

Abstract
Methylmercury (MeHg) is an environmental pollutant that biomagnifies throughout the aquatic food chain, thus representing a toxicological concern for humans subsiding on fish for their dietary intake. Although the developing brain is considered the critical target organ of MeHg toxicity, recent evidence indicates that the cardiovascular system may be the most sensitive in adults. However, data on the mechanisms mediating MeHg-induced cardiovascular toxicity are scarce. Based on the close relationship between cardiovascular disease and dyslipidemia, this study was designed to investigate the effects of long-term MeHg exposure on plasma lipid levels in mice, as well as their underlying mechanisms and potential relationships to MeHg-induced neurotoxicity. Our major finding was that long-term MeHg exposure induced dyslipidemia in rodents. Specifically, Swiss and C57BL/6 mice treated for 21 days with a drinking solution of MeHg (40mg/l, ad libitum) diluted in tap water showed increased total and non-HDL plasma cholesterol levels. MeHg-induced hypercholesterolemia was also observed in low-density lipoprotein receptor knockout (LDLr–/–) mice, indicating that this effect was not related to decreased LDLr-mediated cholesterol transport from blood to other tissues. Although the hepatic synthesis of cholesterol was unchanged, significant signs of nephrotoxicity (glomerular shrinkage, tubular vacuolization, and changed urea levels) were observed in MeHg-exposed mice, indicating that the involvement of nephropathy in MeHg-induced lipid dyshomeostasis may not be ruled out. Notably, Probucol (a lipid-lowering drug) prevented the development of hypercholesterolemia when coadministered with MeHg. Finally, hypercholesterolemic LDLr–/– mice were more susceptible to MeHg-induced cerebellar glial activation, suggesting that hypercholesterolemia in itself may pose a risk factor in MeHg-induced neurotoxicity. Overall, based on the strong and graded positive association between total as well as LDL cholesterol and risk of cardiovascular diseases, our data support the concept of MeHg-induced cardiovascular toxicity.
Key Words: methylmercury, cardiovascular disease, neurotoxicity, low-density lipoprotein receptor, cholesterol, dyslipidemia
Methylmercury (MeHg), an environmental toxic pollutant, is generated by methylation of inorganic mercury, which is catalyzed by sulfate-reducing bacteria in the aquatic environment (Hintelmann, 2010). Once in the aquatic food chain, MeHg bioaccumulates and magnifies, reaching high levels in predatory fish, thus representing a toxicological concern for humans subsiding on fish for their dietary intake (Clarkson et al., 2003). Despite its ubiquitous distribution in tissues, the central nervous system (CNS) represents the main target of MeHg toxicity, especially when exposures occur during the early stages of brain development (Clarkson et al., 2003).
Even though the developing brain has been considered the critical target organ of MeHg toxicity in children, recent evidence indicates that the cardiovascular system may be the most sensitive organ in adults (Choi et al., 2009). In humans, cardiovascular outcomes of MeHg exposure include myocardial infarction (Rissanen et al., 2000), increased blood pressure, heart rate variability, and atherosclerosis (Salonen et al., 2000). Furthermore, MeHg-exposure biomarkers are significantly associated with increased thickness of the intima-media layer of the carotid artery (Choi et al., 2009). Although the mechanisms mediating MeHg-induced cardiovascular dysfunction are not completely understood, epidemiological evidence invokes altered cardiovascular autonomic tone secondary to MeHg-induced neurotoxicity in the brainstem (Grandjean et al., 2004; Murata et al., 2004). Moreover, pro-oxidative events, leading to oxidation of plasma low-density lipoprotein (LDL) (Jin et al., 2012), decreased production of endothelial nitric oxide (de Marco et al., 2010) and endothelium-derived relaxing factor (Ohno et al., 1995) are likely involved in the etiology of MeHg-induced cardiovascular diseases.
Human exposure to MeHg is mainly due to the ingestion of seafood and freshwater fish (Clarkson et al., 2003). High fish intake is also thought to provide protective effects against coronary heart disease (Wall et al., 2010). Accordingly, in humans subsiding on large amounts of fish, the cardiovascular benefits afforded by beneficial nutrients present in fish (i.e., n-3 fatty acids and selenium) seem to be masked, at least in part, by MeHg and other contaminants (Mozaffarian and Rimm, 2006).
Although data on the potential involvement of the renal system in mediating MeHg-induced cardiovascular dysfunction are sparse, the possibility is worthy of consideration. In addition to being a known target organ of MeHg accumulation and toxicity (Yasutake et al., 1997; Zalups, 2000), mortality ratios for nephritis, nephrosis, and nephrotic syndrome are significantly higher in MeHg-intoxicated patients than in the general population (Tamashiro et al., 1984). Furthermore, cardiovascular dysfunction is observed (and represents the main cause of morbidity and mortality) in patients with kidney disease. Of note, dyslipidemia, characterized mainly by increased triglycerides (TGs) and reduced high-density lipoprotein (HDL) levels in plasma, has been observed in renal disease patients (Keane et al., 2011). Moreover, nephrotic syndrome in humans is also known to be associated with plasma lipid disorders, such as hypercholesterolemia and altered plasma fatty acid composition (Fujita et al., 2006).
Because hypercholesterolemia is crucial to the development and progression of atherosclerosis, and is a causative factor of coronary artery disease (Ware, 2008), we hypothesized that cholesterol plays an important role in MeHg-mediated cardiovascular dysfunction. Specifically, we (1) investigated and characterized the occurrence of MeHg-induced dyslipidemic-like profile in mice; (2) analyzed the effects of a lipid-lowering drug against MeHg-induced dyslipidemia, and (3) evaluated the underlying mechanisms leading to MeHg-induced dyslipidemia, such as the involvement of low-density lipoprotein receptors (LDLr), hepatic cholesterol synthesis, and renal damage. In addition, we also investigated whether hypercholesterolemic mice are more susceptible to MeHg-induced neurotoxicity.
MATERIALS AND METHODS
Animals
Male adult Swiss Albino mice (3 months old and 40–50g weight) were provided by the animal facility of the Universidade Federal de Santa Catarina (UFSC, Florianópolis, Brazil). C57BL/6 wild-type mice and LDL receptor knockout (LDLr–/–) mice founders were purchased from Jackson Laboratory (Bar Harbor, ME) and were bred at UFSC. Animals were maintained in groups of four to five animals per cage, under controlled temperature (23°C ± 1°C) and 12-h light cycle (lights on 7:00 a.m.), with free access to food and water. All experimental procedures were in compliance with the guidelines on the animal care of the UFSC Ethics Committee on the Use of Animals (protocol numbers PP00373 and PP00547), which follows the NIH “Principles of Laboratory Animal Care.”
Schedules of MeHg Administration and Experimental Design
Experiment 1.
In order to investigate the effects of MeHg exposure on plasma lipid profile in Swiss mice, MeHg (40mg/l, ad libitum) was diluted in tap water and administered for 28 days via drinking solution (Franco et al., 2009). Liquid consumption was measured daily, giving a MeHg dose of 6.0mg/kg. Parallel groups (control and MeHg) were treated with the lipid-lowering drug Probucol, which was injected once a day (10mg/kg; ip) based on a previous study from our group (Santos et al., 2012). At the end of the experiments (24h after the last injection), the animals were sacrificed and blood was collected by heart puncture for analysis of plasma lipids.
Because fish omega-3 polyunsaturated fatty acids (n-3 PUFA), such as docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA), may counteract some of the deleterious effects of MeHg (Mozaffarian and Rimm, 2006), we performed a parallel experiment on the simultaneous administration of DHA and MeHg to Swiss mice. Animals were treated for 21 days with a drinking solution of MeHg (40mg/l, ad libitum) diluted in tap water (Franco et al., 2009). Liquid consumption was measured daily, giving an Hg dose of 5.6mg/kg. Parallel groups (control and MeHg) were cotreated with DHA (8mg/kg once a day as a DHA:bovine serum albumin [BSA] complex, ip) based on previous study by Kielar et al. (2003). In short, DHA (catalog #D2534; Sigma Chemical Company, St Louis, MO) was dissolved in absolute ethanol (20mg/ml); 50 µl was added dropwise to 1ml of delipidated, endotoxin-free BSA (0.1g/ml, catalog #A7030; Sigma Chemical Company, St. Louis, MO). This mixture was vortexed for 2min and incubated for 2h at 37°C. This yielded a clear solution that was diluted to the appropriate DHA concentration, and 1 cc was injected ip. The DHA dosage was selected based on the fact that similar (or even lower) dosages displayed anti-inflammatory and protective effects in a mouse model of renal ischemic injury (Kielar et al., 2003). In addition, such a dosage was also chosen based on the recommended intake of DHA + EPA for adult humans (0.5g/day; Kris-Etherton et al., 2002), which gave a daily dose of 7.1mg/kg based on a 70-kg person as reference.
Experiment 2.
To investigate the potential role of LDL receptors in the MeHg-induced dyslipidemic-like profile, MeHg (40mg/l of MeHg diluted in tap water) was administered for 21 days via drinking solution to C57BL/6 wild-type and low-density lipoprotein receptor knockout (LDLr–/–) mice (Franco et al., 2009). Moreover, because the kidney is a known target organ of MeHg accumulation, and chronic kidney disease is associated with a highly atherogenic lipid profile, after 21 days of daily administration of MeHg to C57BL/6 wild-type mice, we performed a pathological evaluation of the kidneys and analyzed hepatic cholesterol levels, as well as the levels of plasma urea.
In another set of experiments, we evaluated the temporal relationship between MeHg treatment and glutathione peroxidase (GPx) inhibition, motor impairment and dyslipidemia. To this end, we analyzed locomotor activity in the open field test followed by collection of blood (cardiac puncture) and cerebellum of C57BL/6 wild-type mice 7, 14, or 21 days after daily administration of MeHg. Cholesterol levels and GPx activity were analyzed in the blood and cerebellum, respectively.
Finally, we investigated whether hypercholesterolemic mice are more susceptible to MeHg-induced neurotoxicity. C57BL/6 wild-type mice and LDLr–/– mice were treated for 21 days with a drinking solution of MeHg (40mg/l) diluted in tap water, followed by analyses of GPx activity and glial activation (glial fibrillary acidic protein [GFAP] immunostaining) in the cerebellum.
Open Field
Spontaneous locomotor activity of mice was evaluated in an open field test. The apparatus, made of transparent PVC, had a white floor of 50cm × 50cm and transparent walls (divided by black lines into squares of 10cm × 10cm) and 40-cm high. The experiments were conducted in a dimly lit (7 lx) and sound-isolated room. Each mouse was placed in the center of the open field and the number of squares crossed and vertical activity (rearing) was registered during a 5-min period.
Plasma Lipids and Urea Levels
Animals were food-deprived overnight, anesthetized using ketamine/xylazine (75 and 10mg/kg, respectively, ip), and the blood was collected by cardiac puncture, placed in heparinized tubes and immediately centrifuged at 1000 × g, and the plasma frozen at –80°C. Total cholesterol (TC), HDL cholesterol, TGs, and urea were measured in plasma using the enzymatic kit according to the manufacturer’s instructions (Gold Analisa Diagnóstica Ltda, Minas Gerais, Brazil). The concentration of non-HDL cholesterol was calculated with the equation: LDL + VLDL + IDL = TC – HDL.
Hepatic Cholesterol Levels
Liver cholesterol was extracted by the method of Folch et al. (1957). Ten microliters of supernatant were evaporated to dryness, and the residue was dissolved in 10 µl of dimethylsulfoxide. Next, the cholesterol content was determined using enzymatic kit according to the manufacturer’s instructions (Gold Analisa Diagnóstica Ltda, Minas Gerais, Brazil).
Glutathione Peroxidase Activity
The animals were killed by decapitation and the cerebellum was dissected and homogenized (1:10 wt/vol) in 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid buffer (20mM, pH 7.0). The tissue homogenates were centrifuged at 16,000 × g, at 4°C for 20min, and the supernatants were used for the determination of enzymatic activities. GPx activity was determined indirectly by measuring the consumption of NADPH at 340nm, as previously reported (Farina et al., 2009). GPx uses glutathione (GSH) to reduce tert-butyl hydroperoxide, producing oxidized glutathione (GSSG), which is readily reduced to GSH by glutathione reductase using NADPH as a reducing equivalent donor. Data are expressed as nmol of NADPH oxidized/min/mg protein. The protein content was quantified by the method of Bradford (1976), using BSA as a standard.
Pathological Analyzes
The mice were anesthetized using ketamine/xylazine (75 and 10mg/kg, respectively, ip) and transcardially perfused with PBS containing 4% paraformaldehyde. The kidneys were extracted and kept in the latter solution for 24h. The kidneys were then sectioned and processed using standard histological techniques (i.e., embedded in paraffin, sectioned at 7 µm and stained by standard hematoxylin and eosin [HE] technique). The slides were analyzed using light microscopy (Nikon Eclipse 50i; Nikon, New York, NY).
Immunofluorescence
A total of 20 animals (5 per group) were anesthetized using ketamine/xylazine (75 and 10mg/kg, respectively, ip) and were perfused through the left cardiac ventricle with 40ml of 0.9% saline solution, followed by 40ml of 4% paraformaldehyde in 0.1M PBS, pH 7.4. After perfusion, the brains were removed, postfixed in the same fixative solution for 4h at room temperature and cryoprotected by immersion in 15 and 30% sucrose solution in PBS at 4°C. The brains were then frozen by immersion in cooled isopentane with CO2 and stored in a freezer (–80°C) for later analyses.
Serial coronal sections (40 µm) of cerebellum were obtained with a cryostat (Leica) at –20°C. The free-floating sections were preincubated in 2% BSA diluted in PBS containing 0.1% Triton X-100 (PBS-Tx 0.1%) for 30min. Next, the sections were incubated for 48h at 4°C with polyclonal anti-GFAP from rabbit (DAKO) diluted 1:3000 in PBS-Tx 0.1%. After three washes in PBS, tissue sections were incubated with antirabbit Alexa Fluor 488 (Molecular Probes) diluted 1:500 in PBS-Tx 0.1% for 1h at room temperature. In the negative control, the primary antibody was omitted. Thereafter, the sections were washed three times in PBS, mounted on slides with Fluor Save and covered with coverslips (adapted from Rodrigues et al., 2010).
Immunofluorescence Analysis and Quantification
Images from mouse cerebellum were obtained with an Olympus IX-70 microscope coupled to a digital camera. All lighting conditions and magnifications ( × 200) were held constant. Rectangular areas of interest (region of interest [ROI], 262 pixel2) were set for cortical cerebella layers (molecular, Purkinje, and granular layers) and the immunofluorescence of each ROI was measured with Cell M 2.6 Imaging Software. A total of 10 images (one image per section, TIF format) from each mouse were measured and the data were expressed as arbitrary units (AU, mean ± SEM). Also, several images were obtained with a Confocal Olympus IX-81 microscope.
Statistical Analyses
Statistical analyses were carried out using one- or two-way ANOVA or unpaired t-tests, as indicated in the Results section. Where p values were significant, multiple post hoc comparisons were performed using the Newman-Keuls test. Pearson’s correlation coefficient was used as a measure of the strength of the association between the variables. The accepted level of significance for all tests was p ≤ 0.05. Data are expressed as mean ± SEM. All tests were performed using the STATISTICA software package (StatSoft Inc., Tulsa, OK).
RESULTS
MeHg Induces Dyslipidemia in Mice
Based on the potential relationship between MeHg exposure and cardiovascular disease, our initial goal was to investigate the effects of MeHg exposure on plasma lipid profile in Swiss mice. MeHg was administered to Swiss mice via drinking solution (40mg/l, diluted in tap water, during 28 days) (Farina et al., 2003). Parallel groups were cotreated with the lipid-lowering drug Probucol (10mg/kg; ip) (Santos et al., 2012). Two-way ANOVA indicated a significant main effects for MeHg by Probucol interaction on total plasma cholesterol [F(1, 20) = 17.29, p < 0.05], HDL cholesterol [F(1, 20) = 8.19, p < 0.05], non-HDL cholesterol [F(1, 20) = 12.51, p < 0.05], and TG levels [F(1, 17) = 15.75, p < 0.05]. Subsequent post hoc comparisons revealed significant increases in TC, non-HDL cholesterol, HDL cholesterol, and TG levels in Swiss mice treated with MeHg (p < 0.05). Moreover, concomitant treatment with the lipid-lowering drug Probucol blunted all the MeHg-induced plasma lipid alterations (Figs. 1A–D, respectively). With respect to the parallel study on the simultaneous exposure to MeHg and DHA, no significant effects of DHA on plasma cholesterol and TG levels were observed (data not shown).
Because of the increased total and non-HDL cholesterol levels in the plasma of MeHg-treated Swiss mice, we further investigated the potential involvement of LDLr in this event by using the LDLr–/– mouse, which has the C57Bl/6 genetic background. After 21 days treatment with MeHg (40mg/l, diluted in tap water) (Dietrich et al., 2005), two-way ANOVA indicated significant main effects of genotype (p < 0.05) and MeHg (p < 0.05) on total plasma cholesterol, HDL, and non-HDL cholesterol levels. The absence of significant interaction between genotype and MeHg for total, HDL, and non-HDL cholesterol indicates that MeHg-induced hypercholesterolemia is not related to changes in the LDLr function. With respect to the TG levels, two-way ANOVA indicated significant main effects for genotype by MeHg interaction [F(1, 20) = 6.41, p < 0.05]. Subsequent post hoc comparisons revealed increased TC and non-HDL cholesterol levels in the C57BL/6 wild-type and LDLr–/– mice treated with MeHg compared with their respective controls (Figs. 1E and G, respectively). Post hoc comparisons also revealed increased HDL cholesterol levels and decreased TG levels in the MeHg-treated LDLr–/– mice compared with their respective controls (Figs. 1F and H, respectively).
FIG. 1.

Dyslipidemia induced by chronic MeHg administration in mice.
Swiss mice were treated for 28 days with MeHg (40mg/l diluted in tap water) and/or Probucol. Plasma TC (A), HDL cholesterol (B), non-HDL cholesterol (C), and TG (D) levels are presented as mg/dl. C57BL/6 (wild type or LDLr–/–) mice were treated for 21 days with MeHg (40mg/l diluted in tap water). Plasma TC (E), HDL cholesterol (F), non-HDL cholesterol (G), and TG (H) levels are presented as mg/dl. Data are expressed as mean ± SEM. *p < 0.05 compared with the control group, #p < 0.05 compared with MeHg-treated mice, &p < 0.05 compared with control (non-MeHg treated) LDLr–/– mice (two-way ANOVA followed by Newman-Keuls post hoc test).
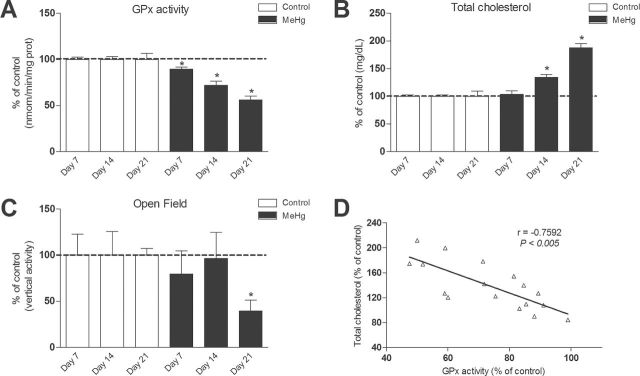
Time Course of MeHg Toxicity in C57BL/6 Mice: Dyslipidemia, Locomotor Deficits and Impaired Cerebellar GPx Activity
We have previously shown that MeHg-induced inhibition of GPx is a key event involved in its neurotoxicity (Franco et al., 2009). Because this study is the first to show that MeHg exposure causes hypercholesterolemia, we evaluated the temporal relationship between MeHg treatment and GPx inhibition, motor impairment, and dyslipidemia. Accordingly, plasma cholesterol levels, cerebellar GPx activity, and motor performance were evaluated in C57BL/6 mice 7, 14, and 21 days after MeHg exposure (40mg/l, diluted in tap water). Corroborating our previous studies (Franco et al., 2009), MeHg-induced inhibition of cerebellar GPx activity was the earliest event, and it was detected within 7 days of MeHg treatment (Fig. 2A). Significant increases in total plasma cholesterol were observed after 14 days of MeHg treatment (Fig. 2B). Locomotor impairment was the most delayed event, and it was detected only 21 days after MeHg treatment (Fig. 2C). Although our data do not permit us to conclude that hypercholesterolemia contributed to the occurrence of motor impairment, it affirms that MeHg-induced plasma cholesterol homeostasis precedes the behavioral change associated with exposure to this metal. Moreover, Pearson’s correlation was used to analyze a possible correlation between cerebellar GPx activity and total plasma cholesterol levels in the MeHg-treated mice after 7, 14, and 21 days of the beginning of MeHg treatment (Fig. 2D). We observed a decrease in the activity of GPx in cerebellum of MeHg-treated mice, and this decrease was negatively correlated with the total plasma cholesterol level (Pearson’s coefficient = –0.7592; p < 0.005).
FIG. 2.
Time-dependent MeHg-induced toxicity in C57BL/6 mice.
C57BL/6 mice were treated with 40mg/l MeHg diluted in tap water for 7, 14, or 21 days. After treatments, cerebellar GPx activity (A), plasma cholesterol levels (B), and vertical activity in the open field test (C) were measured. Data are presented as percent of the respective control and expressed as mean ± SEM (n = 8 animals per group). *p < 0.05 compared with the control group (unpaired Student’s t-test). (D) Scatter plot of the relationship between cerebellar GPx activity and total plasma cholesterol levels. Data of GPx activity are derived from (A) and (B), respectively. Pearson’s coefficient: –0.7592 (p < 0.005).
Chronic MeHg Treatment Induced Renal Toxicity, But Did Not Alter Hepatic Cholesterol Levels
Although LDLr is a key protein in mediating the transport of cholesterol from plasma to tissues, our data (see MeHg Induces Dyslipidemia in Mice section) showed no significant involvement of this receptor in MeHg-induced hypercholesterolemia. Because the kidney is a known target organ of MeHg accumulation, and chronic kidney disease is associated with a highly atherogenic lipid profile, we hypothesized that MeHg-induced renal toxicity could be involved in MeHg-induced dyslipidemia. In agreement with this hypothesis, plasma urea levels (a biomarker for renal function) were significantly decreased in the MeHg-treated group compared with the control group (t = 2.97, p = 0.21) (Fig. 3B). Moreover, histological analyses showed significant changes in the renal tissue of MeHg-treated mice, such as altered renal corpuscle, with shrinkage of glomeruli. In addition, vacuoles were present within epithelial cells of some proximal convoluted tubules, which displayed distorted shape (Fig. 3A). These changes (glomerular shrinkage and tubular vacuolization) were not evident in non-MeHg-treated animals (data not shown). These data point to significant MeHg-induced nephrotoxicity, which could be responsible, at least in part, to the hypercholesterolemia induced by this toxicant (Shoji and Nishizawa, 2006). Because the liver is the main lipogenic organ, we also investigated whether MeHg increased liver cholesterol synthesis. No significant changes in hepatic cholesterol content were observed between MeHg-treated and control animals (t = 1.35, p = 0.21) (Fig. 3C).
FIG. 3.

Chronic MeHg exposure induces renal toxicity, but does not alter hepatic cholesterol levels in C57BL/6 mice.
C57BL/6 mice were treated for 21 days with 40mg/l MeHg diluted in tap water. (A) Photomicrograph of kidney from an MeHg-exposed animal. The image depicts a normal glomerulus (NG), as well as an altered renal corpuscle (arrow) with glomerular shrinkage (original magnification × 100). The inset shows high magnification (× 400): vacuoles (V) within epithelial cells of proximal convoluted tubules. Stain: HE. Serum urea levels (B) and hepatic cholesterol levels (C) are expressed as mean ± SEM (n = 6 animals per group). *p < 0.05 compared with the control group (unpaired Student’s t-test).
LDLr–/– Mice Are More Susceptible to MeHg-Induced Neurotoxicity
Although it is well known that MeHg induces neurotoxicity due to its direct effects on neurons, astrocytes, and microglial cells (Farina et al., 2011a,b; Ni et al., 2011), our data raise the possibility that indirect events (i.e., hypercholesterolemia) may contribute to the neurotoxic effects elicited by MeHg. This hypothesis was posited based on our recent observations (de Oliveira et al., 2011; Moreira et al., forthcoming) on neurochemical and neurobehavioral impairments in hypercholesterolemic animals. Thus, we investigated whether hypercholesterolemic mice are more susceptible to MeHg-induced neurotoxicity. We took advantage of LDLr–/– mice, which present with 2- to 4-fold increase in plasma cholesterol concentration when maintained on a low-fat diet and thus are a useful model for studying the effects of high circulating cholesterol levels (Zadelaar et al., 2007). After 21-day exposure to MeHg (40mg/l in tap water, ad libitum), cerebellar GPx activity and glial activation (GFAP immunostaining) were evaluated as hallmarks of neurotoxicity in C57BL/6 wild-type and LDLr–/– mice. Figure 4 shows abundant astrogliosis in LDLr–/– mice treated with MeHg. A normal astrocytic staining pattern was detected in cell bodies and processes in all the experimental groups in the molecular, Purkinje, and granular layers. In the molecular layer, the glial processes (radial or Bergmann fibers) follow a strict and uniform arrangement, and cell bodies of Bergmann glia could be identified in the Purkinje layer. Bushy astrocytes were found in the granular layer; they are a conventional type of protoplasmic astrocyte that is stellate in shape. Quantitatively, two-way ANOVA indicated a significant effect for MeHg [F(1, 16) = 13.75, p < 0.05] on GFAP expression in the cerebellum. Subsequent post hoc comparisons revealed increased GFAP immunostaining in the MeHg-treated LDLr–/– mice compared with their respective controls (Fig. 4B).
FIG. 4.

Hypercholesterolemic LDLr–/– mice are more susceptible to MeHg-induced glial activation.
C57BL/6 and LDLr–/– mice were treated for 21 days with 40mg/l MeHg diluted in tap water. The serial stack images of GFAP immunofluorescence staining were obtained with an Olympus confocal FV-1000 microscope. The images showed abundant astrogliosis in the LDLr–/– MeHg-treated group (A) in the cerebellar cortical layers: ML (molecular layer), PL (Purkinje layer), and GL (granular layer). Magnification × 40, 0.61 µm of optical stack thickness and seven confocal planes. →, Bergmann fiber; *, blood vessel. Scale bars = 50 µm. (B) Quantification of GFAP immunofluorescence in cerebellar cortex. Data are expressed as mean ± SEM. &p < 0.05 compared with control (non-MeHg treated) LDLr–/– mice (two-way ANOVA followed by Newman-Keuls post hoc test).
Furthermore, two-way ANOVA indicated a significant effect for genotype [F(1, 24) = 5.53, p < 0.05] and for treatment [F(1, 24) = 41.04, p < 0.05] on GPx activity in the cerebellum. Subsequent post hoc comparisons revealed decreased GPx activity in the cerebellum of MeHg-treated C57BL/6 wild-type and LDLr–/– mice compared with their respective control groups (Fig. 5A). Moreover, Pearson’s correlation was used to analyze a possible correlation between cerebellar GPx activity and total plasma cholesterol levels in C57Bl/6 and hypercholesterolemic LDLr–/– mice treated with MeHg (Fig. 5B). We observed that the GPx activity in cerebellum was negatively correlated with total plasma cholesterol levels (Pearson’s coefficient = -0.56; p < 0.005).
FIG. 5.
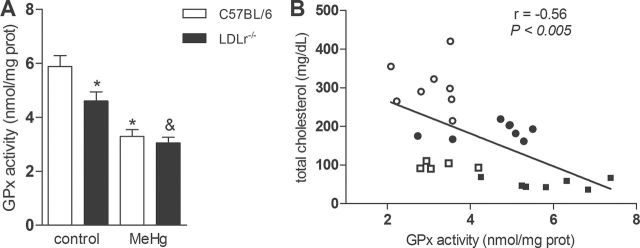
Chronic MeHg exposure decreases GPx activity in the cerebellum of C57BL/6 and LDLr–/– mice.
C57BL/6 and LDLr–/– mice were treated for 21 days with 40mg/l MeHg diluted in tap water. (A) Cerebellar GPx activity is presented as nmol of NADPH oxidized/mg protein/min and expressed as mean ± SEM (n = 7–10 animals per group). *p < 0.05 compared with the control (non-MeHg treated) group, &p < 0.05 compared with control (non-MeHg treated) LDLr–/– mice (two-way ANOVA followed by Newman-Keuls post hoc test). (B) Scatter plot of the relationship between cerebellar GPx activity and total plasma cholesterol levels; data are derived from (A) and (1E), respectively. Squares represent C57BL/6 mice and circles represent LDLr–/– mice. White color represents control and black color represents MeHg-treated animals. Pearson’s coefficient: -0.56 (p < 0.005).
DISCUSSION
Although methylmercury (MeHg) has been recognized for decades as a neurotoxic compound (for review, see Clarkson et al., 2003), recent studies have indicated that it also may promote cardiovascular disease (Choi et al., 2009). Studies on the mechanisms by which MeHg leads to cardiovascular disease have largely focused on deregulation of endothelial nitric oxide (de Marco et al., 2010) and increased oxidation of plasma LDL (Jin et al., 2012) as likely etiologic events. Our present results shed new light on the etiology of MeHg-induced cardiovascular disease by providing novel evidence that MeHg causes hypercholesterolemia in mice (Fig. 1).
Taking into account that the human population is exposed to MeHg primarily due to the ingestion of contaminated fish (Clarkson et al., 2003) and that n-3 PUFA, such as DHA and EPA (present in fish), have long been posited to be efficient compounds in preventing cardiovascular diseases (Leaf, 1990; Smith and Guentzel, 2010), we performed an experiment on the simultaneous administration of DHA and MeHg to Swiss mice to investigate whether DHA can blunt the hypercholesterolemia induced by MeHg. Interestingly, concomitant treatment with DHA did not change MeHg-induced dyslipidemia. Although the beneficial nutrient of fish oil (DHA) was unable to abolish the deleterious (particularly, dyslipidemic) effects of MeHg, our data do not necessarily contradict the growing body of literature showing the beneficial effects of fish n-3 PUFA against MeHg toxicity (Jin et al., 2009; Kaur et al., 2008). Although the DHA dosage used in our protocol (8mg/kg) has been shown to display beneficial (anti-inflammatory and renoprotective) effects in mouse models (Kielar et al., 2003) and is near to the recommended intake of DHA + EPA for adult humans (Kris-Etherton et al., 2002), the potential beneficial effects of a higher dose (or longer treatment period) of DHA on MeHg-induced hypercholesterolemia in our experimental model cannot be ruled out. In fact, the DHA:Hg ratio used in our study is near 1 (8mg/kg DHA:5.6mg/kg Hg), which is very low compared with those ratios found in the muscle of fish (Smith and Guentzel, 2010). Our results do not provide a definitive answer as to whether DHA can antagonize the dyslipidemic effects induced by MeHg. This constitutes a very interesting and broad research field that deserves additional attention. Based on our novel data on the hypercholesterolemic effects of MeHg, it is incumbent that additional studies be designed to evaluate if n-3 PUFA can modulate MeHg-induced dyslipidemia, as well as the formation of atheroma plaque and the development of other hallmarks associated with cardiovascular disease.
Monogenic hypercholesterolemia occurs mainly as a result of impaired receptor-mediated uptake of LDL by the LDLr in the hepatocytes and other tissues (Garg and Simha, 2007). Our experiments in the LDLr–/– knockout mice showed no significant relationship between the LDLr and MeHg-induced hypercholesterolemia (Figs. 1E–H). In view of this “negative” result, we investigated if MeHg might affect liver cholesterol levels, representing the predominant organ for cholesterol synthesis. However, no significant differences in cholesterol levels were noted between hepatic cholesterol levels of control and MeHg-treated mice (Fig. 3C). Conversely, histopathological analyses revealed significant changes in the kidney of MeHg-exposed mice, such as altered renal corpuscle, shrinkage of glomeruli and vacuolization in epithelial cells of proximal convoluted tubules (Fig. 3A). Our results corroborate previous reports on MeHg-induced nephropathology (Eto et al., 1997; Jin et al., 2008). These morphological alterations are also in agreement with the observed decrease in plasma urea nitrogen levels (Fig. 3B) (Jin et al., 2009). Accordingly, decreased plasma urea nitrogen levels in MeHg-treated animals might represent suppressed protein catabolism, and/or increased urinary excretion of urea (Slotkin et al., 1986).
The results of the temporal effects of MeHg on (1) hypercholesterolemia, (2) cerebellar GPx inhibition, and (3) motor impairment are noteworthy (Fig. 2). GPx is a selenoprotein with a critical structural selenol group for its peroxide-reducing activity. Notably, MeHg binds avidly to selenols (with a higher affinity than that to thiols), and selenoproteins have been shown as primary targets of MeHg (Franco et al., 2009). Specifically, our group has shown that GPx is involved in MeHg-induced neurotoxicity, leading to low peroxide detoxification and increased lipid peroxidation (Farina et al., 2009), and mediating MeHg-induced cerebellar ataxia and motor impairment (Carvalho et al., 2008; Farina et al., 2003). Here, the temporal analysis on the effects of MeHg showed that cerebellar GPx inhibition was the earliest event detected (Fig. 2A), and it was significant even when plasma cholesterol levels and motor impairment were unchanged. Hypercholesterolemia was observed 14 days after the beginning of MeHg exposure (Fig. 2B), preceding the MeHg-induced motor impairment (observed only at 21 days after the beginning of treatments) (Fig. 2C). Although no causal relationship can be drawn from these findings, namely that hypercholesterolemia contributed to the occurrence of motor impairment, it is noteworthy that MeHg-induced plasma cholesterol dyshomeostasis precedes the behavioral change. These observations, combined with the fact that hypercholesterolemia induces reactive oxygen species production (Munzel et al., 2010) and oxidative damage (Aytan et al., 2008), raises the question as to whether MeHg-induced hypercholesterolemia could contribute to the development of MeHg-induced neurotoxicity. This question could be answered by evaluating the potential protective/beneficial effects of hypocholesterolemic drugs against MeHg-induced neurotoxicity. In fact, one might posit that MeHg-induced hypercholesterolemia could contribute to the development of MeHg-induced neurotoxicity if hypocholesterolemic drugs afforded neuroprotective effects against MeHg. However, this question is not easy to evaluate experimentally because hypocholesterolemic drugs, such as statins and Probucol possess pharmacological properties other than those exclusively related to their cholesterol lowering effects. For example, statins possess several pleiotropic properties (Greenwood et al., 2006), which are related to their beneficial effects in neurodegenerative diseases (Miida et al., 2007). Probucol possesses anti-inflammatory and antioxidant properties, which may prevent MeHg-induced neurotoxicity per se, absent effects on cholesterol homeostasis. Notably, our group observed protective effects of Probucol against MeHg-induced neurotoxicity (Farina et al., 2009), which were largely related to its antioxidant properties rather than hypocholesterolemic effects. Thus, given the complexities inherent to the discrimination between the lipid-lowering versus pleiotropic properties of Prubucol, it is difficult to confirm that MeHg-induced hypercholesterolemia plays a causal role in the development of MeHg-induced neurotoxicity. Nonetheless, we observed higher astroglial activation, a well-known marker of MeHg-induced neurotoxicity and neuroinflammation (el-Fawal et al., 1996; Toimela and Tahti, 1995) in hypercholesterolemic mice (Fig. 4). This result indicates that higher plasma cholesterol levels may render the CNS more susceptible to MeHg-induced astrogliosis, the latter typically observed during neurotoxic events. Moreover, a negative correlation between cerebellar GPx activity and total plasma cholesterol levels was observed (Figs. 2D and 5B). Taking into account that previous studies point to decrease in the GPx activity in several tissues of hypercholesterolemic mice (de Oliveira et al., 2011; Silva et al., 1995), it is reasonable to posit a relationship between both phenomena.
As previously described, epidemiological evidence indicates a potential relationship between MeHg exposure and adverse cardiovascular effects in man (Choi et al., 2009). Here, we observed that MeHg exposure in mice caused hypercholesterolemia, which is a key contributor to the development of cardiovascular disease. Nonetheless, it is difficult to make a direct comparison/extrapolation from our animal data to those from humans (Choi et al., 2009). In our experimental study with mice, a single MeHg dose (40mg/l, dissolved in drinking water) and a 3- or 4-week exposure period were selected. A daily MeHg dose of 6.0mg/kg body weight (about 4.7mg Hg/kg body weight) was calculated based on the animal’s liquid ingestion. In a previous study, we observed that a similar exposure led to cerebellar Hg levels of 2.6 ppm in adult mice (Franco et al., 2006), which are closely similar to those found in brains obtained at autopsy from Seychellois infants (Myers et al., 1995), indicating the relevance of the exposures to human health concerns. However, with regards to the study by Choi et al. (2009), which specifically delved into the theme of MeHg-induced adverse cardiovascular effects in humans, direct comparisons between exposure rates are difficult to make. The exposure extent in consumers of meat from pilot whale was evaluated, for example, by measuring Hg levels in nail samples (Choi et al., 2009), which reflect the incorporation of Hg that has occurred over ~1 year (Yoshizawa et al., 2002). Intoxication of adult man to MeHg due to seafood intake normally occurs as result of long-term exposures, which are not easily reproduced in experimental rodent models. Accordingly, based on the levels of Hg commonly found in whale meat (~6 ppm; Endo and Haraguchi, 2010) and on the average amount of whale consumed by the Faroese whaling men (around three whale meals per month), it is possible to assume that the Hg dosage used in our experimental approach with mice (4.7mg/kg/day) is at least two orders of magnitude greater than the amount of Hg consumed in the studied population by Choi et al. (2009) study, on a mg/kg b.wt/day basis.
In summary, we demonstrate, for the first time, that long-term MeHg treatment induces dyslipidemia, characterized by increased serum cholesterol levels in mice. Our data are of particular relevance to MeHg-induced cardiovascular disease, which has been recently reported in humans (Choi et al., 2009). Furthermore, we show that hypercholesterolemic mice were more susceptible to MeHg-induced astrogliosis. A putative mechanism for the hypercholesterolemia effect remains to be fully elucidated, but it likely encompasses a noxious oxidative-inflammatory cycle in the neurovasculature that might impact brain areas affected by MeHg.
FUNDING
Brazilian institutions Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq); the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES); the Fundação de Apoio à Pesquisa do Estado de Santa Catarina (FAPESC); Programa de Apoio aos Núcleos de Excelência (PRONEX—Project NENASC); IBN-Net/CNPq, INCT (Instituto Nacional de Ciência e Tecnologia) for Excitotoxicity and Neuroprotection; CNPq-Brazil to C.A.G., A.F.B., R.D.S.P., and M.F.; National Institute of Environmental Health Science (ES R01 07331; ES P30 000267 to M.A.).
ACKNOWLEDGMENTS
The authors thank the Electron Microscopy Center of the Federal University of Rio Grande do Sul for the microscopy analyzes and also thank Mr Henrique Biehl for technical assistance. The authors have no financial or personal conflicts of interest related to this work.
REFERENCES
- Aytan N., Jung T., Tamtürk F., Grune T., Kartal-Ozer N. (2008). Oxidative stress related changes in the brain of hypercholesterolemic rabbits. Biofactors 33 225–236 [DOI] [PubMed] [Google Scholar]
- Bradford M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72 248–254 [DOI] [PubMed] [Google Scholar]
- Carvalho C. M., Chew E. H., Hashemy S. I., Lu J., Holmgren A. (2008). Inhibition of the human thioredoxin system. A molecular mechanism of mercury toxicity. J. Biol. Chem. 283 11913–11923 [DOI] [PubMed] [Google Scholar]
- Choi A. L., Weihe P., Budtz-Jørgensen E., Jørgensen P. J., Salonen J. T., Tuomainen T. P., Murata K., Nielsen H. P., Petersen M. S., Askham J., et al. (2009). Methylmercury exposure and adverse cardiovascular effects in Faroese whaling men. Environ. Health Perspect. 117 367–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson T. W., Magos L., Myers G. J. (2003). The toxicology of mercury–current exposures and clinical manifestations. N. Engl. J. Med. 349 1731–1737 [DOI] [PubMed] [Google Scholar]
- de Marco K. C. Passos C. J. Sertorio J. Tanus-Santos J. E. Barbosa F.Jr. (2010). Environmental exposure to methylmercury is associated with a decrease in nitric oxide production. Basic Clin. Pharmacol. Toxicol. 106 411–415 [DOI] [PubMed] [Google Scholar]
- de Oliveira J., Hort M. A., Moreira E. L., Glaser V., Ribeiro-do-Valle R. M., Prediger R. D., Farina M., Latini A., de Bem A. F. (2011). Positive correlation between elevated plasma cholesterol levels and cognitive impairments in LDL receptor knockout mice: Relevance of cortico-cerebral mitochondrial dysfunction and oxidative stress. Neuroscience 197 99–106 [DOI] [PubMed] [Google Scholar]
- Dietrich M. O., Mantese C. E., Anjos G. D., Souza D. O., Farina M. (2005). Motor impairment induced by oral exposure to methylmercury in adult mice. Environ. Toxicol. Pharmacol. 19 169–175 [DOI] [PubMed] [Google Scholar]
- el-Fawal H. A., Gong Z., Little A. R., Evans H. L. (1996). Exposure to methyl mercury results in serum autoantibodies to neurotypic and gliotypic proteins. Neurotoxicology 17 267–276 [PubMed] [Google Scholar]
- Endo T., Haraguchi K. (2010). High mercury levels in hair samples from residents of Taiji, a Japanese whaling town. Mar. Pollut. Bull. 60 743–747 [DOI] [PubMed] [Google Scholar]
- Eto K., Yasutake A., Miyamoto K., Tokunaga H., Otsuka Y. (1997). Chronic effects of methylmercury in rats. II. Pathological aspects. Tohoku J. Exp. Med. 182 197–205 [DOI] [PubMed] [Google Scholar]
- Farina M., Aschner M., Rocha J. B. (2011a). Oxidative stress in MeHg-induced neurotoxicity. Toxicol. Appl. Pharmacol. 256 405–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina M., Campos F., Vendrell I., Berenguer J., Barzi M., Pons S., Suñol C. (2009). Probucol increases glutathione peroxidase-1 activity and displays long-lasting protection against methylmercury toxicity in cerebellar granule cells. Toxicol. Sci. 112 416–426 [DOI] [PubMed] [Google Scholar]
- Farina M., Frizzo M. E., Soares F. A., Schwalm F. D., Dietrich M. O., Zeni G., Rocha J. B., Souza D. O. (2003). Ebselen protects against methylmercury-induced inhibition of glutamate uptake by cortical slices from adult mice. Toxicol. Lett. 144 351–357 [DOI] [PubMed] [Google Scholar]
- Farina M., Rocha J. B., Aschner M. (2011b). Mechanisms of methylmercury-induced neurotoxicity: Evidence from experimental studies. Life Sci. 89 555–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folch J., Lees M., Sloane Stanley G. H. (1957). A simple method for the isolation and purification of total lipids from animal tissues. J. Biol. Chem. 226 497–509 [PubMed] [Google Scholar]
- Franco J. L., Posser T., Dunkley P. R., Dickson P. W., Mattos J. J., Martins R., Bainy A. C., Marques M. R., Dafre A. L., Farina M. (2009). Methylmercury neurotoxicity is associated with inhibition of the antioxidant enzyme glutathione peroxidase. Free Radic. Biol. Med. 47 449–457 [DOI] [PubMed] [Google Scholar]
- Franco J. L., Teixeira A., Meotti F. C., Ribas C. M., Stringari J., Garcia Pomblum S. C., Moro A. M., Bohrer D., Bairros A. V., Dafre A. L., Santos A. R., Farina M. (2006). Cerebellar thiol status and motor deficit after lactational exposure to methylmercury. Environ. Res. 102, 22–28 [DOI] [PubMed] [Google Scholar]
- Fujita T., Nakamura N., Kumasaka R., Shimada M., Murakami R., Osawa H., Yamabe H., Okumura K. (2006). Comparison of lipid and fatty acid metabolism between minimal change nephrotic syndrome and membranous nephropathy. In Vivo 20(6B)891–893 [PubMed] [Google Scholar]
- Garg A., Simha V. (2007). Update on dyslipidemia. J. Clin. Endocrinol. Metab. 92 1581–1589 [DOI] [PubMed] [Google Scholar]
- Grandjean P., Murata K., Budtz-Jørgensen E., Weihe P. (2004). Cardiac autonomic activity in methylmercury neurotoxicity: 14-year follow-up of a Faroese birth cohort. J. Pediatr. 144 169–176 [DOI] [PubMed] [Google Scholar]
- Greenwood J., Steinman L., Zamvil S. S. (2006). Statin therapy and autoimmune disease: From protein prenylation to immunomodulation. Nat. Rev. Immunol. 6 358–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hintelmann H. (2010). Organomercurials. Their formation and pathways in the environment. Met. Ions Life Sci. 7 365–401 [DOI] [PubMed] [Google Scholar]
- Jin X., Chan H. M., Lok E., Kapal K., Taylor M., Kubow S., Mehta R. (2008). Dietary fats modulate methylmercury-mediated systemic oxidative stress and oxidative DNA damage in rats. Food Chem. Toxicol. 46 1706–1720 [DOI] [PubMed] [Google Scholar]
- Jin X., Hidiroglou N., Lok E., Taylor M., Kapal K., Ross N., Sarafin K., Lau A., De Souza A., Chan H. M., et al. (2012). Dietary selenium (Se) and vitamin E (V(E)) supplementation modulated methylmercury-mediated changes in markers of cardiovascular diseases in rats. Cardiovasc. Toxicol. 12 10–24 [DOI] [PubMed] [Google Scholar]
- Jin X., Lok E., Caldwell D., Mueller R., Kapal K., Liston V., Kubow S., Chan H. M., Mehta R. (2009). Dietary fats altered nephrotoxicity profile of methylmercury in rats. J. Appl. Toxicol. 29 126–140 [DOI] [PubMed] [Google Scholar]
- Kaur P., Heggland I., Aschner M., Syversen T. (2008). Docosahexaenoic acid may act as a neuroprotector for methylmercury-induced neurotoxicity in primary neural cell cultures. Neurotoxicology 29 978–987 [DOI] [PubMed] [Google Scholar]
- Keane W. F., Tomassini J. E., Neff D. R. (2011). Lipid abnormalities in patients with chronic kidney disease. Contrib. Nephrol. 171 135–142 [DOI] [PubMed] [Google Scholar]
- Kielar M. L., Jeyarajah D. R., Zhou X. J., Lu C. Y. (2003). Docosahexaenoic acid ameliorates murine ischemic acute renal failure and prevents increases in mRNA abundance for both TNF-alpha and inducible nitric oxide synthase. J. Am. Soc. Nephrol. 14 389–396 [DOI] [PubMed] [Google Scholar]
- Kris-Etherton P. M., Harris W. S., Appel L. J., and. American Heart Association. Nutrition Committee (2002). Fish consumption, fish oil, omega-3 fatty acids, and cardiovascular disease. Circulation 106 2747–2757 [DOI] [PubMed] [Google Scholar]
- Leaf A. (1990). Cardiovascular effects of fish oils. Beyond the platelet. Circulation 82 624–628 [DOI] [PubMed] [Google Scholar]
- Miida T., Takahashi A., Ikeuchi T. (2007). Prevention of stroke and dementia by statin therapy: Experimental and clinical evidence of their pleiotropic effects. Pharmacol. Ther. 113 378–393 [DOI] [PubMed] [Google Scholar]
- Moreira E. L., de Oliveira J., Nunes J. C., Santos D. B., Nunes F. C., Vieira D. S., Ribeiro-do-Valle R. M., Pamplona F. A., de Bem A. F., Farina M., et al. Age-related cognitive decline in hypercholesterolemic LDL receptor knockout mice (LDLr–/–): Evidence of antioxidant imbalance and increased acetylcholinesterase activity in the prefrontal cortex. J. Alzheimers Dis. (forthcoming) doi: 10.3233/JAD-2012-120541. [DOI] [PubMed] [Google Scholar]
- Mozaffarian D., Rimm E. B. (2006). Fish intake, contaminants, and human health: Evaluating the risks and the benefits. J. Am. Med. Assoc. 296 1885–1899 [DOI] [PubMed] [Google Scholar]
- Munzel T., Gori T., Bruno R. M., Taddei S. (2010). Is oxidative stress a therapeutic target in cardiovascular disease? Eur. Heart J. 31 2741–2748 [DOI] [PubMed] [Google Scholar]
- Murata K., Weihe P., Budtz-Jørgensen E., Jørgensen P. J., Grandjean P. (2004). Delayed brainstem auditory evoked potential latencies in 14-year-old children exposed to methylmercury. J. Pediatr. 144 177–183 [DOI] [PubMed] [Google Scholar]
- Myers G. J., Marsh D. O., Davidson P. W., Cox C., Shamlaye C. F., Tanner M., Choi A., Cernichiari E., Choisy O., Clarkson T. W. (1995). Main neurodevelopmental study of Seychellois children following in utero exposure to methylmercury from a maternal fish diet: Outcome at six months. Neurotoxicology 16 653–664 [PubMed] [Google Scholar]
- Ni M., Li X., Yin Z., Sidoryk-Węgrzynowicz M., Jiang H., Farina M., Rocha J. B., Syversen T., Aschner M. (2011). Comparative study on the response of rat primary astrocytes and microglia to methylmercury toxicity. Glia 59 810–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M., Kishimoto T., Tada M. (1995). The effect of methylmercury (CH3HgCl) on the production of endothelium-derived relaxing factor (EDRF) by cultured human umbilical vascular endothelial cells based on its anti-aggregatory effect on human platelets. Cell Biol. Toxicol. 11 303–311 [DOI] [PubMed] [Google Scholar]
- Rissanen T., Voutilainen S., Nyyssönen K., Lakka T. A., Salonen J. T. (2000). Fish oil-derived fatty acids, docosahexaenoic acid and docosapentaenoic acid, and the risk of acute coronary events: The Kuopio ischaemic heart disease risk factor study. Circulation 102 2677–2679 [DOI] [PubMed] [Google Scholar]
- Rodrigues L., Dutra M. F., Ilha J., Biasibetti R., Quincozes-Santos A., Leite M. C., Marcuzzo S., Achaval M., Gonçalves C. A. (2010). Treadmill training restores spatial cognitive deficits and neurochemical alterations in the hippocampus of rats submitted to an intracerebroventricular administration of streptozotocin. J. Neural Transm. 117 1295–1305 [DOI] [PubMed] [Google Scholar]
- Salonen J. T., Seppänen K., Lakka T. A., Salonen R., Kaplan G. A. (2000). Mercury accumulation and accelerated progression of carotid atherosclerosis: A population-based prospective 4-year follow-up study in men in eastern Finland. Atherosclerosis 148 265–273 [DOI] [PubMed] [Google Scholar]
- Santos D. B., Peres K. C., Ribeiro R. P., Colle D., dos Santos A. A., Moreira E. L., Souza D. O., Figueiredo C. P., Farina M. (2012). Probucol, a lipid-lowering drug, prevents cognitive and hippocampal synaptic impairments induced by amyloid β peptide in mice. Exp. Neurol. 233 767–775 [DOI] [PubMed] [Google Scholar]
- Shoji T., Nishizawa Y. (2006). Plasma lipoprotein abnormalities in hemodialysis patients–Clinical implications and therapeutic guidelines. Ther. Apher. Dial. 10 305–315 [DOI] [PubMed] [Google Scholar]
- Silva E. L., Moriel P., Chang Y. H., Abdalla D. S. (1995). Plasma antioxidant enzymes and oxidized lipoproteins in hypercholesterolemic rabbits. Biochem. Mol. Biol. Int. 36 679–687 [PubMed] [Google Scholar]
- Slotkin T. A., Kavlock R. J., Cowdery T., Orband L., Bartolome M., Gray J. A., Rehnberg B. F., Bartolome J. (1986). Functional consequences of prenatal methylmercury exposure: Effects on renal and hepatic responses to trophic stimuli and on renal excretory mechanisms. Toxicol. Lett. 34 231–245 [DOI] [PubMed] [Google Scholar]
- Smith K. L., Guentzel J. L. (2010). Mercury concentrations and omega-3 fatty acids in fish and shrimp: Preferential consumption for maximum health benefits. Mar. Pollut. Bull. 60 1615–1618 [DOI] [PubMed] [Google Scholar]
- Tamashiro H., Akagi H., Arakaki M., Futatsuka M., Roht L. H. (1984). Causes of death in Minamata disease: Analysis of death certificates. Int. Arch. Occup. Environ. Health 54 135–146 [DOI] [PubMed] [Google Scholar]
- Toimela T. A., Tahti H. (1995). Effects of mercury, methylmercury and aluminium on glial fibrillary acidic protein expression in rat cerebellar astrocyte cultures. Toxicol. In Vitro 9 317–325 [DOI] [PubMed] [Google Scholar]
- Wall R., Ross R. P., Fitzgerald G. F., Stanton C. (2010). Fatty acids from fish: The anti-inflammatory potential of long-chain omega-3 fatty acids. Nutr. Rev. 68 280–289 [DOI] [PubMed] [Google Scholar]
- Ware W. R. (2008). High cholesterol and coronary heart disease in younger men: The potential role of stress induced exaggerated blood pressure response. Med. Hypotheses 70 543–547 [DOI] [PubMed] [Google Scholar]
- Yasutake A., Nakano A., Miyamoto K., Eto K. (1997). Chronic effects of methylmercury in rats. I. Biochemical aspects. Tohoku J. Exp. Med. 182 185–196 [DOI] [PubMed] [Google Scholar]
- Yoshizawa K., Rimm E. B., Morris J. S., Spate V. L., Hsieh C. C., Spiegelman D., Stampfer M. J., Willett W. C. (2002). Mercury and the risk of coronary heart disease in men. N. Engl. J. Med. 347 1755–1760 [DOI] [PubMed] [Google Scholar]
- Zadelaar S., Kleemann R., Verschuren L., de Vries-Van der Weij J., van der Hoorn J., Princen H. M., Kooistra T. (2007). Mouse models for atherosclerosis and pharmaceutical modifiers. Arterioscler. Thromb. Vasc. Biol. 27 1706–1721 [DOI] [PubMed] [Google Scholar]
- Zalups R. K. (2000). Molecular interactions with mercury in the kidney. Pharmacol. Rev. 52 113–143 [PubMed] [Google Scholar]