

Abstract
Fluorine (from “le fluor”, meaning “to flow”) is a second row element of Group 17 in the periodic table. When bound to carbon it forms the strongest bond in organic chemistry to give organofluorine compounds. The scientific field treating them, organofluorine chemistry, started before elemental fluorine itself was isolated. Applying the fruits in academia, industrial organofluorine chemistry has developed over 80 years via dramatic changes during World War II. Nowadays, it provides various materials essential for our society. Recently, it utilizes elemental fluorine itself as a reagent for the introduction of fluorine atoms to organic molecules in leading-edge industries. This paper overviews the historical development of organofluorine chemistry especially from the viewpoint of material industry.
Keywords: organofluorine chemistry, fluorine, fluorination
Introduction—Incunabula of organofluorine chemistry
Alexander Borodin (1833–1887), who is well-known as a composer in classical music, is said to have made the first organofluorine compound.1) More precisely, he carried out the first nucleophilic replacement of a different halogen atom by fluoride (Eq. 1) and reported the results in 1862.2)–4) This was the first example of synthesis of an organofluorine compound by halogen exchange, which is now broadly used in fluorine chemistry and especially in fluorochemical industry for the introduction of fluorine atoms into organic molecules. Borodin was a promising young chemist, and became a professor of chemistry in 1864.
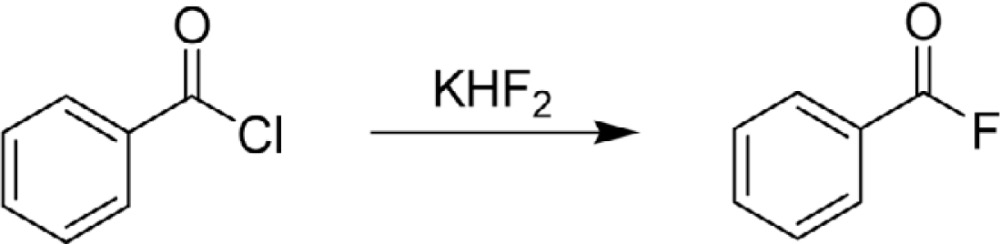 |
[1] |
Actual first synthesis of an organofluorine compound was reported by Dumas et al. in 1835,4) who prepared methyl fluoride from dimethyl sulfate (Eq. 2).
| [2] |
Formation of an aryl carbon–fluorine (C-F) bond was first carried out through diazofluorination by Schmitt et al. in 1870 (though the characterization was wrong), then by Lenz in 1877 (Eq. 3).4)
 |
[3] |
Mineral fluorides were recognized and used as early as 16th century. In 17th century, it was already known that glass was etched when it was exposed to a new acid generated from fluorspar and sulfuric acid.5) The generated acid was called hydrofluoric acid and was characterized by Scheele in 1771.5),6) Then it was eventually realized that hydrofluoric acid contained a previously unknown element, fluorine. Although organofluorine compounds were prepared in mid 19th century, elemental fluorine itself was not isolated at that time.
After continuous efforts by a great number of chemists, elemental fluorine was finally isolated in 1886 by Moissan, who electrolyzed a melt mixture of potassium hydrogen difluoride and hydrogen fluoride.5),7)
Because of hazards and difficulties in handling highly reactive and corrosive reagents, organofluorine chemistry remained relatively undeveloped until 1920’s.
In 1926, carbon tetrafluoride was first isolated from the product of the reaction of fluorine with wood charcoal by French chemists, Lebeau and Damiens,8) and was fully characterized 4 years later by Ruff.9)
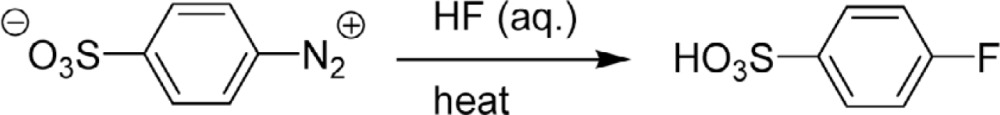
In 1927, Schiemann found an aromatic fluorination methodology:10) diazonium salts of aromatic amines were first prepared, and then decomposed in the presence of fluoroboric acid to give fluorinated aromatic compounds (Eq. 4). This reaction was improved and is still used even now for the manufacture of fluoroaromatic compounds.
| [4] |
Another important synthesis of fluoroaromatic compounds, nucleophilic halogen exchange from Cl to F using KF (Eq. 5), was reported by Gottlieb in 1936.4),11) This was the first example of halogen exchange method used in the fluoroarene synthesis.
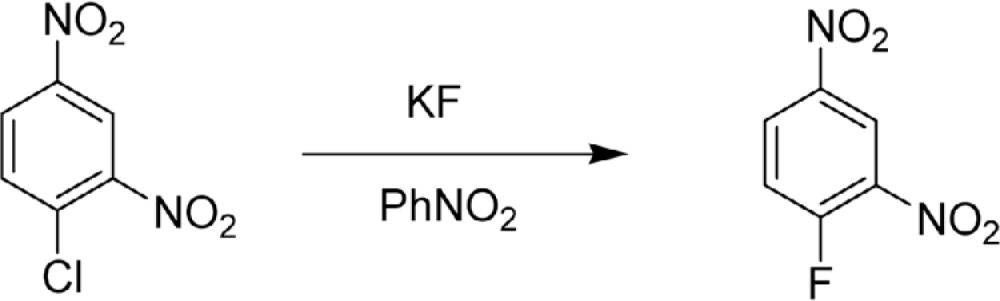 |
[5] |
Aromatic compounds with fluorinated side chain was reported by Swarts in 1898 for the first time.4),12) Benzotrichloride was found to react rapidly with SbF3 (Eq. 6).
| [6] |
This conversion from aromatic –CCl3 to –CF3 was later achieved with HF and reported in 1930’s.13),14)
Reactions above formed the basis of industrial application. The present overview treats mainly the history of material industry in organofluorine chemistry. Pharmaceutical and agrochemical applications are discussed briefly. Detail surveys of them have been provided by several reviews.15)–20) Review books on fluorine chemistry in academic field are also available. 21)–27)
Chlorofluorocarbons—First industrial application
In 1928, General Motors Corporation, manufacturers of refrigerators, appointed McNary, Midgley and Henne to the task of finding inert refrigerants. Until then, available conventional refrigerants had serious drawbacks: some were flammable, others like SO2 were corrosive and toxic, and still others like NH3 combined all three hazards.6) Midgley et al. studied on a fundamental basis, plotting trends of toxicity, flammability, and boiling point on a periodic chart of the element. He concluded that desired boiling range of 0 °C to −40 °C might be achieved by fluoroaliphatic compounds. At first, carbon tetra-fluoride (CF4) seemed appropriate because its boiling point was reported by Moissan as −15 °C28) (though this was incorrect).4) However, feeling that the synthesis of CF4 would be difficult, they decided to select CCl2F2 as their first choice and prepared sample by the reaction of CCl4 and SbF3, a synthesis originally reported by Swarts.4) Midgley gave a dramatic introduction of the new refrigerant by filling his lungs and extinguishing a light candle at a meeting of American Chemical Society in 1930.4),29)
In the meantime, General Motors had approached E. I. du Pont de Nemours & Company (DuPont) with a proposal to manufacture this product.6) The original work was soon supplemented by development studies for the practical production on a commercial scale (Eq. 7).4),6),30)
| [7] |
In 1930, a joint corporation, Kinetic Chemicals Inc., was formed by the two companies. By early 1931, the new product, trademarked Freon®-12, was being produced in commercial quantities. Application of the new compound was successful, and by the end of 1931, Kinetic Chemicals had expanded its facilities, and begun manufacture of anhydrous hydrofluoric acid, a basic raw material (Eq. 8).6)
| [8] |
Expansion during the next few years was continuous, and the product line was expanded as early as 1932 to include Freon®-11 (CCl3F) as well as Freon®-113 (CClF2CCl2F) and Freon®-114 (CClF2CClF2), two of the polychlorofluoroethane derivatives (Eq. 9).4),6),30)–33)
| [9] |
In 1935, this reaction was improved and employed for the synthesis of chlorofluoromethanes, the fluorinated derivatives of chloroform (Eq. 10).6)
| [10] |
First fluoroplastics
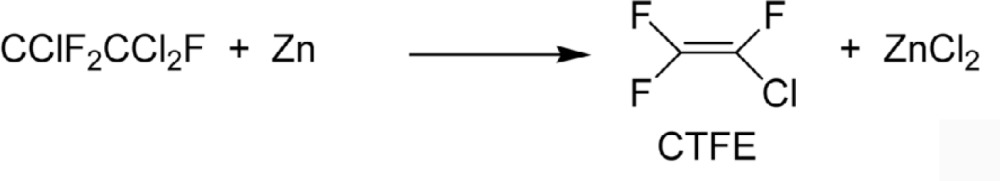
One of these, Freon® -22 (CHClF2), soon gained wide acceptance. This compound later became very important as the precursor of tetrafluoroethylene (TFE). At that time, however, TFE was synthesized by dechlorination of Freon®-114 with zinc (Eq. 11).4),31),32)
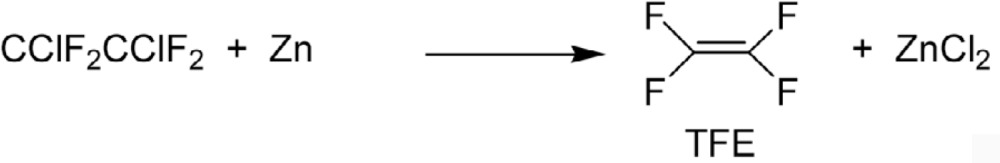 |
[11] |
In 1938, a DuPont chemist Plunkett, who was working with new chlorofluorocarbon gases relating to Freon® refrigerants, discovered polytetrafluoroethylene (PTFE).34) When Plunkett was preparing for an experiment with TFE and attempted to pump the gaseous TFE into hydrochloric acid to chlorinate it, he observed that nothing was coming out. He weighed the cylinder and it was the same as before. Plunkett then cut the cylinder open and discovered a waxy substance formed inside.4) After studying the powder, he found the substance to be heat resistant and chemically inert, and to have very low surface friction so that most other substances would not adhere to it. Plunkett realized that the exact combination of pressure and cold temperature, along with the age of the gas in a cylinder, had allowed the TFE gas molecules to polymerize to give this product PTFE with such potentially useful characteristics. Chemists and engineers in the Central Research Department of DuPont investigated the substance further.
Although PTFE is the best known fluoropolymer nowadays, it was not the first of the fluoroplastics to be prepared. The first one was poly(chlorotrifluoroethylene), PCTFE, the polymer of chlorotrifluoroethylene (CTFE), which was obtained by the dechlorination of Freon®-113 (Eq. 12)31),32) on an experimental scale as early as 1934.35),36)
 |
[12] |
Development during World War II
At that time, fluoropolymers were so expensive to produce that all believed they would never find a market. Their features were striking in fulfilling the requirements of the gaseous diffusion process of the Manhattan project at Columbia University in New York City during World War II and the fluoropolymers were first used as materials that could tolerate fluorine or its derivative uranium hexafluoride (UF6).37)
Even after the isolation of fluorine by Moissan in 1886, the difficulties in research using fluorine discouraged most chemists, and it was not until World War II when development was undertaken. The first large-scale production of fluorine was carried out for the atomic bomb Manhattan project which needed UF6 as a gaseous carrier of uranium to separate the 235U and 238U isotopes of uranium. Uranium tetra-fluoride (UF4), which was prepared from uranium dioxide (UO2) and hydrogen fluoride (HF), was converted to UF6 by reaction with fluorine.6),37),38) Using fluorine chemistry, atomic bombs were constructed and exploded over Hiroshima and Nagasaki, Japan.37)
UF6 was almost as reactive as elemental fluorine. In order to use it in a gas-diffusion plant, a wide range of materials which would not react with UF6 remained to be developed. These would include relatively low molecular weight liquids for coolants; higher molecular weight materials for lubricants; and polymers that could be fabricated into gaskets, valve packings and tubing.6),37) In 1937 and more extensively in 1939, preparative methods and properties of liquid fluorocarbons were disclosed by Simons and Block.39) In 1940, the possibility of use of fluorocarbons as sealants and coolants and as materials that are directly exposed to UF6 was suggested by Simons. A 2 mL sample of liquid fluorocarbon was sent from Simons to Urey at Columbia University and tested to show that it had the desired properties.40) The problem was preparation of fluorocarbons on a large scale. Several methods were subsequently developed.
The first major process was a catalytic fluorination process using fluorine. However, it was found extremely difficult to extend this process to large-scale production.40)
Another and principal method was the metallic fluoride process, which employs cobalt trifluoride (CoF3), in particular.41) Fowler developed this process42), 43) based on the method reported by Ruff.44),45) The fluorides can only be prepared by fluorinating the metal or low valent metal salt, with elemental fluorine. Ruff et al. demonstrated that metal fluorides of higher oxidation state were powerful fluorinating agents and utilized them to prepare a number of fluorocarbons (Scheme 1).
Scheme 1.
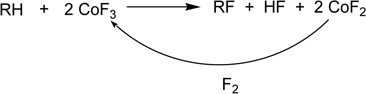
Fluorination with CoF3.
Treatment of a hydrocarbon with CoF3 at 400 °C gives a product where all the hydrogen atoms are replaced by fluorine. Cobalt difluoride (CoF2) is co-produced but is converted with elemental fluorine to cobalt trifluoride.
An entirely different approach was also carried out: the electrochemical fluorination (ECF) process for producing fluorocarbons. ECF was invented by Simons,46) but was not reported until 1949 for security reasons associated with the Manhattan project.
Civilian use of wartime technology
After World War II, Minnesota Mining and Manufacturing Company (3M) acquired the technology of ECF and improved it to a pilot plant level as early as 1947.47) The first commercial plant for the production of fluorocarbons was put into operation in 1951.47)
Further study of ECF was carried out after the launch of fluorocarbons, and product line was extended to production of fluorocarbon derivatives possessing functional groups: perfluoroethers, perfluoroacyl fluorides, perfluoroalkanesulfonyl fluorides, and perfluorinated amines.47),48) Among them, perfluoroacyl fluorides led to the development of 3M’s Scotchgard®, textile finishes.47) It works as an water and oil repellent agent.
Perfluoroalkyl iodides, which are synthesized by the telomerization method (Eq. 13) reported by Haszeldine around 1950,49)–51) are utilized for the same purpose.
| [13 ] |
Further process development on CoF3 fluorination after Manhattan project was carried out at ISC Chemicals Limited,52),53) and led to production of low molecular weight fluorocarbon fluids (Flutec®) later.
The first commercial products of PTFE were sold under the ‘Teflon’ tradename by DuPont in 1948.4) Teflon® has become a familiar household name, recognized worldwide for the superior nonstick properties associated with its use as a coating on cookware and as a soil and stain repellant for fabrics and textile products.
The monomer TFE is manufactured by the method developed in Manhattan project,37),54) namely by the pyrolysis of Freon®-22 (Eq. 14). Product TFE is accompanied by hexafluoropropylene (HFP) when pyrolysis was carried out with steam and alumina (Eq. 15).55),56) HFP is also obtained by pyrolysis of TFE at 850 °C.55)
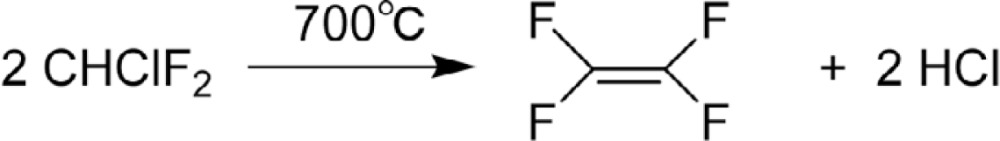 |
[14] |
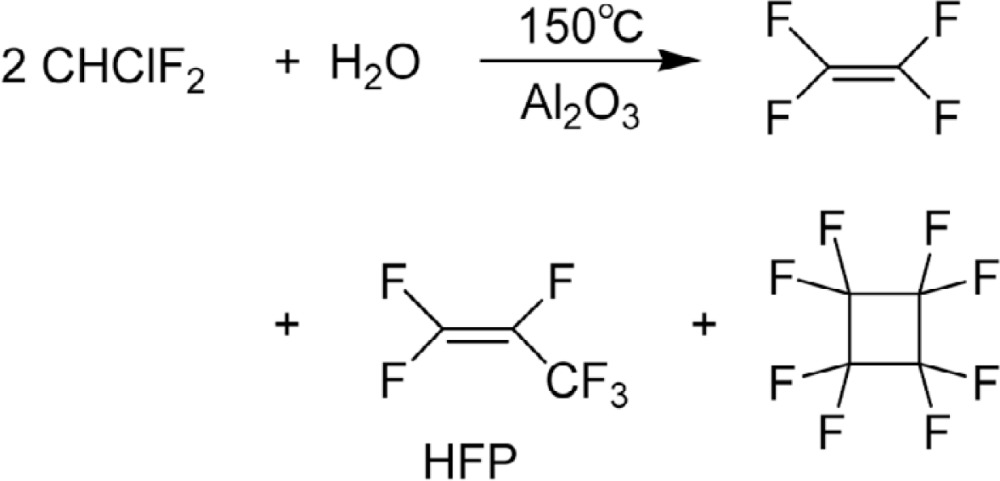 |
[15] |
After World War II, PCTFE, or Kel-F®, was marketed by M. W. Kellogg. It became generally available in the early 1950’s and was taken over by 3M in 1957.36)
In addition to perfluorinated homopolymer, TFE/HFP copolymer (fluorinated ethylene/propylene copolymer: FEP) was studied during the Manhattan project36) and commercialized in 1959 by Du-Pont.57) PTFE is characterized by high melting point, high thermal stability, insolubility, and chemical inertness. However, it cannot be processed by conventional melt techniques. This difficulty was overcome by FEP.
Partially-fluorinated polymers
Polymers with partially-fluorinated monomers, that is, PVdF (poly(vinylidene fluoride)) was developed in DuPont in 1948 and commercialized in 1950.57) Poly(vinyl fluoride) (PVF) was also studied and launched in 1960’s.36) Their films are important in industry, while PVdF is mainly used for water-resistant coating. The monomers are produced by a route shown in Scheme 2.
Scheme 2.
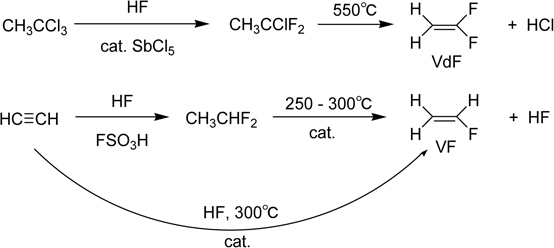
Synthesis of partially-fluorinated ethylene monomers.
The majority of commercial homopolymers nowadays are produced from only four monomers discussed above: tetrafluoroethylene (TFE), chlorotrifluoroethylene (CTFE), vinyl fluoride (VF), and vinylidene fluoride (VdF). Perfluorinated polymers are prepared by a free-radical polymerization reaction in water or in a fluorinated solvent.
Copolymerization of these monomers with hydrocarbon monomers were studied world-wide and commercialized in 1970’s.
Ethylene/TFE copolymer (ETFE): 1972 by Du-Pont and Asahi Glass independently.57)
TFE/propylene copolymer (TFE-P) Aflas®: 1975 by Asahi Glass.57)
ETFE is mainly used for insulation of electric cable, and also provides tough and flexible film which is used for architectural area. TFE-P is one of fluoroelastomers as well as VdF-HFP copolymers.
Functionalized fluoropolymers
On the other hand, it was difficult to copolymerize TFE with perfluorinated monomers other than HFP. However, perfluorinated vinyl ethers were found to react with TFE to give copolymers.58),59) Thus, perfluoroalkoxycopolymer (PFA) was synthesized and commercialized in 1972 by DuPont.57) The monomer, perfluorinated propyl vinyl ether (PPVE) is synthesized from hexafluoropropylene oxide (HFPO)60) as shown in Scheme 3.48),61)
Scheme 3.
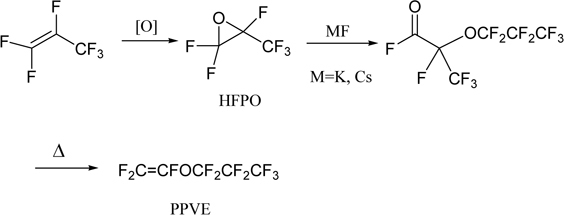
Synthesis of PPVE.
Before PFA was manufactured, HFPO was used as a monomer for anion polymerization in the preparation of perfluoropolyether (PFPE) fluids, which was developed by DuPont and commercialized as Krytox®.62)–64) Direct photo-oxidation of HFP or TFE was also developed for this purpose by Montedison (Fomblin®).63)–65) The products PFPE fluids, which were commercialized in 1960’s, have high chemical and thermal stability and excellent physical properties as lubricants which are highly reliable under very severe conditions.
In 1960’s, NASA used fuel cell for space exploration project including Gemini and Apollo. Nafion®, membranes made of a perfluorinated sulfonic acid ionomer, was first employed as a separator in the fuel cell. It is made of the polymer prepared by copolymerization of TFE and the functional perfluorovinyl ether, which is derived from HFPO as shown in Scheme 4.56)
Scheme 4.
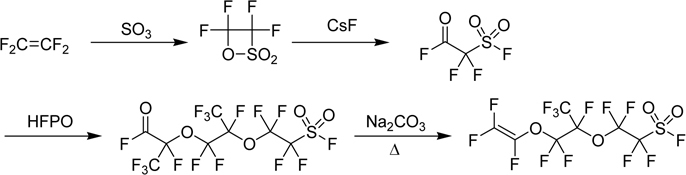
Synthesis of a monomer for ion-exchange membrane.
Organofluorine chemistry in minimization of environmental impact
In the late 1960’s, the chemical business came up against environmental issues. The sea around Kyushu, Japan, turned out to be the site of an outbreak of Minamata Disease, said to be one of the worst cases of mercury poisoning in history. This incident caused deep concern about chemical production processes. In 1968, Japanese government announced its consensus that “Minamata Disease was caused by industrial waste water containing organic mercury.” Restrictions were later placed on the use of inorganic mercury as well for electrochemical production of caustic soda (NaOH). One of the candidates for new mercury-free approach was the “ion-exchange membrane electrolysis method”. The key was a central membrane that allow only specific ions to pass through. When sodium chloride (NaCl) is electrolyzed to produce sodium hydroxide (NaOH), a violent situation to the membrane with strong alkali occurs. Only a membrane made from fluoropolymer could withstand this reaction.
The ion-exchange membrane Nafion® for fuel cell was applied to this purpose.56) Performance of Nafion® was not high enough, however, and a perfluorinated carboxylate polymer membrane was required to obtain a higher concentration of caustic soda. Flemion® (Asahi Glass, Japan) were developed as such an ion exchange membrane,66) and commercially produced since 1978.56),67)
The perfluorinated carboxylic acid membrane consists of a copolymer of TFE and perfluorinated vinyl ether, which has a carboxylic acid group in the side chain. The manufacturing process is shown in Scheme 5.68)
Scheme 5.
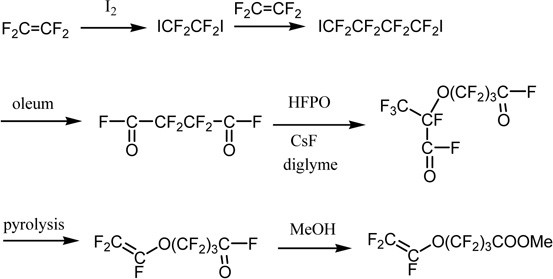
Synthesis of a monomer for perfluorinated carboxylic acid membrane.
The electrolysis process with ion-exchange membrane electrolysis method became common in commercial applications in 1984, and further improved to employ sulfonate-carboxylate laminated polymer membrane eventually achieving 100% proliferation in Japan and 51% worldwide (as of 2007). Moreover, the energy consumption of electrolysis processes was dramatically reduced by developing an advanced electrode.
Common plastics are made from a variety of organic materials, but the use of these materials should be minimized for reducing environmental burden. One way is to ensure products last longer. For example, for outdoor coating products that are exposed to outdoor environment, durable fluorinated materials are used. Lumiflon®, the first solvent-soluble fluoropolymer for coatings, was developed by Asahi Glass,57) which is the material for anti-corrosive coating of various structures such as bridges. It is made of CTFE/non-fluorinated vinyl ether copolymer. 56),69) Although conventional coating materials without fluorine last a few more than 10 years at best, Lumiflon® lasts up to 30 or 40 years. The added fluorinated structure enabled to create surprisingly durable coating materials.
The outstanding resistance to violent chemicals in ion-exchange membranes and the excellent weatherability are derived from high stability of C-F bonds. Any oxidants or chemicals hardly attack the fluorine-substituted carbons.
However, this stability was backfired in chlorofluorocarbons (CFCs). Studies have shown that in the deep blue sky, CFCs and other substances released into the atmosphere are causing damage to the ozone layer.70) In 1985, an ozone hole was discovered over the South Pole. CFCs are so stable substances that they moved far into the upper atmosphere, causing the destruction of the ozone layer. Carbon–chlorine bond cleavage of CFCs by UV light generates radical species, which cause ozone depletion, whereas C-F bonds are stable even in the stratosphere. It was therefore necessary to find alternative CFCs that possess carbon-hydrogen bonds, which are cleaved before they reach the stratosphere.
CFCs had three main applications: Refrigerants for air conditioners and refrigerators, blowing agents for urethane foaming, and precision equipment cleaning agents. Refrigerant CFC-12 (the same compound as Freon®-12: CCl2F2) was replaced by HFC-134a (CF3CH2F), and blowing agent CFC-11 (CCl3F) was substituted by HCFC-141b (CH3CCl2F) and then HFC-245fa (CF3CH2CHF2).71) However, there were no alternatives for cleaning agents CFC-113 (CCl2FCClF2).
The alternative material for CFC-113 was developed by Asahi Glass (HCFC-225ca and cb: ASAHIKLIN® AK-225). The innovative development was assisted by not only conventional experimental chemistry but computer chemistry technologies and the most appropriate alternative to CFC-113 was selected from thousands of candidate materials, then actually synthesized by modified Prins reaction of TFE and CHCl2F (HCFC-21) (Eq. 16).72)–74)
 |
[16] |
Thus, organofluorine chemistry plays important roles in minimization of environmental impact and in production of various materials for industrial use such as thermoplastics, elastomers, membranes, textile finishes, coatings.
Pharmaceuticals, agrochemicals and fine chemicals
Fluorine is the most electronegative element in the periodic table. When bound to carbon, it forms the strongest bonds in organic chemistry. This makes introduction of fluorine attractive for the development of material science. Although highly polarized, the C-F bond gains stability from the resultant electrostatic attraction between C and F atoms.75) Application to materials is mostly based on the properties derived from the fluorine-carbon bonds, which are hardly attacked by any oxidants or chemicals since C-F bonds tolerate various conditions. Therefore, normal metabolism in a living body is easily inhibited as well (block effect) by such strong bonds.
On the other hand, the van der Waals radius of fluorine is similar to that of hydrogen. Accordingly, organofluorine compounds are similar in steric size to non-fluorinated ones but quite different in electronic nature. As a living body can not sterically distinguish fluorinated molecules from the corresponding non-fluorinated one, fluorinated molecules are incorporated into metabolic sequences in a manner similar to that of non-fluorinated one (mimic effect). However, since the C-F bond in the fluorinated molecule resists the metabolism common with the parent compound due to opposite polarization, the normal metabolism in a living body is inhibited to cause various biological effects. This makes fluorine substitution attractive for invention of pharmaceuticals and agrochemicals.15),16)
There are many applications. For examples, inhalation anesthetics15) are synthesized by classical halogen exchange reactions.30)–32) Aryl-CF3 containing herbicides16)–18) are synthesized using a halogen exchange reaction originated in 1930’s (Eq. 6).13),14) Non-steroidal anti-inflammatory drugs (NSAIDs),15),16) anti-bacterial agents19),20) and insecticides18) are synthesized by Balz-Schiemann reaction (Eq. 4)10) or aromatic halogen exchange (HALEX) reaction (Eq. 5).11) The HALEX reaction is extensively performed on a commercial scale to produce aromatic intermediate building blocks of low fluorine content for functional materials such as liquid crystals (LCs).
More sophisticated syntheses are carried out for manufacturing LCs particularly for thin film transistor-liquid crystal display (TFT-LCD),76),77) transparent fluorinated resin such as Cytop®78) for plastic optical fiber (POF)79) and for semi-conductor manufacturing process,80) and a new lubricant, which is synthesized by application of direct fluorination, for a hard disc drive (HDD).81)
Direct fluorination with elemental fluorine
Elemental fluorine was first prepared in small quantities by Henri Moissan in 1886 by electrolysis of anhydrous hydrogen fluoride.5),7) Moissan himself was the first to carry out the reactions between neat fluorine and several organic compounds. However, only decomposed products resulted and, occasionally, explosions occurred.82)
In the early 20th century, it was very difficult to control the reactions with fluorine because of the violence in nature. For example, in 1929, Bancroft and Jones reported explosions during attempted fluorination of benzene and toluene with molecular fluorine.83),84) However, dilution of fluorine by inert gases such as nitrogen came to allow the reactions with fluorine to be carried out safely and efficiently.
In 1931, Bancroft and Wherty tried the fluorination of benzene again using fluorine diluted with nitrogen. The explosion was avoided, though they obtained only tarry products.82),85) Beckmuller attempted the fluorination of several aromatic compounds with fluorine, but also obtained only tars with high fluorine content.82),86)
During World War II, the first sample of fluorocarbons, found to be inert towards UF6, had been made by direct reaction between carbon and elemental fluorine catalyzed by mercury.37) In 1941, Bigelow and Fukuhara reported that direct fluorination of benzene with elemental fluorine in the vapor phase over copper gauze catalyst gave perfluorocyclohexane with only moderate degradation.37),87) This method looked promising for Manhattan project, and was investigated in detail. Although yields were improved up to 58% of perfluorocyclohexane from benzene, CoF3 method was found more suitable to preparation of various fluorocarbons.37)
After World War II, in order to modify the high reactivity of fluorine, the vapor-phase fluorination (“Jet fluorination”) apparatus for direct fluorination was developed by Bigelow and coworkers.88) Jet fluorination seems to be a method of choice for preparation of low-molecular-weight fluorocarbons. It was used on a semi-technical scale for production of perfluoropropane, which is used as a dry (plasma) etchant in microelectronics industry.48)
In 1970, the low temperature gradient fluorination (“LaMar fluorination”) technique was developed by Lagow and Margrave.89),90) In the LaMar fluorination process, substrates are condensed at low temperature into a tube packed with copper turnings through which fluorine, initially highly diluted in either helium or nitrogen, is passed. The concentration of fluorine and the reaction temperature are slowly increased over a period of several days to permit perfluorination. This batch process requires relatively long reaction times to obtain perfluorinated material.
Some years later, Adcock invented a flow version, an aerosol fluorination process.91) The principle is that substrates are absorbed onto the surface of fine sodium fluoride particles in a fluorination apparatus, into which fluorine is introduced and the apparatus is warmed and UV irradiated to complete perfluorination. This process has an advantage that it is a continuous flow method and the control of reaction parameters are easier.
In 1980’s, Scherer, Yamanouchi, and Ono disclosed a practical liquid-phase direct perfluorination method through study on artificial blood substitutes.84),92) The method is quite characteristic in i) inverse addition of a substrate into an inert liquid saturated F2 gas, ii) undiluted 100% fluorine gas is used, and iii) UV irradiation. In fact, in the liquid-phase direct fluorination process, the reactant is injected at a very slow constant rate into an inert fluorocarbon solvent saturated by fluorine. It is pointed out to be very important that a large excess of F2 relative to hydrogen atoms to be replaced in a substrate should be maintained. This method is, however, only suitable for the perfluorination of substrates, such as partially fluorinated ethers and amines, both soluble in a perfluorocarbon solvent and can withstand such vigorous reaction conditions.
Almost the same time, Exfluor Corporation claimed that various ethers and esters are perfluorinated without UV irradiation (Exfluor-Lagow method).90),93) Their methods are based on the essential idea of inverse addition of substrates into an inert liquid dissolving fluorine gas. The Exfluor-Lagow method involves slow addition of both a hydrocarbon substrate and fluorine in excess into a vigorously stirred chlorofluorocarbon (CFC) or a perfluorinated inert solvent. If required, the reaction is accelerated by adding a small quantity of a highly reactive hydrocarbon such as benzene, which reacts spontaneously with fluorine to produce a very high concentration of fluorine radicals that ensure perfluorination of substrates.
The Exfluor-Lagow elemental fluorine process can give products of the liquid-phase fluorination process in high yields. However, reaction solvents are limited to now-regulated CFCs, particularly for direct fluorination of substrates which contain functional group(s) in the structure because of solubility problem.
Partially fluorinated substrates are more stable towards the fluorination process, since solubility increases, and presence of a polyfluoroalkyl group significantly lowers the oxidation potential of the substrates. Consequently, perfluorinated compounds are generally produced in higher yields than the corresponding non-fluorinated compounds. This method was applied to preparation of perfluoro ethers.94),95)
Considering these results, it is suggested that direct fluorination method had almost reached an adequate standard level for monomer synthesis in industry.
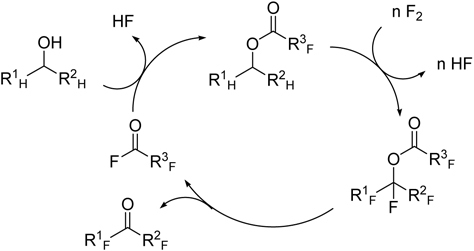
Combination of direct fluorination and organic synthesis
Okazoe et al. attempted at the direct application of Exfluor-Lagow method to the synthesis of the monomer PPVE for perfluoroalkoxy copolymer (PFA) (Scheme 6). However, it was found hard to reproduce the results.96) In some cases, vapor phase reaction partly took place due to high volatility of substrate and led to an explosion. In order to solve this problem, they have developed an entirely new method that involves combination of direct fluorination and organic synthesis, perfluorination of esterified compounds followed by thermolysis, abbreviated as PERFECT (Scheme 7).96) By employing partially-fluorinated ester of high-molecular weight, the dangerous vapor-phase reactions can be suppressed and solubility of substrates in a solvent used in the liquid-phase fluorination can be much improved. This contrasts sharply to that of non-fluorinated compounds. Thus, available perfluoro(alkoxyalkanoyl) fluorides such as perfluoro(2-propoxypropionyl) fluoride can be multiplied by use of the hydrocarbon counterpart alcohols and fluorine gas as raw materials (Scheme 7).
Scheme 6.
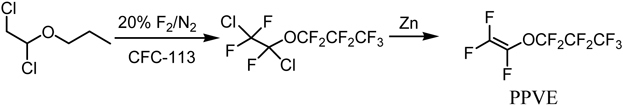
Synthesis of PPVE from a non-fluorinated dichloroethyl ether.
Scheme 7.
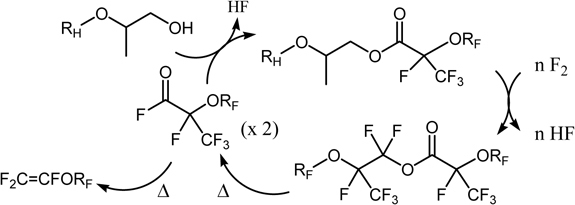
The PERFECT method for synthesis of perfluorinated vinyl ethers.
Subsequently, the possibility of the PERFECT method was extended to synthesis of more general perfluoroacyl fluorides. In the case that the desired perfluoro(alkoxyalkanoyl) fluoride is not readily available, it can be obtained from its hydrocarbon counterpart alcohol and an available perfluoroacyl fluoride (Scheme 8).97)
Scheme 8.
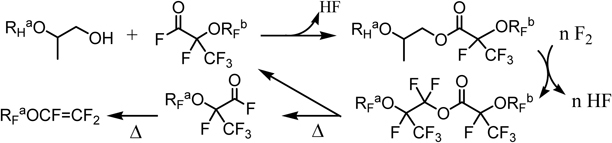
The PERFECT method for synthesis of various perfluorinated acyl fluorides.
The PERFECT method was applied to the synthesis of perfluorinated diacyl fluorides, which are used for the carboxylic acid type ion exchange membrane (Scheme 9).98) Non-fluorinated diols are employed as the substrates.
Scheme 9.
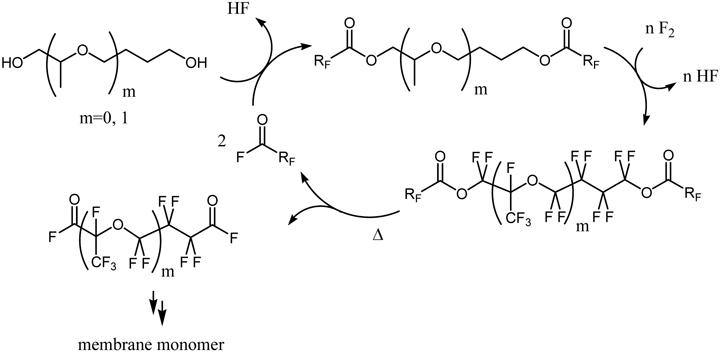
The PERFECT method for synthesis of monomers for carboxylic acid membrane.
Synthesis of monomers for perfluoalkanesulfonic acid type membrane was achieved through direct fluorination of the substrate which possesses a sulfonyl group at the end (Scheme 10).99)
Scheme 10.
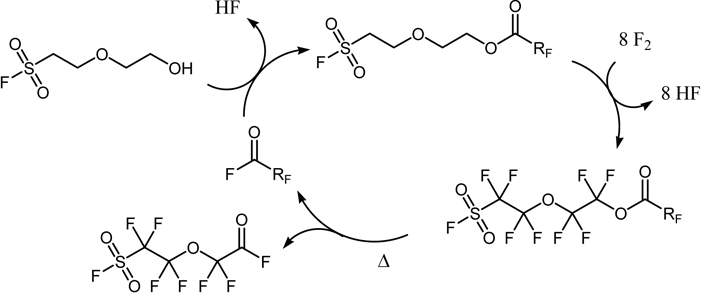
The PERFECT method for synthesis of monomers for perfluorosulfonic acid membrane.
The application of the PERFECT method to non-fluorinated secondary alcohols as a starting material (Scheme 11) enabled to synthesize perfluorinated ketones.100) A precursor polyfluoro ketone for fluoropolymer resists for 157 nm microlithography was synthesized.100)
Scheme 11.
The PERFECT method for synthesis of perfluoro ketones.
Recently, the PERFECT method was successfully applied to polymers as substrates81) and provides manufacturing process of the new lubricant for HDD.
The PERFECT process can provide with tailor-made functionalized materials. Because raw materials are inexpensive hydrocarbons, and the synthesis from hydrocarbon components makes it possible to create entirely new fluorinated compounds at will. Theoretically, by-product of the process is hydrogen only, because HF, which is formed in the process, can be converted electrochemically back to hydrogen and fluorine; fluorine can be used again in the process. Moreover, the PERFECT process dose not use any solvent other than products or intermediates of the process. In that sense, it contributes to reduce environmental burden.
Conclusion
Nowadays, organofluorine compounds are essential materials especially in recent IT, electronics, and medical applications.101) Moreover, organofluorine chemistry plays important roles in minimization of environmental impact. They are based on the history over 80 years. Among them, recent development of direct fluorination with elemental fluorine is promising to prepare versatile materials both in creating comfortable life and in reducing environmental burden.
Profile
Takashi Okazoe was born in Sapporo in 1961. He received his B. of Eng. and M. of Eng. from Kyoto University under the direction of Professor Hitosi Nozaki in 1985 and Professor Kiitiro Utimoto in 1987, respectively. He joined Asahi Glass Co., Ltd. as a researcher at Research Center in 1987 and started study of fluorinated pharmaceutical intermediates and ingredients. There he was promoted to Group Leader in fluorinated materials field in 1999. In 2006, he moved to the head office of AGC Chemicals, Asahi Glass Co., Ltd. and engaged in planning of research and development strategy. He received Technology Award from the Society of Synthetic Organic Chemistry, Japan in 2006, and was awarded the Doctorate degree from Kyoto University in 2009. He is the inventor of over 100 patents as well as the author of academic papers. His current research interests are organofluorine chemistry and industrial synthetic methodology which minimize environmental impact for sustainable society.

References
- 1).Behrman, J. (2006) Borodin. J. Chem. Edu. 83, 1138–1139 [Google Scholar]
- 2).Gordin, M. D. (2006) Facing the magic: how original was Borodin’s chemistry? J. Chem. Edu. 83, 561–565 [Google Scholar]
- 3).Borodin, A. (1862) Facts for serve to the history of fluoride and preparation of benzoyl fluoride. Compt. Rend. 55, 553–556 [Google Scholar]
- 4).Banks, R. E. and Tatlow, J. C. (1986) Synthesis of C-F bonds: the pioneering years, 1835–1940. J. Fluorine Chem. 33, 71–108 [Google Scholar]
- 5).Banks, R. E. (1986) Isolation of fluorine by Moissan: setting the scene. J. Fluorine Chem. 33, 3–26 [Google Scholar]
- 6).Hamilton, J. M.Jr. (1963) The organic fluorochemicals industry. Adv. Fluorine Chem. 3, 117–180 [Google Scholar]
- 7).Flahaut, J. and Viel, C. (1986) The life and scientific work of Henri Moissan. J. Fluorine Chem. 33, 27–42 [DOI] [PubMed] [Google Scholar]
- 8).Lebeau, P. and Damiens, A. (1926) Carbon tetrafluoride. Compt. Rend. 182, 1340–1342 [Google Scholar]
- 9).Ruff, O. and Keim, R. (1930) Reaction products of different types of carbon with fluorine. I. Carbon tetrafluoride (tetrafluoro methane). Z. Anorg. Allgem. Chem. 192, 249–256 [Google Scholar]
- 10).Balz, G. and Schiemann, G. (1927) Aromatic fluorine compounds. I. A new method for their preparation. Chem. Ber. 60, 1186–1190 [Google Scholar]
- 11).Gottlieb, H. B. (1936) The replacement of chlorine by fluorine in organic compounds. J. Am. Chem. Soc. 58, 532–533 [Google Scholar]
- 12).Swarts, F. (1898) Fluorine derivatives of toluene. Bull. Acad. Roy. Belg. 35, 375–420 [Google Scholar]
- 13).Osswald, P., Muller, F. and Steinhauser, F. (1933) Fluorinated hydrocarbons. Deutsche Patent 575593 [Google Scholar]
- 14).Simons, J. H. and Lewis, C. J. (1938) The preparation of benzotrifluoride. J. Am. Chem. Soc. 60, 492 [Google Scholar]
- 15).Filler, R., Kobayashi, Y. and Yagupolskii, L. M. (1993) Organofluorine Compounds in Medicinal Chemistry and Biomedical Applications. Elsevier, Amsterdam [Google Scholar]
- 16).Hiyama, T. (2000) Biologically active organofluorine compounds. InOrganofluorine Compounds, Chemistry and Applications. Springer, Berlin, pp. 137–182 [Google Scholar]
- 17).Kumai, S. (1997) Fluorine-containing agrochemicals and their intermediates. InDevelopment of Fluoro Functional Materials (eds. Yamabe, M. and Matsuo, M.). CMC, Tokyo, pp. 209–230 [Google Scholar]
- 18).Morizawa, Y. (2004) Fluorine-containing agrochemicals. InFusso Kagaku Nyumon (ed. The 155th Committee on Fluorine Chemistry, Japan Society for the Promotion of Science). Sankyo Shuppan, Tokyo, pp. 425–434 [Google Scholar]
- 19).Yasuda, A. (1997) Fluorine-containing pharmaceuticals and their intermediates. InDevelopment of Fluoro Functional Materials (eds. Yamabe, M. and Matsuo, M.). CMC, Tokyo, pp. 182–208 [Google Scholar]
- 20).Matsumura, Y. (2004) Fluorine-containing pharmaceuticals. InFusso Kagaku Nyumon (ed. The 155th Committee on Fluorine Chemistry, Japan Society for the Promotion of Science). Sankyo Shuppan, Tokyo, pp. 407–424 [Google Scholar]
- 21).Hudlicky, M. and Pavlath, A. E. (1995) Chemistry of Organic Fluorine Compounds II. Am. Chem. Soc., Washington [Google Scholar]
- 22).Baasner, B., Hagemann, H. and Tatlow, J. C. (1999) Methoden Org. Chem. (Houben-Weyl), 4th ed., Georg Thieme Verlag, Stuttgart, Vol. E10 [Google Scholar]
- 23).Hiyama, T. (2000) Organofluorine Compounds, Chemistry and Applications. Springer, Berlin [Google Scholar]
- 24).Chambers, R. D. (2004) Fluorine in Organic Chemistry, 2nd ed., Blackwell, Oxford [Google Scholar]
- 25).The 155th Committee on Fluorine Chemistry, Japan Society for the Promotion of Science (2004) Fusso Kagaku Nyumon. Sankyo Shuppan, Tokyo [Google Scholar]
- 26).Kirsh, P. (2004) Modern Fluoroorganic Chemistry: synthesis, reactivity, applications. Wiley-VCH, Weinheim [Google Scholar]
- 27).Uneyama, K. (2006) Organofluorine Chemistry: Blackwell, Oxford [Google Scholar]
- 28).Moissan, H. (1890) Carbon tetrafluoride. Compt. Rend. 110, 951–954 [Google Scholar]
- 29).Midgley, T. and Henne, A. L. (1930) Organic fluorides as refrigerants. Ind. Eng. Chem. 22, 542–545 [Google Scholar]
- 30).Daudt, H. W. (1935) Organic fluorine compound. US Patent 2005706 [Google Scholar]
- 31).Booth, H. S., Mong, W. L. and Burchfield, P. E. (1932) Fluorination of hexachloroethane under pressure. Ind. Eng. Chem. 24, 328–331 [Google Scholar]
- 32).Locke, H. G., Brode, W. R. and Henne, A. L. (1934) Fluorochloroethanes and fluorochloroethylenes. J. Am. Chem. Soc. 56, 1726–1728 [Google Scholar]
- 33).Sheppard, W. A. and Sharts, C. M. (1969) Classical methods of fluorination. InOrganic Fluorine Chemistry. W. A. Benjamin Inc., New York, pp. 78–79 [Google Scholar]
- 34).Plunkett, R. J. (1939) Tetrafluoroethylene polymers. US Patent 2230654
- 35).I. G. Farbenindustrie A.-G. (1936) Manufacture of polymerisation products. Fr. Patent 796026
- 36).Willoughby, B. G. (1982) Non-TFE-based fluoroplastics. InPreparation, Properties, and Industrial Applications of Organofluorine Coupounds (ed. Banks, R. E.). John Wiley & Sons, New York, pp. 201–235 [Google Scholar]
- 37).Goldwhite, H. (1986) The Manhattan project. J. Fluorine Chem. 33, 109–132 [Google Scholar]
- 38).Atherton, M. J. (2000) Following fluorine in nuclear fuel manufacture at BNFL. InFluorine Chemistry at the Millennium: Fascinated by Fluorine(ed. Banks, R. E.). Elsevier, Amsterdam, pp. 1–14 [Google Scholar]
- 39).Simons, J. H. and Block, L. P. (1937) Fluorocarbons. J. Am. Chem. Soc. 59, 1407; [Google Scholar]; Simons, J. H. and Block, L. P. (1939) Fluorocarbons. J. Am. Chem. Soc. 61, 2962–2966 [Google Scholar]
- 40).Brice, T. J. (1950) Fluorocarbons—their properties and wartime development. InFluorine Chemistry (ed. Simmons, J. H.). Academic Press Inc., New York, Vol. 1, pp. 423–462 [Google Scholar]
- 41).Fowler, R. D., Burford, W. B. III, Hamilton, J. M.Jr., Sweet, R. G., Weber, C. E., Kasper, J. S.et al. (1951) Vapor-phase fluorination using cobalt trifluoride. InPreparation, Properties and Technology of Fluorine and Organic Fluoro Compounds (eds. Slesser, C. and Schram, S. R.). McGraw-Hill Book Co., Inc., New York, pp. 349–371 [Google Scholar]
- 42).Fowler, R. D., Burford, W. B. III, Hamilton, J. M. Jr., Sweet, R. G., Weber, C. E., Kasper, J. S.et al. (1947) Synthesis of fluorocarbons. Ind. Eng. Chem. 39, 292–298 [Google Scholar]
- 43).Burford, W. B. III, Fowler, R. D., Hamilton, J. M. Jr., Anderson, H. C., Weber, C. E. and Sweet, R. G. (1947) Pilot plant synthesis. Ind. Eng. Chem. 39, 319–329 [Google Scholar]
- 44).Ruff, O. and Ascher, E. (1929) Fluorides of the eighth group of the periodic system. Z. Anorg. Allgem. Chem. 183, 193–213 [Google Scholar]
- 45).Ruff, O. and Keim, R. (1931) The fluorination of carbon compounds (benzene and tetrachloromethane with iodine pentafluoride tetrachloromethane with fluorine). Z. Anorg. Allgem. Chem. 201, 245–258 [Google Scholar]
- 46).Simons, J. H. (1949) Electrochemical process for the production of fluorocarbons. J. Electrochem. Soc. 95, 47–67 [Google Scholar]
- 47).Abe, T. and Nagase, S. (1982) Electrochemical fluorination (Simons process) as a route to perfluorinated organic compounds of industrial interest. InPreparation, Properties and Industrial Applications of Organofluorine Compounds (ed. Banks, R. E.). Ellis Horwood, Chichester, pp. 19–43 [Google Scholar]
- 48).Banks, R. E. and Tatlow, J. C. (1986) A guide to modern organofluorine chemistry. J. Fluorine Chem. 33, 227–284 [Google Scholar]
- 49).Haszeldine, R. N. (1949) The reaction of fluorocarbon radicals. Part I. The reaction of iodotrifluoromethane with ethylene and tetrafluoroethylene. J. Chem. Soc. 2856–2861
- 50).Haszeldine, R. N. (1953) Reactions of fluorocarbon radicals. Part XII. The synthesis of fluorocarbons and of fully fluorinated iodo-, bromo-, chloroalkanes. J. Chem. Soc. 3761–3768 [Google Scholar]
- 51).Haszeldine, R. N. (1986) Perfluoroalkyl iodides. J. Fluorine Chem. 33, 307–313 [Google Scholar]
- 52).Kingdom, R. J., Bond, G. D. and Linton, W. L. (1972) Improved fluorination process. Brit. Patent 1281822
- 53).Joyner, B. D. (1986) Production and applications of Flutec® fluorocarbons. J. Fluorine Chem. 33, 337–345 [Google Scholar]
- 54).Park, J. D., Benning, A. F., Downing, F. B., Laucius, J. F. and McHarness, R. C. (1947) Synthesis of tetrafluoroethylene. Ind. Eng. Chem. 39, 354–358 [Google Scholar]
- 55).Ukihashi, H. and Hisamune, M. (1969) Process of producing tetrafluoroethylene and hexafluoropropylene. US Patent 3459818
- 56).Hiyama, T. (2000) Fluorine-containing polymers. InOrganofluorine Compounds, Chemistry and Applications. Springer, Berlin, pp. 212–233 [Google Scholar]
- 57).Miyake, H. and Takakura, T. (1997) Fluororesin. InDevelopment of Fluoro Functional Materials (eds. Yamabe, M. and Matsuo, M.). CMC, Tokyo, pp. 46–62 [Google Scholar]
- 58).Feiring, A. E. (1994) Fluoroplastics. InOrganofluorine Chemistry: Principles and Commercial Applications (eds. Banks, R. E., Smart, B. E. and Tatlow, J. C.). Plenum Press, New York, pp. 339–372 [Google Scholar]
- 59).Hintzer, K. and Löhr, G. (1997) Melt processable tetrafluoroethylene—perfluoropropylvinyl ether copolymers (PFA). InModern Fluoropolymers (ed. Scheirs, J.). John Wiley & Sons, Chichester, pp. 223–237 [Google Scholar]
- 60).Millauer, H., Schwertfeger, W. and Siegemund, G. (1985) Hexafluoropropylene oxide—a key compound in organofluorine chemistry. Angew. Chem., Int. Ed. Engl. 24, 161–179 [Google Scholar]
- 61).Fritz, C. G. and Selman, S. (1966) Fluorinated vinyl ethers and their preparation. US Patent 3291843
- 62).Moore, E. P. (1966) Fluorocarbon ethers derived from hexafluoropropylene oxide. US Patent 3250808
- 63).Caporiccio, G. (1986) Perfluoropropylether fluids: properties and applications. J. Fluorine Chem. 33, 314–320 [Google Scholar]
- 64).Itami, Y. (2004) Oil and inert medium. InFusso Kagaku Nyumon (ed. The 155th Committee on Fluorine Chemistry, Japan Society for the Promotion of Science). Sankyo Shuppan, Tokyo, pp. 279–289 [Google Scholar]
- 65).Sianesi, D., Pasetti, A. and Belardinelli, G. (1967) Fluorine-containing polyethers. Brit. Patent 1217871
- 66).Ukihashi, H., Yamabe, M. and Miyake, H. (1987) Polymeric fluorocarbon acids and their applications. Prog. Polym. Sci. 12, 229–270 [Google Scholar]
- 67).Yamabe, M. and Miyake, H. (1994) Fluorinated membranes. InOrganofluorine Chemistry: Principles and Commercial Applications (eds. Banks, R. E., Smart, B. E. and Tatlow, J. C.). Plenum Press, New York, pp. 403–411 [Google Scholar]
- 68).Yamabe, M., Munekata, S., Kaneko, I. and Ukihashi, H. (1999) Synthesis of perfluorinated vinyl ethers having ester group. J. Fluorine Chem. 94, 65–68 [Google Scholar]
- 69).Yamabe, M., Higaki, H. and Kijima, G. (1984) New fluoropolymer coatings. InOrganic Coatings Science and Technology (eds. Parfitt, G. D. and Pastis, A. V.). Marcel Dekker, New York, Vol. 7, pp. 25–39 [Google Scholar]
- 70).Rowland, F. S. and Molina, M. (1974) Stratospheric sink for chlorofluoromethanes. Nature 249, 810–812 [Google Scholar]
- 71).Hiyama, T. (2000) Chlorofluoro-, Hydrochlorofluoro-, Hydrofluorocarbons and Alternatives. InOrganofluorine Compounds, Chemistry and Applications. Springer, Berlin, pp. 192–196 [Google Scholar]
- 72).Paleta, O., Posta, A. and Tesarik, K. (1971) Reaction of tetrafluoroethylene with monofluoromethanes in the presence of aluminum chloride. Coll. Czech. Chem. Commun. 36, 1867–1875 [Google Scholar]
- 73).Morikawa, S., Yoshitake, M., Tatematsu, S. and Tanuma, T. (1990) Preparation of chlorofluoropropanes. Jpn. Patent 02017134
- 74).Okamoto, H. (2004) Cleaning agents. InFusso Kagaku Nyumon (ed. The 155th Committee on Fluorine Chemistry, Japan Society for the Promotion of Science). Sankyo Shuppan, Tokyo, pp. 114–120 [Google Scholar]
- 75).O’Hagan, D. (2008) Understanding organofluorine chemistry. An introduction to the C-F bond. Chem. Soc. Rev. 37, 308–319 [DOI] [PubMed] [Google Scholar]
- 76).Yokokoji, O. (1997) Fluorine-containing liquid crystal. InDevelopment of Fluoro Functional Materials (eds. Yamabe, M. and Matsuo, M.). CMC, Tokyo, pp. 123–134 [Google Scholar]
- 77).Asai, T. (2004) Fluorine-containing liquid crystal. InFusso Kagaku Nyumon (ed. The 155th Committee on Fluorine Chemistry, Japan Society for the Promotion of Science). Sankyo Shuppan, Tokyo, pp. 235–243 [Google Scholar]
- 78).Oharu, K., Sugiyama, N., Nakamura, M. and Kaneko, I. (1991) Preparation and reactions of perfluoro(alkenyl vinyl ethers). Reports Res. Asahi Glass Co., Ltd. 41, 51–58 [Google Scholar]
- 79).Ogawa, G. (2004) Plastic optical fiber. InFusso Kagaku Nyumon (ed. The 155th Committee on Fluorine Chemistry, Japan Society for the Promotion of Science). Sankyo Shuppan, Tokyo, pp. 316–339 [Google Scholar]
- 80).Matsukura, I. (2004) Amorphous fluororesin. InFusso Kagaku Nyumon (ed. The 155th Committee on Fluorine Chemistry, Japan Society for the Promotion of Science). Sankyo Shuppan, Tokyo, pp. 316–339 [Google Scholar]
- 81).Shirakawa, D., Sonoda, K. and Ohnishi, K. (2007) A study on design and synthesis of new lubricant for near contact recording. IEEE Trans. Magn. 43, 2253–2255 [Google Scholar]
- 82).Hatchinson, J. and Sandford, G. (1997) Elemental fluorine in organic chemistry. Top. Curr. Chem. 193, 1–43 [Google Scholar]
- 83).Bancroft, W. D. and Jones, N. C. (1929) Electrolysis with fluorine. Trans. Amer. Electrochem. Soc., 55, 183–198 [Google Scholar]
- 84).Ono, T. (2003) Direct fluorination as a tool for preparing perfluoro compounds: an overview of the liquid-phase direct fluorination. Chimica Oggi. 39–43 (July August). [Google Scholar]
- 85).Bancroft, W. D. and Whearty, S. F. (1931) Aromatic substitution products with fluorine. Proc. Nat. Acad. Sci. USA 17, 183–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86).Beckmuller, W. (1933) Fluorination of organic compounds. III. Action of fluorine on organic compounds. Liebigs Ann. Chem. 506, 20–59 [Google Scholar]
- 87).Fukuhara, N. and Bigelow, L. A. (1941) The vapor phase fluorination of benzene. J. Am. Chem. Soc. 63, 2792–2795 [Google Scholar]
- 88).Tyczkowskyi, E. A. and Bigelow, L. A. (1955) A New Jet Fluorination Reactor. J. Am. Chem. Soc. 77, 3007–3008 [Google Scholar]
- 89).Lagow, R. and Margrave, J. (1979) Direct fluorination: a “new” approach to fluorine chemistry. Prog. Inorg. Chem. 26, 161–210 [Google Scholar]
- 90).Lagow, R. (2000) The discovery of successful direct fluorination synthesis: three era of elemental fluorine chemistry. InFluorine Chemistry at the Millennium: Fascinated by Fluorine (ed. Banks, R. E.). Elsevier, Amsterdam, pp. 283–296 [Google Scholar]
- 91).Adcock, J. L., Horita, K. and Renk, E. B. (1981) Low-temperature fluorination of aerosol suspensions of hydrocarbons utilizing elemental fluorine. J. Am. Chem. Soc. 103, 6937–6947 [Google Scholar]
- 92).Scherer, K. V., Yamanouchi, K. and Ono, T. (1990) A new synthetic approach to perfluorochemicals: liquid phase photo fluorination with elemental fluorine. Part I. J. Fluorine Chem. 50, 47–65 [Google Scholar]
- 93).Bierschenk, T. R., Juhlke, T., Kawa, H. and Lagow, R. J. (1992) Liquid phase fluorination. US Patent 5093432
- 94).Badyal, J. P. S., Chambers, R. D. and Joel, A. K. (2000) Partly fluorinated polyethers as additives for surface modification. J. Fluorine Chem. 60, 297–300 [Google Scholar]
- 95).Chambers, R. D., Joel, A. K. and Rees, A. J. (2000) Fluorination of modified ethers and polyethers. J. Fluorine Chem. 101, 97–105 [Google Scholar]
- 96).Okazoe, T., Watanabe, K., Itoh, M., Shirakawa, D., Murofushi, H., Okamoto, H.et al. (2001) A new route to perfluoro(propyl vinyl ether) monomer: synthesis of perfluoro(2-propoxypropionyl) fluoride from non-fluorinated compounds. Adv. Synth. Catal. 343, 215–219 [Google Scholar]
- 97).Okazoe, T., Watanabe, K., Itoh, M., Shirakawa, D. and Tatematsu, S. (2001) A new route to perfluorinated vinyl ether monomers: synthesis of perfluoro(alkoxyalkanoyl) fluorides from non-fluorinated compounds. J. Fluorine Chem. 112, 109–116 [Google Scholar]
- 98).Okazoe, T., Watanabe, K., Itoh, M., Shirakawa, D., Kawahara, K. and Tatematsu, S. (2005) Synthesis of perfluorinated carboxylic acid membrane monomer by utilizing liquid-phase direct fluorination. J. Fluorine Chem. 126, 521–527 [Google Scholar]
- 99).Okazoe, T., Murotani, E., Watanabe, K., Itoh, M., Shirakawa, D., Kawahara, K.et al. (2004) An entirely new methodology for synthesizing perfluorinated compounds: synthesis of perfluoroalkanesulfonyl fluorides from non-fluorinated compounds. J. Fluorine Chem. 125, 1695–1701 [Google Scholar]
- 100).Okazoe, T., Watanabe, K., Itoh, M., Shirakawa, D., Takagi, H., Kawahara, K.et al. (2007) Synthesis of perfluorinated ketones by utilizing liquid-phase direct fluorination. Bull. Chem. Soc. Jpn. 80, 1611–1616 [Google Scholar]
- 101).Holloway, J. H. (2000) Fluorine, high-tech element for the next century. J. Fluorine Chem. 104, 3–4 [Google Scholar]