

Abstract
Cilia are microtubule-based cellular organelles that are widely distributed in vertebrate tissues. They were first observed hundreds of years ago. Recent studies indicate that this small organelle plays important roles in numerous physiological phenomena, including tissue morphogenesis, signal transduction, determination of left-right asymmetry during development, and adult neurogenesis. Ciliopathies, syndromes resulting from a genetic disorder of cilial components, frequently have complex effects involving many organ systems, owing to the broad distribution of cilia in the body.
Keywords: cilia, ciliopathy, morphogenesis
Introduction
Cilia are small, largely external, cellular organelles that are involved in diverse biological processes. They were first observed on ciliated protozoa over 300 years ago by Dutch microscope maker Antoni van Leeuwenhoek. These hair-like structures protrude from the membrane of ciliates, and their primary role is in cell locomotion. In mammals, these “motile” cilia are located on the cells lining the brain ventricles and tracheal duct, and the female reproductive system; they are also present on sperm cells and at the embryonic ventral node. Dysfunction of the motile cilia can cause hydrocephalus, chronic airway disease, male infertility, and situs inversus (whole-body inversion of asymmetrical structures).1) Although this organelle has a long history, the exciting and rapid progress in understanding its physiological roles, which have been emerging over the past decade, have led to a renewed focus on cilia and ciliary function. In this article, we describe the function and structure of cilia. We then focus on the role of subventricular zone (SVZ) ependymal cilia in the migration of newborn neurons in the adult mammalian brain.
Cilia structure and ciliopathies
The cilia of eukaryotic cells are hair-like structures that extend from the cell surface. They are composed of microtubules and are classified according to their microtubule components and motility into four groups (9+2 motile, 9+2 immotile, 9+0 motile, and 9+0 immotile).1)
The axoneme of 9+2 motile cilia is composed of nine peripheral microtubule doublets and two central single microtubules (central pair) (Fig. 1). It also contains dynein arms, and radial spokes, which are important for motility. The dynein arms, which are bound to the ciliary doublet, enable the microtubules in the axonemes to slide in an ATPase-dependent reaction, which generates ciliary beating.2),3) The peripheral doublets extend from the basal body of the cilium, across the transition zone(Fig. 1), and reach almost to the tip of the cilium. The basal body is a special structure derived from the centriole, and is constructed of nine microtubule triplets without central singlets. A protrusion called the basal foot, which extends from the lateral side of the basal body, indicates the direction in which the polarized cilium will beat. Below the basal body, there is a fibrillary compartment extending to the cell nucleus, called the striated rootlet. The rootlet is not essential for ciliogenesis or the formation of basal bodies, but it is necessary for the long-term stability of the cilia on photoreceptors.4)
Fig. 1.
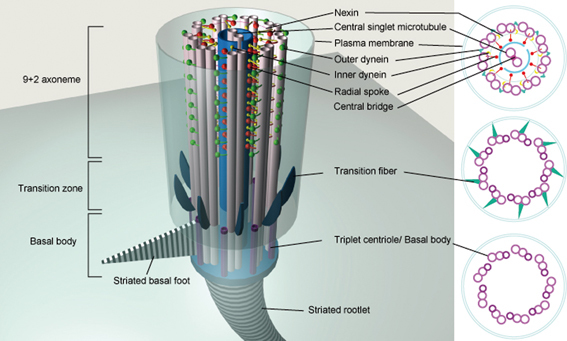
Anatomy of the 9+2 motile cilium. The axoneme, an array of nine microtubule doublets and two central singlets, is the core structure of the 9+2 motile cilium. The transition zone is a structure connecting the axoneme and the basal body. It converts the 9 × 2 axonemal doublet microtubules into the 9 × 3 triplet structure of the basal body. The transition fibers extend from the distal part of the basal body to the plasma membrane. The basal foot is a process that extends laterally from the basal body and is oriented in a consistent direction in polarized ciliated cells. The striated rootlet is a conical banded structure that extends from the proximal end of the basal body to the cell nucleus.
Cilia perform a variety of functions, both by sensing signals from their surroundings and, often, by creating fluid flows. In the embryonic ventral node, which is located at the most posterior portion of the notochordal plate, 9+0 motile monocilia, called nodal cilia, generate the fluid flow that is necessary for the formation of the left-right asymmetry of the body.5)–7) The nodal cilia also sense FGFs, which trigger the secretion of vesicles carrying Sonic hedgehog and retinoid acid, which play critical roles in left-right determination.5) Asymmetric cilia-dependent fluid flow is also found in Kupffer’s vesicle, a likely equivalent of the node, which determines left-right asymmetry in the medaka fish and the zebrafish.6),7)
A single 9+0 immotile, or “primary,” cilium exists in almost every quiescent cell in the body, and some of these cilia can sense signals such as fluid flow or molecular components in their surroundings. 8),9) The renal epithelial monocilia mediate the sensation of shear stress to activate the intracellular Ca2+ channel.9),10) The subsequent increase in intracellular Ca2+ is thought to influence numerous sub-cellular activities that are required for tissue morphogenesis. A defect in the mechanosensory monocilia, as occurs in polycystic kidney disease (PKD), for example, usually leads to abnormal renal cell proliferation and cyst formation.
Vertebrate primary cilia also play an essential role in the transduction of the Hedgehog (Hh) signal, which controls growth, cell-fate decisions, and morphogenesis during development. Several Hh signaling components, including Smoothened and Gli2/3, associate physically with the primary cilium. A defect in the intraflagellar transport (IFT) in cilia causes the loss of the primary cilium and defective Hh signaling. Cilia are also suggested to be associated with hedgehog-associated signaling particles. These data suggest that key steps of the Hh signaling pathway may occur within the cilium (Fig. 2).1),5),11)–13) Recent studies also indicate that a protein located in the ciliary basal body, Inversin, acts as a molecular switch between the canonical and non-canonical Wnt pathways. Inversin interacts with cytosolic but not membrane-bound Dishevelled and promotes its degradation, thus inhibiting β-catenin signaling (Fig. 3).14)
Fig. 2.
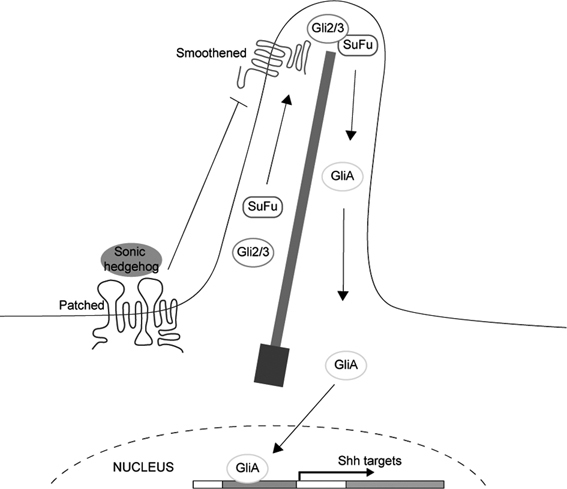
Hedgehog signaling in primary cilia. The intraflagellar transport (IFT) machinery moves Hh proteins to their functional sites, and thus transduces the Hh signal. When the Hh ligand binds to its receptor Patched, the transcription factor Gli and suppressor of fused (SuFu) are transported by IFT to the cilium tip, where Smoothened interacts with SuFu and converts Gli to its active form (GliA). The GliA then enters the cell nucleus to turn on gene expression.
Fig. 3.
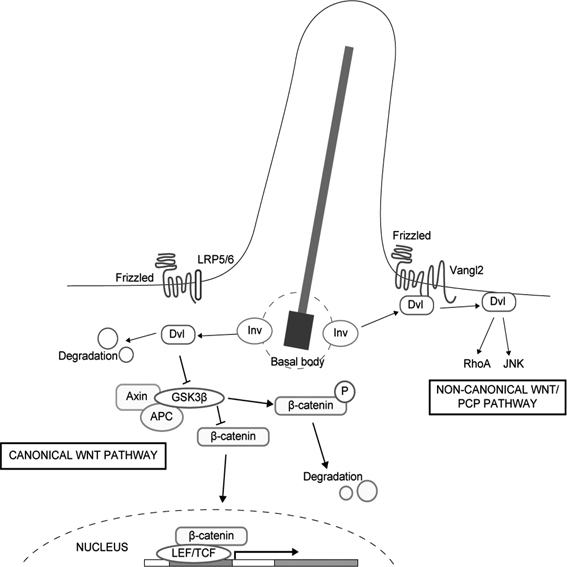
Inversin functions as a switch between the canonical Wnt and non-canonical Wnt/PCP pathways. Inversin (Inv) is localized to the basal body, where it can interact with cytoplasmic but not membranous Dishevelled (Dvl) and promote its degradation. The degradation of cytoplasmic Dishevelled blocks transduction of the canonical Wnt pathway. Membrane-bound Dishevelled then switches on the non-canonical Wnt/PCP signaling.14)
Cilia are classified by their microtubule structure and motility: cilia composed of 9 peripheral doublets and 2 central singlets are “9+2 motile” or “9+2 immotile” cilia; those composed of 9 peripheral doublets without a central pair are “9+0 motile” (nodal cilia) and “9+0 immotile cilia” (other primary cilia) (Fig. 4). 9+2 immotile cilia are distributed in the hair cells of the inner ear and play important roles in auditory transduction.15),16) 9+2 motile ciliated cells overlie the surface of airways, oviducts, and brain ventricles, and transport mucus, the ovum, and cerebrospinal fluid (CSF), respectively. Although their function is still unclear, cilia with a 9+4 axoneme exist on the notochordal plate of the rabbit embryo.17) Since the cilia of the different groups share many common components, a defect in one structural protein usually leads to severe disorders (Table 1).
Fig. 4.
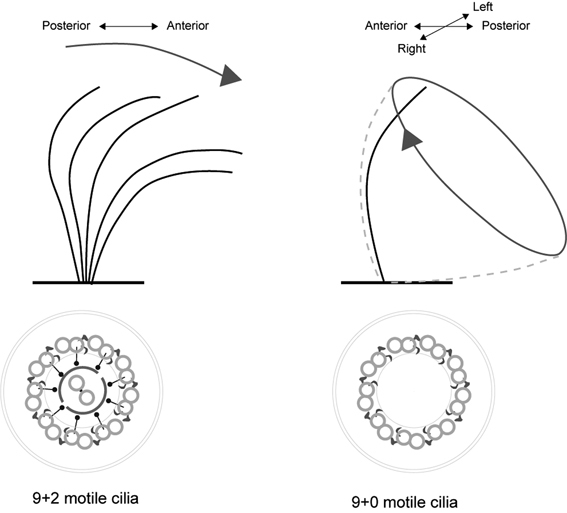
Movement patterns of 9+2 motile cilia and 9+0 motile cilia. The 9+2 motile cilia are found in the lateral ventricle, respiratory ducts, female reproductive system, and sperm. 9+0 motile monocilia are located at the surface of the embryonic ventral node and rotate quickly to create a smooth leftward flow, which determines the left-right asymmetry of the animal body. The generation of the leftward nodal flow is dependent on the posterior tilt of the rotating cilia. Motile cilia beat coordinately along the anterior-posterior axis and generate a directional fluid flow, a phenomenon known as planar cell polarity. The sperm flagellum is a specialized 9+2 structure that gives sperm cells their motility.
Table 1.
| Disease | Gene | Cellular Function | Protein Location |
|---|---|---|---|
| Almström syndrome54),55) | ALMS1 | Ciliogenesis | Basal body |
| Hypersecretory lungs | |||
| Retinitis | |||
| Obesity | |||
| Diabetes mellitus | |||
|
| |||
| Senior-Loken syndrome (type 1, 4, 5, 6)56)–59) | NPHP1 | Uncertain | Cilia, basal body |
| NPHP4 | |||
| Nephronophthisis | NPHP5/IQCB1 | ||
| Progressive eye disease | NPHP6/CEP290 | ||
|
| |||
| Polycystic kidney disease (PKD)60) | PKD1 | Mechanosensing | Cilia |
| PKD2 | |||
| Urinary tract infections | PKHD1 | Unknown | Cilia, basal body |
| Liver and pancreatic cysts | |||
|
| |||
| Primary cilia dyskinesia (PCD), also known as immotile cilia syndrome, or Kartagener’s syndrome61),62) | DNAH5 | Ciliary motility | Outer dynein arms |
| DNAl1 | Ciliary motility | Outer dynein arms | |
| Bronchiectasis | |||
| Chronic sinusitis | |||
| Infertility | |||
| Situs inversus | |||
|
| |||
| Bardet-Biedl syndrome63)–68) | BBS1–12 | Ciliogenesis | Basal body, IFT complex |
| Obesity | |||
| Retinal degeneration | |||
| Polycystic kidney | |||
|
| |||
| Meckel-Gruber syndrome69),70) | Cep290 | Unknown | Basal body |
| MKS1 | Ciliogenesis | Basal body | |
| Brain malformation | MKS3 | Ciliogenesis | Ciliary membrane |
| Polydactyly | |||
| Polycystic kidney | |||
|
| |||
| Oral-Facial-Digital syndrome71) | OFD1 | Ciliogenesis | Basal body |
| Craniofacial abnormality | |||
| Polydactyly | |||
| Polycystic kidney | |||
|
| |||
| Nephronophthisis | NPHP1–9 | Uncertain | Axoneme, basal body |
|
| |||
| Retinitis pigmentosa72) | RPGR | Retinal transport | Basal body |
|
| |||
| Situs inversus73) | DNAH11 | Ciliary motility | Dynein arms |
|
| |||
| Neural tube defect74) | VANGL1 | Uncertain | Uncertain |
| Spinal dysraphisms | |||
Defects in ciliary motility can cause ciliopathies. A defect in the directional beating of the SVZ ependymal cilia results in abnormal CSF flow and causes hydrocephalus in small animals such as mice. However, in humans the dysmotility of ependymal cilia, such as in Kartagener’s syndrome (KS, also known as primary cilia dyskinesia, PCD) rarely causes hydrocephalus, but results in severe symptoms in the respiratory duct and inner ear.18)–20) The differences between humans and rodents are probably related to morphological differences, such as the diameter of the aqueduct. In human PCD patients, if hydrocephalus occurs, it is usually caused by congenital aqueduct closure (which occurs at a rate 83-fold higher than that of the general population).2),21)–24) The high incidence of congenital aqueduct closure indicates that a disturbance of ependymal flow due to dysmotile cilia may cause aqueduct stenosis, thus increasing the risk for hydrocephalus in humans.
The extracellular fluid flow generated by motile cilia has important roles during embryonic development. In mammals, the 9+0 motile monocilia at the surface of the embryonic ventral node move to generate fluid flow. Unlike other motile cilia, such as ependymal cilia, which beat back and forth, the nodal cilia move with a tilted and clockwise rotation that generates a leftward extra-embryonic fluid flow (nodal flow), which is required for the establishment of the left-right asymmetry of the body (Fig. 4).25)–29) About 50% of KS patients develop situs inversus totalis, in which the organs are reversed in a “mirror-image” pattern from that of normal individuals, which is thought to result from the absence of nodal ciliary rotation and failure of the leftward fluid flow. These patients mainly suffer from chronic respiratory infections due to the impaired motility of their respiratory epithelial cilia,18),30) and from male infertility due to sperm immotility.
Ciliary polarity
The function of motile cilia requires that they beat coordinately in the same direction on each cell within a region of tissue. Within a tissue, most cells display several types of polarization. Some cell types with oriented motile cilia are polarized in a plane orthogonal to the epithelial apical-basal axis and generate directional fluid flow. In vertebrate embryos, monocilia are tilted posteriorly and generate a localized net leftward fluid flow over the surface of the ventral node.28),29) This polarity is generally referred to as planar cell polarity (PCP). The signaling pathway that regulates the establishment of PCP was originally identified in Drosophila, and is evolutionarily conserved in vertebrates.31) Most of the core PCP genes, including Frizzled, Strabismus (Stbm, also known as Vang), and Dishevelled, were discovered in genetic studies in Drosophila, in which PCP signals regulate the orientation of hairs on the wings and abdomen and the arrangement of ommatidia in the compound eyes.
Current models propose that in Drosophila epithelial tissues, neighboring cells communicate PCP signals through PCP proteins that are distributed asymmetrically between cells (Fig. 5A).32),33) PCP signaling, which is also called non-canonical Wnt signaling, is activated by the binding of the extracellular ligands Wnt11/5a to the Frizzled family membrane receptors. The intracellular PCP effector Dishevelled binds to Frizzled and/or Stbm and transduces signals through the RhoA and/or JNK signaling cascade to control tissue polarity. Dishevelled can also be bound by Prickle to antagonize the Frizzled/PCP signaling (Fig. 6).34),35) The seven-pass transmembrane protein Flamingo (Fmi) mediates the membrane localization of Stbm and Frizzled.33)
Fig. 5.
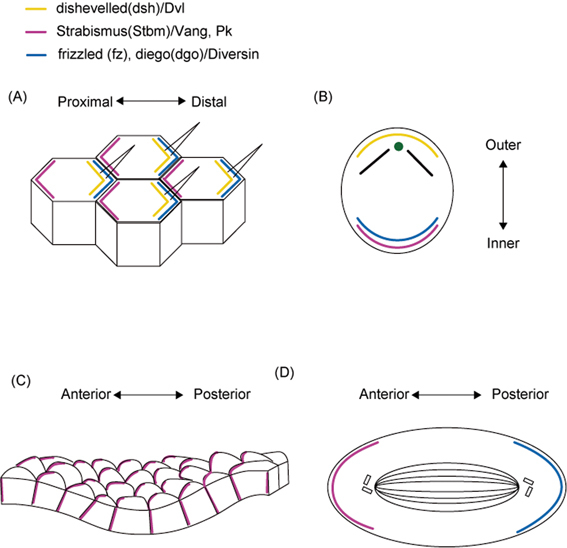
Distribution of the core PCP factors in Drosophila melanogaster and vertebrate cells. (A) Schematic of the core PCP (planar cell polarity) protein localization in Drosophila melanogaster wing cells (A) and mouse sensory hair cells in the cochlea (B). Vangl2 is polarized along the anterior-posterior (A-P) axis in the epidermal basal layer of embryonic mouse skin (C). The asymmetric PCP protein distribution orients the D. melanogaster sensory organ precursor (SOP) cell division (D). Colored lines indicate the location of core PCP proteins. Yellow: Dishevelled; purple: Vang/Vangl2 and Prickle; blue: Frizzled and Diego/Diversin.
Fig. 6.
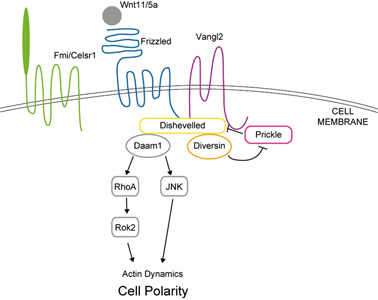
The planar cell polarity pathway (PCP pathway, non-canonical pathway) cascade. The extracellular ligands (Wnt11/5a, etc.) bind to the membrane protein/receptor (Frizzled, Vangl2, Celsr1), then activate the PCP pathway and influence the dynamics of the actin cytoskeleton, thus controlling cell polarity. Core PCP proteins (in color): Frizzled, Vangl2, Fmi/Celsr1, Diversin, Dishevelled, and Prickle. PCP effectors (gray): Daam1, RhoA, Rok2, and JNK.
In mouse, mutants in the PCP protein Vangl2 (also known as Strabismus1) exhibit an open neural tube, defective convergent extension, and disorganized kinocilia in the organ of Corti (Fig. 5B).36)–39) Moreover, recent studies indicated that a mouse Frizzled orthologue, Fz6, controls hair patterning in mice (Fig. 5C). In the mouse embryonic epidermal basal layer and hair germ, the PCP proteins Vangl2 and an Fmi homologue, Celsr1, show anterior-posterior polarization, as reported in Drosophila models, and a defect in these proteins leads to the loss of the global asymmetry of hair follicles.40),41) These findings indicate that PCP genes play important roles during PCP establishment in both invertebrates and vertebrates. Although the relationship between the polarity of motile cilia and the PCP pathway is still unclear, one study showed that Vangl2 is localized to the basal part and transition zone of the motile cilia of airway epithelial cells,38) suggesting that Vangl2 might also influence the polarization of motile cilia. A recent study showed that, in the mucociliary epithelia of the Xenopus laevis embryo, Dishevelled signaling governs both the apical docking and the planar polarization of the basal bodies during the ciliogenesis of motile cilia.42)
Moreover, 9+2 motile cilia not only cause fluid flow, but may also sense signals from the flow (e.g., shear stress) to determine their orientation.43),44) Recent studies showed that ultrastructural defects in the central pair apparatus of motile cilia cause disturbances in both ciliary beating and polarization. 18),21),45) Similarly, in PCD patients, a deficiency of the central pair of 9+2 motile cilia results in the impairment of cilia motility and the loss of ciliary orientation.18) These observations suggest that motile cilia might sense the extracellular fluid flow during ciliogenesis.
Neurogenesis in the subventricular zone
Until recently, it was believed that new neurons could not be generated in the adult mammalian brain. However, recent studies have shown that in restricted areas, new neurons are born throughout adulthood. The first evidence of postnatal neurogenesis in a rodent brain was obtained in 1965, and by the 1990’s it had become generally accepted that adult neurogenesis indeed occurs in mammals, including humans.46),47) Neural stem cells are found in the dentate gyrus of the hippocampus and in the subventricular zone (SVZ) of the lateral ventricles, where they divide to self-renew and to generate new neurons throughout life.
Neurogenesis occurs continuously in the SVZ of postnatal rodent brains. The slowly dividing neural stem cells (type B cells) self-replicate and differentiate into transit-amplifying cells (type C cells) (Fig. 7B). The type C cells proliferate rapidly to give rise to neuroblasts (type A cells).47) The newborn neurons in the SVZ attach to one another and form elongated cell aggregates called “chains,” in which cells migrate tangentially. These neuroblasts then enter a highly restricted route termed the “rostral migratory stream” (RMS) and migrate ∼8 mm (in mice) anteriorly toward the olfactory bulb (OB). Once the chain of migrating neuroblasts reaches the OB, the cells detach and begin to migrate radially to either the deep granular layer or the superficial periglomerular layer of the bulb, and differentiate into GABA-containing and dopaminergic local circuit interneurons (Fig. 7A).48) The regulatory mechanisms underlying such long-distance, directional, and high-speed (about 100 μm/hr) neuronal migration in the mature brain are still unclear.
Fig. 7.
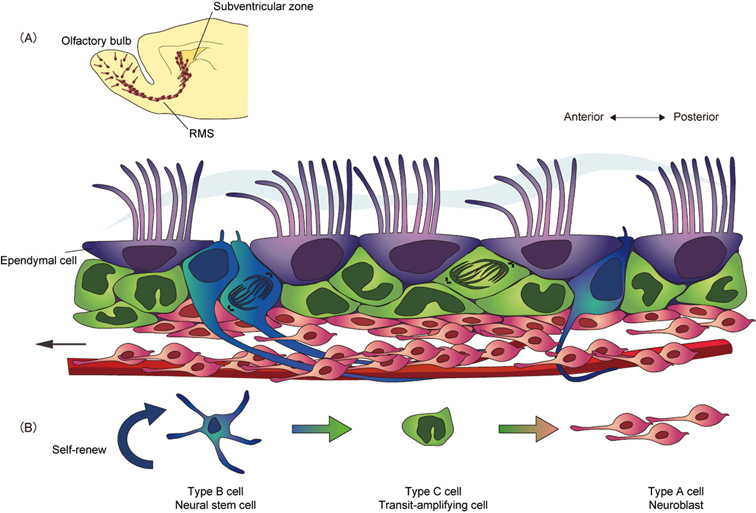
Neuronal migration in the adult rodent brain. (A) Neurogenesis occurs continuously in the postnatal rodent brain. In the subventricular zone (SVZ), neural stem cells (type B cells, blue) generate migratory neuroblasts (type A cells, red) via highly proliferative transit-amplifying cells (type C cells, green). The ciliated ependymal cells (type E cells, purple) line the surface of the ventricle. Neuroblasts migrate along a restricted pathway, the rostral migratory stream (RMS), toward the olfactory bulb (OB). Once the chain of migrating neuroblasts reaches the OB, the cells detach and disperse to either the deep granular layer or the superficial periglomerular layer, and differentiate into interneurons. The cerebrospinal fluid (CSF) flow, created by the beating of polarized ependymal cilia, and the concentration gradient of Slit protein are necessary to orient neuroblast migration. (B) The lineage of SVZ cells.
We recently showed that the migrational direction of newborn neurons corresponds to the direction of CSF flow generated by the SVZ ependymal cilia.49) It is possible that the CSF flow affects the distribution of various proteins, and directs the formation of anterior-posterior concentration gradients in the SVZ, which are important for the control of neuroblast migration. A secreted protein, Slit, is a chemorepulsive molecule that plays an important role in directing the extension and branching of developing sensory axons by regulating the cytoskeletal distribution of the growth cone, via its binding of the Robo receptor. In mutant mice with defective cilia, the concentration gradient of Slit is disturbed, and neuroblast migration occurs in an irregular direction. These results suggest that the CSF flow generated by the polarized ependymal cilia is important for neuronal migration in the adult brain.
Although the ependymal cilia beat coordinately and generate directional fluid flow in adult mammalian brain ventricles, it is still unclear how this polarity is determined. Recent studies indicate that both PCP signaling and fluid flow play important roles in the ciliary polarity of the developing Xenopus larval skin.42),44) These mechanisms may also be involved in the coordinated ciliary beating of SVZ ependymal cells. Ependymal cells are transformed from radial glial cells at the late embryonic stage and generate motile cilia postnatally.50) Since CSF is produced by the choroid plexus, circulated in the lateral ventricles, and absorbed in the anterior part of ventricles, there is fluid flow in the lateral ventricle during the period of differentiation of ependymal cells. These findings raise the possibility that CSF flow is involved in the polarization of the ependymal cilia during development.
Conclusion and discussion
The motile cilia/flagella are complicated cellular organelles that link various physiological functions—even some that precede conception—from the motility of sperm to respiratory tract clearance. In addition, our studies have suggested that the coordinated beating of ependymal cilia plays important roles in neuronal migration and adult neurogenesis. A recent study indicated that the ependymal cells retain their radial glial potential in the adult forebrain, and they can respond to ischemia by giving rise to neurons. 50),51)
Clarification of the molecular mechanisms of the polarization of cilia may help us not only to understand the mechanisms of ciliary development, but also to develop new therapeutic strategies for treating ciliary disorders. Furthermore, elucidating the involvement of ciliary functions in adult neurogenesis and neuronal migration may lead to new ideas for regeneration therapies for the injured adult brain.
Acknowledgments
This work was supported by the Human Frontier Science Program, Ministry of Education, Culture, Sports, Science and Technology and Ministry of Health, Labour and Welfare, the Japan Society for the Promotion of Science, the Toray Science Foundation, the Mitsubishi Foundation, the Japan Science Society, and the Hayashi Memorial Foundation for Female Natural Scientists. S. H. is supported by an Interchange Association, Japan (IAJ) Scholarship.
Abbreviations:
- SVZ
subventricular zone
- CSF
cerebrospinal fluid
- RMS
rostral migratory stream
- OB
olfactory bulb
- PKD
polycystic kidney disease
- Hh
Hedgehog
- PCD
primary cilia dyskinesia
- IFT
intraflagellar transport
- PCP
planar cell polarity
- KS
Kartagener’s syndrome
Biographies
Profile
Shihhui Huang was born in 1981 at Taipei and graduated from National Taiwan University in 2003. She worked at Global biotech INC for one year. In 2004, because of the great interests of neuroscience, she joined the Neural Regeneration Laboratory in Taipei Veterans General Hospital and studied repairs of spinal cord injury for 2 years. In 2008, she joined Prof. Kazunobu Sawamoto’s laboratory at Nagoya City University Graduate School of Medical Sciences as a master course student.

Profile
Yuki Hirota was born in Tokyo, Japan, in 1975, and graduated from Tokyo Metropolitan University in 1997. She received her Ph.D. from Osaka University in 2003 under the supervision of Prof. Hideyuki Okano. After three years of postdoctoral working in Keio University, she started her research on the development of ciliated ependymal cells as an Assistant Professor with Prof. Kazunobu Sawamoto at the Department of Developmental and Regenerative Biology, Nagoya City University Graduate School of Medical Sciences.

Profile
Kazunobu Sawamoto was born in Hokkaido, Japan, in 1967, and received his Ph.D. from The University of Tokyo in 1996 under the supervision of Professor Katsuhiko Mikoshiba. He started his research career as a Research Associate at Professor Hideyuki Okano’s laboratory at University of Tsukuba (1996–1997) and Osaka University (1997–2001). He had the opportunity to study in Professor Arturo Alvarez-Buylla’s laboratory at University of California San Francisco as an Overseas Researcher of Ministry of Education, Culture, Sports, Science and Technology (2001–2003). After returning to Japan, he joined Professor Hideyuki Okano’s laboratory as an Assistant Professor at Keio University (2003–2005). He started his own research group as an Associate Professor at Keio University (2005–2007) and moved to Nagoya City University in 2007 as a Professor of Developmental and Regenerative Biology. He received the Inoue Research Award for Young Scientists (1997), the Sanshikai Award (2004), the Japan Neuroscience Society Young Investigator Award (2004), the Award for Young Investigator of Japan Society for Neurochemistry (2006), the JSPS Prize (2006) and the Young Scientists’ Prize from the Ministry of Education, Culture, Sports, Science and Technology (2007).

References
- 1).Fliegauf, M., Benzing, T. and Omran, H. (2007) When cilia go bad: cilia defects and ciliopathies. Nat. Rev. Mol. Cell Biol. 8, 880–893 [DOI] [PubMed] [Google Scholar]
- 2).Ibanez-Tallon, I., Pagenstecher, A., Fliegauf, M., Olbrich, H., Kispert, A., Ketelsen, U.P.et al. (2004) Dysfunction of axonemal dynein heavy chain Mdnah5 inhibits ependymal flow and reveals a novel mechanism for hydrocephalus formation. Hum. Mol. Genet. 13, 2133–2141 [DOI] [PubMed] [Google Scholar]
- 3).King, S.M. (2000) The dynein microtubule motor. Biochim. Biophys. Acta 1496, 60–75 [DOI] [PubMed] [Google Scholar]
- 4).Yang, J., Gao, J., Adamian, M., Wen, X.H., Pawlyk, B., Zhang, L.et al. (2005) The ciliary rootlet maintains long-term stability of sensory cilia. Mol. Cell Biol. 25, 4129–4137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5).Tanaka, Y., Okada, Y. and Hirokawa, N. (2005) FGF-induced vesicular release of Sonic hedgehog and retinoic acid in leftward nodal flow is critical for left-right determination. Nature 435, 172–177 [DOI] [PubMed] [Google Scholar]
- 6).Okada, Y., Nonaka, S., Tanaka, Y., Saijoh, Y., Hamada, H. and Hirokawa, N. (1999) Abnormal nodal flow precedes situs inversus in iv and inv mice. Mol. Cell 4, 459–468 [DOI] [PubMed] [Google Scholar]
- 7).Kramer-Zucker, A.G., Olale, F., Haycraft, C.J., Yoder, B.K., Schier, A.F. and Drummond, I.A. (2005) Cilia-driven fluid flow in the zebrafish pronephros, brain and Kupffer’s vesicle is required for normal organogenesis. Development 132, 1907–1921 [DOI] [PubMed] [Google Scholar]
- 8).Gerdes, J.M., Davis, E.E. and Katsanis, N. (2009) The vertebrate primary cilium in development, homeostasis, and disease. Cell 137, 32–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9).Nauli, S.M., Alenghat, F.J., Luo, Y., Williams, E., Vassilev, P., Li, X.et al. (2003) Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet. 33, 129–137 [DOI] [PubMed] [Google Scholar]
- 10).Praetorius, H.A. and Spring, K.R. (2001) Bending the MDCK cell primary cilium increases intracellular calcium. J. Membr. Biol. 184, 71–79 [DOI] [PubMed] [Google Scholar]
- 11).Huangfu, D., Liu, A., Rakeman, A.S., Murcia, N.S., Niswander, L. and Anderson, K.V. (2003) Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 426, 83–87 [DOI] [PubMed] [Google Scholar]
- 12).May, S.R., Ashique, A.M., Karlen, M., Wang, B., Shen, Y., Zarbalis, K.et al. (2005) Loss of the retrograde motor for IFT disrupts localization of Smo to cilia and prevents the expression of both activator and repressor functions of Gli. Dev. Biol. 287, 378–389 [DOI] [PubMed] [Google Scholar]
- 13).Kiprilov, E.N., Awan, A., Desprat, R., Velho, M., Clement, C.A., Byskov, A.G.et al. (2008) Human embryonic stem cells in culture possess primary cilia with hedgehog signaling machinery. J. Cell Biol. 180, 897–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14).Simons, M., Gloy, J., Ganner, A., Bullerkotte, A., Bashkurov, M., Kronig, C.et al. (2005) Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat. Genet. 37, 537–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15).Wang, J., Mark, S., Zhang, X., Qian, D., Yoo, S.J., Radde-Gallwitz, K.et al. (2005) Regulation of polarized extension and planar cell polarity in the cochlea by the vertebrate PCP pathway. Nat. Genet. 37, 980–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16).Sobkowicz, H.M., Slapnick, S.M. and August, B.K. (1995) The kinocilium of auditory hair cells and evidence for its morphogenetic role during the regeneration of stereocilia and cuticular plates. J. Neurocytol. 24, 633–653 [DOI] [PubMed] [Google Scholar]
- 17).Feistel, K. and Blum, M. (2006) Three types of cilia including a novel 9+4 axoneme on the notochordal plate of the rabbit embryo. Dev. Dyn. 235, 3348–3358 [DOI] [PubMed] [Google Scholar]
- 18).Afzelius, B.A. (1980) Genetic disorders of cilia. InInternational Cell Biology 1980–1981 (ed. Schweiger, H.G.), Springer-Verlag, Berlin, p. 8 [Google Scholar]
- 19).Afzelius, B.A. (2004) Cilia-related diseases. J. Pathol. 204, 470–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20).Bush, A., Cole, P., Hariri, M., Mackay, I., Phillips, G., O’Callaghan, C.et al. (1998) Primary ciliary dyskinesia: diagnosis and standards of care. Eur. Respir. J. 12, 982–988 [DOI] [PubMed] [Google Scholar]
- 21).Lechtreck, K.F., Delmotte, P., Robinson, M.L., Sanderson, M.J. and Witman, G.B. (2008) Mutations in Hydin impair ciliary motility in mice. J. Cell Biol. 180, 633–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22).Badano, J.L., Mitsuma, N., Beales, P.L. and Katsanis, N. (2006) The ciliopathies: an emerging class of human genetic disorders. Annu. Rev. Genomics Hum. Genet. 7, 125–148 [DOI] [PubMed] [Google Scholar]
- 23).Ibanez-Tallon, I., Gorokhova, S. and Heintz, N. (2002) Loss of function of axonemal dynein Mdnah5 causes primary ciliary dyskinesia and hydrocephalus. Hum. Mol. Genet. 11, 715–721 [DOI] [PubMed] [Google Scholar]
- 24).Picco, P., Leveratto, L., Cama, A., Vigliarolo, M.A., Levato, G.L., Gattorno, M.et al. (1993) Immotile cilia syndrome associated with hydrocephalus and precocious puberty: a case report. Eur. J. Pediatr. Surg. 3 (Suppl. 1), 20–21 [PubMed] [Google Scholar]
- 25).Hirokawa, N., Tanaka, Y., Okada, Y. and Takeda, S. (2006) Nodal flow and the generation of left-right asymmetry. Cell 125, 33–45 [DOI] [PubMed] [Google Scholar]
- 26).Nonaka, S., Shiratori, H., Saijoh, Y. and Hamada, H. (2002) Determination of left-right patterning of the mouse embryo by artificial nodal flow. Nature 418, 96–99 [DOI] [PubMed] [Google Scholar]
- 27).Nonaka, S., Tanaka, Y., Okada, Y., Takeda, S., Harada, A., Kanai, Y.et al. (1998) Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell 95, 829–837 [DOI] [PubMed] [Google Scholar]
- 28).Nonaka, S., Yoshiba, S., Watanabe, D., Ikeuchi, S., Goto, T., Marshall, W.F.et al. (2005) De novo formation of left-right asymmetry by posterior tilt of nodal cilia. PLoS Biol. 3, e268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29).Okada, Y., Takeda, S., Tanaka, Y., Belmonte, J.C. and Hirokawa, N. (2005) Mechanism of nodal flow: a conserved symmetry breaking event in left-right axis determination. Cell 121, 633–644 [DOI] [PubMed] [Google Scholar]
- 30).Afzelius, B.A. (1976) A human syndrome caused by immotile cilia. Science 193, 317–319 [DOI] [PubMed] [Google Scholar]
- 31).Seifert, J.R. and Mlodzik, M. (2007) Frizzled/PCP signalling: a conserved mechanism regulating cell polarity and directed motility. Nat. Rev. Genet. 8, 126–138 [DOI] [PubMed] [Google Scholar]
- 32).Montcouquiol, M., Sans, N., Huss, D., Kach, J., Dickman, J.D., Forge, A.et al. (2006) Asymmetric localization of Vangl2 and Fz3 indicate novel mechanisms for planar cell polarity in mammals. J. Neurosci. 26, 5265–5275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33).Chen, W.S., Antic, D., Matis, M., Logan, C.Y., Povelones, M., Anderson, G.A.et al. (2008) Asymmetric homotypic interactions of the atypical cadherin flamingo mediate intercellular polarity signaling. Cell 133, 1093–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34).Wallingford, J.B. (2006) Planar cell polarity, ciliogenesis and neural tube defects. Hum. Mol. Genet. 15 (2), R227–R234 [DOI] [PubMed] [Google Scholar]
- 35).Michaels, L. and Soucek, S. (1991) Auditory epithelial migration. III. Development of the stratified squamous epithelium of the tympanic membrane and external canal in the mouse. Am. J. Anat. 191, 280–292 [DOI] [PubMed] [Google Scholar]
- 36).Ciruna, B., Jenny, A., Lee, D., Mlodzik, M. and Schier, A.F. (2006) Planar cell polarity signalling couples cell division and morphogenesis during neurulation. Nature 439, 220–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37).Jones, C., Roper, V.C., Foucher, I., Qian, D., Banizs, B., Petit, C.et al. (2008) Ciliary proteins link basal body polarization to planar cell polarity regulation. Nat. Genet. 40, 69–77 [DOI] [PubMed] [Google Scholar]
- 38).Ross, A.J., May-Simera, H., Eichers, E.R., Kai, M., Hill, J., Jagger, D.J.et al. (2005) Disruption of Bardet-Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat. Genet. 37, 1135–1140 [DOI] [PubMed] [Google Scholar]
- 39).Torban, E., Kor, C. and Gros, P. (2004) Van Goghlike2 (Strabismus) and its role in planar cell polarity and convergent extension in vertebrates. Trends Genet. 20, 570–577 [DOI] [PubMed] [Google Scholar]
- 40).Devenport, D. and Fuchs, E. (2008) Planar polarization in embryonic epidermis orchestrates global asymmetric morphogenesis of hair follicles. Nat. Cell Biol. 10, 1257–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41).Guo, N., Hawkins, C. and Nathans, J. (2004) Frizzled6 controls hair patterning in mice. Proc. Natl. Acad. Sci. USA 101, 9277–9281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42).Park, T.J., Mitchell, B.J., Abitua, P.B., Kintner, C. and Wallingford, J.B. (2008) Dishevelled controls apical docking and planar polarization of basal bodies in ciliated epithelial cells. Nat. Genet. 40, 871–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43).Marshall, W.F. and Kintner, C. (2008) Cilia orientation and the fluid mechanics of development. Curr. Opin. Cell Biol. 20, 48–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44).Mitchell, B., Jacobs, R., Li, J., Chien, S. and Kintner, C. (2007) A positive feedback mechanism governs the polarity and motion of motile cilia. Nature 447, 97–101 [DOI] [PubMed] [Google Scholar]
- 45).Wilson, C.W., Nguyen, C.T., Chen, M.H., Yang, J.H., Gacayan, R., Huang, J.et al. (2009) Fused has evolved divergent roles in vertebrate Hedgehog signalling and motile ciliogenesis. Nature 459, 98–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46).Altman, J. and Das, G.D. (1965) Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J. Comp. Neurol. 124, 319–335 [DOI] [PubMed] [Google Scholar]
- 47).Chojnacki, A.K., Mak, G.K. and Weiss, S. (2009) Identity crisis for adult periventricular neural stem cells: subventricular zone astrocytes, ependymal cells or both? Nat. Rev. Neurosci. 10, 153–163 [DOI] [PubMed] [Google Scholar]
- 48).Ghashghaei, H.T., Lai, C. and Anton, E.S. (2007) Neuronal migration in the adult brain: are we there yet? Nat. Rev. Neurosci. 8, 141–151 [DOI] [PubMed] [Google Scholar]
- 49).Sawamoto, K., Wichterle, H., Gonzalez–Perez, O., Cholfin, J.A., Yamada, M., Spassky, N.et al. (2006) New neurons follow the flow of cerebrospinal fluid in the adult brain. Science 311, 629–632 [DOI] [PubMed] [Google Scholar]
- 50).Spassky, N., Merkle, F.T., Flames, N., Tramontin, A.D., Garcia-Verdugo, J.M. and Alvarez-Buylla, A. (2005) Adult ependymal cells are postmitotic and are derived from radial glial cells during embryogenesis. J. Neurosci. 25, 10–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51).Carlen, M., Meletis, K., Goritz, C., Darsalia, V., Evergren, E., Tanigaki, K.et al. (2009) Forebrain ependymal cells are Notch-dependent and generate neuroblasts and astrocytes after stroke. Nat. Neurosci. 12, 259–267 [DOI] [PubMed] [Google Scholar]
- 52).D’Angelo, A. and Franco, B. (2009) The dynamic cilium in human diseases. Pathogenetics 2, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53).Marshall, W.F. (2008) The cell biological basis of ciliary disease. J. Cell Biol. 180, 17–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54).Collin, G.B., Marshall, J.D., Ikeda, A., So, W.V., Russell-Eggitt, I., Maffei, P.et al. (2002) Mutations in ALMS1 cause obesity, type 2 diabetes and neurosensory degeneration in Alstrom syndrome. Nat. Genet. 31, 74–78 [DOI] [PubMed] [Google Scholar]
- 55).Hearn, T., Renforth, G.L., Spalluto, C., Hanley, N.A., Piper, K., Brickwood, S.et al. (2002) Mutation of ALMS1, a large gene with a tandem repeat encoding 47 amino acids, causes Alstrom syndrome. Nat. Genet. 31, 79–83 [DOI] [PubMed] [Google Scholar]
- 56).Helou, J., Otto, E.A., Attanasio, M., Allen, S.J., Parisi, M.A., Glass, I.et al. (2007) Mutation analysis of NPHP6/CEP290 in patients with Joubert syndrome and Senior-Loken syndrome. J. Med. Genet. 44, 657–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57).Otto, E.A., Loeys, B., Khanna, H., Hellemans, J., Sudbrak, R., Fan, S.et al. (2005) Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat. Genet. 37, 282–288 [DOI] [PubMed] [Google Scholar]
- 58).Otto, E., Hoefele, J., Ruf, R., Mueller, A.M., Hiller, K.S., Wolf, M.T.et al. (2002) A gene mutated in nephronophthisis and retinitis pigmentosa encodes a novel protein, nephroretinin, conserved in evolution. Am. J. Hum. Genet. 71, 1161–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59).Caridi, G., Murer, L., Bellantuono, R., Sorino, P., Caringella, D.A., Gusmano, R.et al. (1998) Renal-retinal syndromes: association of retinal anomalies and recessive nephronophthisis in patients with homozygous deletion of the NPH1 locus. Am. J. Kidney Dis. 32, 1059–1062 [DOI] [PubMed] [Google Scholar]
- 60).Ward, C.J., Hogan, M.C., Rossetti, S., Walker, D., Sneddon, T., Wang, X.et al. (2002) The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat. Genet. 30, 259–269 [DOI] [PubMed] [Google Scholar]
- 61).Pennarun, G., Escudier, E., Chapelin, C., Bridoux, A.M., Cacheux, V., Roger, G.et al. (1999) Loss-of-function mutations in a human gene related to Chlamydomonas reinhardtii dynein IC78 result in primary ciliary dyskinesia. Am. J. Hum. Genet. 65, 1508–1519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62).Olbrich, H., Haffner, K., Kispert, A., Volkel, A., Volz, A., Sasmaz, G.et al. (2002) Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left-right asymmetry. Nat. Genet. 30, 143–144 [DOI] [PubMed] [Google Scholar]
- 63).Mykytyn, K., Nishimura, D.Y., Searby, C.C., Shastri, M., Yen, H.J., Beck, J.S.et al. (2002) Identification of the gene (BBS1) most commonly involved in Bardet-Biedl syndrome, a complex human obesity syndrome. Nat. Genet. 31, 435–438 [DOI] [PubMed] [Google Scholar]
- 64).Badano, J.L., Ansley, S.J., Leitch, C.C., Lewis, R.A., Lupski, J.R. and Katsanis, N. (2003) Identification of a novel Bardet-Biedl syndrome protein, BBS7, that shares structural features with BBS1 and BBS2. Am. J. Hum. Genet. 72, 650–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65).Mykytyn, K., Braun, T., Carmi, R., Haider, N.B., Searby, C.C., Shastri, M.et al. (2001) Identification of the gene that, when mutated, causes the human obesity syndrome BBS4. Nat. Genet. 28, 188–191 [DOI] [PubMed] [Google Scholar]
- 66).Chiang, A.P., Beck, J.S., Yen, H.J., Tayeh, M.K., Scheetz, T.E., Swiderski, R.E.et al. (2006) Homozygosity mapping with SNP arrays identifies TRIM32, an E3 ubiquitin ligase, as a Bardet-Biedl syndrome gene (BBS11). Proc. Natl. Acad. Sci. USA 103, 6287–6292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67).Nishimura, D.Y., Swiderski, R.E., Searby, C.C., Berg, E.M., Ferguson, A.L., Hennekam, R.et al. (2005) Comparative genomics and gene expression analysis identifies BBS9, a new Bardet-Biedl syndrome gene. Am. J. Hum. Genet. 77, 1021–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68).Stoetzel, C., Muller, J., Laurier, V., Davis, E.E., Zaghloul, N.A., Vicaire, S.et al. (2007) Identification of a novel BBS gene (BBS12) highlights the major role of a vertebrate-specific branch of chaperonin-related proteins in Bardet-Biedl syndrome. Am. J. Hum. Genet. 80, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69).Kyttala, M., Tallila, J., Salonen, R., Kopra, O., Kohlschmidt, N., Paavola-Sakki, P.et al. (2006) MKS1, encoding a component of the flagellar apparatus basal body proteome, is mutated in Meckel syndrome. Nat. Genet. 38, 155–157 [DOI] [PubMed] [Google Scholar]
- 70).Smith, U.M., Consugar, M., Tee, L.J., McKee, B.M., Maina, E.N., Whelan, S.et al. (2006) The transmembrane protein meckelin (MKS3) is mutated in Meckel-Gruber syndrome and the wpk rat. Nat. Genet. 38, 191–196 [DOI] [PubMed] [Google Scholar]
- 71).Ferrante, M.I., Giorgio, G., Feather, S.A., Bulfone, A., Wright, V., Ghiani, M.et al. (2001) Identification of the gene for oral-facial-digital type I syndrome. Am. J. Hum. Genet. 68, 569–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72).Meindl, A., Dry, K., Herrmann, K., Manson, F., Ciccodicola, A., Edgar, A.et al. (1996) A gene (RPGR) with homology to the RCC1 guanine nucleotide exchange factor is mutated in X-linked retinitis pigmentosa (RP3). Nat. Genet. 13, 35–42 [DOI] [PubMed] [Google Scholar]
- 73).Bartoloni, L., Blouin, J.L., Pan, Y., Gehrig, C., Maiti, A.K., Scamuffa, N.et al. (2002) Mutations in the DNAH11 (axonemal heavy chain dynein type 11) gene cause one form of situs inversus totalis and most likely primary ciliary dyskinesia. Proc. Natl. Acad. Sci. USA 99, 10282–10286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74).Kibar, Z., Torban, E., McDearmid, J.R., Reynolds, A., Berghout, J., Mathieu, M.et al. (2007) Mutations in VANGL1 associated with neural-tube defects. N. Engl. J. Med. 356, 1432–1437 [DOI] [PubMed] [Google Scholar]