

Abstract
Transforming growth factor-β (TGF-β) is a multifunctional cytokine that induces growth arrest, tissue fibrosis, and epithelial-mesenchymal transition (EMT) through activation of Smad and non-Smad signaling pathways. EMT is the differentiation switch by which polarized epithelial cells differentiate into contractile and motile mesenchymal cells. Cell motility and invasive capacity are activated upon EMT. Multiple transcription factors, including δEF1/ZEB1, SIP1/ZEB2, and Snail/SNAI1, are induced by TGF-β–Smad signaling and play critical roles in TGF-β-induced EMT. In addition, both non-Smad signaling activated by TGF-β and cross-talk with other signaling pathways play important roles in induction of EMT. Of these, Ras signaling synergizes with TGF-β-Smad signaling, and plays an important role in the induction of EMT. TGF-β inhibitors prevent invasion and metastasis of advanced cancer through multiple mechanisms, including inhibition of EMT. The discovery of molecules that inhibit TGF-β-induced EMT but not TGF-β-induced growth arrest may be an ideal strategy for treatment of invasion and metastasis of cancer.
Keywords: TGF-β, EMT, E-cadherin, Ras, invasion, metastasis
1. Introduction
Transforming growth factor-β (TGF-β) is a multifunctional cytokine that potently inhibits the proliferation of most types of cells, including epithelial cells, endothelial cells, hematopoietic cells, and lymphocytes.1) TGF-β also induces tissue fibrosis through induction of various extracellular matrix proteins. Recently, numerous studies have revealed that TGF-β stimulates epithelial-mesenchymal transition (EMT) in certain epithelial cells.2) TGF-β has thus been found to have bidirectional functions in the progression of cancer: it acts as a tumor suppressor by inhibition of cell proliferation through repression of c-Myc expression and some cyclin-dependent kinase inhibitors,3) while it functions as a pro-oncogenic factor through stimulation of matrix deposition, perturbation of immune function, and induction of EMT.4) Recent findings have shown that cancer cells, e.g. diffuse-type gastric cancer cells, treated with TGF-β secrete an anti-angiogenic factor thrombospondin-1, and thus that TGF-β acts as an anti-oncogenic factor via effects on the tumor microenvironment.5),6)
EMT is an important step in the invasion and metastasis of cancer, and TGF-β induces progression of cancer through EMT. TGF-β was originally discovered as a cytokine that induces anchorage-independent growth of normal fibroblasts in the presence of epidermal growth factor (EGF),7) and TGF-β-induced EMT-like activity was reported by many investigators even in the early 1990’s. Welch et al. reported that treatment of mammary adenocarcinoma cells with TGF-β1 resulted in acceleration of lung metastasis in a xenograft model, due to increased capacity for extravasation and elevated expression of gelatinolytic activity.8) Similarly, Meth A sarcoma cells expressing active TGF-β1 were shown to be more adhesive in vitro and more tumorigenic in vivo than parental cells.9) Although levels of expression of epithelial and mesenchymal markers were not fully determined in these studies, it is most likely that TGF-β triggered EMT in these cancer cells, and thereby promoted invasion and metastasis of cancer.
EMT occurs in various settings during embryonic development, but is also tightly linked to the pathogenesis of certain disease processes, including progression of fibrosis and cancer. In addition to TGF-β, various extracellular factors, including bone morphogenetic proteins (BMPs), Wnt, Notch ligands, hepatocyte growth factor, platelet-derived growth factor, and fibroblast growth factor, induce EMT in a coordinated and/or sequential fashion.2) Since the roles of EMT during embryonic development and progression of fibrosis have been discussed in other review articles,2),10),11) I will focus my discussion on TGF-β-induced EMT in cancer progression in this review.
2. TGF-β activates Smad and non-Smad signaling pathways
TGF-β binds to type II and type I serine-threonine kinase receptors, termed TβRII and TβRI, respectively. 12),13) TβRII transphosphorylates TβRI, and the latter activates receptor-regulated Smads (R-Smads) (Fig. 1).14) The R-Smads activated by TβRI or activin type I receptors are Smad2 and Smad3, while BMP type I receptors induce phosphorylation of Smad1, 5, and 8.15) Activated R-Smads form complexes with common-partner Smad (co-Smad; Smad4 in mammals), and translocate into the nucleus.14) The R-Smad-co-Smad complexes interact with various transcription factors and transcriptional co-activators or co-repressors, and regulate transcription of target genes.16),17) Smad7 is an inhibitory Smad (I-Smad), which inhibits TGF-β signaling through multiple mechanisms.18) Importantly, Smad7 binds to activated type I receptors and competes with R-Smads for receptor binding, resulting in repression of TGF-β signaling. c-Ski and its related protein SnoN are transcriptional co-repressors, which inhibit the transcriptional activity of R-Smadco-Smad complexes through interaction with Smad2/3 as well as Smad4.19) Expression of Smad7 and c-Ski, induced by TGF-β-Smad signaling, represses TGF-β signaling through negative feedback loops.
Fig. 1.

TGF-β transduces signaling through Smad and non-Smad signaling pathways. Smad pathway (right): TGF-β binds to TβRII and TβRI. TβRII phosphorylates TβRI, which activates Smad2 and Smad3. Activated Smad2/3 form complexes with Smad4, and translocate into the nucleus. The Smad complexes interact with various transcription factors and transcriptional co-activators, and regulate the transcription of target genes. Non-Smad pathways (left): TGF-β signals through multiple intra-cellular signaling cascades other than the Smad pathway. TGF-β receptors activate Erk, JNK, and p38 MAP kinases, PI3 kinase, and small GTPases such as Cdc42 and Rac. TGF-β receptors also bind Par6, induce TβRII kinase to phosphorylate Par6, and recruit Smurf1. Smurf1 then induces ubiquitylation and degradation of substrates such as RhoA.
In addition to the Smad signaling pathways, TGF-β activates various types of non-Smad signaling in certain types of cells (Fig. 1). Among them, it is reported that Erk, c-Jun N-terminal kinase (JNK), and p38 MAP kinases, phosphatidylinsitol-3 (PI3) kinase, and RhoA, Rac1, and Cdc42 GTPases play important roles in TGF-β-induced EMT.2)
3. Process of EMT
EMT is a process in which epithelial cells forming an organized, tightly connected sheet trans-differentiate into disorganized motile mesenchymal cells (Fig. 2).2),10),11) EMT occurs in an orchestrated fashion; one of the earliest events in it involves the disruption of tight junctions that connect epithelial cells, as well as delocalization of tight junction proteins, including zonula occludens 1 (ZO-1), claudin 1, and occludin. Adherens junction complexes, which contain E-cadherin and β-catenin, are also disrupted, and reorganization of the actin cytoskeleton from a cortical location into actin stress fibers anchored to the focal adhesion complexes is observed.
Fig. 2.

Processes of EMT and MET. During the process of EMT, epithelial cells expressing E-cadherin and claudin-1 differentiate into mesenchymal cells expressing N-cadherin, SMA, and FSP1. MET is the inverse process of EMT, in which mesenchymal cells differentiate into epithelial cells (Modified from R.A. Saito et al., Ref. 58).
Upon EMT, apical-basal cell polarity is lost, and cells exhibit a spindle-shaped, fibroblast-like morphology and express mesenchymal markers, including N-cadherin, vimentin, fibronectin, smooth muscle α-actin (SMA), and fibroblast-specific protein-1 (FSP1).20) The resultant mesenchymal cells exhibit a phenotype with altered adhesive properties, increased secretion of extracellular proteases, and altered expression of extracellular matrix proteins and their receptors, leading to increased migratory and invasive capacity.21) Cancer metastasis is accelerated by EMT through stimulation of cell migration and invasion, cell-substrate adhesion, intravasation, and extravasation, as well as cell survival. Indeed, cell survival is important for transit of tumor cells in the blood and lymph systems, and reestablishment at sites of metastasis. Metastasis is also accelerated through immunosuppression during EMT induced by the transcription factor Snail.22) EMT may also accelerate tumor progression through production of cells in the tumor microenvironment, such as cancer-associated fibroblasts (CAFs).
4. Transcriptional program eliciting TGF-β-induced EMT
Several transcription factors, including the zinc-finger factors Snail (also known as SNAI1) and Slug (SNAI2), the two-handed zinc-finger factors of δEF1 family proteins, including δEF1/ZEB1 and Smad-interacting protein 1 (SIP1)/ZEB2, and the basic helix-loop-helix (bHLH) factors Twist and E12/E47, play critical roles in the induction of EMT.2) Although it is currently unclear how each of the transcription factors plays a role in the EMT program, they may act independently in the induction of EMT in certain types of cells, but may also act co-operatively on the promoters of some genes, e.g. the E-cadherin promoter, in other cells. Mouse NMuMG mammary epithelial cells have been widely used to study the mechanisms of induction of EMT by TGF-β.23) TGF-β induces the expression of several transcription factors and transcription regulators involved in EMT, including δEF1, SIP1, and Snail, and represses that of Id proteins in NMuMG cells (Fig. 3).21),24) TGF-β does not affect the expression of E12/E47, and expression of Twist is not detectable in these cells.
Fig. 3.
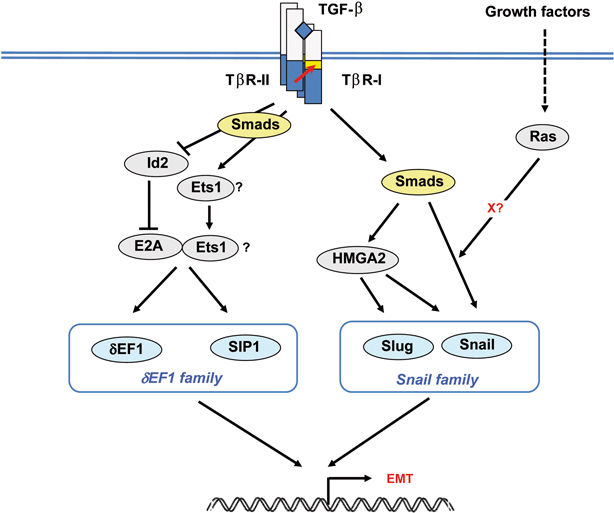
Transcriptional network for TGF-β-mediated induction of EMT. TGF-β induces the expression of several transcription factors involved in EMT, including the δEF1 family proteins (δEF1/ZEB1 and SIP1/ZEB2) and the Snail family proteins (Snail and Slug). Left: TGF-β represses the expression of Id proteins and induces that of Ets1. Id proteins interact with and inhibit the transcriptional activity of E2A proteins. Ets1 acts in cooperative fashion with the E2A proteins released from Id proteins, and is involved in the up-regulation of δEF1 and SIP1. Right: TGF-β induces the expression of Snail in Smaddependent fashion. Ras signaling cooperates with TGF-β signaling in inducing Snail. In addition, HMGA2 is induced by TGF-β-Smad signaling, and induces the expression of Snail and Slug in certain types of cells.
Id (inhibitor of differentiation) proteins have a helix–loop–helix (HLH) domain but lack the basic DNA-binding domain. Id proteins interact with and inhibit the transcriptional activity of bHLH proteins. 25) Four isoforms of Id proteins, i.e. Id1–4, have been identified in mammals, and TGF-β suppresses the expression of Id1, 2, and 3 in NMuMG cells.21),26) Suppression of the expression of Id2 mRNA is observed at 5 hours after the stimulation by TGF-β, preceding the repression of E-cadherin mRNA expression, and continues for more than 24 hours. E12 and E47, two alternatively spliced products of the E2A gene, are bHLH factors which induce a fibroblastic phenotype and suppress the expression of E-cadherin. Through constitutive association with E2A proteins, Id proteins maintain epithelial phenotypes by repressing the function of E2A.
δEF1 and SIP1 are the two-handed zinc-finger factors involved in EMT.27),28) Levels of expression of δEF1 and SIP1 are gradually increased by TGF-β until 24 hours, with profiles of expression reciprocal to that of E-cadherin in NMuMG cells.24) δEF1 and SIP1 repress the transcription of E-cadherin through binding to the E-box region of the E-cadherin promoter. Smads interact with δEF1 and SIP1, and form repressor complexes on the E-cadherin promoter, 27),29) although binding of Smads to δEF1 and SIP1 appears not to be required for their repressor activity.24) In contrast to the repression of E-cadherin, expression of mesenchymal markers, such as N-cadherin and fibronectin, is not affected by SIP1 and δEF1 in NMuMG cells.24)
Snail and Slug are zinc-finger-containing transcription factors and repress E-cadherin expression and induce EMT.30),31) TGF-β induces the expression of Snail in biphasic, Smad-dependent, fashion. It induces greater than 10-fold expression of Snail mRNA by 1 hour in NMuMG cells, following which mild but significant induction is observed at 10–24 hours.24) A similar biphasic profile of Snail induction can be observed in other types of cells.32) In Madin-Darby canine kidney (MDCK) cells, Slug, but neither Snail nor Twist, is induced by TGF-β.33)
Snail suppresses E-cadherin expression by direct binding to the E2-box sequence of the E-cadherin promoter.30) Snail interacts with transcriptional co-repressor Sin3A, and histone deacetylase activity recruited to Snail is essential for repression of the E-cadherin mRNA expression.34) SIP1 and Snail bind to partly overlapping E-cadherin promoter sequences. 27) Snail has also been shown to induce cell survival and movement, while it represses cell proliferation. In addition to TGF-β, Snail is induced by various extracellular signals, including those of the EGF and Notch pathways.34) The activity of Snail is regulated by different phosphorylation events via various kinases, including GSK3β and p21-activated kinase-1 (PAK1). In addition, Snail protein is stabilized by tumor necrosis factor (TNF)-α through activation of the NF-κB pathway.35) LIV1, a breast-cancer associated zinc transporter protein, is required for the nuclear localization of Snail.36)
The transcriptional program of the EMT inducers is regulated by certain upstream transcription factors (Fig. 3). TGF-β induces the expression of Ets1, which functions as an upstream component of δEF1 and SIP1. Since Ets1 acts in cooperative fashion with the E2A proteins, Id proteins are also involved in the suppression of the TGF-β-induced up-regulation of δEF1 and SIP1. The high mobility group factor HMGA2 was identified as a new regulator of EMT. HMGA2 is induced by TGF-β-Smad signaling, and induces the expression of Snail, Slug, and Twist.37),38) In addition, TGF-β induces nuclear translocation of myocardin-related transcription factor-A and -B (MRTF-A and -B; also known as MAL and MKL1/2) in Rho-dependent fashion. MRTFs form complexes with Smad3, and induce the expression of Slug by direct binding to the Slug promoter, leading to execution of EMT in various epithelial cells, including MDCK and NMuMG cells.33)
MicroRNAs (miRNAs) are short non-coding RNAs of 20–22 nucleotides, which are implicated in tumor progression. MiRNAs repress gene expression by interaction of their seed sequences with complementary sequences located in the 3’ UTR of target mRNAs. Emerging evidence indicates that miR-200 family members regulate the process of EMT by targeting δEF1 and SIP1, and that their expression is down-regulated in cells undergoing EMT. Loss of miR-200 results in acquisition of the EMT phenotype, while re-expression of miR-200 leads to reversal of the process of EMT.39)
5. Non-Smad signaling
Both Smad and non-Smad signaling appear to be important for full development of EMT (Fig. 1). Although the involvement of Smad4 in EMT is controversial, key transcriptional events of EMT are mediated, at least in part, by the Smad signaling pathways.20),40),41) Analyses of certain fibrotic diseases using Smad3-null mice have revealed the essential role of Smad3 in the process of EMT.42) However, there is some evidence that non-Smad signaling also participates in the process of EMT. Direct interaction between the TGF-β receptors and the polarity complex that regulates epithelial polarization has been demonstrated. TβRI is localized in the tight junction through interaction with the PAK network and occludin.43) Activated TβRII induces phosphorylation of the tight junction protein Par6, which in turn recruits Smurf1, an E3 ubiquitin ligase. Smurf1 then stimulates ubiquitylation and degradation of RhoA, leading to disorganization of tight junctions and loss of cell polarity.
6. Synergism between TGF-β and Ras signaling in EMT
Various signals, including integrin, Notch, Wnt, TNF-α, and EGF signals, have been reported to cooperate or synergize with TGF-β signaling and stimulate tumor invasion and metastasis.2),22) Synergism between TGF-β and Ras signaling has been the most extensively investigated. Oft et al.44) reported that when transformed by mutant Ras, mammary epithelial EpH4 cells undergo EMT by TGF-β1 and exhibit a fibroblast-like and invasive and metastatic phenotype resistant to the growth-inhibitory activity of TGF-β1. Using specific small-molecule inhibitors blocking the MAP kinase or PI3 kinase pathway, Janda et al. reported that hyperactivation of the Raf-MAP kinase pathway synergizes with TGF-β signaling and induces EMT, and accelerates the tumorigenesis and metastasis of EpH4 mammary epithelial cells, while activation of PI3 kinase results in tumorigenesis but not metastasis via stimulation of scattering of cells and their survival.45) Vogelmann et al. reported that Ras and PI3 kinase appear to activate Src-family tyrosine kinases, leading to phosphorylation of α- and β-catenin, destabilization of E-cadherin/catenin complexes, and disruption of the adherens junctions.46) It is, however, unknown which transcription factor(s) are involved in the cooperation of TGF-β- and Ras-induced EMT.
Induction of Snail by TGF-β occurs only in cooperation with active Ras signaling in some types of cells, including MDCK cells and pancreatic carcinoma Panc-1 cells (Fig. 3).32),34) Constitutively active Ras dramatically enhances the induction of Snail by TGF-β, while expression of other targets of TGF-β, e.g. Smad7 and plasminogen activator inhibitor-1, is not affected by Ras signaling in Panc-1 cells.32) This finding is of interest, since Smad2 and Smad3 are phosphorylated by MAP kinases and/or other kinases at linker regions, resulting in prevention of nuclear accumulation of Smads. Interestingly, phosphorylation at the linker region of Smad2/3 does not affect the synergistic induction of Snail by TGF-β and Ras. Neither Erk/JNK MAP kinases nor PI3 kinase is involved in this synergistic effect between TGF-β and Ras in Panc-1 cells, while Snail is transcriptionally induced by TGF-β in MAP kinase- and PI3 kinase-dependent fashion in MDCK cells.34) Thus, Ras and TGF-β-Smad signaling selectively cooperate in the induction of Snail, which occurs independently of phosphorylation at the linker region of R-Smads by Ras signaling.
7. Endothelial-mesenchymal transition (EndMT)
Blood vessels are lined by endothelial cells and surrounded by mural cells (i.e. pericytes or smooth muscle cells). Endothelial cells have been shown to differentiate into mesenchymal cells during development, and this process is termed endothelial-mesenchymal (or mural) transition (EndMT).47) During the process of EndMT, expression of VE-cadherin, platelet-endothelial cell adhesion molecule 1 (PECAM1), and claudin-5 is reduced, while that of SMA and other mesenchymal markers is induced (Fig. 4). Similar to EMT, EndMT is regulated by various signaling pathways, including TGF-β, Wnt, and Notch. Indeed, TGF-β is required at the onset of EndMT to initiate endocardial cushion tissue formation in mouse embryonic heart.48) In addition, several in vitro studies have shown that TGF-β induces the differentiation of endothelial cells into mural cells.49),50) Although EndMT induced by TGF-β plays essential roles in cardiovascular development, it is implicated in certain types of pathology, e.g. cardiac fibrosis and cancer progression. 47) Activated fibroblasts induced by EndMT may play an important role in cancer progression as a source of CAFs.
Fig. 4.

EMT and EndMT. During the process of EMT, epithelial cells forming an organized, tightly connected sheet trans-differentiate into disorganized, motile mesenchymal cells (upper panel; see also Fig. 2). During the process of EndMT, endothelial cells expressing VE-cadherin, PECAM-1, and claudin-5 differentiate into mesenchymal (or mural) cells expressing SMA and FSP1 (lower panel) (courtesy of Drs. Takuya Shirakihara and Hajime Mihira).
Embryonic stem cell (ESC)-derived vascular progenitor cells differentiate into both endothelial and mural cells in vitro.51),52) Recently, Kokudo et al.53) demonstrated that TGF-β and activin are capable of inducing differentiation of endothelial cells derived from mouse ESCs into mural cells. TGF-β decreases the expression of claudin-5, and increases that of mural markers, including SMA, SM22α, and calponin. Expression of Snail is induced by TGF-β, and, similar to the process of EMT, Snail is essential for TGF-β-induced EndMT. Other transcription factors involved in EMT, e.g. Slug, SIP1, and δEF1, are not induced by TGF-β in mouse ESC-derived endothelial cells. Snail is also induced in human umbilical vein endothelial cells by TGF-β.53) Although only Snail appears to be involved in EndMT of mouse ESC-derived endothelial cells, TGF-β induces differentiation of neural crest cells into smooth muscle cells through the activity of δEF1.54) Thus, transcription factors involved in EMT may also play crucial roles in EndMT, and EndMT in different tissues may be regulated by a particular subset of transcriptional activators and/or repressors.
8. Is MET inducible by TGF-β inhibition?
EMT is most often transient and reversible, and mesenchymal-epithelial transition (MET), the inverse process of EMT, occurs under certain physiological and pathological conditions. However, the signals that induce MET are largely unknown.
Thyroid transcription factor-1 (TTF-1, the product of Nkx2.1 gene) is a homeodomain-containing transcription factor, and serves as a master regulator for lung morphogenesis.55) Frequent expression of it is reported in lung adenocarcinoma (72.1%),56) and TTF-1 positivity has been reported to be a good prognostic marker in non-small cell lung cancer.57) Saito et al.58) reported that TTF-1 antagonizes TGF-β-induced EMT and restores epithelial phenotype in lung adenocarcinoma cells. TTF-1 antagonizes the effect of TGF-β, presumably through interaction with Smads,59) and represses the expression of TGF-β-target genes, including Snail and Slug. Decrease in the expression of TTF-1 accelerates TGF-β-induced EMT. Moreover, TGF-β represses the expression of TTF-1, and TTF-1 represses that of TGF-β2, the TGF-β isoform expressed in distal bronchial airway.58) These findings suggest that TTF-1 exhibits a tumor-suppressive effect with abrogation of response of adenocarcinoma cells to TGF-β, and that modulation of TTF-1 expression can be a novel therapeutic strategy for treatment of lung adenocarcinoma.
Inhibition of TGF-β signaling by TGF-β antagonists blocks EMT and induces MET, leading to prevention of cancer metastasis in vivo. JygMC(A) mouse mammary carcinoma cells spontaneously metastasize to lung and liver. Inhibition of TGF-β signaling by systemic administration of adenoviruses carrying Smad7 or c-Ski cDNA resulted in prevention of metastasis of JygMC(A) cells.60) Interestingly, growth of primary tumors inoculated subcutaneously in mice was not significantly affected by administration of Smad7- or c-Ski-adenoviruses. High-throughput Western blot analysis demonstrated increased expression of major components of adherens junctions and tight junctions, and decrease in migratory and invasive capacities of breast cancer cells infected with Smad7-adenovirus.60) TGF-β also induces the survival of mammary carcinoma cells through expression of transcription factor DEC1, which is suppressed by TGF-β antagonists.61) These findings thus strongly suggest that inhibition of TGF-β signaling leads to prevention of cancer metastasis by inhibition of the process of EMT.
9. Conclusion and perspective
Several TGF-β antagonists, including small-molecule inhibitors that inhibit the kinase activity of TβRI and monoclonal antibody to TGF-β, have been generated, and have been reported to inhibit metastasis of cancer through multiple mechanisms. 62)–64) Regulation of immune function and inhibition of EMT may be among the most important mechanisms for prevention of metastasis by TGF-β antagonists.63),64) As described, recent findings have suggested that TGF-β induces growth arrest and EMT through partially distinct pathways. It is thus important to discover new drug targets that block EMT, i.e. the tumor-progression arm of the TGF-β signaling pathway, without affecting its growth-inhibitory activity, i.e. the tumor-suppression arm.
Acknowledgments
I would like to thank the members of the Department of Molecular Pathology, Graduate School of Medicine, University of Tokyo. This work was supported by KAKENHI (Grants-in-aid for Scientific Research) on Priority Areas “New strategies for cancer therapy based on advancement of basic research” from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Abbreviations:
- TGF-β
transforming growth factor-β
- EMT
epithelial-mesenchymal transition
- EGF
epidermal growth factor
- BMP
bone morphogenetic protein
- TβR
TGF-β receptor
- R-Smad
receptor-regulated Smad
- co-Smad
common-partner Smad
- I-Smad
inhibitory Smad
- JNK
c-Jun N-terminal kinase
- PI3
phosphatidylinsitol-3
- ZO-1
zonula occludens 1
- SMA
smooth muscle α-actin
- FSP1
fibroblast-specific protein-1
- SIP1
Smad-interacting protein 1
- bHLH
basic helix-loop-helix
- Id
inhibitor of differentiation
- HLH
helix–loop–helix
- MDCK
Madin-Darby canine kidney
- PAK1
p21-activated kinase-1
- TNF
tumor necrosis factor
- MRTF
myocardin-related transcription factor
- miRNA
microRNA
- EndMT
endothelial-mesenchymal transition
- PECAM1
platelet-endothelial cell adhesion molecule 1
- ESC
embryonic stem cell
- MET
mesenchymal-epithelial transition
- TTF-1
thyroid transcription factor-1
Profile
Kohei Miyazono was born in Saga, Japan, in 1956, and received his M.D. degree at the University of Tokyo in 1981, and D.M.S. at the same university in 1988. He started to work at the Department of Hematology at the University of Tokyo, and then moved to Dr. Carl-Henrik Heldin’s lab in Uppsala, Sweden in 1985. After going back to Japan between 1988–1990, he moved to Sweden again, and became assistant and associate member at the Ludwig Institute for Cancer Research, Uppsala, Sweden (between 1990 and 1995). He returned to Japan, and became the Chief of the Department of Biochemistry, the Cancer Institute of Japanese Foundation for Cancer Research in 1995. In 2000, he was appointed to his current position as a full professor at the University of Tokyo. Kohei Miyazono received Incitement Award of the Japanese Cancer Association (1996), Erwin von Bälz Prize (1997), Honorary Doctor of Medicine of Uppsala University, Sweden (1999), Princess Takamatsu Cancer Research Award (2000), Academic Award of the Mochida Memorial Foundation (2003), Inoue Prize for Science (2006), Medical Award of the Japan Medical Association (2006), and the Takeda Foundation Award (2008). His research interest is mechanisms of action of the TGF-β family factors and their relation to development of cancer, mechanisms of angiogenesis and lymphangiogenesis, and studies on tumor suppressor genes.
During the past 10 years, his laboratory has studied the signaling mechanisms of TGF-β and bone morphogenetic proteins. Kohei Miyazono was the Vice President of the Japanese Cancer Association (2007–2008), and actively working for Cancer Science and Journal of Biochemistry as an editor, and for Oncogene and Journal of Biological Chemistry as a member of the editorial board.

References
- 1).Schilling, S.H., Hjelmeland, A.B., Rich, J.N. and Wang, X.F. (2008) TGF-β: A multipotential cytokine. InThe TGF-β Family (eds. Derynck, R. and Miyazono, K.). Cold Spring Harbor Laboratory Press, New York, pp. 45–78 [Google Scholar]
- 2).Moustakas, A. and Heldin, C.-H. (2007) Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci. 98, 1512–1520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3).Yagi, K., Furuhashi, M., Aoki, H., Goto, D., Kuwano, H., Sugamura, K.et al. (2002) c-myc is a downstream target of Smad pathway. J. Biol. Chem. 277, 854–861 [DOI] [PubMed] [Google Scholar]
- 4).Roberts, A.B. and Wakefield, L.M. (2003) The two faces of transforming growth factor β in carcinogenesis. Proc. Natl. Acad. Sci. USA 100, 8621–8623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5).Komuro, A., Yashiro, M., Iwata, C., Morishita, Y., Johansson, E., Matsumoto, Y.et al. (2009) Diffuse-type gastric carcinoma: Progression, angiogenesis, and transforming growth factor-β signaling. J. Natl. Cancer Inst. 101, 592–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).Kiyono, K., Suzuki, H.I., Morishita, Y., Komuro, A., Iwata, C., Yashiro, M.et al. (2009) c-Ski overexpression promotes tumor growth and angiogenesis through inhibition of transforming growth factor-β signaling in diffuse-type gastric carcinoma. Cancer Sci. 100, 1809–1816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7).Moses, H.L. and Roberts, A.B. (2008) The discovery of TGF-β: A historical perspective. InThe TGF-β Family (eds. Derynck, R. and Miyazono, K.). Cold Spring Harbor Laboratory Press, New York, pp. 1–28 [Google Scholar]
- 8).Welch, D.R., Fabra, A. and Nakajima, M. (1990) Transforming growth factor β stimulates mammary adenocarcinoma cell invasion and metastatic potential. Proc. Natl. Acad. Sci. USA 87, 7678–7682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9).Chang, H.L., Gillett, N., Figari, I., Lopez, A.R., Palladino, M.A. and Derynck, R. (1993) Increased transforming growth factor β expression inhibits cell proliferation in vitro, yet increases tumorigenicity and tumor growth of Meth A sarcoma cells. Cancer Res. 53, 4391–4398 [PubMed] [Google Scholar]
- 10).Thiery, J.P. and Sleeman, J.P. (2006) Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 7, 131–142 [DOI] [PubMed] [Google Scholar]
- 11).Derynck, R. and Akhurst, R.J. (2007) Differentiation plasticity regulated by TGF-β family proteins in development and disease. Nat. Cell Biol. 9, 1000–1004 [DOI] [PubMed] [Google Scholar]
- 12).Lin, H.Y., Wang, X.F., Ng-Eaton, E., Weinberg, R.A. and Lodish, H.F. (1992) Expression cloning of the TGF-β type II receptor, a functional transmembrane serine/threonine kinase. Cell 68, 775–785 [DOI] [PubMed] [Google Scholar]
- 13).Franzén, P., ten Dijke, P., Ichijo, H., Yamashita, H., Schulz, P., Heldin, C.-H.et al. (1993) Cloning of a TGFβ type I receptor that forms a heteromeric complex with the TGF β type II receptor. Cell 75, 681–692 [DOI] [PubMed] [Google Scholar]
- 14).Heldin, C.-H., Miyazono, K. and ten Dijke, P. (1997) TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature 390, 465–471 [DOI] [PubMed] [Google Scholar]
- 15).Miyazawa, K., Shinozaki, M., Hara, T., Furuya, T. and Miyazono, K. (2002) Two major Smad pathways in TGF-β superfamily signaling. Genes Cells 7, 1191–1204 [DOI] [PubMed] [Google Scholar]
- 16).Nishihara, A., Hanai, J.-I., Okamoto, N., Yanagisawa, J., Kato, S., Miyazono, K.et al. (1998) Role of p300, a transcriptional coactivator, in signaling of TGF-β. Genes Cells 3, 613–623 [DOI] [PubMed] [Google Scholar]
- 17).Akiyoshi, S., Inoue, H., Hanai, J.-I., Kusanagi, K., Nemoto, N., Miyazono, K.et al. (1999) c-Ski acts as a transcriptional co-repressor in TGF-β signaling through interaction with Smads. J. Biol. Chem. 274, 35269–35277 [DOI] [PubMed] [Google Scholar]
- 18).Miyazono, K. (2008) Regulation of TGF-β family signaling by inhibitory Smads. InThe TGF-β Family (eds. Derynck, R. and Miyazono, K.). Cold Spring Harbor Laboratory Press, New York, pp. 363–388 [Google Scholar]
- 19).Miyazono, K., Maeda, S. and Imamura, T. (2006) Smad transcriptional co-activators and co-repressors. InSmad Signaling (eds. ten Dijke, P. and Heldin, C.-H.). Springer, Heidelberg, pp. 277–293 [Google Scholar]
- 20).Valcourt, U., Kowanetz, M., Niimi, H., Heldin, C.-H. and Moustakas, A. (2005) TGF-β and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol. Biol. Cell 16, 1987–2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21).Kondo, M., Cubillo, E., Tobiume, K., Shirakihara, T., Fukuda, N., Suzuki, H.et al. (2004) A role for Id in the regulation of TGF-β-induced epithelial-mesenchymal trans-differentiation. Cell Death Differ. 11, 1092–1101 [DOI] [PubMed] [Google Scholar]
- 22).Kudo-Saito, C., Shirako, H., Takeuchi, T. and Kawakami, Y. (2009) Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell 15, 195–206 [DOI] [PubMed] [Google Scholar]
- 23).Miettinen, P.J., Ebner, R., Lopez, A.R. and Derynck, R. (1994) TGF-β induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J. Cell Biol. 127, 2021–2036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24).Shirakihara, T., Saitoh, M. and Miyazono, K. (2007) Differential regulation of epithelial and mesenchymal markers by δEF1 proteins in epithelial mesenchymal transition induced by TGF-β. Mol. Biol. Cell 18, 3533–3544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25).Miyazono, K. and Miyazawa, K. (2002) Id: a target of BMP signaling. Sci STKE. 2002, PE40. [DOI] [PubMed] [Google Scholar]
- 26).Kowanetz, M., Valcourt, U., Bergstrom, R., Heldin, C.-H. and Moustakas, A. (2004) Id2 and Id3 define the potency of cell proliferation and differentiation responses to transforming growth factor β and bone morphogenetic protein. Mol. Cell. Biol. 24, 4241–4254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27).Comijn, J., Berx, G., Vermassen, P., Verschueren, K., van Grunsven, L., Bruyneel, E.et al. (2001) The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol. Cell 7, 1267–1278 [DOI] [PubMed] [Google Scholar]
- 28).Postigo, A.A. (2003) Opposing functions of ZEB proteins in the regulation of the TGFβ/BMP signaling pathway. EMBO J. 22, 2443–2452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29).Vandewalle, C., Comijn, J., De Craene, B., Vermassen, P., Bruyneel, E., Andersen, H.et al. (2005) SIP1/ZEB2 induces EMT by repressing genes of different epithelial cell-cell junctions. Nucleic Acids Res. 33, 6566–6578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30).Cano, A., Pérez-Moreno, M.A., Rodrigo, I., Locascio, A., Blanco, M.J., del Barrio, M.G.et al. (2000) The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2, 76–83 [DOI] [PubMed] [Google Scholar]
- 31).Batlle, E., Sancho, E., Francí, C., Domínguez, D., Monfar, M., Baulida, J.et al. (2000) The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2, 84–89 [DOI] [PubMed] [Google Scholar]
- 32).Horiguchi, K., Shirakihara, T., Nakano, A., Imamura, T., Miyazono, K. and Saitoh., M. (2009) Role of Ras signaling in the induction of Snail by transforming growth factor-β. J. Biol. Chem. 284, 245–253 [DOI] [PubMed] [Google Scholar]
- 33).Morita, T., Mayanagi, T. and Sobue, K. (2007) Dual roles of myocardin-related transcription factors in epithelial mesenchymal transition via slug induction and actin remodeling. J. Cell Biol. 179, 1027–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34).Barrallo-Gimeno, A. and Nieto, M.A. (2005) The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development 132, 3151–3161 [DOI] [PubMed] [Google Scholar]
- 35).Wu, Y., Deng, J., Rychahou, P.G., Qiu, S., Evers, B.M. and Zhou, B.P. (2009) Stabilization of snail by NF-κB is required for inflammation-induced cell migration and invasion. Cancer Cell 15, 416–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36).Yamashita, S., Miyagi, C., Fukada, T., Kagara, N., Che, Y.S. and Hirano, T. (2004) Zinc transporter LIVI controls epithelial-mesenchymal transition in zebrafish gastrula organizer. Nature 429, 298–302 [DOI] [PubMed] [Google Scholar]
- 37).Thuault, S., Valcourt, U., Petersen, M., Manfioletti, G., Heldin, C.-H. and Moustakas, A. (2006) Transforming growth factor-β employs HMGA2 to elicit epithelial-mesenchymal transition. J. Cell Biol. 174, 175–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38).Thuault, S., Tan, E.J., Peinado, H., Cano, A., Heldin, C.-H. and Moustakas, A. (2008) HMGA2 and Smads co-regulate SNAIL1 expression during induction of epithelial-to-mesenchymal transition. J. Biol. Chem. 283, 33437–33446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39).Cano, A. and Nieto, M.A. (2008) Non-coding RNAs take centre stage in epithelial-to-mesenchymal transition. Trends Cell Biol. 18, 357–359 [DOI] [PubMed] [Google Scholar]
- 40).Deckers, M., van Dinther, M., Buijs, J., Que, I., Löwik, C., van der Pluijm, G.et al. (2006) The tumor suppressor Smad4 is required for transforming growth factor β-induced epithelial to mesenchymal transition and bone metastasis of breast cancer cells. Cancer Res. 66, 2202–2209 [DOI] [PubMed] [Google Scholar]
- 41).Takano, S., Kanai, F., Jazag, A., Ijichi, H., Yao, J., Ogawa, H.et al. (2007) Smad4 is essential for down-regulation of E-cadherin induced by TGF-β in pancreatic cancer cell line PANC-1. J. Biochem. 141, 345–351 [DOI] [PubMed] [Google Scholar]
- 42).Sato, M., Muragaki, Y., Saika, S., Roberts, A.B. and Ooshima, A. (2003) Targeted disruption of TGF-β1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J. Clin. Invest. 112, 1486–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43).Ozdamar, B., Bose, R., Barrios-Rodiles, M., Wang, H.R., Zhang, Y. and Wrana, J.L. (2005) Regulation of the polarity protein Par6 by TGFβ receptors controls epithelial cell plasticity. Science 307, 1603–1609 [DOI] [PubMed] [Google Scholar]
- 44).Oft, M., Peli, J., Rudaz, C., Schwarz, H., Beug, H. and Reichmann, E. (1996) TGF-β1 and Ha-Ras collaborate in modulating the phenotypic plasticity and invasiveness of epithelial tumor cells. Genes Dev. 10, 2462–2477 [DOI] [PubMed] [Google Scholar]
- 45).Janda, E., Lehmann, K., Killisch, I., Jechlinger, M., Herzig, M., Downward, J.et al. (2002) Ras and TGFβ cooperatively regulate epithelial cell plasticity and metastasis: dissection of Ras signaling pathways. J. Cell Biol. 156, 299–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46).Vogelmann, R., Nguyen-Tat, M.D., Giehl, K., Adler, G., Wedlich, D. and Menke, A. (2005) TGFβ-induced down-regulation of E-cadherin-based cell-cell adhesion depends on PI3-kinase and PTEN. J. Cell Sci. 118, 4901–4912 [DOI] [PubMed] [Google Scholar]
- 47).Potenta, S., Zeisberg, E. and Kalluri, R. (2008) The role of endothelial-to-mesenchymal transition in cancer progression. Br. J. Cancer 99, 1375–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48).Nakajima, Y., Miyazono, K., Kato, M., Takase, M., Yamagishi, T. and Nakamura, H. (1997) Extracellular fibrillar structure of latent TGFβ binding protein-1: Role in TGFβ-dependent endothelial-mesenchymal transformation during endocardial cushion tissue formation in mouse embryonic heart. J. Cell Biol. 136, 193–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49).Arciniegas, E., Sutton, A.B., Allen, T.D. and Schor, A.M. (1992) Transforming growth factor β promotes the differentiation of endothelial cells into smooth muscle-like cells in vitro. J. Cell Sci. 103, 521–529 [DOI] [PubMed] [Google Scholar]
- 50).Ishisaki, A., Hayashi, H., Li, A.J. and Imamura, T. (2003) Human umbilical vein endothelium-derived cells retain potential to differentiate into smooth muscle-like cells. J. Biol. Chem. 278, 1303–1309 [DOI] [PubMed] [Google Scholar]
- 51).Yamashita, J., Itoh, H., Hirashima, M., Ogawa, M., Nishikawa, S., Yurugi, T.et al. (2000) Flk1-positive cells derived from embryonic stem cells serve as vascular progenitors. Nature 408, 92–96 [DOI] [PubMed] [Google Scholar]
- 52).Watabe, T., Nishihara, A., Mishima, K., Yamashita, J., Shimizu, K., Miyazawa, K.et al. (2003) TGF-β receptor kinase inhibitor enhances growth and integrity of embryonic stem cell-derived endothelial cells. J. Cell Biol. 163, 1303–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53).Kokudo, T., Suzuki, Y., Yoshimatsu, Y., Yamazaki, T., Watabe, T. and Miyazono, K. (2008) Snail is required for TGFβ-induced endothelial-mesenchymal transition of embryonic stem cell-derived endothelial cells. J. Cell Sci. 121, 3317–3324 [DOI] [PubMed] [Google Scholar]
- 54).Nishimura, G., Manabe, I., Tsushima, K., Fujiu, K., Oishi, Y., Imai, Y.et al. (2006) δEF1 mediates TGF-β signaling in vascular smooth muscle cell differentiation. Dev. Cell 11, 93–104 [DOI] [PubMed] [Google Scholar]
- 55).Minoo, P., Su, G., Drum, H., Bringas, P. and Kimura, S. (1999) Defects in tracheoesophageal and lung morphogenesis in Nkx2.1-/-mouse embryos. Dev. Biol. 209, 60–71 [DOI] [PubMed] [Google Scholar]
- 56).Fujita, J., Ohtsuki, Y., Bandoh, S., Ueda, Y., Kubo, A., Tojo, Y.et al. (2003) Expression of thyroid transcription factor-1 in 16 human lung cancer cell lines. Lung Cancer 39, 31–36 [DOI] [PubMed] [Google Scholar]
- 57).Tan, D., Li, Q., Deeb, G., Ramnath, N., Slocum, H.K., Brooks, J.et al. (2003) Thyroid transcription factor-1 expression prevalence and its clinical implications in non-small cell lung cancer: a high-throughput tissue microarray and immunohistochemistry study. Hum. Pathol. 34, 597–604 [DOI] [PubMed] [Google Scholar]
- 58).Saito, R.A., Watabe, T., Horiguchi, K., Kohyama, T., Saitoh, M., Nagase, T.et al. (2009) Thyroid transcription factor-1 inhibits transforming growth factor-β-mediated epithelial-to-mesenchymal transition in lung adenocarcinoma cells. Cancer Res. 69, 2783–2791 [DOI] [PubMed] [Google Scholar]
- 59).Minoo, P., Hu, L., Zhu, N., Borok, Z., Bellusci, S., Groffen, J.et al. (2008) SMAD3 prevents binding of NKX2.1 and FOXA1 to the SpB promoter through its MH1 and MH2 domains. Nucleic Acid Res. 36, 179–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60).Azuma, H., Ehata, S., Miyazaki, H., Watabe, T., Maruyama, O., Imamura, T.et al. (2005) Effect of Smad7 expression on metastasis of mouse mammary carcinoma JygMC(A) cells. J. Natl. Cancer Inst. 97, 1734–1746 [DOI] [PubMed] [Google Scholar]
- 61).Ehata, S., Hanyu, A., Hayashi, M., Aburatani, H., Kato, Y., Fujime, M.et al. (2007) Transforming growth factor-β promotes survival of mammary carcinoma cells through induction of antiapoptotic transcription factor DEC1. Cancer Res. 67, 9694–9703 [DOI] [PubMed] [Google Scholar]
- 62).Ehata, S., Hanyu, A., Fujime, M., Katsuno, Y., Fukunaga, E., Goto, K.et al. (2007) Ki26894, a novel transforming growth factor-β type I receptor kinase inhibitor, inhibits in vitro cell motility and invasion and in vivo bone metastasis of a human breast cancer cell line. Cancer Sci. 98, 127–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63).Nam, J.S., Terabe, M., Mamura, M., Kang, M.J., Chae, H., Stuelten, C.et al. (2008) An anti-transforming growth factor β antibody suppresses metastasis via cooperative effects on multiple cell compartments. Cancer Res. 68, 3835–3843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64).Nam, J.S., Terabe, M., Kang, M.J., Chae, H., Voong, N., Yang, Y.A.et al. (2008) Transforming growth factor β subverts the immune system into directly promoting tumor growth through interleukin-17. Cancer Res. 68, 3915–3923 [DOI] [PMC free article] [PubMed] [Google Scholar]