

Abstract
The asymmetric localisation of core planar polarity proteins at apicolateral junctions is required to specify cell polarity in the plane of epithelia. This asymmetric distribution of the core proteins is proposed to require amplification of an initial asymmetry by feedback loops. In addition, generation of asymmetry appears to require the regulation of core protein levels, but the importance of such regulation and the underlying mechanisms is unknown. Here we show that ubiquitylation acts through more than one mechanism to control core protein levels in Drosophila, and that without this regulation cellular asymmetry is compromised. Levels of Dishevelled at junctions are regulated by a Cullin-3-Diablo/Kelch ubiquitin ligase complex, the activity of which is most likely controlled by neddylation. Furthermore, activity of the deubiquitylating enzyme Fat facets is required to maintain Flamingo levels at junctions. Notably, ubiquitylation does not alter the total cellular levels of Dishevelled or Flamingo, but only that of the junctional population. When junctional core protein levels are either increased or decreased by disruption of the ubiquitylation machinery, their asymmetric localisation is reduced and this leads to disruption of planar polarity at the tissue level. Loss of asymmetry by altered core protein levels can be explained by reference to feedback models for amplification of asymmetry.
Keywords: Planar polarity, PCP, Ubiquitination, Neddylation, Dishevelled, Drosophila
INTRODUCTION
Polarisation of cells in the plane of an epithelium is essential for morphogenesis and depends on a group of core planar polarity proteins (the ‘core proteins’), the function of which is conserved among diverse animal species. The core proteins localise asymmetrically within cells, and this asymmetric localisation regulates downstream processes such as polarised cell rearrangements, oriented cell divisions and the production of uniformly oriented arrays of structures such as hairs and cilia (McNeill, 2010; Goodrich and Strutt, 2011; Gray et al., 2011; Vichas and Zallen, 2011). The function of the core proteins has been best studied in the Drosophila wing. Here, the core proteins Frizzled (Fz), Dishevelled (Dsh) and Diego (Dgo) localise at the distal cell edge, whereas Strabismus (Stbm, also known as Van Gogh) and Prickle (Pk) localise proximally and Flamingo (Fmi, also known as Starry night) localises at both edges. This leads to the formation of an actin-rich trichome at the distal cell edge.
A key question is how the asymmetric localisation of core proteins is achieved. Correct asymmetry depends on the activity of all the other core proteins, and it is thought that an initial asymmetry on the proximodistal (PD) axis caused by an upstream cue is amplified by feedback interactions between the core proteins (Tree et al., 2002; Amonlirdviman et al., 2005; Le Garrec et al., 2006; Meinhardt, 2007). Feedback loops operate as bistable switches, enhancing the initially weak PD bias in core protein distribution, such that core proteins ultimately show high asymmetry in their localisation to the proximal and distal cell edges, and low levels of localisation on the anterior-posterior (AP) cell edges. Feedback could be caused by either positive or negative protein interactions: for instance, a possible positive interaction would be clustering of asymmetric complexes of the same polarity (Strutt et al., 2011), whereas inhibition between proximal and distal complex components would constitute a negative interaction (Tree et al., 2002; Jenny et al., 2005).
Interestingly, asymmetrically localised core proteins are not uniformly localised on the PD cell membranes but are instead organised in discrete puncta (Aigouy et al., 2010; Strutt et al., 2011), and the presence and size of these puncta correlate with the degree of asymmetry. We have previously provided evidence that they form by a two-step mechanism: first, a stable complex forms with Fz and Fmi on one side of the junctions and Stbm and Fmi on the other; second, the cytoplasmic components cause these stable complexes to congregate into discrete membrane subdomains (Strutt et al., 2011). Therefore, the activity of the cytoplasmic proteins appears to be a crucial feature of the feedback loops necessary to generate asymmetry.
It also appears that overall core protein levels must be regulated, as overexpression of the cytoplasmic proteins (Dsh, Pk and Dgo) causes excessive accumulation of the other core proteins at junctions and a loss of asymmetry (Feiguin et al., 2001; Tree et al., 2002; Bastock et al., 2003). In theory, both positive- and negative-feedback interactions require that core protein levels are modulated. For example, an excess of one or more core proteins could disrupt both positive interactions (by causing clustering to spread beyond the required domain) and negative interactions (by an excess of inhibition excluding competitor proteins from an increased proportion of membrane domains). Despite this, the degree to which asymmetry is dependent on the levels of core proteins at junctions has not been studied. In addition, it is not known whether it is the levels of all or of just some of the core proteins that must be regulated.
One mechanism by which cellular levels of proteins can be regulated is ubiquitylation, which can lead to either the proteasomal degradation of cytoplasmic proteins or the targeting of transmembrane proteins for internalisation and degradation in the lysosome (Hershko and Ciechanover, 1998; Traub and Lukacs, 2007; Clague et al., 2012). Fz and Dsh also act in canonical Wnt signalling and, interestingly, are known to be regulated by ubiquitylation in this context. In flies, Fz levels are modulated by the deubiquitylating enzyme dUBPY (Mukai et al., 2010), and in vertebrates Dsh homologues are regulated by ubiquitylation pathways involving KLHL12 and Cyld (Angers et al., 2006; Tauriello et al., 2010). However, no studies have described a role for ubiquitylation in regulating Fz and Dsh levels in planar polarity. Only one ubiquitylation pathway affecting planar polarity has been reported, which involves recruitment of murine Smurf2 to junctions by phosphorylated Dsh, leading to local degradation of Pk (Narimatsu et al., 2009). Other mechanisms by which ubiquitylation might regulate core protein levels and asymmetry in vivo are yet to be identified.
Ubiquitylation of target proteins requires the sequential action of a cascade of E1, E2 and E3 enzymes (Hershko and Ciechanover, 1998). Ubiquitin is directly conjugated to the E2 enzyme via E1 enzyme activity, and the E3 ligase then transfers ubiquitin to a lysine residue within the target protein. In addition to ubiquitylation, proteins can be modified by the conjugation of several other ubiquitin-like molecules. Nedd8 is one such molecule, and it is attached to substrates using a similar E1, E2 and E3 enzyme cascade (Rabut and Peter, 2008). Neddylation can alter protein stability or activity, and the best characterised targets of neddylation are Cullin E3 ligases. Neddylation promotes recruitment of the E2 enzyme to Cullins, and cycles of neddylation and deneddylation are required for proper functioning of Cullins (Wu et al., 2005).
Here, we characterise two distinct ubiquitylation pathways, which act through the core proteins Dsh and Fmi. Both loss of ubiquitylation and increased ubiquitylation alter core protein levels at junctions and reduce core protein asymmetry. Furthermore, we show that ubiquitylation does not affect the entire cellular pool of Fmi and Dsh, but only the junctional population.
MATERIALS AND METHODS
Fly stocks and genetics
Fly stocks are described in FlyBase. fzP21, stbm6, pkpk-sple13, dshV26, dgo380, fafBX4, fafF08, lqf7F1 and kelDE1 are null alleles. Cul-3gft2 is a loss-of-function allele and Nedd8AN015, fzN21 and fzJ22 are hypomorphs. faf7F1 is a semi-viable EMS-induced hypomorphic allele (this work) and dorIR-33733 is a GD RNAi line (VDRC).
Transgenes used were ArmP-fmi-EGFP (Strutt et al., 2011) and ActP-FRT-polyA-FRT-dsh-ECFP (Strutt and Strutt, 2007). UAS-Myc-dbo, ActP-FRT-polyA-FRT-Myc-dbo and ActP-FRT-polyA-FRT-faf-EGFP express Myc-tagged Dbo and Faf tagged at its C-terminus with EGFP using the vectors pUAST and pActP-FRT-polyA-FRT. The dboΔ25.1 knockout is a deletion of the entire open reading frame by homologous recombination using the pRK2 vector as described (Huang et al., 2008). dbo small hairpin RNAi (shRNAi) lines contain 21-mer sequences of dbo (CGAGCGTTATGATCCACAAAC and CACTGAACTAAATATGCTACG) in pVALIUM-20 (Ni et al., 2011) and do not overlap with each other or dboIR-105407.
Mitotic clones were induced using the FLP/FRT system and Ubx-FLP. Clones of ActP-FRT-polyA-FRT-Myc-dbo were made using Ubx-FLP and Δ2-3. Overexpression of UAS-Myc-dbo or RNAi lines used the GAL4/UAS system with ptc-GAL4. For pupal wing stainings, larvae expressing Ubc12, Uba3 and Roc1a RNAi were raised at 18°C, and shifted to 29°C at 0 hours after prepupa formation (APF), whereas larvae expressing Cul-3, dbo, kel and faf RNAi were raised at 25°C and shifted to 29°C at 0 hours APF.
Transgenics were generated by Bestgene, Genetivision and Genetic Services.
RNAi screening
For the E2 screen, 32 genes encoding E2 ubiquitin ligases were identified by the presence of the ubiquitin conjugating enzyme E2 domain (IPR000608) within a predicted open reading frame. This correlates with the number of D. melanogaster genes analysed in a study of the evolution of the E2 gene family in metazoans (Michelle et al., 2009). The initial screen was carried out using GD lines from the VDRC or RNAi lines from NIG-FLY, but KK lines were subsequently screened when they became available.
BTB-encoding genes were selected based on the presence of a BTB/POZ domain (IPR013069). Additional RNAi lines for genes containing a BTB/Kelch-associated domain (IPR011705), Kelch repeat type 1 (IPR006652), Kelch repeat type 2 (IPR011498) or Kelch-type beat propeller (IPR015915) were also screened.
In the adult wing screens, RNAi lines were crossed to MS1096-GAL4 virgins at 29°C. Male wings were mounted, unless the insertion was on the X chromosome, or as stated. For the pupal wing screens, RNAi lines were crossed to ptc-GAL4 and raised at 18°C until 0 hours APF when white prepupae were collected and aged at 29°C for 26 hours before dissection and immunostaining with antibodies against Fmi and E-cadherin (Ecad; Shotgun - FlyBase).
Immunolabelling
Pupal wings were dissected at 28 hours APF at 25°C or at 25-26 hours at 29°C and imaged as previously (Strutt, 2001). Primary antibodies used were 1.5 μg/ml mouse anti-Fmi 74 [DSHB (Usui et al., 1999)], 1/300 rabbit anti-Fz (Bastock and Strutt, 2007), 1/1000 rat anti-Stbm (Strutt and Strutt, 2008), 1/1000 rat anti-Dsh (Strutt et al., 2006), 1/20 rat anti-Ecad [DSHB (Oda et al., 1994)], 1/2500 guinea pig anti-Senseless (Sens) (Nolo et al., 2000), 1/100 rabbit anti-Distalless (Dll) (Panganiban et al., 1995), 1/400 mouse anti-β-gal (Promega), 1/4000 rabbit anti-β-gal (Cappel), 1/4000 rabbit anti-GFP (Abcam) and 40 μg/ml mouse anti-Myc 9E10 (DSHB). A rabbit anti-Dsh serum (1/1000) was directed against amino acids 480-623, and a rat anti-Pk antibody (1/25) was directed against amino acids 514-769 and affinity purified. Phalloidin-A568 (1/100) was from Molecular Probes.
Biochemistry and western analysis
Venus-Cul3, Myc-Dbo and Myc-Kelch fusions were made in pAVW and pAMW Gateway vectors and also shuttled into pcDNA3.1 (Invitrogen). Myc-Dbo deletions are of amino acids 72-169 (DboΔBTB), 174-276 (DboΔBACK), 323-end (DboΔKR) and 370-end (DboΔKR2-6) and are in pAc5.1 (Invitrogen). Dsh-ECFP is in pAc5.1 and EGFP-Dsh is in pEGFP-C1 (Clontech).
For Cul-3-Dbo pulldowns, S2 cell lysates were made in RIPA buffer [50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% NP40, 0.5% Na deoxycholate, 0.1% SDS, 1× protease inhibitor cocktail (Roche)]. Pulldowns used goat anti-Myc agarose (Abcam). For Dbo-Dsh pulldowns, S2 or COS-7 cells were treated with 10 μM MG132 for 5 hours prior to making lysates in IP buffer [20 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5% Triton X-100, 1 mM Na3VO4, 5 mM NaF, 1× protease inhibitor cocktail (Roche)]. Immunoprecipitations used rabbit anti-GFP serum (Abcam) and protein G Sepharose (Xerxes). Westerns were probed with 1/2000 rabbit anti-GFP (Abcam), 1/1000 mouse anti-GFP JL8 (Clontech) or 0.2 μg/ml mouse anti-Myc 9E10 (DSHB).
For pupal wing westerns, 28-hour pupal wings were dissected into sample buffer, and one pupal wing equivalent was loaded per lane. Westerns were probed with 1.5 μg/ml mouse anti-Fmi 74 (DSHB), 1/200 rabbit anti-Dsh (this work), or 1/5000 Actin AC-40 mouse antibody (Sigma), and imaged on a UVIprochemie gel documentation system (UVItec). Bands from westerns of three biological replicates were quantitated in ImageJ (NIH).
Quantitation of protein levels and asymmetry at junctions
RNAi lines were expressed using the MS1096-GAL4 driver. Wings were fixed and immunostained in parallel, and the same region of the wing was imaged at the same magnification and settings. To measure asymmetry, a 400×400 pixel region was selected and ImageJ used to mark a five pixel line on all junctions (∼200 per wing). The mean intensity and angle of each junction were measured, and a background value (non-junctional staining) subtracted. Excel (Microsoft) was used to bin angles into two categories: >45° (PD junctions) and <45° (AP junctions) from horizontal. The mean intensity of staining at PD and AP junctions was then calculated. Measurements were taken from eight wings of each genotype and the significance was determined using an unpaired t-test.
To compare absolute levels of junctional protein, a threshold value for wild-type images was empirically determined that highlighted the junctions, such that 4-5% of the image was above the threshold. The same threshold value was applied to the Ubc12, Cul-3 and faf images. Overall intensity was mean intensity above the threshold (with background subtracted) multiplied by area above the threshold. Measurements were taken from at least eight wings of each genotype and the significance was determined using an unpaired t-test.
RESULTS
Regulation of core proteins by ubiquitylation and neddylation
In a genetic screen for enhancers of a hypomorphic fz phenotype in the Drosophila eye (Strutt and Strutt, 2003), we identified a new allele of fat facets (faf) (supplementary material Fig. S1A-D), which encodes a deubiquitylating enzyme (Huang et al., 1995). Ommatidia in faf mutant eye clones often contain extra photoreceptors due to defective Notch-Delta signalling (Overstreet et al., 2004), but in ommatidia with normal numbers of photoreceptor cells planar polarity defects were also observed (supplementary material Fig. S1E,F). Furthermore, in clones of cells lacking faf activity in the pupal wing, levels of all core proteins at apicolateral junctions were reduced (Fig. 1A-E), whereas levels of adherens junction proteins were unaffected (supplementary material Fig. S1G). In photoreceptor recruitment, Faf acts by deubiquitylating the Epsin Liquid facets (Lqf) (Chen et al., 2002). However, loss of lqf did not cause a decrease in the levels of any of the core proteins (Fig. 1F; data not shown), suggesting that the effect of faf on core proteins was independent of lqf.
Fig. 1.

Regulation of core proteins in Drosophila by ubiquitylation and neddylation. (A-E) fafBX4 clones. (F) lqf7F1 clone. (G-N) Ubc12IR-7573R-3 (G-L), Uba3IR-17139 (M) and Roc1aIR-32399 (N), expressed with ptc-GAL4. (O,P) Nedd8AN015 clones. Immunostaining is for Fmi (green in A,F,G,M-O), Stbm (green in B,H), Fz (green in C,I), Dsh (green in D,J,P), Pk (green in E,K) and Ecad (green in L). Clones are marked by loss of β-gal (A-F) or GFP (O,P) in red. The band of stronger staining in L is the wing vein. Note the poor proliferation and small cells in Nedd8 clones (O,P). The small lower clone in P does not show an increase in Dsh staining and cell size is normal, probably because it was induced late and there is perdurance of Nedd8 activity. Yellow bars mark the ptc-GAL4 domain. Scale bar: 20 μm.
To identify ubiquitylation pathways acting on the core proteins, we carried out an in vivo RNAi screen. We focused on E2 enzymes, as there are only 32 E2s in the Drosophila genome (Michelle et al., 2009), whereas there is a single ubiquitin E1 enzyme and several hundred putative E3 ligases. RNAi lines against a single E2, Ubc12, showed the reciprocal phenotype to that of faf in the pupal wing: an increase in core protein levels at apicolateral junctions (Fig. 1G-L; supplementary material Table S1).
Ubc12 does not encode a conventional ubiquitin E2, but the E2 for the related small modifying protein Nedd8 (Rabut and Peter, 2008). Other components of the Nedd8 pathway were therefore examined for effects on core protein levels. RNAi against the Nedd8 E1 subunit Uba3 and the Nedd8 E3 subunit Roc1a also showed an increase in core protein levels (Fig. 1M,N; supplementary material Table S2), as did mutant clones of Nedd8 itself (Fig. 1O,P).
A Cul-3-BTB E3 ubiquitin ligase complex regulates core protein levels and is the likely target of neddylation
One or more of the core proteins could be directly neddylated, or the effect of the Nedd8 pathway could be indirect via neddylation of another target. The best characterised substrates of Nedd8 are Cullin E3 ubiquitin ligases (Rabut and Peter, 2008). RNAi against Cullin-3 (Cul-3), but none of the other Cullins, caused an increase in core protein levels at junctions, as did Cul-3 null mutant clones (Fig. 2A-D; supplementary material Table S3). As Cul-3 is known to be modified by Nedd8 in flies (Wu et al., 2005), this suggests that Cul-3 is the likely target of Nedd8 in this context.
Fig. 2.
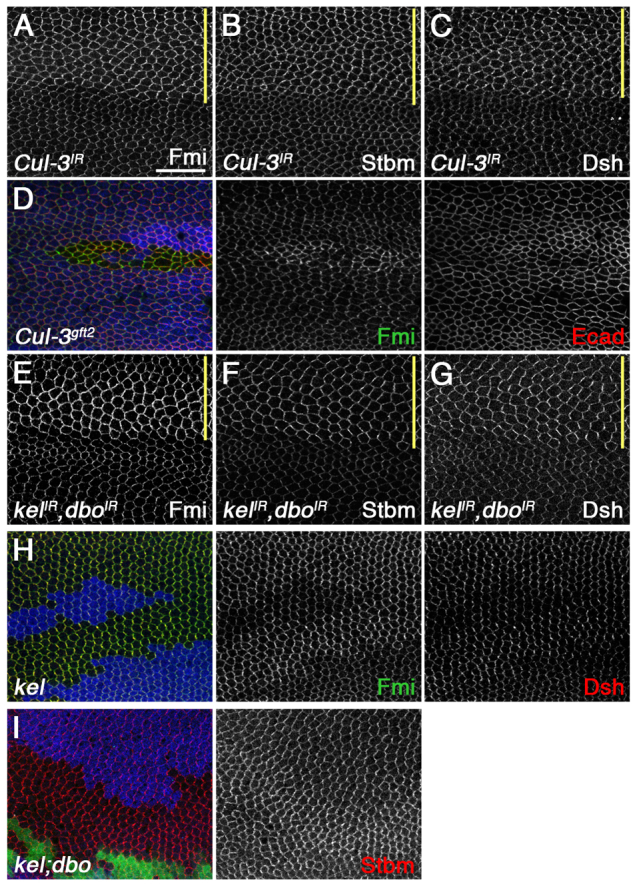
A Cul-3-BTB protein complex regulates core protein levels. (A-C) ptc-GAL4/Cul-3IR-109415. (D) Cul-3gft2 clone. (E-G) ptc-GAL4/dboIR-105407; kelIR-JF01768/UAS-Dcr2. (H) kelDE1 clone. Immunostaining is for Fmi (A,D,E,H), Stbm (B,F), Dsh (C,G,H) and Ecad (D). Clones are marked by loss of β-gal (blue). Note the poor proliferation in the Cul-3gft2 clone (D). (I) Pupal wing with kelDE1 clones [marked by loss of GFP (green) staining] and overlapping dboΔ25.1 clones [marked by loss of β-gal (blue)], stained for Stbm (red): most of the tissue is mutant for kel, and there is an increase in Stbm staining in tissue that is also mutant for dbo. Yellow bars mark the ptc-GAL4 domain. Scale bar: 20 μm.
Cul-3 E3 ligase subunits act through substrate-specific partners of the BTB (BR-C, ttk and bab) family (Petroski and Deshaies, 2005). Screening of BTB proteins in the pupal wing (supplementary material Table S4) revealed that knockdown of diablo (dbo) and kelch (kel), which encode closely related Kelch family BTB proteins, caused subtle increases in core protein levels at apical junctions (supplementary material Fig. S2A-C). Simultaneous knockdown of dbo and kel caused a robust increase in core protein levels at junctions, suggesting that these two proteins act redundantly (Fig. 2E-G; compare with supplementary material Fig. S2A,C). kel null mutant animals are viable but female sterile (Schüpbach and Wieschaus, 1989), with no defects in trichome polarity (data not shown). However, core protein levels at junctions increased in kel mutant clones, consistent with the RNAi phenotype (Fig. 2H). We ruled out off-target effects for dbo in two ways. First, we generated two independent small hairpin RNAi (shRNAi) lines (Ni et al., 2011): both showed an increase in core protein levels at junctions when co-expressed with a kel RNAi line, but not when expressed alone (supplementary material Fig. S2D-G). Second, we knocked out the dbo open reading frame by homologous recombination: like kel mutants, dbo mutants were viable, with no trichome polarity phenotype (data not shown). No increase in core protein levels was observed in dbo mutant clones, possibly owing to perdurance of protein (supplementary material Fig. S2H), but induction of overlapping dbo and kel clones revealed that loss of both kel and dbo caused a greater increase in core protein levels than loss of kel alone (Fig. 2I).
Cul-3 binds to Kel in tissue culture (Hudson and Cooley, 2010): we confirmed this, finding that both Dbo and Kel were binding partners of Cul-3 in Drosophila S2 cells (Fig. 3B). Cul-3 binds to BTB proteins via their BTB and BACK domains (Petroski and Deshaies, 2005). Consistent with this, the Dbo-Cul-3 interaction depended on the BTB and BACK domains of Dbo, but not the Kelch repeats (Fig. 3C).
Fig. 3.

Ubiquitylation regulates core protein asymmetry. (A) Myc-Dbo, showing the position of the BTB and BACK domains and the Kelch repeats (KR). Beneath are the deletion constructs used in the pulldown experiments. (B,C) Western blots showing pulldown of Venus-Cul-3 by Myc-tagged Dbo and Kelch (B) and the full-length (FL) and deleted forms of Myc-Dbo (C). Loading of input is 10% of pulldown. Molecular mass (kDa) is shown to the left. Anti-GFP recognises Venus-Cul-3. DboΔBTB and DboΔBTB no longer bind to Cul-3, whereas DboΔKR still binds but only weakly, suggesting that deletion of the Kelch repeats might perturb the secondary structure of the BTB/BACK domains. (D,E) Mean intensity of Fmi staining on proximodistal (PD) junctions (>45° from horizontal) relative to anterior-posterior (AP) junctions (<45° from horizontal) in wings expressing wIR-30033, Ubc12IR-7375R-3, Cul-3IR-109415 and fafIR-2956 using MS1096-GAL4 (D) or in wings from w1118 and kelDE1; dboΔ25.1 double-mutant flies (E). Asymmetry was scored below vein 4 (see supplementary material Fig. S3A-G). As MS1096-GAL4 is more strongly expressed dorsally, asymmetry in these wings was scored on the dorsal surface only. Error bars indicate s.e.m.; **P<0.01. (F,G) kelDE1; dboΔ25.1 double-mutant wing (F) and adult eye section (G). Trichome swirls are only seen below vein 4, and ∼1% of ommatidia are mispolarised. Arrow points to a mispolarised ommatidium.
Regulation of core protein levels at junctions via ubiquitylation pathways is required for robust asymmetry
Although defects in ubiquitylation alter core protein levels at junctions, it is not clear whether this is important for asymmetry and trichome polarity. We therefore quantitated core protein asymmetry in pupal wings in which ubiquitylation was altered. For Ubc12, Cul-3 and faf, RNAi lines were expressed ubiquitously in the wing using the MS1096-GAL4 driver: this caused clear alterations in core protein levels compared with a control RNAi line, similar to that seen using ptc-GAL4 (supplementary material Fig. S3A-E). Overall asymmetry was reduced by 35-50% when protein levels at junctions were increased by expression of Ubc12 and Cul-3 RNAi (Fig. 3D; supplementary material Fig. S3A-C) or in kel; dbo double-mutant wings (Fig. 3E; supplementary material Fig. S3F,G), or when protein levels were decreased by loss of faf activity (Fig. 3D; supplementary material Fig. S3D,E). In addition, cell packing was disrupted (supplementary material Fig. S3H); this might be a consequence of reduced asymmetry of the core proteins (Classen et al., 2005), or it could be due to effects of altered ubiquitylation that are independent of core proteins. Notably, cell packing was essentially normal in faf wings, even though core protein asymmetry was reduced, suggesting that the change in core protein asymmetry is not an indirect result of defects in cell packing. Thus, tight regulation of junctional core protein levels appears to be necessary for maximal cellular asymmetry.
It is known that the mechanisms leading to trichome placement are very robust, and trichomes can form at the correct cell edge in wings in which there is little or no visible asymmetry of core protein localisation, such as in pk or dgo mutants (Strutt and Strutt, 2007), apparently due to downstream amplifying mechanisms. Consistent with this, loss of faf activity delayed trichome formation (supplementary material Fig. S3I), but trichome orientation was normal (supplementary material Fig. S3J). However, kel; dbo double mutants showed localised trichome swirling in the wing (Fig. 3F) and occasional defects in ommatidial polarity in the eye (Fig. 3G). Knockdown of Ubc12 and Cul-3 caused pleiotropic effects, preventing analysis of trichome polarity defects. However, expression of Ubc12 RNAi using ptc-GAL4 caused trichomes to point towards the ptc-GAL4 domain (supplementary material Fig. S3K), indicative of misoriented cell polarity.
Neddylation and ubiquitylation pathways target Dsh and Fmi
Abnormal ubiquitylation causes an increase or decrease of all the core proteins at junctions. The most probable scenario is that one core protein is the target of the ubiquitylation or deubiquitylation pathways and altered levels of this protein drive a similar alteration in the others. Likely targets of Dbo/Kel are the cytoplasmic proteins Dsh, Pk and Dgo, as their overexpression is known to cause accumulation of all the other core proteins at junctions (Feiguin et al., 2001; Tree et al., 2002; Bastock et al., 2003). Furthermore, in vertebrate canonical Wnt signalling, Dsh levels are regulated by a Cul-3-BTB E3 ubiquitin ligase complex (Angers et al., 2006), making Dsh a plausible substrate of a similar complex in planar polarity signalling in Drosophila.
Notably, loss of pk or dgo activity did not affect the ability of Ubc12 or Cul-3 RNAi to cause an accumulation of core proteins at junctions (compare supplementary material Fig. S4C,D and S4G,H with Fig. 2A and Fig. 1G, respectively; supplementary material Table S5). However, the increase in core protein levels at junctions was suppressed when dshV26 clones were induced in wings expressing either Ubc12 or Cul-3 RNAi (Fig. 4A,B), or in dshV26; kelDE1 double-mutant clones (Fig. 4C). Fz and Stbm are required for normal recruitment of Dsh to the plasma membrane (Axelrod, 2001; Shimada et al., 2001; Bastock et al., 2003). Loss of their activities also suppressed the effects of Cul-3, Ubc12 and kel; dbo RNAi (supplementary material Fig. S4A,B,E,F,J,K, Table S5), again consistent with Dsh being the target.
Fig. 4.

Dsh is the target of Cul-3-Dbo/Kel. (A-D) dshV26 FRT19A/FRT19A; ptc-GAL4/+; Ubc12IR-7375R-3, Ubx-FLP/+ (A), dshV26 FRT19A/Ubx-FLP FRT19A; ptc-GAL4/Cul-3IR-109415 (B), dshV26 FRT19A/Ubx-FLP FRT19A; kelDE1 FRT40/arm-lacZ FRT40 (C) and dshV26 FRT19A/Ubx-FLP FRT19A; ptc-GAL4/+; fafIR-2956/+ (D). Wings are stained for Stbm (green), Dsh (red) and β-gal (blue, C); yellow bars mark the ptc-GAL4 domain. Compare Stbm levels in the same wing in the presence or absence of dsh activity. (A,B) Stbm no longer accumulates in dsh clones. (C) In dsh; kel double-mutant tissue (arrow), Stbm no longer accumulates compared with kel mutant tissue (loss of blue), and levels are similar to those upon loss of dsh alone (arrowhead). (D) The dsh clone crosses the ptc-GAL4 boundary, and where fafIR is expressed (above the arrow) Stbm levels are decreased. Scale bar: 20 μm. (E-G) Western blots showing pulldown of Myc-tagged Dbo and Kelch by GFP-tagged Dsh in S2 cells (E,G) and COS-7 cells (F). Loading of input is 1% of pulldown. Molecular mass (kDa) is shown to the left.
Interestingly, whereas loss of fz and dsh completely suppressed the increase in core protein levels in wings expressing Cul-3 RNAi (Fig. 4B; supplementary material Fig. S4A), there was still a residual increase in core protein levels in wings expressing Ubc12 RNAi (supplementary material Fig. S4E,I). This suggests that Ubc12 might also affect core proteins by a second mechanism that is independent of Cul-3 and Dsh. For example, core proteins could be regulated via another Cullin homologue, or by direct neddylation of core proteins (Jones et al., 2008).
By contrast, loss of dsh activity did not alter the reduction of core protein levels caused by faf RNAi, indicating that Dsh is not the target of Faf (Fig. 4D; supplementary material Table S5). Likewise, core protein levels were still reduced when faf RNAi was expressed in fz, stbm, pk or dgo mutant backgrounds (supplementary material Fig. S4L-O, Table S5). Therefore, Faf is acting upstream of the other core proteins, consistent with it acting at the level of Fmi itself, either targeting Fmi directly or acting via an unknown adaptor protein. Note that because Fmi is normally required to recruit all of the other core proteins to junctions (Feiguin et al., 2001; Shimada et al., 2001; Strutt, 2001; Tree et al., 2002; Bastock et al., 2003), we were unable to directly determine whether Fmi is required for the effect of faf knockdown on the other core proteins.
The effects of Cul-3-Dbo/Kel and Faf on core protein levels could be mediated by direct regulation of Dsh and Fmi protein levels or via transcriptional regulation. Tagged forms of Dsh and Fmi expressed under ubiquitous promoters also increased or decreased when Cul-3 or faf RNAi was expressed, suggesting that their effect is post-transcriptional (supplementary material Fig. S5A,B).
To test whether Dsh could be a direct target of Dbo/Kel, we expressed tagged Dsh and Dbo or Kel in S2 cells, and found that Dbo co-immunoprecipitated with Dsh (Fig. 4E), although only poorly, suggesting a weak or transient interaction. BTB domain proteins bind their substrates via the Kelch repeats (Petroski and Deshaies, 2005), and, consistent with the Dbo-Dsh interaction being specific, Dbo lacking all but one of its Kelch repeats no longer binds Dsh (Fig. 4G). Kel was not pulled down with Dsh in S2 cells, possibly owing to its poor expression (Fig. 4E), but both Dbo and Kel were pulled down with Dsh in COS-7 cells (Fig. 4F).
Ubiquitylation targets junctional Dsh for degradation
We next asked whether alterations in core protein levels at junctions reflect a change in the total cellular amount of protein or whether the ubiquitylation machinery acts specifically on a junctional population. Interestingly, although Dsh levels at junctions were increased several fold when Ubc12 or Cul-3 RNAi was expressed throughout the wing (see quantitation in supplementary material Fig. S6A-C,H), western blotting revealed that overall Dsh levels did not change (Fig. 5A,B). Overall Dsh levels were also unaltered in kel; dbo double-mutant wings (Fig. 5C,D; see also supplementary material Fig. S6D,E,I). Thus, ubiquitylation specifically regulates Dsh levels at junctions.
Fig. 5.

Ubiquitylation targets junctional Dsh and Fmi for degradation. (A-H) Western blots probed for Dsh (A,C,E) or Fmi (G) and Actin (A,C,E,G) and quantitation of Dsh (B,D,F) and Fmi (H) levels, normalised to Actin loading control. Extracts are from male pupal wings expressing wIR-30033, Ubc12IR-7375R-3, Cul-3IR-109415, fafIR-2956 or UAS-Myc-dbo with MS1096-GAL4 (A,E,G), or from w1118 or kel; dbo pupal wings (C). The Dsh blot shows two bands, which correspond to hyperphosphorylated and unphosphorylated forms of Dsh (Yanagawa et al., 1995). Quantitation is from western blots of three biological replicates. Error bars indicate s.e.m.; NS, not significant; *P=0.02. (I-K) ptc-GAL4, UAS-Myc-dbo adult wing (K) or pupal wings (I,J) stained for Dsh (green), Ecad (red in I), Fmi (red in J) and Myc (blue in I). Yellow bars mark the ptc-GAL4 domain. (L,M) dorIR-33733/w; ptc-GAL4/+ (L) or dorIR-33733/w; ptc-GAL4/+; fafIR-2956/+ (M) pupal wings stained for Fmi (green) and Dsh (red). Images are within the ptc-GAL4 domain. Scale bars: 20 μm in I,J; 5 μm in L,M.
We then investigated whether expression of Dbo is sufficient to alter Dsh levels in vivo. Expression of UAS-Myc-dbo using the ptc-GAL4 driver caused strong trichome swirling in the adult wing (Fig. 5K) and a strong reduction in the levels of Dsh at junctions in pupal wings (Fig. 5I). Interestingly, expression of UAS-Myc-dbo throughout the wing caused a significant decrease in total Dsh levels (Fig. 5E,F), consistent with Dbo not only redistributing Dsh from junctions to the cytoplasm but also targeting Dsh for degradation.
We then considered how the ubiquitylation machinery might recognise junctional Dsh. One possibility is that the Dbo/Kel E3 ligase subunits are localised to junctions; however, expression of Myc-Dbo at low levels revealed a diffuse localisation, with only a slight enrichment in plasma membrane puncta that do not colocalise with core proteins (supplementary material Fig. S7A). Instead, we think it more likely that junctional Dsh is marked for ubiquitylation by post-translational modification, as is frequently the case for substrates of Cullin E3 ligases (Petroski and Deshaies, 2005) (see Discussion).
Surprisingly, decreased Dsh at junctions was accompanied by a reduction in Fmi levels (Fig. 5J), suggesting that when Dsh is removed from junctions by Dbo/Kel it can also promote, either directly or indirectly, the internalisation of Fmi. This is in line with previous observations that Fmi levels increase in a dsh mutant background (Shimada et al., 2001; Strutt and Strutt, 2008). One possibility is that Dsh acts as an endocytic adaptor for Fmi; however, we were unable to detect any colocalisation of Fmi and Dsh in intracellular vesicles (supplementary material Fig. S7B,C), implying that any association between them during Fmi internalisation would be transient (see Discussion). Blocking lysosomal maturation with mutations in deep orange (dor) results in intracellular accumulation of Fmi (Strutt and Strutt, 2008), consistent with some population of Fmi normally being sent to the lysosome for degradation. Knockdown of dor did not however cause any accumulation of Dsh (supplementary material Fig. S7D), suggesting that Dsh is not degraded in the lysosome, and must instead be proteasomally degraded.
Loss of Faf increases lysosomal degradation of Fmi
We next investigated whether overall Fmi levels are altered in wings expressing faf RNAi. Again, overall Fmi levels showed negligible change even though Fmi levels at junctions were substantially decreased (Fig. 5G,H; supplementary material Fig. S6F,G,J), suggesting that only the junctional population of Fmi is susceptible to regulation by the ubiquitylation machinery. The simplest model is that Fmi is targeted for internalisation by ubiquitylation, and that the deubiquitylating activity of Faf allows Fmi to return to the plasma membrane. Importantly, simultaneous knockdown of faf and dor causes more extensive accumulation of Fmi in intracellular vesicles than knockdown of dor alone (Fig. 5L,M), consistent with a failure of Fmi deubiquitylation normally promoting lysosomal degradation of Fmi.
We also investigated whether Faf localises with Fmi on endosomes. EGFP-tagged Faf showed a diffuse cytoplasmic distribution and we were unable to see any colocalisation with Fmi (supplementary material Fig. S7E,F).
Ubiquitylation of Dsh by Cul-3-Dbo/Kel does not modulate canonical Wingless signalling
In vertebrate embryos, loss of the Dbo/Kel-related protein KLHL12 increases canonical Wingless (Wg) signalling, presumably by stabilising Dsh (Angers et al., 2006). By contrast, reduced ubiquitylation did not increase overall levels of Dsh in fly wings (Fig. 5A-D). Consistent with this, we also saw no ectopic Wg signalling in wing discs expressing Cul-3 or kel; dbo RNAi (Fig. 6A-D). Furthermore, there was no reduction in Wg signalling when Dbo was overexpressed (Fig. 6E,F), despite the striking effect on Dsh levels at junctions (Fig. 5I,J) and the reduction in overall Dsh levels (Fig. 5E,F).
Fig. 6.

Cul-3, Dbo and Kel do not regulate canonical Wg signalling. (A-F) ptc-GAL4/Cul-3IR-109415 (A,B), ptc-GAL4/dboIR-105407; kelIR-JF01768/UAS-Dcr2 (C,D) and ptc-GAL4/UAS-Myc-dbo (E,F) wing discs, stained for Sens (green or white in A,C,E) or Dll (green or white in B,D,F). ptc-GAL4 domain is marked by Fmi (red in A-D) or Myc (red in E,F). Note that Cul-3IR wing discs are distorted, so Fmi staining is uneven. Wg signalling promotes Sens and Dll expression, and is unaltered. Scale bar: 20 μm.
DISCUSSION
In this study we have identified two ubiquitylation pathways that control the levels of planar polarity proteins at junctions, acting at the level of Dsh and Fmi. Mutations in ubiquitylation pathway components show that both increased and decreased levels of core proteins at junctions result in decreased asymmetry, suggesting that protein levels must be finely tuned in order for asymmetric localisation to be maximised.
With regard to adult planar polarity patterning, the decrease in asymmetry we observed does not lead to severe phenotypes in the wing and eye (Fig. 3F,G). However, it is well established that both tissues have downstream amplifiers to ensure a clearly polarised outcome, as correct polarisation can be achieved with little or no visible core protein asymmetry (Strutt and Strutt, 2007). However, in more dynamic contexts in which such downstream amplifiers might not exist or have time to act, such a reduction of core protein asymmetry would be expected to have severe effects on the final polarity of the tissue.
It is widely accepted that core protein polarity is produced by amplification of a weak initial cue by feedback loops. Such feedback loops could be caused by positive protein interactions, which promote the local accumulation of core proteins of the same species, or by negative interactions that inhibit their accumulation. The actual contribution of positive- or negative-feedback interactions is currently unknown, although mechanisms for both have been proposed (Tree et al., 2002; Jenny et al., 2005; Strutt et al., 2011). In practice, both are likely to operate, as amplification by positive interactions alone might lead to an uninhibited spread of polarised domains (Gierer and Meinhardt, 1972). Notably, one of the simplest forms of negative-feedback interaction is to limit the supply of substrate and we propose that this is the role of the pathways that we have characterised here.
Our observations can be simply explained in the light of such models. If an amplification system operates for local clustering of polarised core protein complexes [as we previously proposed (Strutt et al., 2011)], then the presence of excess core proteins in the junctions will lead to the excess growth of polarised domains and a possible reduction in the degree of overall cellular asymmetry. Conversely, a reduced level of core proteins will result in less efficient clustering of polarised protein complexes (as proteins of the same species would meet less often), again leading to a reduction in cellular asymmetry (Fig. 7A).
Fig. 7.

Models for regulation of core protein levels at junctions by ubiquitylation. (A) Regulation of asymmetry by core protein levels. Green/orange semicircles represent plasma membrane puncta of asymmetrically localised Fz/Stbm complexes. Proximal is to the left and distal to the right. Under normal conditions, feedback loops ensure that Fz is mostly distal, and Stbm is mostly proximal within each cell. An excess of core proteins at junctions driven by loss of Dsh ubiquitylation leads to suboptimal feedback amplification, such that the domain of polarised complexes spreads too far around the cell periphery. Conversely, too little Fmi at junctions results in asymmetric molecular complexes failing to meet and therefore either clustering (feedback caused by positive protein interactions) or mutual inhibition (feedback caused by negative interactions) is abrogated. (B) Regulation of Dsh and Fmi levels by Dbo/Kel and Faf. Dsh is recruited to junctions by Fz and phosphorylated, and this correlates with clustering of asymmetric complexes of the same orientation. Dbo/Kel recognises junctional Dsh, and ubiquitylates excess Dsh leading to its removal from junctions and degradation, most likely by the proteasome (pale blue). Removal of Dsh from junctions also leads to endocytosis of Fmi. An unknown E3 ligase also promotes Fmi internalisation and lysosomal targeting, either by direct ubiquitylation of Fmi (as shown) or of an adaptor protein. Faf normally deubiquitylates some proportion of this internalised Fmi (or a Fmi adaptor) and allows Fmi to be recycled to the plasma membrane.
The cytoplasmic core proteins (Dsh, Pk and Dgo) appear to be of particular importance in the clustering of asymmetric complexes (Strutt et al., 2011), and overexpression of any of them appears to cause excessive clustering (Feiguin et al., 2001; Tree et al., 2002; Bastock et al., 2003). Our data suggest that Dsh is a direct target of a Cul-3-Dbo/Kel ubiquitin ligase complex, and the ubiquitylation and consequent removal of Dsh from junctions is thus a mechanism by which local Dsh levels are regulated.
Interestingly, in ubiquitylation pathway mutants, total cellular levels of Dsh are unaltered, even though levels at apical junctions increase several fold. This suggests that, in this context, ubiquitylation is a specific regulatory event at junctions. We do not know how the ubiquitylation machinery recognises junctional Dsh. One attractive possibility is that phosphorylated Dsh is the target for the Dbo/Kel E3 ligase. Phosphorylation is commonly used as a signal for the recruitment of ubiquitin ligases to their substrates (Petroski and Deshaies, 2005), and Dsh recruitment to junctions correlates with its hyperphosphorylation (Axelrod, 2001; Shimada et al., 2001). Interestingly, in kel; dbo double-mutant wings there is a small but significant increase in the upper, hyperphosphorylated Dsh band at the expense of the lower, unphosphorylated form (Fig. 5C; supplementary material Fig. S6K). Thus, loss of ubiquitylation could lead to an excessive accumulation of hyperphosphorylated Dsh at junctions, consistent with the proposal that ubiquitylation pathways normally act to remove hyperphosphorylated Dsh.
We note that ubiquitylation apparently does not act to remove the total population of hyperphosphorylated junctional Dsh, and so we speculate that either all of the Dsh at junctions is not phosphorylated on the relevant sites to trigger ubiquitylation, or that some of the hyperphosphorylated population of Dsh is protected. Biochemical analyses to directly show that hyperphosphorylated Dsh is specifically ubiquitylated have not proved feasible given the difficulty of obtaining large quantities of tissue of the relevant stage and the small proportion of cellular Dsh that is likely to be modified at any particular time. Similarly, experiments in cell lines are problematic as there is no suitable system for generating a polarised junctional population of Drosophila Dsh in culture, and again the relevant proportion of the total cellular population of Dsh would be very small.
It has been reported previously that, in dsh mutant clones, Fmi levels, but not Fz levels, increase (Shimada et al., 2001; Strutt and Strutt, 2008). This is indicative of a role for Dsh in removing some population of Fmi from junctions, possibly one that is not stably incorporated into asymmetric complexes. We now present further evidence for this role of Dsh, as overexpression of Myc-Dbo causes not only removal of the junctional population of Dsh but also the removal of Fmi. It remains to be determined whether Dsh co-traffics with Fmi, and whether this is the normal mode of removal of Dsh from junctions; however, it is interesting to note that in vertebrates Dsh has been reported to act as an endocytic adapter (Chen et al., 2003; Yu et al., 2007), and in some contexts such adaptors are marked for internalisation by ubiquitylation (reviewed by Traub and Lukacs, 2007). Nevertheless, we were unable to detect colocalisation of Fmi and Dsh in intracellular vesicles, suggesting that if Dsh does act as an adaptor for Fmi and is internalised with it, then Dsh must rapidly dissociate from Fmi before Fmi enters sorting endosomes. Interestingly, if this were the case, then Fmi (or another associated protein) would also have to be ubiquitylated if Fmi were to be subsequently targeted to the lysosome (Clague et al., 2012).
Fmi is known to accumulate in late endosomes when lysosomal targeting is blocked (Strutt and Strutt, 2008), consistent with it being internalised by a ubiquitin-dependent mechanism. Here, we identify the deubiquitylating enzyme Faf as a key regulator of junctional levels of Fmi. Furthermore, we show that loss of faf enhances the intracellular accumulation of Fmi when lysosomal targeting is blocked, suggesting that failure to deubiquitylate Fmi (or an adaptor) causes excess degradation of Fmi. As Fmi levels at junctions decrease in faf mutants, this suggests that recycling of Fmi is essential to maintain sufficient Fmi at junctions. However, we do not know whether Fmi is a direct ubiquitylation target as we were unable to co-immunoprecipitate Fmi and Faf in S2 cells due to their poor expression.
To summarise, we propose a model in which protein levels at junctions are regulated at multiple levels (Fig. 7B). Junctional, phosphorylated Dsh promotes clustering of asymmetric complexes. The level of Dsh at junctions is regulated by Dbo/Kel so as to prevent excess clustering, with ubiquitylated Dsh being removed from junctions and targeted for proteasomal degradation. Dsh could also act as an adaptor in order to remove excess Fmi that is not in complexes from junctions. Fmi is itself also a direct or indirect target of ubiquitylation, and this leads to internalisation of Fmi and targeting of Fmi to the lysosome. Recycling of this population of Fmi to the plasma membrane is promoted by activity of the deubiquitinase Faf.
Interestingly, in vertebrates Dsh has also been reported to be a target of a Cul-3-BTB E3 ligase. However, loss of KLHL12, a vertebrate BTB-Kelch family protein, causes gain-of-function Wnt signalling defects but no planar polarity defects (Angers et al., 2006). Nevertheless, planar polarity defects are seen when KLHL12 is overexpressed, probably owing to excess degradation of Dsh. By contrast, we do not detect any gain-of-function Wnt signalling phenotypes following a reduction in Cul-3-Dbo/Kel activity in flies, consistent with the unaltered total cellular levels of Dsh. Furthermore, even though total Dsh levels are significantly reduced when Dbo is overexpressed, this again does not noticeably affect Wnt signalling, suggesting that the remaining Dsh is sufficient for Wnt signalling activity. This suggests that the Cul-3-Kel/Dbo ubiquitylation mechanism plays a specific role in planar polarity in flies.
Supplementary Material
Acknowledgments
We thank the Bellen, Carroll, Chien, Fischer, Hong and Skeath labs, The Bloomington Stock Center, Developmental Studies Hybridoma Bank (DSHB), BioServ UK, Vienna Drosophila RNAi Center (VDRC), National Institute of Genetics (NIG) and Drosophila RNAi Screening Center (DRSC) for fly stocks, antibodies and vectors.
Footnotes
Funding
This work was supported by a Wellcome Trust Senior Fellowship to D.S. and the Medical Research Council (MRC). Confocal facilities were provided by the Wellcome Trust and Yorkshire Cancer Research. Deposited in PMC for immediate release.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.089656/-/DC1
References
- Aigouy B., Farhadifar R., Staple D. B., Sagner A., Röper J.-C., Jülicher F., Eaton S. (2010). Cell flow reorients the axis of planar polarity in the wing epithelium of Drosophila. Cell 142, 773–786 [DOI] [PubMed] [Google Scholar]
- Amonlirdviman K., Khare N. A., Tree D. R. P., Chen W.-S., Axelrod J. D., Tomlin C. J. (2005). Mathematical modeling of planar cell polarity to understand domineering nonautonomy. Science 307, 423–426 [DOI] [PubMed] [Google Scholar]
- Angers S., Thorpe C. J., Biechele T. L., Goldenberg S. J., Zheng N., MacCoss M. J., Moon R. T. (2006). The KLHL12-Cullin-3 ubiquitin ligase negatively regulates the Wnt-beta-catenin pathway by targeting Dishevelled for degradation. Nat. Cell Biol. 8, 348–357 [DOI] [PubMed] [Google Scholar]
- Axelrod J. D. (2001). Unipolar membrane association of Dishevelled mediates Frizzled planar cell polarity signaling. Genes Dev. 15, 1182–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastock R., Strutt D. (2007). The planar polarity pathway promotes coordinated cell migration during Drosophila oogenesis. Development 134, 3055–3064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastock R., Strutt H., Strutt D. (2003). Strabismus is asymmetrically localised and binds to Prickle and Dishevelled during Drosophila planar polarity patterning. Development 130, 3007–3014 [DOI] [PubMed] [Google Scholar]
- Chen X., Zhang B., Fischer J. A. (2002). A specific protein substrate for a deubiquitinating enzyme: Liquid facets is the substrate of Fat facets. Genes Dev. 16, 289–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W., ten Berge D., Brown J., Ahn S., Hu L. A., Miller W. E., Caron M. G., Barak L. S., Nusse R., Lefkowitz R. J. (2003). Dishevelled 2 recruits beta-arrestin 2 to mediate Wnt5A-stimulated endocytosis of Frizzled 4. Science 301, 1391–1394 [DOI] [PubMed] [Google Scholar]
- Clague M. J., Coulson J. M., Urbé S. (2012). Cellular functions of the DUBs. J. Cell Sci. 125, 277–286 [DOI] [PubMed] [Google Scholar]
- Classen A. K., Anderson K. I., Marois E., Eaton S. (2005). Hexagonal packing of Drosophila wing epithelial cells by the planar cell polarity pathway. Dev. Cell 9, 805–817 [DOI] [PubMed] [Google Scholar]
- Feiguin F., Hannus M., Mlodzik M., Eaton S. (2001). The ankyrin repeat protein Diego mediates Frizzled-dependent planar polarization. Dev. Cell 1, 93–101 [DOI] [PubMed] [Google Scholar]
- Gierer A., Meinhardt H. (1972). A theory of biological pattern formation. Kybernetik 12, 30–39 [DOI] [PubMed] [Google Scholar]
- Goodrich L. V., Strutt D. (2011). Principles of planar polarity in animal development. Development 138, 1877–1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray R. S., Roszko I., Solnica-Krezel L. (2011). Planar cell polarity: coordinating morphogenetic cell behaviors with embryonic polarity. Dev. Cell 21, 120–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A., Ciechanover A. (1998). The ubiquitin system. Annu. Rev. Biochem. 67, 425–479 [DOI] [PubMed] [Google Scholar]
- Huang Y., Baker R. T., Fischer-Vize J. A. (1995). Control of cell fate by a deubiquitinating enzyme encoded by the fat facets gene. Science 270, 1828–1831 [DOI] [PubMed] [Google Scholar]
- Huang J., Zhou W., Watson A. M., Jan Y. N., Hong Y. (2008). Efficient ends-out gene targeting in Drosophila. Genetics 180, 703–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson A. M., Cooley L. (2010). Drosophila Kelch functions with Cullin-3 to organize the ring canal actin cytoskeleton. J. Cell Biol. 188, 29–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenny A., Reynolds-Kenneally J., Das G., Burnett M., Mlodzik M. (2005). Diego and Prickle regulate Frizzled planar cell polarity signalling by competing for Dishevelled binding. Nat. Cell Biol. 7, 691–697 [DOI] [PubMed] [Google Scholar]
- Jones J., Wu K., Yang Y., Guerrero C., Nillegoda N., Pan Z. Q., Huang L. (2008). A targeted proteomic analysis of the ubiquitin-like modifier nedd8 and associated proteins. J. Proteome Res. 7, 1274–1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Garrec J. F., Lopez P., Kerszberg M. (2006). Establishment and maintenance of planar epithelial cell polarity by asymmetric cadherin bridges: a computer model. Dev. Dyn. 235, 235–246 [DOI] [PubMed] [Google Scholar]
- McNeill H. (2010). Planar cell polarity: keeping hairs straight is not so simple. Cold Spring Harb. Perspect. Biol. 2, a003376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinhardt H. (2007). Computational modelling of epithelial patterning. Curr. Opin. Genet. Dev. 17, 272–280 [DOI] [PubMed] [Google Scholar]
- Michelle C., Vourc’h P., Mignon L., Andres C. R. (2009). What was the set of ubiquitin and ubiquitin-like conjugating enzymes in the eukaryote common ancestor? J. Mol. Evol. 68, 616–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai A., Yamamoto-Hino M., Awano W., Watanabe W., Komada M., Goto S. (2010). Balanced ubiquitylation and deubiquitylation of Frizzled regulate cellular responsiveness to Wg/Wnt. EMBO J. 29, 2114–2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narimatsu M., Bose R., Pye M., Zhang L., Miller B., Ching P., Sakuma R., Luga V., Roncari L., Attisano L., et al. (2009). Regulation of planar cell polarity by Smurf ubiquitin ligases. Cell 137, 295–307 [DOI] [PubMed] [Google Scholar]
- Ni J. Q., Zhou R., Czech B., Liu L. P., Holderbaum L., Yang-Zhou D., Shim H. S., Tao R., Handler D., Karpowicz P., et al. (2011). A genome-scale shRNA resource for transgenic RNAi in Drosophila. Nat. Methods 8, 405–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolo R., Abbott L. A., Bellen H. J. (2000). Senseless, a Zn finger transcription factor, is necessary and sufficient for sensory organ development in Drosophila. Cell 102, 349–362 [DOI] [PubMed] [Google Scholar]
- Oda H., Uemura T., Harada Y., Iwai Y., Takeichi M. (1994). A Drosophila homolog of cadherin associated with armadillo and essential for embryonic cell-cell adhesion. Dev. Biol. 165, 716–726 [DOI] [PubMed] [Google Scholar]
- Overstreet E., Fitch E., Fischer J. A. (2004). Fat facets and Liquid facets promote Delta endocytosis and Delta signaling in the signaling cells. Development 131, 5355–5366 [DOI] [PubMed] [Google Scholar]
- Panganiban G., Sebring A., Nagy L., Carroll S. (1995). The development of crustacean limbs and the evolution of arthropods. Science 270, 1363–1366 [DOI] [PubMed] [Google Scholar]
- Petroski M. D., Deshaies R. J. (2005). Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 6, 9–20 [DOI] [PubMed] [Google Scholar]
- Rabut G., Peter M. (2008). Function and regulation of protein neddylation. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep. 9, 969–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schüpbach T., Wieschaus E. (1989). Female sterile mutations on the second chromosome of Drosophila melanogaster. I. Maternal effect mutations. Genetics 121, 101–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada Y., Usui T., Yanagawa S., Takeichi M., Uemura T. (2001). Asymmetric colocalization of Flamingo, a seven-pass transmembrane cadherin, and Dishevelled in planar cell polarization. Curr. Biol. 11, 859–863 [DOI] [PubMed] [Google Scholar]
- Strutt D. I. (2001). Asymmetric localization of frizzled and the establishment of cell polarity in the Drosophila wing. Mol. Cell 7, 367–375 [DOI] [PubMed] [Google Scholar]
- Strutt H., Strutt D. (2003). EGF signaling and ommatidial rotation in the Drosophila eye. Curr. Biol. 13, 1451–1457 [DOI] [PubMed] [Google Scholar]
- Strutt D., Strutt H. (2007). Differential activities of the core planar polarity proteins during Drosophila wing patterning. Dev. Biol. 302, 181–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strutt H., Strutt D. (2008). Differential stability of flamingo protein complexes underlies the establishment of planar polarity. Curr. Biol. 18, 1555–1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strutt H., Price M. A., Strutt D. (2006). Planar polarity is positively regulated by casein kinase Iepsilon in Drosophila. Curr. Biol. 16, 1329–1336 [DOI] [PubMed] [Google Scholar]
- Strutt H., Warrington S. J., Strutt D. (2011). Dynamics of core planar polarity protein turnover and stable assembly into discrete membrane subdomains. Dev. Cell 20, 511–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauriello D. V., Haegebarth A., Kuper I., Edelmann M. J., Henraat M., Canninga-van Dijk M. R., Kessler B. M., Clevers H., Maurice M. M. (2010). Loss of the tumor suppressor CYLD enhances Wnt/beta-catenin signaling through K63-linked ubiquitination of Dvl. Mol. Cell 37, 607–619 [DOI] [PubMed] [Google Scholar]
- Traub L. M., Lukacs G. L. (2007). Decoding ubiquitin sorting signals for clathrin-dependent endocytosis by CLASPs. J. Cell Sci. 120, 543–553 [DOI] [PubMed] [Google Scholar]
- Tree D. R. P., Shulman J. M., Rousset R., Scott M. P., Gubb D., Axelrod J. D. (2002). Prickle mediates feedback amplification to generate asymmetric planar cell polarity signaling. Cell 109, 371–381 [DOI] [PubMed] [Google Scholar]
- Usui T., Shima Y., Shimada Y., Hirano S., Burgess R. W., Schwarz T. L., Takeichi M., Uemura T. (1999). Flamingo, a seven-pass transmembrane cadherin, regulates planar cell polarity under the control of Frizzled. Cell 98, 585–595 [DOI] [PubMed] [Google Scholar]
- Vichas A., Zallen J. A. (2011). Translating cell polarity into tissue elongation. Semin. Cell Dev. Biol. 22, 858–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J. T., Lin H. C., Hu Y. C., Chien C. T. (2005). Neddylation and deneddylation regulate Cul1 and Cul3 protein accumulation. Nat. Cell Biol. 7, 1014–1020 [DOI] [PubMed] [Google Scholar]
- Yanagawa S.-i., van Leeuwen F., Wodarz A., Klingensmith J., Nusse R. (1995). The dishevelled protein is modified by wingless signaling in Drosophila. Genes Dev. 9, 1087–1097 [DOI] [PubMed] [Google Scholar]
- Yu A., Rual J. F., Tamai K., Harada Y., Vidal M., He X., Kirchhausen T. (2007). Association of Dishevelled with the clathrin AP-2 adaptor is required for Frizzled endocytosis and planar cell polarity signaling. Dev. Cell 12, 129–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.