trx1, Myc; NES-Trx1, HA; and Trx2, V5; Fig. 3C).
FIG. 3.

Increased expression of nuclei-targeted Trx1 sensitizes cells to death by low-dose Cd. Two days after transfection, HeLa cells expressed with vector control (VC), nuclei-targeted Trx1 (NLS-Trx1), cytoplasm-targeted Trx1 (NES-Trx1), and mitochondria-targeted Trx2 were examined for Cd (0.5µM, 18h) affected cell survival by microscopy (A) and WST-1 (B). Results are means ± SEM for at least three separate experiments. *p < 0.05 vs. VC. Trx1 localized expression was verified by immunocytochemistry probed with antibodies (ab) against myc (VC and NLS-Trx1), HA (NES-Trx1), and V5 (Trx2) epitopes followed by Cy3-conjugated ab (C). Cy3 orange fluorescence detection indicates localized expression of Trx as expected (C).
Redox Active Trx1 in Nuclei Plays A Role in Cd-Induced Cell Death
To define nuclear Trx1 function in potentiation of Cd-induced cytotoxicity, we examined whether a dominant-negative Trx1 targeted to nuclei affected Cd-induced cell death. Expressions of both WT and dominant negative (mutation of cysteine 35 to serine 35) forms of Trx1 in nuclei were confirmed by immunofluorescence and Western blot analyses (Fig. 4A). Specifically, redox Western blot analysis showed that WT Trx1 (NLS-wt Trx1) maintains redox activity upon dithiothreitol (DTT) and H2O2 treatments for reducing and oxidizing reagents, respectively (Fig. 4A); whereas the dominant negative form binds to other proteins and does not produce typical redox Western blots (Zhang et al., 2007). Using these forms of NLS-Trx1 (wt and dn), we evaluated Cd-induced cell toxicity. Results showed that low-dose Cd caused toxicity in the vector control (viable cells remaining, VC: 0.5µM Cd, 85.0±4.0%; 1.0µM Cd, 61.3±6.4; Figs. 4B and 4C). Consistent with results shown in Figure 3, Cd-induced cell death was potentiated by increased WT Trx1 in nuclei (viable cells remaining, NLS-wt Trx1: 0.5µM Cd, 61.1±5.8%; 1.0µM Cd; 44.3±2.3%; Figs. 4B and 4C), whereas no significant difference was observed by NLS-Trx1 expression without Cd treatment (indicated as −Cd in Fig. C). In contrast, targeting catalytically inactive, dominant negative Trx1 to nuclei did not affect cell death compared with vector controls (viable cells remaining, NLS-dn Trx1: 0.5µM Cd, 87.3±2.9%; 1.0µM Cd, 54.2±3.0%; Figs. 4B and 4C). These results show that the redox activity of nuclear-targeted Trx1 contributes to Cd-induced cell death.
FIG. 4.

Redox active Trx1 in nuclei is critical for Cd-induced cell death. Cells transfected with VC, NLS-wt Trx1, or NLS-dn Trx were exposed to Cd and analyzed for cell survival as described in Figure 2. Nuclei-targeted expressions of both WT and DN Trx1 were verified by immunofluorescence examination (A, top, the primary ab; myc for wt and dn Trx1, HA for NES-Trx1, the secondary ab; FITC-labeled ab for NLS-wt Trx1 and NES-Trx1, Cy3-labeled ab for NLS-dn Trx1). Expressions of WT and DN, and redox responsiveness of WT were examined by Western blotting and redox Western blotting (Go and Jones, 2009; Halvey et al., 2005), respectively. Cell survival affected by Cd was examined by WST-1 assay. Data represent the mean ± SE of eight determinations. *p < 0.05.
Increased Redox-Active Trx1 in Nuclei Potentiates Cd-Stimulated NF-κB Activity
As shown in Fig. 1, Cd stimulated nuclear translocation of p65 subunit of NF-κB concurrently with Trx1 nuclear translocation (Figs. 1B and 1C). We used an NF-κB reporter assay to determine effects of increased nuclear Trx1 on Cd-dependent NF-κB activation. Results showed that Cd increased NF-κB activity and that this was substantially increased by a targeted increase in nuclear Trx1 (Fig. 5). To determine whether redox activity of nuclei-targeted Trx1 is required for Cd-induced cell death, HeLa cells were cotransfected with NLS-dn Trx1 (DN); unlike the stimulation of activity by NLS-wt Trx1, NLS-dn Trx1 showed little activation of NF-κB (0.5µM Cd: VC, 3.6±1.2; NLS-wt Trx1, 33.2±2.5; NLS-dn Trx1, 11.0±1.1; 1.0µM Cd: VC, 4.4±0.4; NLS-wt Trx1, 42.3±2.2; and NLS-dn Trx1, 15.1±1.4; Fig. 5A). We also examined this in NRK 52 cells, a cell line derived from normal rat kidney. Similar effects were found (0.5µM Cd: VC, 1.2±0.2; NLS-wt Trx1, 4.7±0.3; NLS-dn Trx1, 2.3±0.4; 1.0µM Cd: VC, 1.5±0.2; NLS-wt Trx1, 7.2±0.8; and NLS-dn Trx1, 3.2±0.4; Fig. 5B).
FIG. 5.

Increased expression of redox active Trx1 in nuclei potentiated Cd-stimulated NF-κB activation. HeLa cells were cotransfected with plasmids containing NF-κB-luciferase, lacZ, and either empty vector (VC), NLS-wt Trx1, NLS-dn Trx1, NES-Trx1 or Trx2 (A). After 30h, the cells were treated with Cd (0.5, 1.0µM) or none for 18h. Cells were washed with PBS, lysed, and luciferase and β-galactosidase activities were measured. Fold increase was calculated as the ratio of luciferase activity in cells transfected with given plasmids with Cd treatments to that in vector control with none (Cd; 0), in all cases normalized to β-galactosidase activity. (B) NRK 52 cells derived from rat kidney were transfected with indicated plasmid and luciferase activity. Luciferase activity was measured by following procedures described above. Results are shown as mean ± SE; n = 8. *p < 0.05.
Following this further, we examined whether increased thioredoxin in other subcellular compartments stimulated Cd toxicity. Cells transfected with constructs to increase thioredoxin in other subcellular locations (cytoplasm [NES-Trx1] and mitochondria [Trx2]) were examined for Cd-stimulated NF-κB luciferase (NF-κB-Luc) activation (Fig. 5A). Results showed that increased thioredoxin in these compartments did not increase NF-κB activity (0.5µM Cd: VC, 3.6±1.2; NES-Trx1, 3.6±2.3; Trx2, 3.1±2.5; 1.0µM Cd: VC, 4.4±0.4; NES-Trx1, 8.4±0.8; and Trx2, 5.6±2.2). Together, these results show that the redox-dependent potentiation of Cd-induced NF-κB activity specifically depends upon increased redox-active nuclear Trx1.
Actin Structure and Organization but Not Zinc (Zn) Plays A Role in Cd-Stimulated Trx1 Nuclear Translocation and NF-κB Activation
Our results so far show that cell toxicity from low-dose Cd involves Trx1 nuclear translocation and that increased nuclear Trx1 stimulates NF-κB activation. Because Cd toxicity is associated with a broad range of diseases, it is important to identify mechanisms that could potentially block/lessen Cd-induced cell damage. Among numerous studies related to Cd toxicity, the antagonizing role of Zn in Cd toxicity has been well documented (Claverie et al., 2000; Jacquillet et al., 2006). Therefore, we examined the role of Zn in low-dose Cd-stimulated Trx1 nuclear translocation and NF-κB activation. The data show that Zn (1µM) had no effect on Cd-induced Trx1 nuclear translocation and NF-κB activation (Figs. 6A and C). Additionally, Zn itself did not stimulate nuclear translocation of Trx1 compared with that by Cd (Fig. 6A). However, Zn concentration studies were not performed and effects might be observable if higher Zn concentrations were used, that is, Zn/Cd > 2. In contrast, disruption of actin polymerization by cytochalasin D (Cyt D) inhibited Cd-induced nuclear translocation of Trx1 (Fig. 5B, top). Cells preincubated with Cyt D (0.1µM, 30min), the same condition that does not affect Cd uptake by HeLa cells (Fig. 2) also inhibited Cd-stimulated activation of NF-κB as examined by EMSA (0.5µM Cd, 2.8±0.3; 0.5µM Cd + Cyt D, 1.7±0.5). Furthermore, Cyt D significantly blocked nuclear Trx1-potentiated NF-κB activation as examined by the luciferase reporter (Fig. 6C). Taken together, these results indicate that actin-associated cell structure and organization is an essential regulator for Cd-induced cytotoxicity by controlling Trx1 nuclear location and NF-κB activation.
FIG. 6.

Actin structure and organization but not zinc (Zn) plays a role in Cd-stimulated Trx1 nuclear translocation and NF-κB activation. (A) Cells exposed to Cd (0.5µM), Zn (1.0µM), or Cd (0.5µM) plus Zn (1.0µM) for 2h were fractionated to isolate cytoplasmic and nuclear fractions to determine nuclear translocation of Trx1 from cytoplasm by Western blotting. As described in Figure 1, fractions were confirmed by marker proteins, lamin and β-actin for nuclei and cytoplasm, respectively (A). Densitometry was performed to quantitate the nuclear Trx1 band intensity and was expressed as fold increase over control (none). Data in bar charts represent means ± SE; n = 3. (B) cells treated with cytochalasin D (Cyt D, 0.1µM) prior to Cd treatment (0.5µM) were fractionated to isolate nuclei. Nuclear fraction was analyzed to determine Trx1 nuclear translocation by Western blotting (B, top) and DNA-binding activity of NF-κB by EMSA (B, bottom). Bands indicated as NF-κB bound DNA probe show activity of NF-κB. Densitometry values shown as fold difference were obtained from measuring relative intensities compared with that in control without Cd or Cyt D treatment. Lanes 1–5 are as follows: 1, NF-κB probe alone; 2, cells treated with Cd (cold NF-κB probe incubation followed by labeled NF-κB probe incubation); 3, cells with no treatment; 4, cells treated with Cd; 5, cells treated with Cyt D prior to Cd.
Cd-Stimulated Nuclear Localization of Trx1 in Mouse Kidney and Increased Cd-Stimulated NF-κB Activation in NLS-Trx1 Tg Mice
Numerous studies show that chronic and acute exposure to Cd critically affect the kidney (Garrett et al., 2011; Smith et al., 1980). To evaluate the potential in vivo relevance of the cell studies, we examined nuclear function in kidneys of mice 6h after treatment with 10mg/kg Cd. Consistent with the HeLa cell data, Western blot analysis of the nuclear fraction of mouse kidney showed that Cd-stimulated nuclear translocation of Trx1 (Fig. 7A). NLS-Trx1 Tg mice with increased Trx1 in cell nuclei (Go et al., 2011) and littermate WT mice were then used as an in vivo model to test effects of nuclear Trx1 on Cd (10mg/kg)-stimulated NF-κB activation. As shown in Figure 7B, WT mice exposed to Cd had increased NF-κB activity (Fig. 7B WT: Cd 0, 100±11.6%; Cd 10mg/kg, 128.9±5.0%). Consistent with cell data, NLS-Trx1 Tg mouse had greater Cd-induced NF-κB activation than WT littermates (Fig. 7B Tg: Cd 0, 82.6±7.3%; Cd 10mg/kg, 152.5±11.1%). The mRNA levels of MT1 and MT2 in both WT and Tg kidneys were substantially increased by Cd with no observable difference between WT and Tg [Fig. 7C MT1: 1.6±0.04 (WT −Cd), 41.1±5.2 (WT +Cd), 3.5±0.5 (Tg −Cd), 38.5±3.2 (Tg +Cd); MT2: 0.07±0.005 (WT −Cd), 5.1±0.9 (WT +Cd), 0.1±0.09 (Tg −Cd), 6.4±0.5 (Tg +Cd)].
FIG. 7.
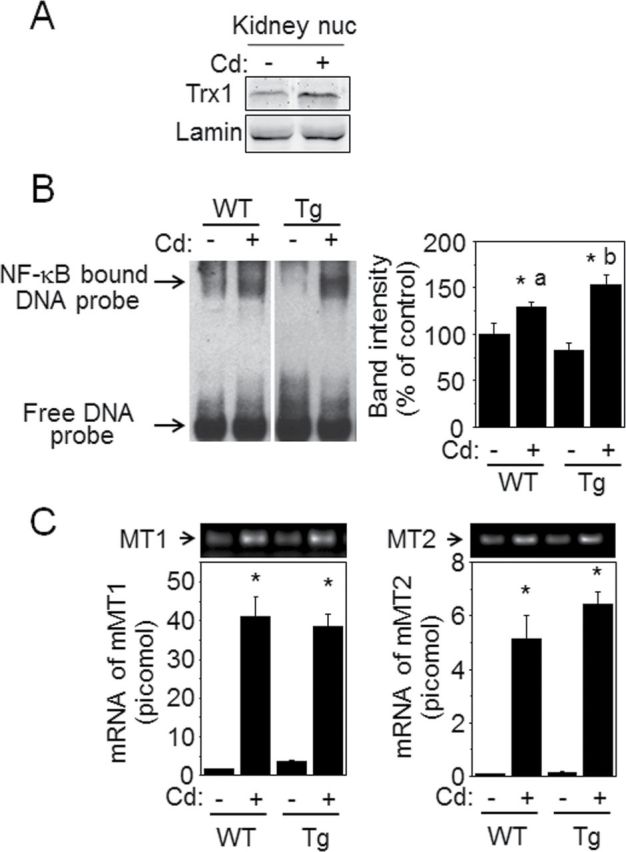
Low level of Cd-induced NF-κB activation in kidney was potentiated by NLS-Trx1 Tg mouse. Kidney tissues obtained from WT and NLS-Trx1 transgenic mice exposed to Cd (10mg/kg) or saline for 6h were used for subcellular fractionation and mRNA isolation. Nuclear fraction was examined for Trx1 nuclear translocation by Western blotting (A) and DNA-binding activity of NF-κB by EMSA (B). (B) Bands indicated as NF-κB-bound DNA probe show activity of NF-κB and intensities of bands were measured as described in Figure 5. Representative results obtained from six mice for each group are shown. *p < 0.05 for WT without Cd. “a” and “b” columns in bar graph (B) show that these are significantly different (p < 0.05). (C) mRNA isolated from kidney tissue was converted to cDNA for qRT-PCR. cDNA was analyzed for MT1 and MT2 by qRT-PCR. Results of mRNA levels for MT1 and MT2 in bar graph are shown as means ± SE. *p < 0.05.
DISCUSSION
Cd affects many cellular functions; however, little is known about the mechanisms responsible for different toxicity and cellular responses among organ systems. In this study, we examined the mechanism of low-dose Cd-induced cell toxicity. Although precise extrapolation to human exposures to Cd is difficult, the low concentrations (0.5, 1.0µM) correspond to minimally toxic or nontoxic amounts that are thought to activate cellular defense mechanisms rather than leading to unrecoverable tissue damage (Gaddipati et al., 2003; Kundu et al., 2009). Cd concentrations (0.5, 1µM) were selected because these levels are low relative to the LC50 for HeLa (32µM; Othumpangat et al., 2005). These low concentrations of Cd activated defense signaling as shown by increased mRNA levels of protective MTs, and GSH was not significantly altered. However, under these conditions, mRNA levels of Bax, a proapoptotic molecule, and cell death were elevated. Although the increases in these toxicity indicators were small, the concomitant activation of protective and toxicity signaling indicates that the altered subcellular localization of Trx1 and increased activation of NF-κB could represent cell defense and/or cell toxicity responses.
Previous research shows that redox states of thioredoxins in different subcellular locations are maintained differently and that the redox sensitivities of thioredoxins to stress signaling also differ depending on its compartmental location. Previous data show that mitochondrial Trx2 normally is more reduced and more sensitive to oxidation than cytoplasmic or nuclear Trx1 (Chen et al., 2006; Hansen et al., 2006). Nuclei also have relatively reduced redox potential but Trx1 in nuclei is resistant to oxidation (Chen et al., 2006; Go et al., 2007; Halvey et al., 2005; Hansen et al., 2006). These observations suggest that disruption of redox-dependent coordination in organelles could be a key mechanism in cellular toxicity.
The present studies show that Cd at low concentrations can disrupt the redox regulatory system in nuclei due to stimulation of nuclear Trx1 translocation. The high transfection efficiency of HeLa cells enabled us to manipulate the abundance of Trx1 in nuclei, compare effects of redox active and inactive forms of Trx1 in nuclei, and compare effects to results obtained by expression of thioredoxins in different subcellular compartments Trx1 (i.e., nuclei, NLS-Trx1; cytoplasm, NES-Trx1; and mitochondria, MLS-Trx2, Trx2). In vivo kidney data using WT and NLS-Trx1 Tg mice exposed to Cd (Fig. 7) supported the findings from HeLa cells showing that Cd increased nuclear Trx1 and that increased nuclear Trx1 contributed to proinflammatory signaling. Such mouse models could be very useful for additional studies of Cd-stimulated pathologic events in other organ systems that are targets for Cd toxicity.
Our study shows that low doses of Cd do not alter redox states of cellular glutathione/glutathione disulfide (GSH/GSSG) or extracellular cysteine/cystine (Cys/CySS), whereas Trx1 translocation to nuclei occurs under these conditions. We infer from these observations that low-level Cd affects the signaling mechanism involving Trx1-associated molecules and their subcellular localization without affecting the GSH system; however, we have not explicitly ruled out redistribution of GSH. Such redistribution, if it occurs, could also be mechanistically important because targeted increases in nuclear H2O2 generation by an NLS-d-amino acid oxidase fusion protein caused increased nuclear protein S-glutathionylation without detectable oxidation of nuclear Trx1 (Halvey et al., 2007). Recent studies establish a critical role for nuclear GSH (Diaz Vivancos et al., 2010), and future studies are needed to test effects of low level Cd on this system.
Results showing that interruption of actin structure affected nuclear translocation of Trx1 supports recent research showing that direct interaction of Trx1 with actin plays a critical role in preventing apoptotic cell death (Zschauer et al., 2011). The present finding that nuclear translocation of Trx1 is dependent upon actin polymerization suggests that the known pleiotropy of low-dose Cd toxicity could occur as a consequence of interactions of compartment-specific redox effects of Cd in the cytoplasm and nucleus with other cell-specific redox-signaling mechanisms, for example, in inflammation, vascular remodeling, or renewal of cell populations, that depend upon the structure and function of the cytoskeleton. Cd-elevated nuclear translocation of Trx1 from cytoplasm is likely a consequential effect after releasing from actin, apparently as an alternate fate to ubiquitination and degradation as shown by Zschauer et al. (2011). Such release could also be associated with stimulation of cell death signaling by ASK-1, as previously shown for Cd (Hansen et al., 2006), and future studies of dose responses in different cell and organ systems will be required to determine relative contributions of these mechanisms at low doses.
Nuclear translocation of Trx1 is closely associated with transcription factor activation because numerous transcription factors function in a redox-sensitive manner. A stimulatory effect of Cd on NF-κB activation has been shown previously at high concentrations of Cd (> 10μM) (Freitas and Fernandes, 2011; Hyun et al., 2007). In this study, we show that Cd-stimulated nuclear translocation of Trx1 is associated with increased activity of NF-κB. Although the experiments do not test whether Cd-induced cell death is due to increased activation of NF-κB, increased activation of NF-κB is likely followed by cellular inflammatory responses such as increased inflammatory genes, cytokines, and adhesion molecules based upon previous studies (Freitas and Fernandes, 2011; Go et al., 2011). Therefore, these data suggest that Cd-stimulated nuclear translocation of Trx1 could be sufficient to affect nuclear transcriptional functions of other redox-sensitive transcription factors, for example, HIF-1α, p53, glucocorticoid receptor, estrogen receptor, and Nrf-2, which are critical for physiology and pathophysiology by controlling cell growth, proliferation, and death. The converse is also potentially important, namely, that increased inflammation could potentiate Cd toxicity. Substantial evidence indicates that chronic low-level inflammation contributes to cardiovascular disease, diabetes, neurodegeneration, and other disease processes; hence, the results suggest a need to evaluate whether low-dose Cd potentiates these disease processes by increasing the nuclear Trx1 redox system.
In summary, the present findings show that disruption of cellular redox control by increasing Trx1 in cell nuclei potentiates low-dose Cd toxicity in HeLa cells. Protective MT was upregulated, and NF-κB activity, proapoptotic Bax and cell death, were increased by low concentrations of Cd without detectable changes in cellular GSH/GSSG or extracellular Cys/CySS redox states. Translocation of Trx1 and p65 NF-κB into nuclei was stimulated by low-dose Cd, and cell death and NF-κB activity were elevated due to increased nuclear Trx1. Thus, the results show that translocation of Trx1 into nuclei is an important event contributing to low-dose Cd-induced toxicities. The improved understanding of the compartmentalized nature of redox signaling in toxicity suggests new avenues to explore toxicologic risks of low levels of environmental exposures and also to explore Trx1-actin interactions and nuclear Trx1 translocation as potential therapeutic targets to prevent or delay adverse effects of Cd.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
National Institute of Environmental Health Sciences (ES011195, ES009047).
Supplementary Material
REFERENCES
- Arai R. J., Masutani H., Yodoi J., Debbas V., Laurindo F. R., Stern A., Monteiro H. P. (2006). Nitric oxide induces thioredoxin-1 nuclear translocation: Possible association with the p21Ras survival pathway. Biochem. Biophys. Res. Commun. 348: 1254–1260. [DOI] [PubMed] [Google Scholar]
- Bartosiewicz M., Penn S., Buckpitt A. (2001). Applications of gene arrays in environmental toxicology: Fingerprints of gene regulation associated with cadmium chloride, benzo(a)pyrene, and trichloroethylene. Environ. Health Perspect. 109: 71–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Cai J., Jones D. P. (2006). Mitochondrial thioredoxin in regulation of oxidant-induced cell death. FEBS Lett. 580: 6596–6602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claverie C., Corbella R., Martín D., Díaz C. (2000). Protective effects of zinc on cadmium toxicity in rodents. Biol. Trace Elem. Res. 75: 1–9. [DOI] [PubMed] [Google Scholar]
- Diaz Vivancos P., Wolff T., Markovic J., Pallardó F. V., Foyer C. H. (2010). A nuclear glutathione cycle within the cell cycle. Biochem. J. 431: 169–178. [DOI] [PubMed] [Google Scholar]
- Freitas M., Fernandes E. (2011). Zinc, cadmium and nickel increase the activation of NF-κB and the release of cytokines from THP-1 monocytic cells. Metallomics 3: 1238–1243. [DOI] [PubMed] [Google Scholar]
- Gaddipati J. P., Rajeshkumar N. V., Grove J. C., Maharaj S. V., Centeno J. A., Maheshwari R. K., Jonas W. B. (2003). Low-dose cadmium exposure reduces human prostate cell transformation in culture and up-regulates metallothionein and MT-1G mRNA. Nonlinearity Biol. Toxicol. Med. 1: 199–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett S. H., Somji S., Sens M. A., Zhang K., Sens D. A. (2011). Microarray analysis of gene expression patterns in human proximal tubule cells over a short and long time course of cadmium exposure. J. Toxicol. Environ. Health Part A 74: 24–42. [DOI] [PubMed] [Google Scholar]
- Go Y. M., Jones D. P. (2009). Thioredoxin redox Western analysis Curr. Protoc. Toxicol. 41: 1–12. [DOI] [PubMed] [Google Scholar]
- Go Y. M., Jones D. P. (2010). Redox control systems in the nucleus: Mechanisms and functions. Antioxid. Redox Signal. 13: 489–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go Y. M., Kang S. M., Roede J. R., Orr M., Jones D. P. (2011). Increased inflammatory signaling and lethality of influenza H1N1 by nuclear thioredoxin-1. PLOS ONE 6: e18918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go Y. M., Park H., Koval M., Orr M., Reed M., Liang Y., Smith D., Pohl J., Jones D. P. (2010). A key role for mitochondria in endothelial signaling by plasma cysteine/cystine redox potential. Free Radic. Biol. Med. 48: 275–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go Y. M., Ziegler T. R., Johnson J. M., Gu L., Hansen J. M., Jones D. P. (2007). Selective protection of nuclear thioredoxin-1 and glutathione redox systems against oxidation during glucose and glutamine deficiency in human colonic epithelial cells. Free Radic. Biol. Med. 42: 363–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurel Z., Ozcelik D., Dursun S. (2007). Apoptotic rate and metallothionein levels in the tissues of cadmium- and copper-exposed rats. Biol. Trace Elem. Res. 116: 203–217. [DOI] [PubMed] [Google Scholar]
- Halvey P. J., Hansen J. M., Johnson J. M., Go Y. M., Samali A., Jones D. P. (2007). Selective oxidative stress in cell nuclei by nuclear-targeted D-amino acid oxidase. Antioxid. Redox Signal. 9: 807–816. [DOI] [PubMed] [Google Scholar]
- Halvey P. J., Watson W. H., Hansen J. M., Go Y. M., Samali A., Jones D. P. (2005). Compartmental oxidation of thiol-disulphide redox couples during epidermal growth factor signalling. Biochem. J. 386(Pt 2):215–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen J. M., Zhang H., Jones D. P. (2006). Differential oxidation of thioredoxin-1, thioredoxin-2, and glutathione by metal ions. Free Radic. Biol. Med. 40: 138–145. [DOI] [PubMed] [Google Scholar]
- Hirota K., Murata M., Sachi Y., Nakamura H., Takeuchi J., Mori K., Yodoi J. (1999). Distinct roles of thioredoxin in the cytoplasm and in the nucleus. A two-step mechanism of redox regulation of transcription factor NF-kappaB. J. Biol. Chem. 274: 27891–27897. [DOI] [PubMed] [Google Scholar]
- Holmgren A. (1985). Thioredoxin. Annu. Rev. Biochem. 54: 237–271. [DOI] [PubMed] [Google Scholar]
- Hyun J. S., Satsu H., Shimizu M. (2007). Cadmium induces interleukin-8 production via NF-kappaB activation in the human intestinal epithelial cell, Caco-2. Cytokine 37: 26–34. [DOI] [PubMed] [Google Scholar]
- Jacquillet G., Barbier O., Cougnon M., Tauc M., Namorado M. C., Martin D., Reyes J. L., Poujeol P. (2006). Zinc protects renal function during cadmium intoxication in the rat. Am. J. Physiol. Renal Physiol. 290: F127–F137. [DOI] [PubMed] [Google Scholar]
- Jones D. P., Liang Y. (2009). Measuring the poise of thiol/disulfide couples in vivo. Free Radic. Biol. Med. 47: 1329–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaassen C. D., Liu J., Choudhuri S. (1999). Metallothionein: An intracellular protein to protect against cadmium toxicity. Annu. Rev. Pharmacol. Toxicol. 39: 267–294. [DOI] [PubMed] [Google Scholar]
- Kundu S., Sengupta S., Chatterjee S., Mitra S., Bhattacharyya A. (2009). Cadmium induces lung inflammation independent of lung cell proliferation: A molecular approach. J. Inflamm. (Lond). 6: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J., Jin T., Nordberg G., Nordberg M. (2001). Metallothionein gene expression in peripheral lymphocytes from cadmium-exposed workers. Cell Stress Chaperones 6: 97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordberg G. F., Herber R. F. M., Alessio L. (1992). Cadmium in the Human Environment: Toxicity and Carcinogenicity. International Agency for Research on Cancer, Lyon, France. [Google Scholar]
- Oh S. H., Lee B. H., Lim S. C. (2004). Cadmium induces apoptotic cell death in WI 38 cells via caspase-dependent Bid cleavage and calpain-mediated mitochondrial Bax cleavage by Bcl-2-independent pathway. Biochem. Pharmacol. 68: 1845–1855. [DOI] [PubMed] [Google Scholar]
- Othumpangat S., Kashon M., Joseph P. (2005). Eukaryotic translation initiation factor 4E is a cellular target for toxicity and death due to exposure to cadmium chloride. J. Biol. Chem. 280: 25162–25169. [DOI] [PubMed] [Google Scholar]
- Qu W., Diwan B. A., Reece J. M., Bortner C. D., Pi J., Liu J., Waalkes M. P. (2005). Cadmium-induced malignant transformation in rat liver cells: Role of aberrant oncogene expression and minimal role of oxidative stress Int. J. Cancer 114: 346–355. [DOI] [PubMed] [Google Scholar]
- Schroeder P., Popp R., Wiegand B., Altschmied J., Haendeler J. (2007). Nuclear redox-signaling is essential for apoptosis inhibition in endothelial cells–important role for nuclear thioredoxin-1. Arterioscler. Thromb. Vasc. Biol. 27: 2325–2331. [DOI] [PubMed] [Google Scholar]
- Smirnova I. V., Bittel D. C., Ravindra R., Jiang H., Andrews G. K. (2000). Zinc and cadmium can promote rapid nuclear translocation of metal response element-binding transcription factor-1. J. Biol. Chem. 275: 9377–9384. [DOI] [PubMed] [Google Scholar]
- Smith T. J., Anderson R. J., Reading J. C. (1980). Chronic cadmium exposures associated with kidney function effects. Am. J. Ind. Med. 1: 319–337. [DOI] [PubMed] [Google Scholar]
- Stoica A., Katzenellenbogen B. S., Martin M. B. (2000). Activation of estrogen receptor-alpha by the heavy metal cadmium. Mol. Endocrinol. 14: 545–553. [DOI] [PubMed] [Google Scholar]
- Templeton D. M., Liu Y. (2010). Multiple roles of cadmium in cell death and survival. Chem. Biol. Interact. 188: 267–275. [DOI] [PubMed] [Google Scholar]
- Toledano M. B., Leonard W. J. (1991). Modulation of transcription factor NF-kappa B binding activity by oxidation-reduction in vitro. Proc. Natl. Acad. Sci. U.S.A. 88: 4328–4332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. Department of Health and Human Services (2011). Report on Carcinogens 12thed. U.S. Department of Health and Human Services, Research Triangle Park, NC. [Google Scholar]
- Waalkes M. P. (2003). Cadmium carcinogenesis. Mutat. Res. 533: 107–120. [DOI] [PubMed] [Google Scholar]
- Zang Y., Odwin-Dacosta S., Yager J. D. (2009). Effects of cadmium on estrogen receptor mediated signaling and estrogen induced DNA synthesis in T47D human breast cancer cells. Toxicol. Lett. 184: 134–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Go Y. M., Jones D. P. (2007). Mitochondrial thioredoxin-2/peroxiredoxin-3 system functions in parallel with mitochondrial GSH system in protection against oxidative stress. Arch. Biochem. Biophys. 465: 119–126. [DOI] [PubMed] [Google Scholar]
- Zschauer T. C., Kunze K., Jakob S., Haendeler J., Altschmied J. (2011). Oxidative stress-induced degradation of thioredoxin-1 and apoptosis is inhibited by thioredoxin-1-actin interaction in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 31: 650–656. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.