

Abstract
Several alkylanilines with structures more complex than toluidines have been associated epidemiologically with human cancer. Their mechanism of action remains largely undetermined, and there is no reported evidence that it replicates that of multicyclic aromatic amines even though the principal metabolic pathways of P450-mediated hydroxylation and phase II conjugation are very similar. As a means to elucidate their mechanisms of action, lethality and mutagenicity in the adenine phosphoribosyltransferase (aprt +/−) gene induced in several Chinese hamster ovary cell types by 2,6- and 3,5-dimethylaniline (2,6-DMA, 3,5-DMA) and their N- and ring-hydroxyl derivatives (N-OH-2,6-DMA, N-OH-3,5-DMA, 2,6-DMAP, 3,5-DMAP) were assessed. Dose-response relationships were determined in the parental AA8 cell line, its repair-deficient UV5 subclone and other repair-deficient 5P3NAT2 or -proficient 5P3NAT2R9 subclones engineered to express mouse cytochrome P4501A2 (CYP1A2) and human N-acetyltransferase (NAT2), and also in AS52 cells harboring the bacterial guanine-hypoxanthine phosphoribosyltransferase (gpt) gene. Mutations in the gpt gene of AS52 cells were characterized and found to be dominated by G:C to A:T and A:T to G:C transitions. Separately, treatment of AS52 cells with N-OH-2,6-DMA, N-OH-3,5-DMA, 2,6-DMAP, 3,5-DMAP, and 3,5-DMAP led to intracellular production of reactive oxygen species (ROS) for at least 24h after removal of the mutagens in every case. Using the comet assay, DNA strand breaks were observed in a dose-dependent manner in AS52 cells when treated with each of the four N-OH-2,6-DMA, N-OH-3,5-DMA, 2,6-DMAP, and 3,5-DMAP derivatives. Comparative evaluation of the results indicates that the principal mechanism of mutagenic action is likely to be through redox cycling of intracellularly bound aminophenol/quinone imine structures to generate ROS rather than through formation of covalent DNA adducts.
Key Words: dimethylaniline, alkylanilines, reactive oxygen species, CHO cells, mutations, DNA strand breaks, comet assay
Monocyclic aromatic amines present a longstanding and enduring challenge for those who seek to understand their molecular mechanisms of carcinogenic activity. Polycyclic aromatic amines are now well understood to be activated through P450-catalyzed oxidation of the amino group, subsequent esterification of the resultant N-hydroxylamine, and heterolysis of the N–O bond to produce a reactive nitrenium ion that forms covalent adducts with DNA bases, leading to presumably carcinogenic genetic damage (Beland and Kadlubar, 1990; Kadlubar, 1994). This and other possible reaction paths discussed below are shown in Scheme 1. Given that monocyclic aromatic amines also undergo extensive N-oxidation in vivo (McCarthy et al., 1985; Son et al., 1980), it might be expected that their mechanism of action is similar. In exploring this possibility, we and others have shown that the acyl esters of numerous monocyclic aromatic N-hydroxylamines react with DNA bases in vitro to form covalent adducts (Cui et al., 2007; Famulok and Boche, 1989; Gonçalves et al., 2001; Jones and Sabbioni, 2003; Marques et al., 1996; Meier and Boche, 1990). Detection of the same adducts in animals treated with the parent amines, however, has thus far proven elusive, leading us to consider whether other mechanisms might prevail.
Scheme 1.
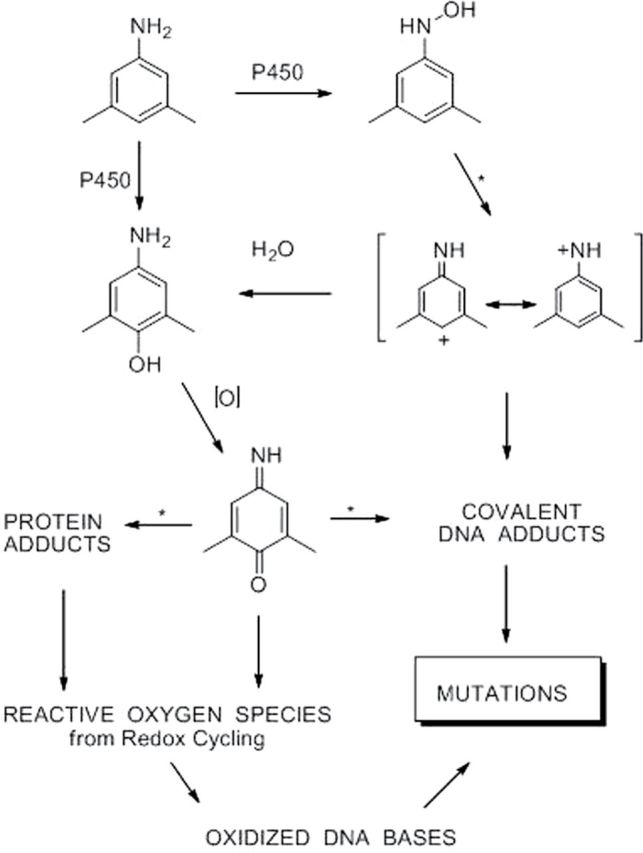
Three pathways to mutagenesis by dimethylanilines. These are shown for the 3,5-isomer and pertain equally to the 2,6-isomer. Arrows marked with * indicate reactions that are known to be or are potentially subject to catalysis. 1. The N-hydroxylamine undergoes N–O bond heterolysis, catalyzed by acyl- or sulfo-transferase activity that forms an unstable N-O- ester. The intermediate highly reactive nitrenium ion thus formed reacts with a DNA base to produce a mutagenic adduct. 2. The aminophenol produced by P450-catalyzed hydroxylation of the aniline or by nucleophilic attack of H2O on the appropriate resonance form of the nitrenium ion is oxidized to its quinone imine form. The electrophilic quinone imine undergoes nucleophilic addition by a DNA base yielding a mutagenic adduct. Chemical studies (Adams and Schowalter, 1952; Irving and Gutmann, 1961) have demonstrated that N-acylated or sulfonylated quinone imines readily undergo 1,2- and 1,4-substitution reactions with amine nucleophiles. 3. Quinone imines either directly or in protein-bound form undergo redox cycling to generate reactive oxygen species that oxidize guanine residues.
There is good reason to question whether a nitrenium ion mechanism is important for the monocyclic amines. Kinetic studies have demonstrated that the stability of nitrenium ions increases greatly with extended conjugation and addition of electronegative substituents to the basic benzene ring structure (Novak et al., 1995). Thus, polycyclic aromatic amines form nitrenium ions with lifetimes sufficiently long to permit reaction with various nucleophiles in the presence of water (Novak and Kennedy, 1995). Nitrenium ions formed by monocyclic aromatic amines have rate constants for reaction with solvent water that approach diffusion controlled rates and thus are limited in their ability to react with cellular nucleophiles. These considerations do not rule out DNA adduct formation by monocyclic nitrenium ions because it has been established that the free nitrenium ion derived from 2,6-DMA can form under aqueous conditions (Fishbein and McClelland, 1987). They do, however, indicate that yields relative to those of polycyclic nitrenium ions will be substantially lower.
We have thus considered whether the other major product of hydroxylation, the aminophenol, has the ability to damage DNA in a manner consistent with mutagenesis and carcinogenesis. The direct mode of action would be electrophilic attack at nucleophilic DNA bases but aminophenols have little electrophilic character. Upon two-electron oxidation to quinone imines, however, they become electrophilic and thus capable of undergoing Michael addition reactions and nucleophilic addition at the keto and imino carbon centers (Eyer, 1994). p-Benzoquinone, a close structural analog of oxidized p-aminophenols, is known to form DNA adducts in vitro (Guliaev et al., 2004), so it is not unreasonable to expect similar reactivity. To our knowledge, though, no DNA adducts of any quinone imines have yet been described.
Evidence for carcinogenicity or other signs of genotoxicity of aminophenols would strengthen the hypothesis that the oxidized form, the quinone imine, can form DNA adducts or otherwise induce mutagenic damage to the DNA bases. p-Amino- phenol itself is regarded as noncarcinogenic (http://ec.europa.eu/health/ph_risk/committees/04_sccp/docs/sccp_o_00e.pdf).
Certain substituted aminophenols have, however, been indirectly associated with genotoxicity (Hill et al., 1997). These are derived from an oncogenic class of chloroacetanilide herbicides that incorporate dimethylaniline and ethylmethylaniline into their structure.
The oncogenic link arises not only because the herbicides are metabolized in vivo to the p-aminophenols and their oxidized quinone imine form (Jefferies et al., 1998) but also because the quinone imines are relatively potent inducers of sister chromatid exchange (Hill et al., 1997). These findings suggest, additionally, that substitution of the aromatic ring with alkyl groups is an important determinant of biological activity.
An alternative mechanism to DNA adduct formation by quinone imines is that they react preferentially with one or more proteins and that the resultant products exert a genetic effect indirectly. This hypothesis is supported by the findings of Jefferies et al. (1998) of thioether products of the diethyl- and ethylmethylaminophenols in livers of rats after administration of either the amines or the aminophenols. Other support comes from studies of hydroquinone/benzoquinone, which is structurally a very close analog of the aminophenol/imino quinone redox pair. Quinones have been shown to react directly with proteins through thiol addition (Slaughter and Hanzlik, 1991). They also react extensively with glutathione to form multi-glutathion-S-yl addition products such as 2,3,5-tris-(glutathion-S-yl)hydroquinone (TGHQ), (Hill et al., 1993). These thioether adducts of benzoquinone can also alkylate proteins through an addition-elimination mechanism rather than the simple addition that characterizes quinone reactivity (Li et al., 2005). Hydroquinone is a nephrotoxin and its glutathion-S-yl adducts exhibit multiple genetic effects (Lau et al., 2001; Patel et al., 2003; Weber et al., 2001; Yang et al., 2005; Yoon et al., 2001). Quinone imines likewise form thioether addition products with glutathione and by analogy might be expected to have similar genotoxic effects (Jefferies et al., 1998; Klos et al., 1992; Martínez-Cabot et al., 2005).
Thioether addition products of benzoquinone are effective generators of reactive oxygen species (ROS), and this might be one mechanism by which protein adducts could elicit a genetic response (Towndrow et al., 2000). TGHQ produces mutation spectra in human and bacterial cells that are similar to those reported for hydroxyl radical–induced mutations (Jeong et al., 1999). In addition to indirect DNA damage, TGHQ has demonstrable effects on expression of genes relevant to carcinogenesis (Patel et al., 2003) and has been shown to induce apoptosis (Yang et al., 2005). Considering the structural homology between TGHQ and quinone imine thioethers, it is reasonable to expect the latter to be a source of ROS as well.
This report describes our initial efforts to evaluate the relative contributions of the most likely pathways to genetic damage by monocyclic aromatic amines through detection and analysis of genetic changes in Chinese Hamster Ovary (CHO) cells grown in culture. 2,6-DMA and 3,5-DMA are its focus because both have been implicated in an epidemiologic study as possible human bladder carcinogens (Gan et al., 2004) and 2,6-DMA is a rodent carcinogen.
MATERIALS AND METHODS
Caution. The following chemicals are hazardous and should be handled carefully: 2,6-DMA, 2,6-DMAP, N-OH-2,6-DMA, 3,5-DMA, 3,5-DMAP, and N-OH-3,5-DMA.
Sources. Reagents and cell culture materials were purchased from the following sources: cell culture materials, Lonza (Walkersville, MD); fetal bovine serum (FBS), Atlanta Biological; 2,6-DMA, 8-azaadenine (8-AA), 6-thioguanine (6-TG), dimethyl sulfoxide (DMSO), NADP, and DL-isocitric acid, N-acetyl cysteine (NAC), Sigma Chemical Co., St Louis, MO; and 3,5-DMA, Acros Organics (Geel, Belgium).
Synthesis of N-hydroxyl metabolites. N-OH-2,6-DMA was synthesized in 50% yield by reduction of 1,3-dimethyl-2-nitrobenzene with zinc dust in aqueous NH4Cl according to a published procedure (Kamm, 1941). The product was obtained as light yellow needles and characterized by mass spectro metry (calcd. for [C8H11NO]H+, 138.0913; found, 138.0913) and 1H-NMR (300 MHz, DMSO-d6, 25°C): δ 2.27 (s, 6H), 6.84 (dd, 7.5 Hz, 1H), 6.92 (d, 7.5 Hz, 2H), 7.16 (d, 2.2 Hz, 1H), 8.08 (d, 2.2 Hz, 1H). N-OH-3,5-DMA was similarly synthesized in 70% yield from 5-nitro-m-xylene to give a crystalline product of light yellow needles. MS: calcd. for [C8H11NO]H+, 138.0913; found, 138.0913. 1H-NMR: δ 2.17 (s, 6H), 6.37 (bs, 1H), 6.44 (m, 2H), 8.10 (d, 2.2 Hz, 1H), 8.19 (d, 2.2 Hz, 1H).
Synthesis of aminophenols. 2,6-DMAP was synthesized in two steps by coupling 3,5-dimethylphenol with the diazonium ion formed by diazotization of sulfanilic acid and reduction of the azo dye with sodium hydrosulfite as described previously (Gan et al., 2001) following the procedure of Albert (1954) with minor modifications. The same approach was used to generate 3,5-DMAP: a mixture of sulfanilic acid (1.73g, 10 mmol), Na2CO3 (0.53g, 5 mmol), and H2O (10ml) was heated to 60°C with stirring. After all the sulfanilic acid was dissolved, the solution was cooled in an ice bath to 15°C. A solution of sodium nitrite (0.74g, 10.7 mmol) in H2O (2ml) was then added drop wise. The resulting solution was poured at once into aqueous hydrochloric acid (14.1ml, 5.6% wt/vol) in an ice bath and the mixture was allowed to stand for 15min. It was then added to a solution of 2,6-dimethylphenol (1.22g, 10 mmol) in aqueous NaOH (2.2g, 55 mmol in 20ml H2O) at 5°C. The dark red reaction mixture was stirred well and allowed to stand for 1h at room temperature. It was then heated to 60°C and to it was gradually added aqueous sodium hydrosulfite (1% wt/vol) until yellow crystals precipitated from colorless solution. After standing 15min at 50°C, the yellow suspension was cooled to 20°C and filtered. The filtrate was washed with 1% sodium hydrosulfite aqueous solution and dried under vacuum. Yield: 82%. MS: calculated for [C8H11NO]H+, 138.0913; found 138.0910. 1H-NMR (500 MHz, DMSO-d6, 25°C): δ 7.17 (s, 1H), 6.15 (m, 2H), 4.3 (s, 2H), 2.02 (s, 6H).
Cell survival and mutation at the aprt locus. AA8 and UV5 CHO cells were purchased from ATCC. Two derivative cell lines 5P3NAT2 and 5P3NAT2R9, which are functionally heterozygous at the aprt locus, were generously provided by Dr J. S. Felton (Lawrence Livermore National Laboratory). The repair-deficient (RD) 5P3NAT2 and -proficient (RP) 5P3NAT2R9 cells both express the cDNAs of the mouse CYP1A2 and human NAT2 genes but differ in repair capability. Details concerning the construction and characterization of these cell lines were described previously (Wu et al., 2003). Prior to each experiment, cells were incubated for 2 days in medium containing CAAT (10µM cytidine, 100µM adenine, 1µM aminopterin, and 17.5µM thymidine) followed by 2–5 days in TAC (thymidine, adenine, and cytidine) medium to reduce the background aprt − mutant frequency. All cells were routinely maintained by monolayer culture in α-Minimal Essential Medium (MEM) containing L-glutamine supplemented with penicillin 100 units/ml, streptomycin 100 µg/ml, and 10% heat-inactivated FBS (complete MEM) in a humidified atmosphere with 5% CO2 at 37°C.
The parental amines, 2,6- and 3,5-DMA, and their N-hydroxyl metabolites dissolved in DMSO were added to exponentially growing cells in 100-mm tissue culture dishes containing 0.5×106 cells in 10ml of complete MEM. The cell lines expressing NAT2 (both 5P3NAT2 and 5P3NAT2R9) were exposed to 0–1000μM of parent compounds for 48h in complete MEM or N-hydroxyl metabolites for 1h in serum-free (SF) MEM. Control cultures were treated with the same volume of vehicle (0.1% DMSO) for 48h or 1h. Concentrations of 2,6- and 3,5-DMA and their metabolites used for mutagenicity experiments were established based on MTT cytotoxicity assays (data not shown). Following treatment, cells were allowed to recover for 24h before determining survival by trypan blue exclusion and maintained in complete MEM thereafter. Previous studies showed that a relative survival of about 30% after chemical exposure facilitated optimum estimates of mutant frequency (Thilly, 1985). Triplicate cultures were exposed to determine mutagenic potencies of 2,6- and 3,5-DMA and their N-hydroxyl metabolites. Seven days after treatment, 6×105 cells from each group were placed in 100ml selective medium containing 80 µg/ml 8-AA and plated at 6×104 cells/10ml/100-mm dish for determination of mutagenicity after 14 days. For determining plating efficiency, dishes were seeded with 200 cells/10ml/100-mm dish in triplicate and incubated for 14 days in the absence of selecting agent.
Cell survival and mutation at the aprt locus with and without ascorbic acid or NAC. AA8 and UV5 cells were adapted to Ham’s F-12 medium supplemented with 100 units/ml penicillin, 100 μg/ml streptomycin, and 10% heat-inactivated FBS (complete Ham’s) medium without ascorbate. The cells were seeded at 1×106 per well of six-well plates and incubated overnight in complete Ham’s medium prior to the treatment. Cells were exposed to N-OH-2,6-DMA (5, 10, 25, 50, 100, and 250μM) or 2,6-DMAP (5, 10, 25, 50, and 100μM) ± 5mM NAC, and N-OH-3,5-DMA (5, 10, 25, 50, 100, and 250μM) or 3,5-DMAP (5, 10, 25, 50, and 100 μM) ± ROS scavengers (5mM NAC or 50 μg/ml ascorbate) for 1h. After treatment, the cells were washed twice with SF Ham’s medium and incubated in complete Ham’s medium for additional 24h prior to determining cell survival. Mutation assay was performed as described above after 7 days of phenotypic expression.
Cell survival and gpt mutagenesis in AS52 cells. CHO AS52 cells, kindly provided by Dr Helga Stopper (University of Würzburg, Germany), were cultured in Ham’s medium supplemented with 100 units/ml penicillin, 100 μg/ml streptomycin, and 10% heat-inactivated FBS (complete Ham’s) in a humidified atmosphere with 5% CO2 at 37°C. The medium was changed routinely, and cells were subcultured when confluence reached about 90%. Cultures were cleansed of pre-existing gpt mutants by culturing in MPA medium (10 µg/ml mycophenolic acid [MPA], 250 μg/ml xanthine, 22 μg/ml adenine, 11 μg/ml thymidine, and 1.2 μg/ml aminopterin) for 7 days followed by recovery medium enriched with xanthine (11.5 μg/ml), adenine (3 μg/ml), and thymidine (1.2 μg/ml) for 3 days.
AS52 cells were placed in six-well plates at a density of 0.5×106 cells per well the day before treatment. Cells were cultured for 5h at 37°C in SF Ham’s medium containing 2,6-DMA or 3,5-DMA (0–1000μM), with or without 5mM NAC, and a human liver S9 (BD Gentest) preparation comprising 16 μl S9 (440 μg S9 protein) and 65 μl sterile-filtered core mixture (25mg/ml NADP, 45mg/ml DL-isocitric acid) per milliliter of SF Ham’s medium. At the end of the treatment period, cells were washed and placed in complete Ham’s medium. For dosing with metabolites of 2,6- and 3,5-DMA, cells were seeded at 1×106 per well of six-well plates and incubated overnight in complete Ham’s medium prior to treatment. The cells were washed two times with SF Ham’s medium and exposed to N-OH-2,6-DMA (5,10, 25, 50, 100, and 250μM) or 2,6-DMAP (5, 10, 25, 50, and 100μM), and N-OH-3,5-DMA (5, 10, 25, 50, 100, and 250μM) or 3,5-DMAP (5, 10, 25, 50, and 100μM) ± 5mM NAC for 1h in SF Ham’s. After the treatment, the cells were processed as above.
AS52 cell viability was determined 24h after the treatments with trypan blue exclusion, and the cells were maintained in complete Ham’s medium for 7 days for phenotypic expression. The level of cell survival was normalized to the negative control and presented as percentage of control. For the gpt mutagenicity measurement, after 7 days incubation, 5×105 cells from each group were placed in 100ml complete Ham’s medium containing selection agent 6-TG (10µM) and plated at 5×104 cells/10ml/100-mm dish for determination of mutagenicity. For plating efficiency analysis, 2500 cells in 50ml complete Ham’s medium from each dose were seeded in 100-mm dishes at a density of 500 cells/10ml. After 14 days incubation, colonies were stained with 0.5% crystal violet in 50% methanol/water for 5 mins, rinsed, and counted. The spontaneous mutant frequency was determined using the negative control (DMSO treated).
DNA extraction, PCR amplification, and molecular analysis of gpt mutants. Single unstained gpt mutant colonies were identified and transferred to 24-well plates and grown to approximately 2×106 mutant cells. Genomic DNA was extracted from each mutant using GenElute mammalian genomic DNA miniprep kit (Sigma). Amplification of the genomic DNA was performed in two rounds of nested PCR in a PTC-200 DNA Engine Thermal Cycler (Bio-Rad, Hercules, CA). One microgram of template DNA was used to run the first round with 10 µl 10×PCR buffer, 2 µl dNTP mix, 0.5 µl taq polymerase, 73.5 µl sterile water, 0.2 µl each 25mM forward (bases −199 to −181; 5′-AAGCTTGGACACAAGACAG-3′) and reverse (bases 520 to 540; 5′-CCAGAATACTTACTGGAAAC-3′) primers (IDT) and amplified with a PCR profile of 94°C: 1min, 30 cycles of 94°C: 1min, 47°C: 1min, 72°C: 1min, and a final extension of 72°C for 7min. The product from this reaction was filtered using a Centricon 50 concentrator and resuspended in 100 µl sterile water to avoid nonspecific binding with remaining primers, and a 10 µl aliquot was used as template in the second round of PCR using nested primers (bases −23 to −4; 5′-ATAAACAGGCTGGGACACTT-3′ and bases 460 to 470; 5′-AGTGCCAGGCGTTGAAAAGA-3′). The PCR conditions were the same in the second round as in the first round reaction, except that the annealing temperature was 52°C. The quantity of gpt gene amplification was analyzed by electrophoresis on 0.8% agarose gels stained with ethidium bromide. The 0.5kb PCR product was cut out and purified for DNA sequence analysis using the QIAquick Gel Extraction Kit (Qiagen). DNA sequencing was carried out by the Dana-Farber/Harvard Cancer Center DNA Resource using the primer sets: 5′-ATAAACAGGCTGGGACACTT-3′ and 5′-AGTGCCAGGCGTTGAAAAGA-3′.
Quantification of intracellular ROS generation. Intracellular ROS detection studies were performed using a Cm-H2DCFDA ROS detection kit (Molecular Probes/Invitrogen). AS52 cells were placed in six-well plates at a density of 1×106 cells per well the day before treatment, washed two times with SF Ham’s medium, and exposed for 1h to various concentrations of N-OH-2,6-DMA, 2,6-DMAP, N-OH-3,5-DMA, or 3,5-DMAP in the presence or absence of 5mM NAC in SF Ham’s medium, wells were washed two times with SF Ham’s medium. Cells were then incubated in complete Ham’s medium for 24h at 37°C, after which they were washed with PBS, treated with trypsin-versene for 5min and suspended in 1ml/well SF Ham’s medium. Aliquots of 100 μl cell suspension from each dose were pipetted into 96-well plates and mixed with 10 μl Hank’s Buffered Salt Solution (HBSS) containing Cm-H2DCFDA (final concentration 25μM) activated by preincubation at 37°C for 30min. ROS generation was measured immediately with an HTS 7000 Plus Bio Assay micro-reader (485nm excitation, 530nm emission; PerkinElmer Life Sciences). ROS levels generated by 1×106 viable treated cells were expressed as percentage of ROS produced by an equal number of viable negative control cells.
CometChip and alkaline comet assay. The alkaline comet assay, used to detect total DNA strand breaks, was performed on the CometChip using the protocol described by Wood et al. (2010). Molten 1% normal melting point agarose (Invitrogen) was poured on top of a sheet of GelBond film (Lonza). The polydimethylsiloxane mold with microposts used to form the microwells was placed into the agarose and removed after the agarose gelled. The gelled agarose with microwells was sandwiched between a glass substrate and a bottomless 96-well plate (Greiner BioOne) and sealed with mechanical force to create the multiwall version of the comet platform, the CometChip.
AS52 cells placed in 100-mm dishes at a density of 1×107 cells were exposed to N-OH-2,6-DMA (50 and 100μM), 2,6-DMAP (10, 25, and 50μM), N-OH-3,5-DMA (50 and 100μM), or 3,5-DMAP (10, 25, and 50μM) ± 5mM NAC using the method described above. This set of treatments was repeated on two additional occasions to provide a set of three independent experiments. After 24 h incubation, 100 μl of cells (106 cells/ml) were pipetted into each of the agarose 96 wells. The bottomless 96-well plate form was then removed, and the gel was covered with 1% low melting point agarose (Invitrogen). After overnight lysis, the CometChips were placed into an electrophoresis chamber filled with alkaline unwinding buffer (0.3M NaOH and 1mM Na2EDTA) for 40min at 4°C. Electrophoresis was performed at the same temperature with the same buffer for 30min at 1V/cm and a current of 300 mA. The chips were then neutralized twice for 15min in fresh buffer (0.4M Tris-HCl at pH 7.5) at 4°C. After neutralization, the CometChips were stained with SYBR Gold (Invitrogen) according to the manufacturer’s instructions for the fluorescence imaging. Graphs were captured using a Nikon 80i upright microscope coupled with an automatic scanning stage and analyzed using the Guicometanalyzer, custom software written in MATLAB (The Mathworks) by Wood et al. (2010). The results generated by the software showed percentage of tail DNA, which represented the level of DNA damage. The cells treated with 100μM H2O2 are the positive control.
In each treatment group, 50–150 comet images were collected and analyzed. However, cells treated with 50μM 3,5-DMAP and 100μM N-OH-3,5-DMA were highly damaged, and viability was less than 30% as indicated in Figure 1. Hence, fewer comets were collected (20–70 comets per experiment) due to the constraint of fewer viable cells.
FIG. 1.

Survival and mutagenicity of AS52 cells treated with 3,5-DMAP, N-OH-3,5-DMA, and 3,5-DMA + S9. Protection against cell killing by ascorbate and NAC is readily apparent when cells were treated with aminophenol (upper left panel) or N-hydroxylamine (lower left) but much less so when the amine was activated by exogenous S9 (lower right). Protection against mutagenicity (upper right) is less pronounced and even reversed with amine plus S9 treatment. Observed mutant frequencies were corrected by subtraction of the vehicle control value (typically, 1.7×10−5) before plotting.
Statistical analysis. Linear regression analysis was used to evaluate the relationship between dose and ROS production in treated cells as measured by fluorescence intensity. Student’s t-test was used for analysis of differences observed in the Comet assays. Statistical analysis of gpt mutants was performed using Fisher’s exact method.
RESULTS
Toxicity and Mutagenicity: Cell Culture Assays
Five cell lines were used to assay toxicity and mutagenicity of 2,6-DMA and 3,5-DMA and their N-hydroxy and p-hydroxy derivatives in vitro. Three of these are derived from one parental cell line AA8 and, together with the parent, were used comparatively to evaluate the effects of –OH substitution and the role of nucleotide excision repair (NER). Scheme 2 shows the derivation and gives the NER status and added metabolic properties of the four cell lines. The fifth cell line is AS52. It was used in this study to investigate ROS production and DNA single-strand break production as discussed in the following sections. Survival and mutagenicity data are considered here in relation to data from the other cell lines because they reveal the striking and critical influence of ascorbic acid.
Scheme 2.
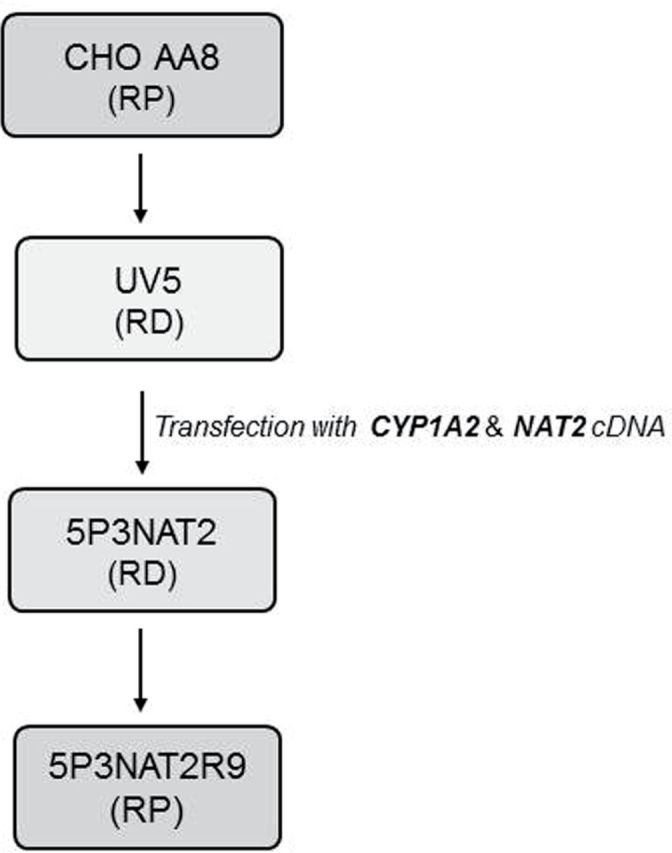
Derivation of the various cell lines related to the AA8 line. Adapted from Wu et al. (2003). RD, repair deficient; RP, repair proficient.
Preliminary studies revealed no material differences between the two DMA isomers; consequently, the more comprehensive studies described below were conducted only with 3,5-DMA and its hydroxylated derivatives. Not all permutations of cell line, test compound, and antioxidant were undertaken. Instead, specific combinations were chosen based on their potential to be informative regarding the roles of excision repair, metabolic capacity, and oxidation potential. The choices were guided to a considerable extent by the relative potencies observed in preliminary studies, which are reflected in the results shown in Figure 1 for AS52 cells. Aminophenols were far more toxic and induced mutations at lower concentrations than N-hydroxylamines, and both hydroxy derivatives were more toxic than the anilines activated by exogenous S9 or by metabolically competent cells.
Oxidation potential as an important determinant of susceptibility to the toxic and mutagenic effects of the DMAs and their metabolites emerged as the result of testing AS52 cells after a body of data was collected from experiments with the AA8 line and its derivatives. AS52 cells proved to be an order of magnitude more sensitive, and the difference was traced to the absence of ascorbate in the growth medium used for these cells, which was different from the medium used for the others. Figure 1 shows survival curves and mutagenicity results for AS52 cells using 3,5-DMA with exogenous S9 activation, N-OH-3,5-DMA, and 3,5-DMAP with and without ascorbate or NAC. The data for 3,5-DMAP in this cell line, with and without ascorbate, can be compared with data obtained with both the AA8 and UV5 cells (Fig. 2). When the latter two are grown without ascorbate, toxicity and mutagenicity are very similar to that observed with the AS52 cells: 50% survival occurs in the range 10–25µM 3,5-DMAP, and there is a mutagenic response. Restoration of ascorbate to the medium increases survival—in all three cell lines—and reduces mutagenicity in the AA8 and UV5 cells. (Mutagenicity in AS52 cells in the presence of ascorbate was not tested.) The effect of added NAC is also protective in all three cell lines except when the treatment was 3,5-DMA plus S9 (see Discussion section).
FIG. 2.
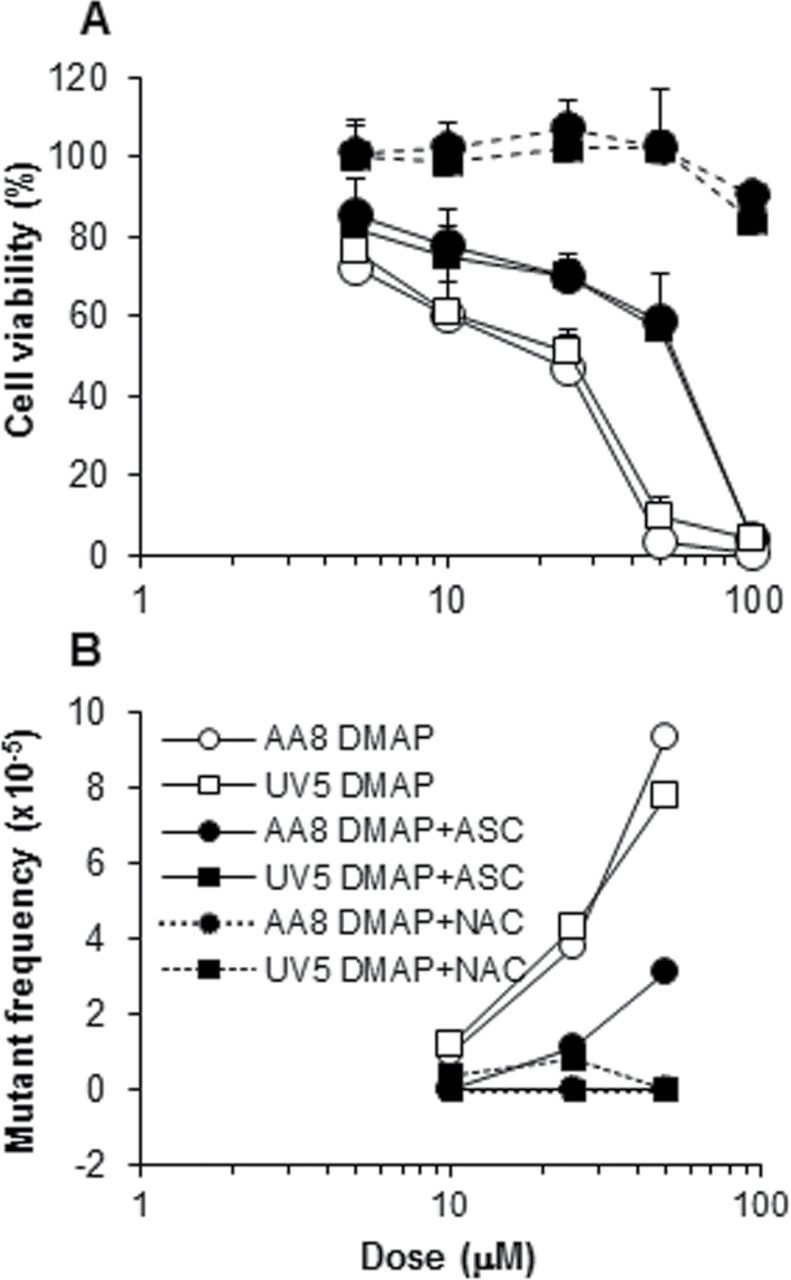
Survival and mutagenicity of AA8 and UV5 cells treated with 3,5-DMAP. No difference in survival is apparent when cells were grown without ascorbate nor in the presence of ascorbate or NAC. In the absence of ascorbate, both cell lines become vulnerable to mutagenicity, which is largely abrogated in the presence of ascorbate or NAC. Vehicle control mutant frequency (typically, 2.9×10–5) subtracted before plotting.
Figure 2 directly compares the AA8 and UV5 cell lines, which differ in NER status, as treated with 3,5-DMAP and ascorbate, NAC, or neither. There appears to be no difference in survival between the two lines under any of the three treatments nor is there any difference in the mutagenic response in the absence of ascorbate or NAC. A possible differential response can be seen in the mutagenic response in the presence of ascorbate, which is not consistent with the idea that NER deficiency would increase mutagenic susceptibility. Overall, the data provide no evidence for a role for NER. More extensive experiments with the 5P3NAT2 and 5P3NAT2R9 cells might be seen as providing evidence for a possible role for NER (Fig. 3). Here, both the 3,5-DMAP and 3,5-DMA mutagenicity curves appear to show greater upslope for the repair-deficient 5P3NAT2 cells than for 5P3NAT2R9 cells. However, more extensive studies would be required to demonstrate that the apparent difference is meaningful.
FIG. 3.

Survival and mutagenicity of NAT2 cells treated with 3,5-DMAP, N-OH-3,5-DMA, and 3,5-DMA. This cell line expresses P450 1A2 and could thus treated with 3,5-DMA without exogenous S9 for the extended period of 48h. These cells also express N-acetyltransferase so the absence of a strong mutagenic response to N-OH-3,5-DMA suggests that the classic aromatic amine mechanism of DNA adduct formation is unimportant for this monocyclic amine. Vehicle control mutant frequency (typically, NAT2, 3.3×10−5; NAT2R9, 4.4×10−5) subtracted before plotting.
gpt Mutations in AS52 Cells
AS52 cells were used to characterize mutations formed in the gpt gene. They are useful for quantitative assessment of mutagenesis and molecular characterization of both point mutations and complex genomic rearrangements affecting the same mutational target gene (Tindall and Stankowski, 1989). These cells carry a single copy of the bacterial gpt gene functionally expressed using the SV40 early promoter and stably integrated into the CHO genome. The xprt/gpt locus is functionally analogous to the mammalian HPRT locus, and mutant clones can be phenotypically selected as 6-TG resistant colonies. Such colonies may arise as a result of point mutations or rearrangements affecting the integrity of the gpt gene or affecting gene expression. The site of genomic integration of the gpt locus in AS52 cells appears also to allow the recovery of complex genomic rearrangements (Tindall and Stankowski, 1989).
In all, mutated gpt sequences were obtained from 221 independent clones. These included 25 sequences resulting from spontaneous mutations in untreated cells and 30–35 sequences from cells subjected to each of six treatment regimens: 2,6-DMA and 3,5-DMA activated by exogenous human hepatic S9 and the chemically synthesized N- and p-hydroxyl derivatives of those two anilines. Single base pair substitutions, insertions, deletions, and multiple sequence changes were all observed. Results are compiled in Table 1.
TABLE 1.
Mutations in the gpt Gene in AS52 cells
| Type of mutation | No. of mutations (% of total) | ||||||
|---|---|---|---|---|---|---|---|
| Control | 2,6-DMA | 2,6-N-OH | 2,6-DMAP | 3,5-DMA | 3,5-N-OH | 3,5-DMAP | |
| Single base pair substitutions | 12 (48) | 20 (57) | 17 (53) | 17 (57) | 19 (54) | 19 (63) | 24 (71) |
| Transversion | |||||||
| G:C → T:A | 6 (26) | 3 (9) | 0 (0) | 3 (10) | 4 (11) | 8 (27) | 13 (38) |
| G:C → C:G | 4 (16) | 2 (6) | 1 (3) | 2 (7) | 1 (3) | 0 (0) | 2 (6) |
| A:T → T:A | 2 (8) | 2 (6) | 3 (9) | 4 (13) | 2 (6) | 0 (0) | 1 (3) |
| A:T → C:G | 0 (0) | 1 (3) | 0 (0) | 0 (0) | 1 (3) | 0 (0) | 3 (9) |
| Transition | |||||||
| G:C → A:T | 0 (0) | 9a (26) | 12a (38) | 7a (23) | 4 (11) | 6a (20) | 2 (6) |
| A:T → G:C | 0 (0) | 3 (9) | 1 (3) | 1 (3) | 7a (20) | 5b (17) | 3 (9) |
| Insertion | 4 (16) | 12 (34) | 8 (25) | 6 (20) | 6 (17) | 5 (17) | 4 (11) |
| Deletion | |||||||
| 1 base pair | 5 (20) | 1 (3) | 2 (6) | 0 (0) | 4 (11) | 0 (0) | 5 (14) |
| 2 base pair | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 5 (15) | 0 (0) | 0 (0) |
| 3 base pair | 3 (12) | 1 (3) | 0 (0) | 7 (23) | 0 (0) | 0 (0) | 0 (0) |
| Multiple changes | 1 (4) | 1 (3) | 5 (16) | 0 (0) | 1 (3) | 6 (20) | 1 (3) |
| Total | 25 | 35 | 32 | 30 | 35 | 30 | 34 |
aSignificantly different from control frequency (p < 0.05, Fisher’s exact test, two tailed).
b p < 0.05, one-tailed test.
Single base pair substitutions were the most commonly occurring genetic change, comprising over 50% of mutational events in every case of treatment (48% spontaneous). G:C to A:T transitions were significantly increased by every form of 2,6-DMA and accounted for 40–70% of all substitutions. No such predominance was observed with the 3,5 dimethyl isomer. A:T to G:C transitions were most abundant when cells were treated with 3,5-DMA + S9, comprising 37% of all substitutions (p < 0.05). N-OH-3,5-DMA significantly increased both possible transition mutations. Though not significantly elevated relative to the control, G:C to T:A transversions were induced very selectively by 3,5-DMAP; they all occurred at a single position in the genome. Notably, 6 (out of 8 total) G:C to T:A transversions induced by N-OH-3,5-DMA also occurred at the same position.
ROS and DNA Strand Breaks
Direct chemical evidence that ROS are generated within cells by hydroxylated anilines was obtained using a fluorometric assay that responds to the presence of intracellular oxidants (Jakubowski and Bartosz, 2000). In this assay, a reduced, acetylated form of fluorescein is taken up by cells and becomes sequestered when esterases remove the acyl groups, leaving the previously lipophilic dye in an ionized state. Removal of the acyl groups also leaves the dye susceptible to oxidation, after which it becomes fluorescent. The assay is not specific for the chemical nature of the oxidant.
Persistence of ROS generation following removal of mutagen from the medium was assessed in AA8 and UV5 cells using 3,5-DMAP as shown in Figure 4, which also shows the protective effect of NAC. ROS production and cytoxicity were determined as a function of dose, cell type, and absence/presence of NAC immediately after exchange of the dosing medium and at 24h. There was clearly no dimunition of ROS after 24h; instead, the data indicate higher generation at this later time point. These experiments also confirmed earlier results that NER status was not a factor in determining cell survival so subsequent experiments were conducted using AS52 cells, and ROS generation was determined at 24h.
FIG. 4.
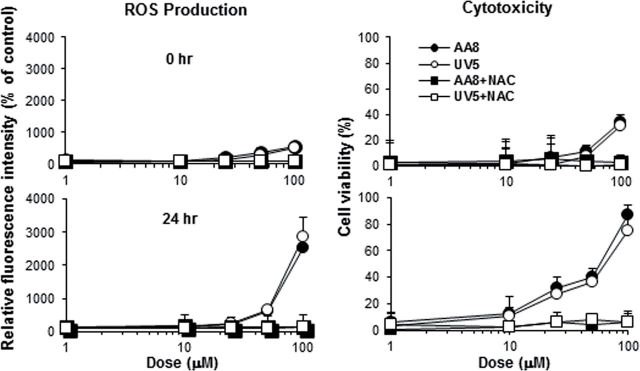
Intracellular production of ROS by 3,5-DMAP in AA8 and UV5 cells. ROS were detected as increased fluorescence after addition of a reduced dichlorofluorescein indicator dye. Cells were treated with test compounds for 1h, washed to remove residual free mutagen, and either assayed immediately (0h) or grown for 24h in fresh medium before assay (24h).
Dose-response data are shown in Figure 5 for the N- and ring-hydroxylated anilines. Not shown are data from experiments with the anilines + S9 because these did not produce a positive dose-response. In the absence of exogenous NAC, there was a highly significant dose-response for each of the four mutagens. In the presence of NAC, no significant dose-response was observed with any of the test compounds except for N-OH-3,5-DMA. (The dose-response to N-OH-3,5-DMA in the presence of NAC, although statistically significant, was nevertheless much lower than in the absence of NAC). The strong ability of the aminophenols of both isomers at concentrations up to 25µM to generate ROS is particularly noteworthy; ROS levels induced by these compounds exceeded those induced by 100µM concentrations of the N-hydroxylamines by several fold.
FIG. 5.
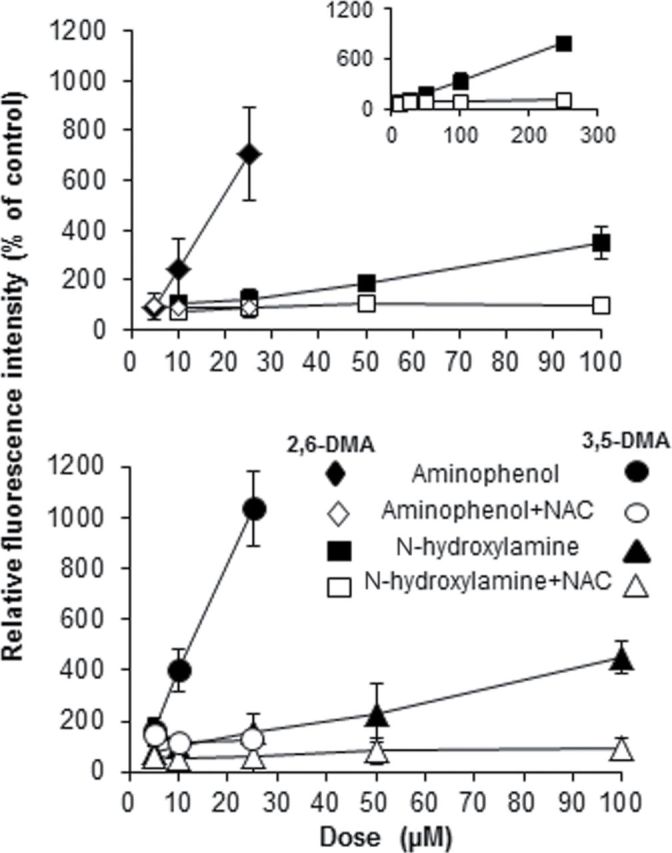
Intracellular production of ROS by hydroxylated dimethylanilines and inhibition by NAC. AS52 cells were treated with test compounds for 1h, washed to remove residual free mutagen, and grown in complete Ham’s medium for an additional 24h in the absence (solid symbols) or presence (open symbols) of 5mM NAC. Treatment with 2,6- and 3,5-DMA + S9 did not generate a significant response and are not shown. All dose-responses in the absence of NAC were statistically significant by linear regression analysis (p < 0.02). In the presence of NAC, only N-OH-3,5-DMA produced a significant dose-response (p < 0.01).
The comet assay was used inferentially to assess the impacts of ROS generation as reflected in the formation of DNA strand breaks. As performed, the assay detected total strand breaks and alkali-labile sites. NAC was used as an inhibitor of ROS generation. Data in Figure 6 are presented as percentage of DNA present in the comet tail in the assay, which provide an estimate of total strand breakage. All four mutagens produced a dose-related increase in strand breaks. The differences from negative control were statistically significant for 2,6-DMAP at 50µM (the highest level tested), for 3,5-DMAP at 25 and 50µM, and for N-OH-3,5-DMA at 100µM (highest level). The observed increase for N-OH-2,6-DMA at the highest level (250µM) was more than double the control but did not reach statistical significance. Wherever differences from the negative control were observed to be significant, the corresponding differences from treatment in the presence of NAC were also significant. In the absence of NAC, the two aminophenols elicited a greater response than did the corresponding hydroxylamines, the differences being in the range of four to fivefold. There were also differences between the two dimethyl isomers. The 2,6-DMAP and N-OH-2,6-DMA generally induced fewer strand breaks than their 3,5 counterparts. For example, 50µM concentration of 2,6-DMAP induced about two thirds the amount of strand breaks as did 3,5-DMAP; comparison of the 100µM doses gives a similar ratio. For the most part, NAC was an effective inhibitor of strand breaks although it did not always maintain tail fraction at the same value as negative controls.
FIG. 6.

Formation of DNA strand breaks by hydroxylated dimethylanilines. Strand breaks were detected using a microarray version of the comet assay (Wood et al., 2010). Each point represents the average of three independent experiments and each experiment includes 50 to 150 determinations. Cells treated with 50µM 3,5-DMAP demonstrated low viability, and only 20–50 comets were collected and analyzed from each experiment; however, a total of more than 100 determinations were obtained from three replicates. Standard error is plotted as error bars on the graph. Results noted with * differ from the negative control and from the same treatment in the presence of NAC by Student’s t-test with p < 0.05.
Information produced by these two measures, production of ROS as determined by oxidation of an intracellular fluorescent probe and generation of DNA strand breaks, is highly congruent. As noted above, the 2,6 isomers were less effective than the 3,5 isomers in eliciting DNA strand breaks by a factor of about two thirds. The ratios of the slopes of the dose-responses for ROS generation are very similar: 0.71 in the case of the aminophenols and 0.79 for the hydroxylamines. The ratios of the slopes of the dose-responses for the two different hydroxyl derivatives (aminophenol: hydroxylamine) are 10.3 (2,6 isomer) and 11.3 (3,5 isomer), which approximate the ratio estimated for strand-break induction.
DISCUSSION
Among arylamines occurring in the environment, 4-aminobiphenyl (4-ABP) is classified as a known human carcinogen (IARC, 1972), and how it induces DNA damage and mutagenesis in various experimental systems has been extensively studied (Beland and Kadlubar, 1990; Kadlubar, 1994). Results argue convincingly for mechanisms involving formation of bulky covalent adducts produced by electrophilic attack of a nitrenium ion intermediate on DNA bases. Specific examples of in vivo results and results from cell culture studies that support this conclusion can be found in Chen et al. (2005) and Besaratinia et al. (2002).
No similar body of data exists regarding the monocyclic aromatic amines. The experiments described here were designed to assess the relative contributions of the bulky, covalent adduct mechanism described above for 4-ABP and two other biologically plausible pathways shown in Scheme 1. Covalent adducts arising from exposure to ring-hydroxylated anilines assumes that 2-electron oxidation occurs in vivo to yield quinoneimines and that these are responsible for adduction. No DNA adducts of quinoneimines have yet been produced synthetically, but their protein adducts are well documented (Eyer, 1994; Irving and Gutmann, 1961; Jefferies et al., 1998), and it is reasonable to assume that they can react with DNA bases as has been shown with quinones. Finally, the aminophenol/quinoneimine redox couple is certain to generate ROS in the presence of appropriate biological electron acceptors and donors as has been shown for 2- and 4-aminophenol in yeast (Brennan and Schiestl, 1997). As argued below, our results do not rule out the possible participation of any of these three pathways but, in our view, they strongly suggest that oxidative damage is the major contributor to mutagenesis in these cell-based assays regardless of whether cells are treated with N- or ring-hydroxylated anilines.
The effects of ascorbate and NAC on toxicity and mutagenicity are key to elucidating the likely mechanism(s) of action of the DMAs and their hydroxylated derivatives. Both were protective when cells were treated with ring- and N-hydroxylated 3,5-DMA, reducing toxicity and mutagenicity. Effectiveness of NAC appears to be equal to or greater than the effectiveness of ascorbate with regard to DMAP, which may reflect the fact that NAC can scavenge DMQI through Michael reaction and scavenge ROS or modulate the oxidation state of the cell, whereas protection by ascorbate is likely limited to redox effects. That these two antioxidants have very similar effects when cells were treated with aminophenol or N-hydroxylamine, with the survival and mutagenicity curves shifted well to the right for the latter, is consistent with hydroxylamine rearrangement to aminophenol as its principal mechanism of action, especially considering that N-hydroxylamines are sensitive to further oxidation of the N–O group to form nitroso compounds that react further to a variety of products. Ascorbate actually inhibits such oxidation (Turesky et al., 1991) and would therefore enhance any pathway dependent on further nonoxidative transformation of the N-hydroxyanilines, such as O-esterification.
The failure of NAC to protect against mutagenicity when AS52 cells were treated with 3,5-DMA + S9 is notable and, together with the results of treating the metabolically competent 5P3NAT2 and 5P3NAT2R9 cells with 3,5-DMA, which then gave a greater mutagenic response than with either hydroxylated derivative, provides additional mechanistic insight. These two treatment regimens undoubtedly produce lower levels of active metabolites at any point in time than occurred when cells were treated with aminophenol or N-hydroxylamine, and exposure would occur for as much as 6h (AS52 cells) or 48h (NAT2 cells). In contrast, the hydroxylated DMAs were present as a bolus dose for 1h. These observations can be readily explained by assuming that toxic response reflects primarily peak dose, whereas mutagenesis reflects integrated dose of toxin. The validity of this assumption depends, in part, on mechanisms underlying toxicity and mutagenicity. Quinone imines are known to be highly reactive with proteins (Eyer, 1994), a probable mechanism of toxicity reasonably assumed to be proportional to peak dose. Other results in this report demonstrate persistent intracellular generation of ROS and a pattern of DNA damage consistent with ROS activity. Oxidative DNA damage is therefore a plausible mechanism of mutagenicity, which would be more integrative than protein damage if DNA repair is slow relative to recovery from protein damage. This possible divergence of toxic and mutagenic mechanisms is fully consistent with the results of treatments with parent amine.
Dose-dependent generation of ROS in cells treated with hydroxylamines or aminophenols (Figs. 4 and 5) confirms that both metabolites of both anilines are capable of producing ROS intracellularly and that the aminophenols are far more potent. Results from the comet assays indicate that the levels of ROS detected by the fluorescence assay are sufficient to elicit considerable genetic damage in the form of DNA strand breaks. Presumably, at these levels, mutations are also induced. A notable aspect of the ROS data is that ROS generation is higher at 24h than immediately following a 1h treatment with mutagen. It is highly unlikely that after 24h the ROS generators are any longer free compounds. Consistent with findings that quinones become bound to proteins, we are concurrently developing further detailed evidence that the quinone imines of 2,6- and 3,5-DMA also are bound intracellularly through reaction with proteins. Depending on the length of time that the redox activity of the bound compounds persists, protein binding may provide a mechanism for greatly potentiating the cumulative genotoxicity of the quinone imine metabolites.
In the present experiments, establishing a central role for quinoneimines does not reveal their mechanism(s) of action. They are electrophilic compounds and could, in principle, form covalent DNA adducts. Do the types of mutations observed provide any insight into this possibility? Under any of the treatment conditions used, more than 50% of mutations were base substitutions that occurred mostly at G:C pairs, producing G:C to T:A transversions and G:C to A:T transitions. Generally speaking, this pattern of mutation is broadly consistent with oxidative damage (Neeley and Essigmann, 2006; Wang et al., 1998). Molecular mechanisms of mutagenesis and carcinogenesis induced by ROS have been extensively studied by exposing DNA targets to ionizing radiation or to ROS produced by stimulated leucocytes (Breimer, 1990; Newcomb and Loeb, 1998; Reid and Loeb, 1986). For example, treatment of human HL60 neutrophils with tumor promoters stimulates production primarily of O2 •− and H2O2, which in the presence of free iron produce HO• via the Haber-Weiss reaction. In representative experiments, mutant frequency was increased sixfold in the lacZα gene of M13mp2 plasmid transfected into SOS-induced Escherichia coli after coincubation with stimulated cells; most mutations (96%) induced were single base substitutions, a preponderance of which occurred opposite guanine residues, leading to equal numbers of G to T and G to C transversions. Studies in which mutations induced in the supF gene in a shuttle vector exposed to H2O2 with or without Fe/EDTA to generate HO• demonstrated that exposure of the vector prior to transfection to H2O2 did not induce mutations, but exposure to HO• caused a combination of base substitutions and deletions. When cells carrying the vector were exposed to H2O2 (presumably leading to intracellular production of HO•), the frequency of base substitution-deletion mutations was increased. Virtually, all base substitutions were located at G:C base pairs, remarkably only in the 3′ bases of TC:AG or CC:GG sequences; G:C to A:T transitions were by far the predominant mutation observed. In contrast, when the plasmid was exposed to hydroxyl radicals before transfection into cells, the base changes observed were predominantly located at AT base pairs.
Overall, our results are generally indicative that generation of ROS by aminophenol/quinone imine metabolites mediates most of the mutagenic activity attributable to both of the predominant hydroxylated metabolites of the two alkylanilines investigated. All the hydroxylated anilines were shown to produce ROS in cells long after exposure. All induced DNA strand breaks, determined as the total of single- and double-strand breaks. Ascorbate and NAC were strongly protective in every case. The kinds of mutations produced and their relative abundances reflect the results of others regarding oxidative damage to DNA. And finally, the lack of any observable influence of NER status on cytoxicity and mutagenicity is in line with previous results comparing AA8 and UV5 cells when treated with H2O2 or alkylating agents (Johansson et al., 2004).
In summary, we note that there may be some evidence for covalent modification of DNA by the metabolites of these two dimethylanilines as a mechanism of genotoxicity but that on balance the evidence appears to favor generation of ROS as the predominant mechanism.
FUNDING
National Institute of Environmental Health Sciences (P01-ES006052); National Institute of Environmental Health Sciences (ES02109) grant, and the National Institute of Environmental Health Sciences (NIEHS) Genes, Environment, and Health Initiative Grant U01-ES016045.
REFERENCES
- Adams R., Schowalter K. A. (1952). Quinone imides. X. Addition of amines to p-quinonedibenzenesulfonimide J. Am. Chem. Soc. 74 2597–2602 [Google Scholar]
- Albert H. E. (1954). Some new amino alkylphenols J. Am. Chem. Soc. 76 4985–4988 [Google Scholar]
- Beland F. A., Kadlubar F. F. (1990). Metabolic activation and DNA adducts of aromatic amines and nitroaromatic hydrocarbons. In Handbook of Experimental Pharmacology. Chemical Carcinogenesis and Mutagenesis (Cooper C. S., Grover P. L., Eds.), Vol. 94/I, pp. 267–325 Springer-Verlag; Berlin: [Google Scholar]
- Besaratinia A., Bates S. E., Pfeifer G. P. (2002). Mutational signature of the proximate bladder carcinogen N-hydroxy-4-acetylaminobiphenyl: Inconsistency with the p53 mutational spectrum in bladder cancer. Cancer Res. 62 4331–4338 [PubMed] [Google Scholar]
- Breimer L. H. (1990). Molecular mechanisms of oxygen radical carcinogenesis and mutagenesis: The role of DNA base damage Mol. Carcinog. 3 1082–1086 [DOI] [PubMed] [Google Scholar]
- Brennan R. J., Schiestl R. H. (1997). Aniline and its metabolites generate free radicals in yeast. Mutagenesis 12 215–220 [DOI] [PubMed] [Google Scholar]
- Chen T., Mittelstaedt R. A., Beland F. A., Heflich R. H., Moore M. M., Parsons B. L. (2005). 4-Aminobiphenyl induces liver DNA adducts in both neonatal and adult mice but induces liver mutations only in neonatal mice. Int. J. Cancer 117 182–187 [DOI] [PubMed] [Google Scholar]
- Cui L., Sun H. L., Wishnok J. S., Tannenbaum S. R., Skipper P. L. (2007). Identification of adducts formed by reaction of N-acetoxy-3,5-dimethylaniline with DNA. Chem. Res. Toxicol. 20 1730–1736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyer P. (1994). Reactions of oxidatively activated arylamines with thiols: Reaction mechanisms and biologic implications. An overview. Environ. Health Perspect. 102(Suppl. 6)123–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Famulok M., Boche G. (1989). Formation of N-(deoxyguanosin-8-yl) aniline in the in vitro reaction of N-acetoxyaniline with deoxyguanosine and DNA Angew. Chem. Int. Ed. Engl. 28 468–469 [Google Scholar]
- Fishbein J. C., McClelland R. A. (1987). Azide ion trapping of the intermediate in the Bamberger rearrangement. Lifetime of a free nitrenium ion in aqueous solution J. Am. Chem. Soc. 109 2824–2825 [Google Scholar]
- Gan J., Skipper P. L., Tannenbaum S. R. (2001). Oxidation of 2,6-dimethylaniline by recombinant human cytochrome P450s and human liver microsomes. Chem. Res. Toxicol. 14 672–677 [DOI] [PubMed] [Google Scholar]
- Gan J., Skipper P. L., Gago-Dominguez M., Arakawa K., Ross R. K., Yu M. C., Tannenbaum S. R. (2004). Alkylaniline-hemoglobin adducts and risk of non-smoking-related bladder cancer. J. Natl. Cancer Inst. 96 1425–1431 [DOI] [PubMed] [Google Scholar]
- Gonçalves L. L., Beland F. A., Marques M. M. (2001). Synthesis, characterization, and comparative 32P-postlabeling efficiencies of 2,6-dimethylaniline-DNA adducts. Chem. Res. Toxicol. 14 165–174 [DOI] [PubMed] [Google Scholar]
- Guliaev A. B., Hang B., Singer B. (2004). Structural insights by molecular dynamics simulations into specificity of the major human AP endonuclease toward the benzene-derived DNA adduct, pBQ-C. Nucleic Acids Res. 32 2844–2852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill A. B., Jefferies P. R., Quistad G. B., Casida J. E. (1997). Dialkylquinoneimine metabolites of chloroacetanilide herbicides induce sister chromatid exchanges in cultured human lymphocytes. Mutat. Res. 395 159–171 [DOI] [PubMed] [Google Scholar]
- Hill B. A., Kleiner H. E., Ryan E. A., Dulik D. M., Monks T. J., Lau S. S. (1993). Identification of multi-S-substituted conjugates of hydroquinone by HPLC-coulometric electrode array analysis and mass spectrometry Chem. Res. Toxicol. 6 459–469 [DOI] [PubMed] [Google Scholar]
- IARC (1972). 4-Aminobiphenyl. In IARC Monographs on the Evaluation of Carcinogenic Risk of Chemicals to Humans, Vol. 1, pp. 74–79 International Agency for Research on Cancer; Lyon, France: [Google Scholar]
- Irving C. C., Gutmann H. R. (1961). Protein binding of model quinone imides. III. Preparation of Ne-(1-hydroxy-2-acetamido-4-fluorenyl)-DL-lysine J. Org. Chem. 26 1859–1861 [Google Scholar]
- Jakubowski W., Bartosz G. (2000). 2,7-dichlorofluorescin oxidation and reactive oxygen species: What does it measure? Cell Biol. Int. 24 757–760 [DOI] [PubMed] [Google Scholar]
- Jefferies P. R., Quistad G. B., Casida J. E. (1998). Dialkylquinonimines validated as in vivo metabolites of alachlor, acetochlor, and metolachlor herbicides in rats. Chem. Res. Toxicol. 11 353–359 [DOI] [PubMed] [Google Scholar]
- Jeong J. K., Wogan G. N., Lau S. S., Monks T. J. (1999). Quinol-glutathione conjugate-induced mutation spectra in the supF gene replicated in human AD293 cells and bacterial MBL50 cells. Cancer Res. 59 3641–3645 [PubMed] [Google Scholar]
- Johansson F., Allkvist A., Erixon K., Malmvärn A., Nilsson R., Bergman A., Helleday T., Jenssen D. (2004). Screening for genotoxicity using the DRAG assay: Investigation of halogenated environmental contaminants. Mutat. Res. 563 35–47 [DOI] [PubMed] [Google Scholar]
- Jones C. R., Sabbioni G. (2003). Identification of DNA adducts using HPLC/MS/MS following in vitro and in vivo experiments with arylamines and nitroarenes. Chem. Res. Toxicol. 16 1251–1263 [DOI] [PubMed] [Google Scholar]
- Kadlubar F. F. (1994). DNA adducts of carcinogenic aromatic amines. In DNA Adducts: Identification and Biological Significance. IARC Scientific Publications No. 125 (Hemminki K., Dipple A., Shuker D. E. G., Kadlubar F. F., Segerbäck D., Bartsch H., Eds.), pp. 199–216 International Agency for Research on Cancer; Lyon, France: [PubMed] [Google Scholar]
- Kamm O. (1941). β-Phenylhydroxylamine. In Organic Syntheses, Collective Vol. 1, p. 445 Wiley, New York, NY. [Google Scholar]
- Klos C., Koob M., Kramer C., Dekant W. (1992). p-aminophenol nephrotoxicity: Biosynthesis of toxic glutathione conjugates. Toxicol. Appl. Pharmacol. 115 98–106 [DOI] [PubMed] [Google Scholar]
- Lau S. S., Monks T. J., Everitt J. I., Kleymenova E., Walker C. L. (2001). Carcinogenicity of a nephrotoxic metabolite of the “nongenotoxic” carcinogen hydroquinone. Chem. Res. Toxicol. 14 25–33 [DOI] [PubMed] [Google Scholar]
- Li W. W., Heinze J., Haehnel W. (2005). Site-specific binding of quinones to proteins through thiol addition and addition-elimination reactions. J. Am. Chem. Soc. 127 6140–6141 [DOI] [PubMed] [Google Scholar]
- Marques M. M., Mourato L. L., Santos M. A., Beland F. A. (1996). Synthesis, characterization, and conformational analysis of DNA adducts from methylated anilines present in tobacco smoke. Chem. Res. Toxicol. 9 99–108 [DOI] [PubMed] [Google Scholar]
- Martínez-Cabot A., Morató A., Messeguer A. (2005). Synthesis and stability studies of the glutathione and N-acetylcysteine adducts of an iminoquinone reactive intermediate generated in the biotransformation of 3-(N-phenylamino)propane-1,2-diol: Implications for toxic oil syndrome. Chem. Res. Toxicol. 18 1721–1728 [DOI] [PubMed] [Google Scholar]
- McCarthy D. J., Waud W. R., Struck R. F., Hill D. L. (1985). Disposition and metabolism of aniline in Fischer 344 rats and C57BL/6 X C3H F1 mice. Cancer Res. 45 174–180 [PubMed] [Google Scholar]
- Meier C., Boche G. (1990). N-Aryl-O-(α-aminoacyl)hydroxylamine: Modellreaktionen mit desoxyguanosin, guanosin und 5’-guanosinmonophosphat zur aktivierung monocyclischer aromatischer amine (z.B. phenacetin) zu ultimaten carcinogenen Chem. Ber. 123 1699–1705 [Google Scholar]
- Neeley W. L., Essigmann J. M. (2006). Mechanisms of formation, genotoxicity, and mutation of guanine oxidation products. Chem. Res. Toxicol. 19 491–505 [DOI] [PubMed] [Google Scholar]
- Newcomb T. G., Loeb L. A. (1998). Mechanisms of mutagenicity of oxidatively-modified bases. In Molecular Biology of Free Radicals in Human Diseases (Aruoma O. I., Halliwell B., Eds.), pp. 139–166 OICA International, London; [Google Scholar]
- Novak M., Kennedy S. A. (1995). Selective trapping of N-acetyl-N-(4-biphenylyl)nitrenium and N-acetyl-N-(2-fluorenyl)nitrenium ions by 2’-deoxyguanosine in aqueous solution J. Am. Chem. Soc. 117 574–575 [Google Scholar]
- Novak M., Kahley M. J., Lin J., Kennedy S. A., James T. G. (1995). Involvement of free nitrenium ions, ion pairs, and preassociation trapping in the reactions of ester derivatives of N-arylhydroxylamines and N-arylhydroxamic acids in aqueous solution J. Org. Chem. 60 8294–8304 [Google Scholar]
- Patel S. K., Ma N., Monks T. J., Lau S. S. (2003). Changes in gene expression during chemical-induced nephrocarcinogenicity in the Eker rat. Mol. Carcinog. 38 141–154 [DOI] [PubMed] [Google Scholar]
- Reid T. M., Loeb L. A. (1986). Mutagenic specificity of oxygen radicals produced by human leukemia cells Cancer Res. 52 1082–1086 [PubMed] [Google Scholar]
- Slaughter D. E., Hanzlik R. P. (1991). Identification of epoxide- and quinone-derived bromobenzene adducts to protein sulfur nucleophiles. Chem. Res. Toxicol. 4 349–359 [DOI] [PubMed] [Google Scholar]
- Son O. S., Everett D. W., Fiala E. S. (1980). Metabolism of o-[methyl-14C]toluidine in the F344 rat. Xenobiotica 10 457–468 [DOI] [PubMed] [Google Scholar]
- Thilly W. G. (1985). Dead cells don’t form mutant colonies: A serious source of bias in mutation assays. Environ. Mutagen. 7 255–258 [DOI] [PubMed] [Google Scholar]
- Tindall K. R., Stankowski L. F., Jr (1989). Molecular analysis of spontaneous mutations at the gpt locus in Chinese hamster ovary (AS52) cells. Mutat. Res. 220 241–253 [DOI] [PubMed] [Google Scholar]
- Towndrow K. M., Mertens J. J., Jeong J. K., Weber T. J., Monks T. J., Lau S. S. (2000). Stress- and growth-related gene expression are independent of chemical-induced prostaglandin E(2) synthesis in renal epithelial cells. Chem. Res. Toxicol. 13 111–117 [DOI] [PubMed] [Google Scholar]
- Turesky R. J., Lang N. P., Butler M. A., Teitel C. H., Kadlubar F. F. (1991). Metabolic activation of carcinogenic heterocyclic aromatic amines by human liver and colon. Carcinogenesis 12 1839–1845 [DOI] [PubMed] [Google Scholar]
- Wang D., Kreutzer D. A., Essigmann J. M. (1998). Mutagenicity and repair of oxidative DNA damage: Insights from studies using defined lesions. Mutat. Res. 400 99–115 [DOI] [PubMed] [Google Scholar]
- Weber T. J., Huang Q., Monks T. J., Lau S. S. (2001). Differential regulation of redox responsive transcription factors by the nephrocarcinogen 2,3,5-Tris(glutathion-S-yl)hydroquinone. Chem. Res. Toxicol. 14 814–821 [DOI] [PubMed] [Google Scholar]
- Wood D. K., Weingeist D. M., Bhatia S. N., Engelward B. P. (2010). Single cell trapping and DNA damage analysis using microwell arrays. Proc. Natl. Acad. Sci. U.S.A. 107 10008–10013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R. W., Panteleakos F. N., Felton J. S. (2003). Development and characterization of CHO repair-proficient cell lines for comparative mutagenicity and metabolism of heterocyclic amines from cooked food. Environ. Mol. Mutagen. 41 7–13 [DOI] [PubMed] [Google Scholar]
- Yang M. Y., Lau S. S., Monks T. J. (2005). 2,3,5-tris(Glutathion-S-yl)hydroquinone (TGHQ)-mediated apoptosis of human promyelocytic leukemia cells is preceded by mitochondrial cytochrome c release in the absence of a decrease in the mitochondrial membrane potential. Toxicol. Sci. 86 92–100 [DOI] [PubMed] [Google Scholar]
- Yoon H. S., Monks T. J., Walker C. L., Lau S. S. (2001). Transformation of kidney epithelial cells by a quinol thioether via inactivation of the tuberous sclerosis-2 tumor suppressor gene. Mol. Carcinog. 31 37–45 [DOI] [PubMed] [Google Scholar]