

Abstract
A fundamental process in biology is the de novo formation and morphogenesis of polarized tubules. Although these processes are essential for the formation of multiple metazoan organ systems, little is known about the molecular mechanisms that regulate them. In this study, we have characterized several steps in tubule formation and morphogenesis using the mouse kidney as a model system. We report that kidney mesenchymal cells contain discrete Par3-expressing membrane microdomains that become restricted to an apical domain, coinciding with lumen formation. Once lumen formation has been initiated, elongation occurs by simultaneous extension and additional de novo lumen generation. We demonstrate that lumen formation and elongation require afadin, a nectin adaptor protein implicated in adherens junction formation. Mice that lack afadin in nephron precursors show evidence of Par3-expressing membrane microdomains, but fail to develop normal apical-basal polarity and generate a continuous lumen. Absence of afadin led to delayed and diminished integration of nectin complexes and failure to recruit R-cadherin. Furthermore, we demonstrate that afadin is required for Par complex formation. Together, these results suggest that afadin acts upstream of the Par complex to regulate the integration and/or coalescence of membrane microdomains, thereby establishing apical-basal polarity and lumen formation/elongation during kidney tubulogenesis.
Keywords: Kidney development, Lumen, Tubulogenesis, Afadin (Mllt4), Polarity, Nectin, Cadherin, Par complex
INTRODUCTION
Formation and elongation of epithelial tubules is essential for the structure and function of many organs. Although general principles for mammalian tubulogenesis have been described, tubules and their associated lumens form by diverse mechanisms specific to each organ (Datta et al., 2011). In the kidney, epithelial tubules develop from cell types of distinct embryonic origins using different cellular mechanisms (Little et al., 2010). The ureteric bud forms as an outgrowth of a pre-existing tubule and undergoes many rounds of branching to form the renal collecting system (Cebrián et al., 2004). By contrast, the metanephric mesenchyme becomes compacted to form a pretubular aggregate and undergoes a mesenchymal-to-epithelial transition, forming a sphere of polarized epithelia with de novo generation of a central lumen. This sphere, called the renal vesicle, elongates to form a primordial tubule called the S-shaped body, ultimately giving rise to a nephron. Each developing nephron tubule will fuse with the tip of an adjacent ureteric bud tubule. A schematic of these stages is illustrated in Fig. 1A. The cellular and molecular mechanisms that underlie the spatial and temporal formation of de novo lumen formation in developing nephrons are largely unknown. Indeed, much of our knowledge of renal epithelial lumen formation and morphogenesis is derived from cell culture models that may not accurately represent the process in vivo (Datta et al., 2011; Schlüter and Margolis, 2009).
Fig. 1.

Par-3-containing membrane domains exist prior to lumen formation. (A) General schematic of lumen formation in developing nephrons. Condensed mesenchyme (CM) undergoes compaction to form the pretubular aggregate (PA). The PA then becomes a polarized epithelial sphere called the renal vesicle (RV) that contains a central lumen. The RV forms a primordial tubule, the S-shaped body (SB), the lumen of which is continuous with the ureteric bud (UB) lumen. RV, black; RV lumen, red; UB, green; UB lumen, blue. (B) Quantification of stages of nephrogenesis at E14.5 using NCAM to identify nephron precursors and aPKC to identify lumens. Results are mean±s.d. from three mice. PA, pretubular aggregate; RV, renal vesicle; ERV, extended renal vesicle; SB, S-shaped body. (C-G) Localization of Par3 (green), aPKC (blue or white, as indicated) and NCAM (red) in E14.5 kidneys is shown. (C) In condensed mesenchyme, Par3-containing domains colocalize with NCAM at the cell membrane. (D) In pretubular aggregates, some Par3-containing domains segregate from NCAM. (E-F″) A primitive renal vesicle with two foci containing Par3 (E,E′) and aPKC (E′) is shown. An arrow indicates the larger focus; an arrowhead indicates the smaller focus. NCAM marks the basolateral domain. (F-F″) A 3D reconstruction (5 μm thickness) of E. (F) Merged image; (F′) Par3; (F″) aPKC. (G) A later stage (mature) renal vesicle shows that Par3 is now concentrated at apical junctions while aPKC remains apical. The two circular cells (asterisks) are dividing; others are more elongated. Results are representative of sections from three mice. Scale bars: 5 μm.
Regulation of adhesion between cells impacts many aspects of kidney development (Marciano et al., 2011; Perantoni, 1999). Adherens junctions are comprised of at least two families of adhesion receptors, cadherins and nectins, that interact with each other and signal mainly through their intracellular adaptors, the catenins and afadin (Mllt4 - Mouse Genome Informatics), respectively (Gumbiner, 2005; Nishimura and Takeichi, 2009; Tachibana et al., 2000; Takai et al., 2008a). Although cadherins play key roles in junction formation, in vitro data suggest that nectin/afadin complexes act upstream of cadherins, initiating junction formation and recruiting cadherins to cellular junctions (Honda et al., 2003a; Honda et al., 2003b). Nectin/afadin complexes are also required for tight junction formation, which occurs subsequent to adherens junction formation (Fukuhara et al., 2002a; Fukuhara et al., 2002b; Ooshio et al., 2010). The nectin family consists of nectins 1 to 4, which interact on the surface of the same cell to form cis-dimers, and then interact with nectin dimers in trans on the apposing cell to form cell-cell adhesions (Harrison et al., 2012; Narita et al., 2011; Rikitake et al., 2012; Satoh-Horikawa et al., 2000). Afadin, initially identified as an F-actin binding protein, is the main intracellular regulator of nectins 1 to 3 (Mandai et al., 1997; Takahashi et al., 1999), and an effector of the small GTPase Rap1 (Boettner and Van Aelst, 2009). It directly interacts with nectins via binding of its PDZ domain to the C-terminal residues of nectins. Afadin promotes nectin-nectin binding both in cis and trans (Kurita et al., 2011). In addition, afadin may promote interactions with F-actin through its interactions with α-catenin, ponsin, ADIP, LMO7 and vinculin (Takai et al., 2008b). Mice that lack afadin have defects in gastrulation and disorganization of the ectoderm (Ikeda et al., 1999). More recently, conditional deletion of afadin from intestinal epithelia has shown that afadin is essential for recruiting nectins to apical junctions and for maintaining barrier function (Tanaka-Okamoto et al., 2011).
In the current work, we identify afadin as a key regulator of de novo lumen formation and elongation in the nephron. Our characterization of afadin gene mutants reveals that de novo tubule formation in the mouse kidney is regulated by the establishment of pre-apical membrane microdomains on multiple adjacent cells that eventually coalesce or integrate to form a continuous lumen. Afadin is required for this coalescence, and mice that lack afadin do not form a continuous lumen in nephrons. This study represents the first high-resolution subcellular characterization of de novo lumen formation in the mouse kidney that can act as a model for future studies on tubule formation. Furthermore, it defines a previously unappreciated role for afadin in one specific step in this process.
MATERIALS AND METHODS
Animals
We crossed Afadinfl/fl (floxed afadin) females to Afadinfl/+; Pax3-creTg males (Beaudoin et al., 2012) to obtain Afadinfl/fl; Pax3-creTg mutant mouse embryos. Using the same strategy, we generated Afadinfl/fl; FoxD1-creTg (Humphreys et al., 2008) and Afadinfl/fl; Six2-eGFP-creTg mice (Kobayashi et al., 2008). Mice were maintained on mixed genetic backgrounds and genotyped by standard PCR. Procedures were performed according to UTSW IACUC-approved guidelines.
Histology and immunofluorescence
For paraffin wax-embedded sections, kidneys (E14.5, P0, 4 weeks) were fixed in 4% paraformaldehyde in PBS overnight at 4°C, embedded in paraffin wax and sectioned at 5 μm. Antigen retrieval was performed with Trilogy (Cell Marque). Non-paraffin sections were fixed for 2 hours in 4% PFA/PBS or Nakane fixative (periodate-lysine-paraformaldehyde) (McLean and Nakane, 1974), permeabilized with 0.3% Triton X-100/PBS (PBST) and blocked with 10% donkey sera/PBST. Sections were incubated with primary antibodies overnight (4°C), then with fluorophore-conjugated secondary antibodies and mounted with Prolong Gold (Invitrogen).
Antibodies
All primary antibodies were used 1:100 unless stated otherwise: afadin (Sigma, A0224), nectin 1 (MBL, D146-3), nectin 2 (MBL, D083-3), nectin 3 (MBL, D084-3 and a gift from Y. Takai, Osaka University, Japan), NCAM (DSHB, 5B8; GeneTex H28-123) (Dodd et al., 1988), R-cadherin (1:50, DSHB, MRCD5) (Matsunami and Takeichi, 1995), Six2 (1:200, a gift from A. McMahon, Harvard University, MA, USA) (Kobayashi et al., 2008), Meis1/2 (Santa Cruz Biotechnology, sc-10599), laminin (Sigma, 9393), Jag1 (Sigma, A37872), Lef1 (Cell Signaling), WT1 (Millipore, 05-753), Epha4 (BD Biosciences, 610471), Par6b (SCBT, sc-67392), Par3 (Millipore, 07-330), aPKCζ (SCBT, C-20) and phalloidin 647 (Invitrogen, 42008A). Secondary antibodies were purchased from Jackson ImmunoResearch (PA, USA).
Imaging and statistical analysis
Confocal imaging was performed on a Zeiss LSM510 META laser scanning confocal microscope. Immunofluorescence and light microscopy were performed with a Nikon TE300 inverted fluorescence microscope (AxioCam HRc camera and AxioVision 4.5 software). Images were analyzed using ImageJ software. Images were minimally processed and resampled to 300 dpi using Adobe Photoshop. The 3D reconstruction of a z-stack was performed with the LSM510 3-D module by generating 32 images, each separated by a 4° rotation. All data shown are mean±s.d. Statistical significance was tested using two-way ANOVA or unpaired two-tailed Student’s t-test, as indicated.
Electron microscopy
Dissected embryonic kidneys at E14.5 were fixed in 4% paraformaldehyde and 1% glutaraldehyde in 0.1 M sodium cacodolate for 2 hours, and then fixed with 2.5% glutaraldehyde in the same buffer. Kidneys were post-fixed in 1% buffered OsO4, en bloc stained in 2% uranyl acetate, dehydrated and embedded in EMbed-812 resin. Sections were cut on a Leica EM UC6 ultramicrotome and stained with 2% uranyl acetate and lead citrate. Images were acquired on a FEI Tecnai G2 Spirit.
RESULTS
Lumen formation during nephron development
Discrete stages of nephrogenesis have been broadly categorized based on anatomical shape, beginning with the condensed mesenchyme, pretubular aggregate, renal vesicle, comma-shaped body and, finally, the S-shaped body. A detailed characterization of nephron lumen formation, however, is lacking. To gain a better understanding of this process, we imaged primordial nephrons three-dimensionally and re-characterized stages of nephron formation based on anatomical shape, lumen formation, expression of polarity proteins, and fusion of the nephron tubule and ureteric bud. These studies were performed at embryonic day 14.5 (E14.5), as this age contains all stages of nephron precursors.
In early nephrogenesis, the condensed mesenchymal cells undergo compaction to form a sphere of cells called the pretubular aggregate, which lacks a central lumen (Saxén and Sariola, 1987). To determine when cells become polarized during the process of tubule formation, as well as confirm that pretubular aggregates truly lack any lumen (and are not renal vesicles sectioned through a non-luminal plane), we turned to 3D imaging. We immunostained kidneys with antibodies to aPKC, an apical polarity protein that delineates lumens, and NCAM, a plasma membrane protein whose distribution is relatively uniform in mesenchymal cells but becomes restricted to the basolateral surface when lumen formation occurs. We generated z-stacks of all nephron structures within sections and quantified the percentage of each structure (n=3 embryos) (Fig. 1B). Interestingly, we identified pretubular spheres of compacted mesenchymal cells adjacent to a UB stalk and branching tip that did not have a lumen or apical-basal polarity, as indicated by the absence of apical recruitment of aPKC, and uniform, rather than basolateral distribution, of NCAM. We defined these structures as pretubular aggregates (supplementary material Table S1), which accounted for 8±4% of all nephron structures (Fig. 1B).
To examine the molecular events that precede nephron lumen formation in vivo, we asked whether apical polarity proteins could be detected prior to lumen formation. We examined condensed mesenchyme for the apical polarity protein Par3 (Pard3 - Mouse Genome Informatics) and found, surprisingly, that Par3 was located at the cell surface in small clusters, interspersed with NCAM (Fig. 1C). In pretubular aggregates, more surface clusters of Par3 were noted, some of which had little or no NCAM (Fig. 1D; supplementary material Table S1). This implies that plasma membrane delivery of Par3 may precede exclusion of NCAM from membrane microdomains, and suggests these Par3-enriched membrane domains might be apical domain precursors, or ‘pre-apical domains’, similar to what is observed in in vitro lumen formation models (Bryant et al., 2010).
We next questioned whether de novo lumen formation occurs via generation of numerous microlumens that expand and fuse to form larger lumens, as in pancreatic tubulogenesis (Kesavan et al., 2009; Villasenor et al., 2010), or via generation of a single apical focus that expands thereafter. As renal vesicles are thought to have a single lumen, we predicted there would be a single apical focus. As lumen formation was initiated, we observed cell aggregates with one or two apical foci containing Par3 and aPKC in which one focus often appeared larger than the other, perhaps reflecting later lumen maturity (Fig. 1E,E′; 1F-F″ are a 3D projection of 1E′). Given these findings, we defined the primitive renal vesicle (PRV) as having one or two small apical foci containing apical aPKC and Par3, and lacking NCAM (supplementary material Table S1). In addition to colocalization of Par3 with aPKC at the apical surface, Par3 was also present at apical junctions and in surrounding cell surface clusters. At the next stage, the structures contained a single expanded, ovoid lumen with central clearing. In these structures, Par3 was concentrated at apical cell-cell junctions with little at the apical surface, whereas aPKC remained apical (Fig. 1G; supplementary material Table S1). We refer to these structures as mature renal vesicles (MRV). From our static images, we could not determine whether the two foci present in some primitive renal vesicles would merge to form a single lumen or whether each would form a lumen in two separate renal vesicles. We did not observe two adjacent mature renal vesicles with expanded lumens, which suggested the former. However, previous data have shown that a single ureteric bud branch may be associated with more than one nephron, supporting the latter possibility (Cebrián et al., 2004).
After the initial stages of de novo lumen formation, there are several ways in which a lumen might elongate. We present three possible models for such morphogenetic processes in nephrons as Fig. 2A. Macroscopically, a lumen could extend from a preexisting lumen (1), generate additional de novo lumens that subsequently fuse (2) or combine extension and de novo lumen generation (3). We examined aPKC-labeled 3D projections of confocal z-stacks at each stage of nephron lumen formation beginning with the primitive renal vesicle (Fig. 2B-I′). At the mature renal vesicle stage, the lumen had expanded to form an open, ovoid lumen (Fig. 2C). In the next stages, which we called the ‘extended renal vesicle’ stages 1-5 (ERV1-5), we first observed a small distal extension of the RV lumen towards the UB tip (ERV1, Fig. 2D), which subsequently became more elongated (ERV2, Fig. 2E). Surprisingly, ERV lumen extension did not continue until it joined with the UB, but rather a small discreet lumen formed in the distal segment (ERV3, Fig. 2F). Several additional lumens were generated de novo (ERV4, Fig. 2G), which could be seen as separate lumens when the 3D projection was rotated (compare Fig. 2G′ with 2G″) or a depth plot of the projection was performed (Fig. 2G″′). These lumens began to coalesce, but had not yet fused with the ureteric bud lumen at ERV 5 (Fig. 2H,H′). Ultimately, the nephron-UB lumen became continuous at the S-shaped body (SB) stage (Fig. 2I-I′), as has recently been reported by others (Kao et al., 2012). We conducted these experiments using sections fixed with either 4% paraformaldehyde or Nakane (not shown), suggesting these lumen morphologies are unlikely to be an artefact of fixation. Together, these results suggest that elongation of the nephron lumen occurs via extension as well as de novo lumen formation, consistent with model 3 in Fig. 2A.
Fig. 2.

Three-dimensional nephron lumen elongation. (A) Models of nephron lumen elongation. The renal vesicle (RV) lumen may extend towards the ureteric bud (UB) lumen (1), additional de novo lumens may form and coalesce (2), or a combination of extension and de novo lumen generation may exist (3). (B-I′) Three-dimensional reconstructions from confocal z-stacks of immunostaining with aPKC (green) and NCAM (not shown). aPKC marks the apical surface, thereby demarcating the lumen. (B,C) A primitive renal vesicle (B) (PRV) has one or two small lumens that expand to form a mature RV with a single expanded lumen (C). (D) The lumen extends distally towards the ureteric bud lumen (UBL) in stage 1 of the extended renal vesicle (ERV1). (E,F) Lumen extension progresses (ERV2) (E), and then de novo lumen formation (arrow) occurs in the distal or connecting segment of ERV3 (F). (G) Additional distinct lumens (arrows) form in the connecting segment (ERV4). (G′,G″) High magnification of the connecting region shows separation between lumens (arrows) at 0° (G′) and 148° (G″) rotation. (G″′) A depth plot of 0-5 μm illustrates that one of the lumens (asterisks) is not at the same depth as neighboring lumens, which emphasizes its unique origin. (H,H′) Connecting segment lumens begin to coalesce in ERV5, yet the ERV5 lumen is not fused to the UBL. The ERV5 lumen appears fused to the UBL when viewed at 0° rotation (H), but is clearly separate when the 3D reconstruction is rotated to 56° (arrow, H′). (I,I′) As the proximal lumen curves to form an S-shaped body (SB) there is one continuous lumen (arrows). A 3D SB at 0° (I) and 136° (I′) rotation is shown. Results are representative of sections from six mice. Scale bars: 10 μm in B-I′.
Afadin is required for nectin clustering and lumen formation in renal vesicles
To allow for nascent lumen generation and interconnections between extending lumens and de novo-forming lumens, nectin- and cadherin-based cell contacts presumably must be generated and then subsequently reorganized. Indeed, nectins 1 to 3 are expressed during kidney development (Brakeman et al., 2009; Satoh-Horikawa et al., 2000), but their role in tubulogenesis is not known. We first analyzed the cellular distribution of nectins 1 to 3 and afadin, an adaptor protein known to bind and regulate nectins 1 to 3 (Takai et al., 2008a), during early nephrogenesis (Fig. 3). The nectins were faintly detected in condensed mesenchyme and pretubular aggregates, although nectin 3 had a few cell-surface clusters (not shown). Nectins 1 to 3 were all localized to the apical surface in primitive renal vesicles (not shown) and restricted to apical lateral junctions in mature renal vesicles (Fig. 3E,E′,G,G′,I,I′). Nectins 2 and 3 were also localized to apical lateral junctions in the ureteric bud (Fig. 3G,I). Although afadin was not detected in condensed mesenchyme or pretubular aggregates (not shown), it was localized apically in primitive renal vesicles (Fig. 3B) and was restricted to apical lateral cell junctions in mature renal vesicles (Fig. 3A,B′,C,C′ show the lumen en face), as well as ureteric bud (Fig. 3A). Thus, the presence of afadin is associated with the concentration of nectins at sites associated with adherens junction formation.
Fig. 3.

Afadin is required for nectin clustering in renal vesicles. (A-J) Localization of afadin (A-D′), nectin 1 (E-F′), nectin 2 (G-H′) and nectin 3 (I-J″′) in E14.5 kidneys from control and mutant are indicated (green). NCAM (red) delineates nephron precursors and their derivatives; phalloidin (blue) stains F-actin. (A) Afadin is expressed in ureteric bud (UB) and renal vesicles (RV). (B,B′) High magnification of a primitive (B) and mature (B′) RV is shown. (C,C′) Imaging of the lumen en face in mature RVs shows afadin at apical lateral cell junctions and NCAM at the basolateral surface in mature RVs. (D,D′) Mutant kidneys lack afadin in PA/RV. (E-F′) Nectin 1 is expressed in RV. In mutants (F,F′), nectin 1 is not recruited to an apical surface in pretubular aggregates/renal vesicles (PA/RV). (G-J) Nectin 2 and 3 are expressed at the apical lateral surface of UB and RV. In mutants, nectin 2 and 3 are not recruited to an apical surface in PA/RV. (J′-J″′) The inset in J′ (J″,J″′) shows nectin 3 (green) at discrete cell surface patches in mutant PA/RV. All mutant panels show absence of a central clearing or lumen in PA/RVs. (K,L) Hematoxylin and Eosin-stained (K) and Toluidine Blue-stained (L) sections from control (Afadinfl/fl) and mutant (Afadinfl/fl; Pax3-cre) kidneys at E14.5. Arrows in L indicate PA/RV. Lumens are absent in mutants. Broken lines indicate UB and PA/RV. All results are representative of sections from three mice. Scale bars: 5 μm in B-C′; 10 μm in A,D-J′; 50 μm in L.
As one approach to examining the role of nectin-based cellular adhesion in renal vesicle formation, we conditionally deleted afadin using Pax-3 cre (Beaudoin et al., 2012; Li et al., 2000) because this Cre line induces early and efficient recombination in the metanephric mesenchyme and its epithelial derivatives, including renal vesicles (Cheng et al., 2007; Marciano et al., 2011). At E14.5, Afadinfl/fl; Pax3-creTg mice (hereafter, afadin mutants) had slightly smaller kidneys compared with littermate controls (Afadinfl/fl) (Fig. 3K). Because afadin mutants died in late embryogenesis from non-renal defects, we focused our analysis at E14.5. Sections stained with Toluidine Blue showed that afadin mutants had structures similar to pretubular aggregates, but none had a central clearing characteristic of renal vesicles (Fig. 3L). As later figures will demonstrate that these mutant structures showed gene expression and differentiation consistent with the renal vesicle stage, despite this luminal defect, we hereafter refer to these abnormal structures as mutant pretubular aggregates/renal vesicles. In afadin mutants, afadin was absent from these pretubular aggregates/renal vesicles and from their nephron derivatives, but not from ureteric bud, as expected (Fig. 3D,D′). Afadin mutants had impaired recruitment of nectins 1-3 to an apical surface (Fig. 3F,F′,H,H′,J,J′). High-magnification images of nectin 3 in afadin mutants showed localization at numerous small surface patches (Fig. 3J″,J″′), suggesting afadin is required for organizing and coalescing nectins at the cell membrane into larger clusters. Additionally, the cells of mutant pretubular aggregates/renal vesicles did not exhibit the wedge-shape of cells in control renal vesicles (Fig. 3D′,F′,H′,J′, compare with 3A,E′,G′,I′). Collectively, these results suggest that afadin mutants fail to establish apical-basal polarity and lumen formation.
We used our categorization of nephron and lumen stages (supplementary material Table S1) to ascertain at what stage lumen formation failed. Quantification of these results showed that only 20% of mutant pretubular aggregates/renal vesicles had evidence of lumen initiation and 0% had an expanded lumen, compared with 81% and 60% in controls, respectively (Fig. 4A). Even at the later ERV stage, nearly half of mutants lacked lumen initiation. By the S-shaped body stage, 3D reconstructions of these structures showed controls had formed a single continuous lumen, but mutants had small discontinuous lumens even in late S-shaped bodies (Fig. 4B,L,M, compare with 4K). The defect in lumen formation was not a generalized failure to progress through nephrogenesis because S-shaped bodies were observed at similar frequencies in mutant and control (n=3; P>0.05, not shown). We qualitatively confirmed the luminal defect in afadin mutants by directly visualizing nephron lumens with transmission electron microscopy (Fig. 4C-D′). In controls, luminal space and electron-dense material (Fig. 4C′, arrows) consistent with apical junctional complexes were observed in renal vesicles. Mutant pretubular aggregates/renal vesicles lacked a central lumen (Fig. 4D) and electron-dense material at cell junctions (Fig. 4D′, arrows). Together, these results demonstrate that afadin is crucial for lumen initiation and elongation.
Fig. 4.
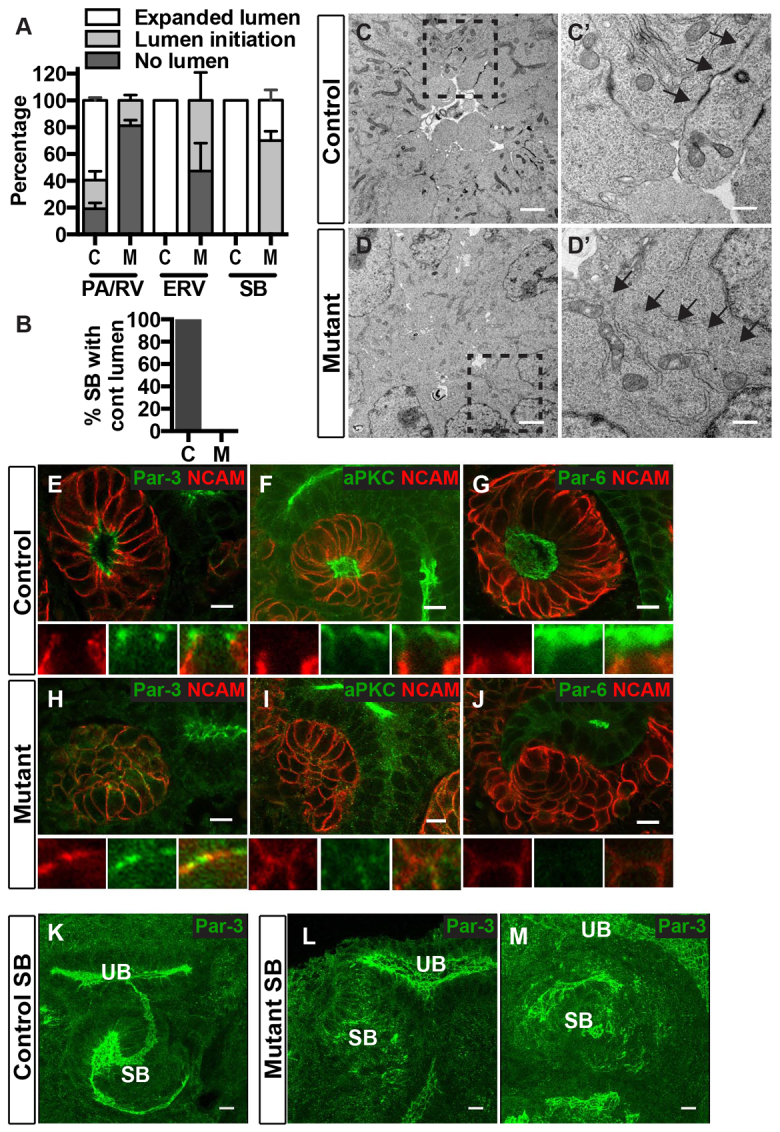
Afadin is required for lumen generation and for Par complex formation in renal vesicles. (A,B) Quantification of lumen stage in controls (Afadinfl/fl) and afadin mutants (Afadinfl/fl; Pax3-cre). Lumen stage (no lumen, lumen initiation and expanded lumen) was assessed by immunostaining with aPKC, NCAM and F-actin for each stage of nephrogenesis. Mutants had an increased percentage of both PA/RVs and ERVs lacking lumens, and a decreased percentage of PA/RV, ERV and SB with expanded lumens. (B) Quantification of continuous lumen formation in SB of controls (C) and mutants (M). For A and B, data are mean±s.d. from three controls and three mutants. P<0.001 with two-way ANOVA. (C,D) Transmission electron micrographs of lumen in renal vesicles of control (C) and mutant (D). (C′,D′) The insets in C,D, respectively. Arrows highlight cell-cell junctions. Results are representative of three experiments. (E-J) Localization of Par3 (E,H), aPKC (F,I) and Par6 (G,J) in E14.5 kidneys from control and mutant mice are indicated (green). NCAM (red) delineates RVs. (E-G) Par3 (E) localizes to apical lateral junctions, whereas aPKC (F) and Par6 (G) are apical in control mature RVs. (H-J) In mutant PA/RVs, Par3 localizes to membrane patches with and without NCAM (H), but a single apical surface is lacking (n=3). (I,J) In mutants, aPKC (I) localizes to small puncta and Par6 (J) is not visualized. (K-M) 3D reconstructions of confocal z-stacks immunostained with anti-Par3 from control (K) and mutant SBs (L,M). M shows a very late stage SB. Results are representative of sections from at least two mice. Abbreviations: PA, pretubular aggregate; RV, renal vesicle; ERV, extended renal vesicle; SB, S-shaped body. Scale bars: 2 μm in C,D; 0.5 μm in C′,D′; 10 μm in E-M.
Because Pax3-cre induces cre-mediated recombination in nephron precursors as well as renal stroma, we wanted to ensure that the luminal defects of afadin mutants were caused by cell-autonomous defects in nephrons, rather than defective stromal development or signaling. Immunostaining with markers of nephron progenitors (Six2) and stromal cells (Meis1) showed a similar pattern of distribution in controls (supplementary material Fig. S1A-A″) and mutants (supplementary material Fig. S1B-B″). Six2 and Meis1 were not co-expressed within the same cells and neither compartment seemed expanded at the expense of the other in the mutant. We generated mice lacking afadin solely in the stromal compartment using FoxD1-cre (Humphreys et al., 2008; Yu et al., 2009), and these mice had normal nephron lumen formation at E14.5 and P0 (supplementary material Fig. S1C,C′). Furthermore, we tested the Six2-eGFP-cre line (Kobayashi et al., 2008), which results in cre-mediated excision in kidneys solely within nephron progenitors, and found qualitatively similar, albeit somewhat less penetrant, defects in lumen formation (n=3 at E14.5; n=3 at P0) (supplementary material Fig. S1D,D′), as well as mildly reduced kidney size (supplementary material Fig. S1E,F). These results clearly demonstrate that afadin has a cell-autonomous role in regulating apical polarity and lumen formation in nephrons. Unlike the Afadinfl/fl; Pax3-cre mutants, the Afadinfl/-; Six2-eGFP-cre mice were viable; however, they showed small kidneys with dysplastic, dilated tubules and severe glomerulosclerosis at 4 weeks of age (4/4 mutants) (supplementary material Fig. S1G,H).
Afadin is crucial for integrating pre-apical membrane microdomains and for Par complex formation
Because of the lumen-forming defects observed in afadin mutants, we investigated the developmental stages and underlying molecular mechanisms requiring afadin activity. Par complex proteins (Par3/aPKC/Par6/Cdc42) are known to be essential for apical membrane formation (Suzuki and Ohno, 2006), and our own results demonstrated that Par3-containing ‘pre-apical domains’ exist prior to lumen formation. We analyzed localization of polarity proteins Par3, aPKC and Par6 in renal vesicles of controls and mutants. As discussed earlier, Par3 colocalized with aPKC at apical surfaces of primitive renal vesicles, but was concentrated at apical junctions in mature renal vesicles (Fig. 4E). Both aPKC and Par6 were distributed across the apical surfaces of renal vesicles, whereas NCAM was restricted to the basolateral surfaces in a non-overlapping distribution with Par3 and aPKC/Par6 (Fig. 4F,G). In afadin mutants, Par3 was dispersed in patches at the cell membranes, and many of these patches segregated from NCAM (Fig. 4H). This immunostaining pattern is analogous to the ‘pre-apical domains’ of control pretubular aggregates (Fig. 1D), demonstrating that afadin facilitates integration or coalescence of these domains prior to apical polarity. Additionally, renal vesicles of afadin mutants had greatly diminished immunostaining of Par6, and to a lesser extent aPKC, at the cell surfaces. The localization of aPKC appeared dispersed in small puncta within cells and at plasma membranes (Fig. 4I), whereas cell cortex staining of Par6 was absent (Fig. 4J). The absence of colocalized Par complex constituents in afadin mutants implies that afadin acts upstream of the Par complex and is required for recruitment and/or clustering of aPKC and Par6. As Par3 interactions with aPKC and Par6 are essential for generating polarity (Horikoshi et al., 2009; McCaffrey and Macara, 2009), these results further suggest that afadin may promote lumen initiation through Par complex formation.
Afadin segregates nectins from aPKC in developing nephrons
During lumen initiation, aPKC colocalized with nectin 2 (in addition to Par3) at developing apical surfaces of primitive renal vesicles (supplementary material Fig. S2A-C, related to Fig. 4). As with Par3, maturation of renal vesicles led to redistribution of nectin 2 to apical junctional complexes, basal to aPKC (supplementary material Fig. S2D-F). Afadin mutants demonstrated limited aPKC and nectin 2 recruitment in pretubular aggregates/renal vesicles (supplementary material Fig. S2G-I). However, at the S-shaped body stage, afadin mutants ultimately formed small, discontinuous lumens with reduced levels of aPKC and nectin 2. Despite this, there was no evidence of redistribution of nectin 2 to apical junctional complexes as in controls, even in the few open lumens that formed (supplementary material Fig. S2J-L). These results suggest that afadin plays a central role localization of nectin 2 and its segregation from aPKC.
Afadin is necessary for R-cadherin and actin recruitment to form apical domains
In vitro studies have suggested that afadin is necessary for recruitment of cadherins to apical junctions in epithelia (Ooshio et al., 2007), but in vivo studies of intestinal epithelial development have not shown similar results (Tanaka-Okamoto et al., 2011). R-cadherin expression is upregulated in renal vesicles (Dahl et al., 2002), and it may be the predominant cadherin expressed at this stage (Brunskill et al., 2008). In developing nephrons, immunostaining with anti-R-cadherin showed widespread R-cadherin in pretubular aggregates (Fig. 5A). By the mature renal vesicle stage, it was highly concentrated at apical junctional complexes with F-actin and also present at cell-cell contact surfaces on the lateral membranes (Fig. 5B,C). In mutants, R-cadherin levels appeared similar to those in controls, but the protein was comparatively evenly distributed at cell-cell contact surfaces (Fig. 5D-F). Additionally, there was complete disruption of the F-actin network and its colocalization with R-cadherin (Fig. 5F′,F″). Quantification demonstrated that no afadin mutant pretubular aggregates/renal vesicles (0/28, n=3) had enrichment of R-cadherin adjacent to nascent lumens compared with 100% of controls (27/27, n=3, P<0.001, unpaired two-tailed Student’s t-test). This demonstrates that afadin is necessary for the recruitment or stabilization of R-cadherin at forming apical junctions and localization of F-actin at these junctions.
Fig. 5.

Afadin is required for cadherin recruitment and ECM organization. Localization of R-cadherin (green), F-actin (stained with phalloidin; red in C,F; blue in G,H), NCAM (red) and laminin (green) are shown in E14.5 kidneys from control (Afadinfl/fl) and mutant (Afadinfl/fl; Pax3-cre) mice. (A-C″) Control PAs (A) have widespread R-cadherin; RVs (B,C) have R-cadherin enrichment at apical, lateral cell junctions that colocalizes with F-actin. F-actin is enriched both apically and apicolaterally. (D-F″) In mutants, R-cadherin is relatively uniform at the cell membrane in PAs (D) and PA/RVs (E,F). F-actin is irregularly distributed. (G-H′) Laminin does not separate the SB and UB tubules in controls (G) or mutants (H), indicating SB-UB fusion (marked by dotted lines). (G″,H″) Although laminin is strictly basal in control SBs, mutant SBs have additional ectopic laminin deposits that colocalize with foci of F-actin (H″, compare with G″). (G″′,H″′) Insets in G″, H″ respectively. (I) Quantification of ectopic laminin deposition in afadin mutants (1/31 RVs and 9/41 ERV/SBs) and controls (0/31 RVs, 0/40 ERV/SBs) show increased ectopic laminin in mutant ERV/SBs. Data are mean±s.d. from three controls and three mutants. *P<0.03 with unpaired two-tailed Student’s t-test. PA, pretubular aggregate; RV, renal vesicle; ERV, extended renal vesicle; SB, S-shaped body; UB, ureteric bud. Scale bars: 5 μm in A-F″; 10 μm in G-G″,H-H″.
Afadin is not required for SB-UB tubule fusion
Primitive and mature renal vesicles are separated from ureteric bud by a basal lamina comprising laminin and other ECM proteins. Part of the ECM surrounding the UB tip and the distal region of the renal vesicle becomes degraded as the nephron tubule and ureteric tubule are joined, prior to the S-shaped body stage (Georgas et al., 2009). We asked whether lumen formation/elongation and/or the establishment of apical-basal polarity are necessary for fusion of these two structures. We immunostained with anti-laminin and, as expected, S-shaped bodies were not separated from the ureteric bud/tubule by laminin at the nephron-ureteric tubule interface in controls (Fig. 5G-G″). Surprisingly, afadin mutant S-shaped bodies also lacked laminin at the nephron-ureteric tubule interface (Fig. 5H-H″), suggesting that fusion of nephron-ureteric tubules had occurred to form a single continuous epithelium despite the abnormal polarity and lumen of the mutant S-shaped body. These results indicate that fusion of nephron-ureteric tubules occurs in an afadin-independent manner and suggest that lumen and tubule formation are not strictly interdependent.
Although nephron-ureteric tubule fusion occurred in afadin mutants, disorganization of the ECM was evident. Ectopic laminin deposition occurred rarely in mutant pretubular aggregates/renal vesicles (1/31, n=3), but was present in 23% (9/41, n=3; P<0.03, unpaired two-tailed Student’s t-test) of mutant extended renal vesicles and S-shaped bodies (Fig. 5I). These ectopic foci of laminin colocalized with F-actin at sites lacking NCAM (Fig. 5H-H″′, where 5H″′ is the inset of 5H″; compare with 5G″′). Because these foci generally appeared at later stages of nephrogenesis, rather than at the time of lumen initiation, it suggests that deficits in nephron cellular polarity may secondarily impair polarized laminin deposition into the basolateral ECM. This may act in a feed-forward loop to disrupt polarized ECM signaling, which normally helps orient apical domain and lumen position (Yu et al., 2005; Rasmussen et al., 2012), thereby promoting the formation of multiple lumens observed in mutant S-shaped bodies.
Afadin is not required for proximal-distal patterning of the renal vesicle
One possible explanation for why afadin mutants do not form proper lumens is that cells comprising pretubular aggregates failed to undergo early steps of differentiation or patterning in order to become renal vesicles and S-shaped bodies. In healthy kidneys, mature nephrons are segmented and patterned along their proximal-distal length into renal corpuscles, proximal tubules, the loop of Henle, distal tubules and connecting tubules. This proximal-distal patterning of the nephron begins at the renal vesicle stage (Georgas et al., 2009), whereby the expression of some genes is restricted to either the cells of the proximal or distal renal vesicle. By the S-shaped stage, further segmentation has occurred, and numerous genes show restricted expression to either the proximal, mid- or distal S-shaped body. We sought to examine whether afadin was required for the production of protein from these genes and whether their restricted domains would be intact.
In renal vesicles, WT1 protein was produced proximally (Fig. 6A,A’) whereas Lef1 (Fig. 6A′) and EphA4 (Fig. 6E,E′) were produced distally. In S-shaped bodies, WT1 was restricted to the proximal domain (Fig. 6C,C′), Jag1 to the mid-domain (Fig. 6I,I′), and Lef1 (Fig. 6C′), EphA4 (Fig. 6G,G′) and HNF1β (not shown) to the mid- and distal domains. All of these proteins were produced in a similar pattern in mutants (Fig. 6B,B′D,D′,F,F′,H,H′,J,J′) although the morphology of the S-shaped tubules was somewhat abnormal. Together, these results suggest that afadin is not required for early differentiation of renal vesicles. Therefore, luminal defects in afadin mutants are not likely to be secondary to impaired differentiation. Importantly, the results also imply that normal polarity and lumen formation are not necessary for initial steps of segment-specific differentiation and proximal-distal patterning of the S-shaped body.
Fig. 6.

Afadin is not required for proximal-distal patterning in nephron precursors. Localization of segment-specific proteins in E14.5 kidneys from controls (Afadinfl/fl) and mutants (Afadinfl/fl; Pax3-cre) are indicated. NCAM delineates the renal vesicle (RV) and S-shaped body (SB). Distal (D), mid- (M) and proximal (P) regions are marked. (A-B′) In RVs, WT1 (green) is proximal whereas Lef1 (red) is distal in both control and mutant. (C-D′) In SBs, WT1 (green) is proximal, whereas Lef1 (red) is mid-SB and distal SB in control and mutant. (E-H′) Epha4 (green) is produced in the distal RV and mid-SB in control and mutant. (I-J′) Jag1 (red) localizes to mid-SB in control and mutant. Results are representative of sections from at least two mice. Scale bars: 10 μm.
DISCUSSION
There is little high-resolution 3D data on how developing nephrons establish polarity and undergo de novo lumen formation and morphogenesis to form a continuous lumen. Many of the general concepts that describe the cellular and molecular mechanisms underlying lumen formation have been derived from cell culture models or 2D images (Datta et al., 2011; Schlüter and Margolis, 2009). In this study, we provide a novel paradigm for de novo lumen formation and elongation in developing nephrons (Fig. 7), and identify afadin as a key regulator of these processes.
Fig. 7.

New model of nephron lumen formation. In pretubular aggregates (PA), Par3-containing pre-apical domains (red) are present in numerous membranes. These domains reorganize and coalesce in an afadin-dependent manner to form one or two small apical foci/lumen in primitive renal vesicles (PRV). Par3 is redistributed to apical lateral cell junctions (not illustrated) and the lumen expands in the mature renal vesicle (MRV). Next, the expanded lumen (red) extends towards the ureteric bud lumen (blue) in the extended renal vesicle (ERV) and then additional de novo lumens form in the distal segment. In the late ERV, these additional distal lumens coalesce, probably in an afadin-dependent manner, whereas the proximal region of the lumen extends. Finally, the extended lumen joins the ureteric bud lumen at the S-shaped body stage (SB), where a pronounced S-shaped lumen is visible.
Our results demonstrate that Par3-containing domains exist at the plasma membrane in condensed mesenchyme. These domains, some of which appear linear, are sparsely distributed along plasma membranes still containing NCAM, suggesting an early stage of membrane organization preceding apical-basal polarity. This stage is analogous to the apical membrane initiation site (AMIS) described in cell culture that contains proteins that later segregate to apical cell-cell junctions or the basolateral domain (Bryant et al., 2010). At the next stage, the pretubular aggregate stage, some (but not all) of these Par3-containing domains have segregated from NCAM. These domains, which we have referred to as pre-apical domains, are analogous to the pre-apical patch (PAP) that has been described in vitro in which basolateral membrane proteins are removed from a forming apical domain (Bryant et al., 2010). Although there are remarkable similarities between what we observe in vivo and what has been described previously in vitro, one main difference is that there are numerous pre-apical domains found within a single pretubular aggregate. This is probably due to the greater number of cells involved in vivo. Therefore, an additional level of complexity probably exists, in which individual pre-apical domains eventually integrate to form a single apical domain across multiple cells. We postulate that this integration occurs through lateral diffusion and adhesion (both in cis and trans) of adjacent domains and/or through endosomal membrane sorting. Pre-apical domains could also serve as docking sites for polarized membrane traffic and define domain boundaries for junction formation.
Lumen formation requires several interdependent cellular processes, including the assembly of intercellular junctional complexes. Both nectins and cadherins are the main adhesive receptors at adherens junctions. Our data demonstrate that the nectin-adaptor protein afadin is required for the integration of pre-apical domains to form a single, continuous, apical surface and lumen. Afadin mutants show reduced cell-surface clustering of nectins, suggesting that afadin regulates nectin-nectin adhesion, as demonstrated in vitro (Kurita et al., 2011; Takai et al., 2008a). Nectin binding in cis and trans could stabilize adhesion between intra- and intercellular pre-apical domains, thereby leading to early identification of a single apical domain.
Recent in vitro data suggest that nectin/afadin complexes act upstream of cadherins, initiating junction formation and recruiting cadherins to cellular junctions (Honda et al., 2003a; Honda et al., 2003b). Our in vivo data support these findings, demonstrating that afadin is required to recruit and/or stabilize R-cadherin at apical junctional complexes. A recent study of afadin deletion in intestinal epithelia failed to show similar defects in cadherin recruitment, although nectin localization was perturbed (Tanaka-Okamoto et al., 2011). These contradictions of our own data may be due to differences in the timing and efficacy of afadin removal, as well as to functional differences in intestinal epithelia.
In addition to the role of afadin in nectin clustering and cadherin recruitment, we show that afadin is essential for Par complex formation during lumen initiation. Afadin mutants fail to localize Par6, and to a lesser extent aPKC, to the plasma membrane with Par3. Additionally, in vitro assays show that nectins 1 and 3 bind Par3 directly (Takekuni et al., 2003), and nectins are capable of activating and recruiting the Cdc42/Par/aPKC complex (Fukuhara et al., 2004; Fukuyama et al., 2005; Komura et al., 2008). Together, these results suggest that nectin/afadin complexes act upstream of Par complex formation in vivo, placing them as one of the earliest indicators of epithelial polarity. Because Par complex proteins are essential for establishing apical domains (McCaffrey and Macara, 2012; Suzuki and Ohno, 2006), these results further suggest that afadin may promote lumen initiation through its regulation of Par complex formation.
After lumen initiation, we show that nephrons form a continuous lumen using a mechanism that combines extension of the renal vesicle lumen towards the ureteric bud tip as well as discrete de novo lumen generation within the most distal region of the extended renal vesicle. Interestingly, there does not appear to be direct lumen extension from ureteric bud tips. Ultimately, we find the nephron-ureteric bud lumen becomes continuous at the S-shaped body stage, which is consistent with recently published data (Kao et al., 2012). Our data also show that whereas afadin is required for lumen initiation in renal vesicles, at later stages many developing nephrons devoid of afadin will eventually form numerous small lumens. It is likely that some of the pre-apical domains in afadin mutants expand to form small lumens, but these lumens do not coalesce or interconnect with other such lumens to form a single continuous apical surface and lumen. Additionally, these small lumens fail to mature, with persistent nectin 2/aPKC colocalization. Together, these results suggest that afadin not only promotes and accelerates de novo lumen formation, but is also required for continuous lumen formation. Afadin may provide signals that are essential for proper positioning of the apical membrane and/or cell-cell junctions, leading to the integration of multiple apical domains. Although this is likely to be due in part to defective cell-cell interactions, our data show that afadin mutants have ectopic laminin deposition at later stages of nephrogenesis, suggesting secondary defects in polarized cell-ECM signaling. Defects in cell-ECM signaling can lead to defective positioning of intercellular junctions and lumens (Tseng et al., 2012; Yu et al., 2005). Thus, defective cell-ECM signaling may secondarily promote multiple discontinuous lumens in S-shaped bodies.
Our analysis of mutant mice that are defective in lumen generation has yielded important insights about the relationship between lumen and tubule formation. Specifically, we demonstrate that proper apical polarity and lumen formation are not required for proximal-distal patterning along the length of the nephron precursor tubule. Additionally, lumen formation is not required for fusion of the nephron precursor tubule to the ureteric bud, suggesting that lumen formation and the morphogenetic events required for fusion of tubules are not strictly interdependent.
In summary, we demonstrate that de novo lumen formation in developing nephrons is preceded by the formation of Par3-containing pre-apical domains and proceeds via afadin-dependent formation of a single lumen. Once initial lumen formation has occurred, the renal vesicle lumen extends by adding on to the existing lumen as well as fusing multiple discontinuous lumens into one continuous structure. Our observations reveal a novel mechanism for lumen formation and morphogenesis in vivo in which afadin plays a central role via its recruitment of polarity and junctional proteins. Future in vivo and in vitro work will be required to gain further insight into renal epithelial lumen generation.
Note added in proof
While this manuscript was in press, a study by Choi et al. (Choi et al., 2013) showed that afadin is essential for establishing apical basal polarity in Drosophila.
Supplementary Material
Acknowledgments
We thank Tom Carroll for manuscript review; David Bryant and Michael Shiloh for invaluable discussions; Jonathan Epstein and Andrew McMahon for providing mice; the Developmental Studies Hybridoma Bank for the MRCD5 (R-cadherin) cells and antibody; Yoshimi Takai for nectin 3 antibody; and Laurie Mueller and Chris Gilpin (UTSW Electron Microscopy Core) for assistance with electron microscopy.
Footnotes
Funding
This work was supported by the National Institutes of Health (NIH) [DK081668 to D.K.M. and P30DK079328 (O’Brien Center grant)]. Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.087957/-/DC1
References
- Beaudoin G. M., 3rd, Schofield C. M., Nuwal T., Zang K., Ullian E. M., Huang B., Reichardt L. F. (2012). Afadin, a Ras/Rap effector that controls cadherin function, promotes spine and excitatory synapse density in the hippocampus. J. Neurosci. 32, 99–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettner B., Van Aelst L. (2009). Control of cell adhesion dynamics by Rap1 signaling. Curr. Opin. Cell Biol. 21, 684–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brakeman P. R., Liu K. D., Shimizu K., Takai Y., Mostov K. E. (2009). Nectin proteins are expressed at early stages of nephrogenesis and play a role in renal epithelial cell morphogenesis. Am. J. Physiol. 296, F564–F574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunskill E. W., Aronow B. J., Georgas K., Rumballe B., Valerius M. T., Aronow J., Kaimal V., Jegga A. G., Yu J., Grimmond S., et al. (2008). Atlas of gene expression in the developing kidney at microanatomic resolution. Dev. Cell 15, 781–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant D. M., Datta A., Rodríguez-Fraticelli A. E., Peränen J., Martín-Belmonte F., Mostov K. E. (2010). A molecular network for de novo generation of the apical surface and lumen. Nat. Cell Biol. 12, 1035–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cebrián C., Borodo K., Charles N., Herzlinger D. A. (2004). Morphometric index of the developing murine kidney. Dev. Dyn. 231, 601–608 [DOI] [PubMed] [Google Scholar]
- Cheng H. T., Kim M., Valerius M. T., Surendran K., Schuster-Gossler K., Gossler A., McMahon A. P., Kopan R. (2007). Notch2, but not Notch1, is required for proximal fate acquisition in the mammalian nephron. Development 134, 801–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi W., Harris N. J., Sumigray K. D., Peifer M. (2013). Rap1 and Canoe/afadin are essential for establishment of apical-basal polarity in the Drosophila embryo. Mol. Biol. Cell (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl U., Sjödin A., Larue L., Radice G. L., Cajander S., Takeichi M., Kemler R., Semb H. (2002). Genetic dissection of cadherin function during nephrogenesis. Mol. Cell. Biol. 22, 1474–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta A., Bryant D. M., Mostov K. E. (2011). Molecular regulation of lumen morphogenesis. Curr. Biol. 21, R126–R136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd J., Morton S. B., Karagogeos D., Yamamoto M., Jessell T. M. (1988). Spatial regulation of axonal glycoprotein expression on subsets of embryonic spinal neurons. Neuron 1, 105–116 [DOI] [PubMed] [Google Scholar]
- Fukuhara A., Irie K., Nakanishi H., Takekuni K., Kawakatsu T., Ikeda W., Yamada A., Katata T., Honda T., Sato T., et al. (2002a). Involvement of nectin in the localization of junctional adhesion molecule at tight junctions. Oncogene 21, 7642–7655 [DOI] [PubMed] [Google Scholar]
- Fukuhara A., Irie K., Yamada A., Katata T., Honda T., Shimizu K., Nakanishi H., Takai Y. (2002b). Role of nectin in organization of tight junctions in epithelial cells. Genes Cells 7, 1059–1072 [DOI] [PubMed] [Google Scholar]
- Fukuhara T., Shimizu K., Kawakatsu T., Fukuyama T., Minami Y., Honda T., Hoshino T., Yamada T., Ogita H., Okada M., et al. (2004). Activation of Cdc42 by trans interactions of the cell adhesion molecules nectins through c-Src and Cdc42-GEF FRG. J. Cell Biol. 166, 393–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuyama T., Ogita H., Kawakatsu T., Fukuhara T., Yamada T., Sato T., Shimizu K., Nakamura T., Matsuda M., Takai Y. (2005). Involvement of the c-Src-Crk-C3G-Rap1 signaling in the nectin-induced activation of Cdc42 and formation of adherens junctions. J. Biol. Chem. 280, 815–825 [DOI] [PubMed] [Google Scholar]
- Georgas K., Rumballe B., Valerius M. T., Chiu H. S., Thiagarajan R. D., Lesieur E., Aronow B. J., Brunskill E. W., Combes A. N., Tang D., et al. (2009). Analysis of early nephron patterning reveals a role for distal RV proliferation in fusion to the ureteric tip via a cap mesenchyme-derived connecting segment. Dev. Biol. 332, 273–286 [DOI] [PubMed] [Google Scholar]
- Gumbiner B. M. (2005). Regulation of cadherin-mediated adhesion in morphogenesis. Nat. Rev. Mol. Cell Biol. 6, 622–634 [DOI] [PubMed] [Google Scholar]
- Harrison O. J., Vendome J., Brasch J., Jin X., Hong S., Katsamba P. S., Ahlsen G., Troyanovsky R. B., Troyanovsky S. M., Honig B., et al. (2012). Nectin ectodomain structures reveal a canonical adhesive interface. Nat. Struct. Mol. Biol. 19, 906–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda T., Shimizu K., Fukuhara A., Irie K., Takai Y. (2003a). Regulation by nectin of the velocity of the formation of adherens junctions and tight junctions. Biochem. Biophys. Res. Commun. 306, 104–109 [DOI] [PubMed] [Google Scholar]
- Honda T., Shimizu K., Kawakatsu T., Yasumi M., Shingai T., Fukuhara A., Ozaki-Kuroda K., Irie K., Nakanishi H., Takai Y. (2003b). Antagonistic and agonistic effects of an extracellular fragment of nectin on formation of E-cadherin-based cell-cell adhesion. Genes Cells 8, 51–63 [DOI] [PubMed] [Google Scholar]
- Horikoshi Y., Suzuki A., Yamanaka T., Sasaki K., Mizuno K., Sawada H., Yonemura S., Ohno S. (2009). Interaction between PAR-3 and the aPKC-PAR-6 complex is indispensable for apical domain development of epithelial cells. J. Cell Sci. 122, 1595–1606 [DOI] [PubMed] [Google Scholar]
- Humphreys B. D., Valerius M. T., Kobayashi A., Mugford J. W., Soeung S., Duffield J. S., McMahon A. P., Bonventre J. V. (2008). Intrinsic epithelial cells repair the kidney after injury. Cell Stem Cell 2, 284–291 [DOI] [PubMed] [Google Scholar]
- Ikeda W., Nakanishi H., Miyoshi J., Mandai K., Ishizaki H., Tanaka M., Togawa A., Takahashi K., Nishioka H., Yoshida H., et al. (1999). Afadin: A key molecule essential for structural organization of cell-cell junctions of polarized epithelia during embryogenesis. J. Cell Biol. 146, 1117–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao R. M., Vasilyev A., Miyawaki A., Drummond I. A., McMahon A. P. (2012). Invasion of distal nephron precursors associates with tubular interconnection during nephrogenesis. J. Am. Soc. Nephrol. 23, 1682–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesavan G., Sand F. W., Greiner T. U., Johansson J. K., Kobberup S., Wu X., Brakebusch C., Semb H. (2009). Cdc42-mediated tubulogenesis controls cell specification. Cell 139, 791–801 [DOI] [PubMed] [Google Scholar]
- Kobayashi A., Valerius M. T., Mugford J. W., Carroll T. J., Self M., Oliver G., McMahon A. P. (2008). Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell 3, 169–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komura H., Ogita H., Ikeda W., Mizoguchi A., Miyoshi J., Takai Y. (2008). Establishment of cell polarity by afadin during the formation of embryoid bodies. Genes Cells 13, 79–90 [DOI] [PubMed] [Google Scholar]
- Kurita S., Ogita H., Takai Y. (2011). Cooperative role of nectin-nectin and nectin-afadin interactions in formation of nectin-based cell-cell adhesion. J. Biol. Chem. 286, 36297–36303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Chen F., Epstein J. A. (2000). Neural crest expression of Cre recombinase directed by the proximal Pax3 promoter in transgenic mice. Genesis 26, 162–164 [DOI] [PubMed] [Google Scholar]
- Little M., Georgas K., Pennisi D., Wilkinson L. (2010). Kidney development: two tales of tubulogenesis. Curr. Top. Dev. Biol. 90, 193–229 [DOI] [PubMed] [Google Scholar]
- Mandai K., Nakanishi H., Satoh A., Obaishi H., Wada M., Nishioka H., Itoh M., Mizoguchi A., Aoki T., Fujimoto T., et al. (1997). Afadin: A novel actin filament-binding protein with one PDZ domain localized at cadherin-based cell-to-cell adherens junction. J. Cell Biol. 139, 517–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciano D. K., Brakeman P. R., Lee C. Z., Spivak N., Eastburn D. J., Bryant D. M., Beaudoin G. M., 3rd, Hofmann I., Mostov K. E., Reichardt L. F. (2011). p120 catenin is required for normal renal tubulogenesis and glomerulogenesis. Development 138, 2099–2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunami H., Takeichi M. (1995). Fetal brain subdivisions defined by R- and E-cadherin expressions: evidence for the role of cadherin activity in region-specific, cell-cell adhesion. Dev. Biol. 172, 466–478 [DOI] [PubMed] [Google Scholar]
- McCaffrey L. M., Macara I. G. (2009). The Par3/aPKC interaction is essential for end bud remodeling and progenitor differentiation during mammary gland morphogenesis. Genes Dev. 23, 1450–1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaffrey L. M., Macara I. G. (2012). Signaling pathways in cell polarity. Cold Spring Harb. Perspect. Biol. 4, a009654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean I. W., Nakane P. K. (1974). Periodate-lysine-paraformaldehyde fixative. A new fixation for immunoelectron microscopy. J. Histochem. Cytochem. 22, 1077–1083 [DOI] [PubMed] [Google Scholar]
- Narita H., Yamamoto Y., Suzuki M., Miyazaki N., Yoshida A., Kawai K., Iwasaki K., Nakagawa A., Takai Y., Sakisaka T. (2011). Crystal structure of the cis-dimer of nectin-1: implications for the architecture of cell-cell junctions. J. Biol. Chem. 286, 12659–12669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura T., Takeichi M. (2009). Remodeling of the adherens junctions during morphogenesis. Curr. Top. Dev. Biol. 89, 33–54 [DOI] [PubMed] [Google Scholar]
- Ooshio T., Fujita N., Yamada A., Sato T., Kitagawa Y., Okamoto R., Nakata S., Miki A., Irie K., Takai Y. (2007). Cooperative roles of Par-3 and afadin in the formation of adherens and tight junctions. J. Cell Sci. 120, 2352–2365 [DOI] [PubMed] [Google Scholar]
- Ooshio T., Kobayashi R., Ikeda W., Miyata M., Fukumoto Y., Matsuzawa N., Ogita H., Takai Y. (2010). Involvement of the interaction of afadin with ZO-1 in the formation of tight junctions in Madin-Darby canine kidney cells. J. Biol. Chem. 285, 5003–5012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perantoni A. O. (1999). Cell adhesion molecules in the kidney: from embryo to adult. Exp. Nephrol. 7, 80–102 [DOI] [PubMed] [Google Scholar]
- Rasmussen J. P., Reddy S. S., Priess J. R. (2012). Laminin is required to orient epithelial polarity in the C. elegans pharynx. Development 139, 2050–2060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rikitake Y., Mandai K., Takai Y. (2012). The role of nectins in different types of cell-cell adhesion. J. Cell Sci. 125, 3713–3722 [DOI] [PubMed] [Google Scholar]
- Satoh-Horikawa K., Nakanishi H., Takahashi K., Miyahara M., Nishimura M., Tachibana K., Mizoguchi A., Takai Y. (2000). Nectin-3, a new member of immunoglobulin-like cell adhesion molecules that shows homophilic and heterophilic cell-cell adhesion activities. J. Biol. Chem. 275, 10291–10299 [DOI] [PubMed] [Google Scholar]
- Saxén L., Sariola H. (1987). Early organogenesis of the kidney. Pediatr. Nephrol. 1, 385–392 [DOI] [PubMed] [Google Scholar]
- Schlüter M. A., Margolis B. (2009). Apical lumen formation in renal epithelia. J. Am. Soc. Nephrol. 20, 1444–1452 [DOI] [PubMed] [Google Scholar]
- Suzuki A., Ohno S. (2006). The PAR-aPKC system: lessons in polarity. J. Cell Sci. 119, 979–987 [DOI] [PubMed] [Google Scholar]
- Tachibana K., Nakanishi H., Mandai K., Ozaki K., Ikeda W., Yamamoto Y., Nagafuchi A., Tsukita S., Takai Y. (2000). Two cell adhesion molecules, nectin and cadherin, interact through their cytoplasmic domain-associated proteins. J. Cell Biol. 150, 1161–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K., Nakanishi H., Miyahara M., Mandai K., Satoh K., Satoh A., Nishioka H., Aoki J., Nomoto A., Mizoguchi A., et al. (1999). Nectin/PRR: an immunoglobulin-like cell adhesion molecule recruited to cadherin-based adherens junctions through interaction with Afadin, a PDZ domain-containing protein. J. Cell Biol. 145, 539–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai Y., Ikeda W., Ogita H., Rikitake Y. (2008a). The immunoglobulin-like cell adhesion molecule nectin and its associated protein afadin. Annu. Rev. Cell Dev. Biol. 24, 309–342 [DOI] [PubMed] [Google Scholar]
- Takai Y., Miyoshi J., Ikeda W., Ogita H. (2008b). Nectins and nectin-like molecules: roles in contact inhibition of cell movement and proliferation. Nat. Rev. Mol. Cell Biol. 9, 603–615 [DOI] [PubMed] [Google Scholar]
- Takekuni K., Ikeda W., Fujito T., Morimoto K., Takeuchi M., Monden M., Takai Y. (2003). Direct binding of cell polarity protein PAR-3 to cell-cell adhesion molecule nectin at neuroepithelial cells of developing mouse. J. Biol. Chem. 278, 5497–5500 [DOI] [PubMed] [Google Scholar]
- Tanaka-Okamoto M., Hori K., Ishizaki H., Itoh Y., Onishi S., Yonemura S., Takai Y., Miyoshi J. (2011). Involvement of afadin in barrier function and homeostasis of mouse intestinal epithelia. J. Cell Sci. 124, 2231–2240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng Q., Duchemin-Pelletier E., Deshiere A., Balland M., Guillou H., Filhol O., Théry M. (2012). Spatial organization of the extracellular matrix regulates cell-cell junction positioning. Proc. Natl. Acad. Sci. USA 109, 1506–1511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villasenor A., Chong D. C., Henkemeyer M., Cleaver O. (2010). Epithelial dynamics of pancreatic branching morphogenesis. Development 137, 4295–4305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J., Carroll T. J., Rajagopal J., Kobayashi A., Ren Q., McMahon A. P. (2009). A Wnt7b-dependent pathway regulates the orientation of epithelial cell division and establishes the cortico-medullary axis of the mammalian kidney. Development 136, 161–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W., Datta A., Leroy P., O’Brien L. E., Mak G., Jou T. S., Matlin K. S., Mostov K. E., Zegers M. M. (2005). Beta1-integrin orients epithelial polarity via Rac1 and laminin. Mol. Biol. Cell 16, 433–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.