

Abstract
The protozoan parasite Trypanosoma brucei is the causative agent of African sleeping sickness, and there is an urgent unmet need for improved treatments. Parasite protein kinases are attractive drug targets, provided that the host and parasite kinomes are sufficiently divergent to allow specific inhibition to be achieved. Current drug discovery efforts are hampered by the fact that comprehensive assay panels for parasite targets have not yet been developed. Here, we employ a kinase-focused chemoproteomics strategy that enables the simultaneous profiling of kinase inhibitor potencies against more than 50 endogenously expressed T. brucei kinases in parasite cell extracts. The data reveal that T. brucei kinases are sensitive to typical kinase inhibitors with nanomolar potency and demonstrate the potential for the development of species-specific inhibitors.
The protozoan parasite Trypanosoma brucei is transmitted by the bite of an infected Tsetse fly and causes African sleeping sickness, which is also known as Human African Trypanosomasis (HAT). The disease is invariably fatal if left untreated and results in upward of 10,000 deaths each year in sub-Saharan Africa.1T. brucei has a complex digenetic lifecycle between the insect vector and mammalian host, and the ability to adapt to these environments is essential to its survival and virulence. During early stages of infection the clinically relevant bloodstream form of the parasite proliferates in the blood and lymph of the human host and then in the second stage enters the cerebrospinal fluid and brain, resulting in coma and death. Current treatments are expensive, toxic, and difficult to administer, leaving an urgent unmet need for improved therapeutic agents.2
Protein kinases play key roles in the control of growth and cell signaling and are a major target of the pharmaceutical industry. Parasite protein kinases have been proposed as attractive targets for drug discovery as such efforts can “piggy-back” on the extensive knowledge of the development of inhibitors against human protein kinases.3 In the case of T. brucei, bioinformatic analysis of the genome has identified 176–182 putative protein kinases on the basis of sequence similarity, the majority of which can be placed within well-recognized kinase groups (Supplementary Table S1).4,5 Efforts to determine the detailed biological role of T. brucei protein kinases are ongoing, although knock-down by RNA interference has provided evidence of the essential nature of a significant number of protein kinases.6 However, the rationale to develop drugs to target the T. brucei kinome poses a conundrum: if mammalian and parasite protein kinases are sufficiently similar to be identified and classified on the basis of sequence similarity and are inhibited by typical inhibitors, will parasite kinase inhibitors lack host–parasite specificity? Conversely, if the kinases are sufficiently different that host–parasite specificity can be readily obtained, will they be inhibited by typical inhibitors of mammalian kinases? In other words, we need to consider the similarity of the chemical space that parasite and mammalian protein kinase inhibitors occupy, rather than the similarity in protein kinase sequence.
One way to probe the inhibitor chemical space is to profile inhibitor activity against both the mammalian and parasite kinomes. Such profiling is often achieved using in vitro activity assays against a panel of recombinant protein kinases,7 but there is no such panel available for the T. brucei kinome; indeed only a handful of active T. brucei kinases have been recombinantly expressed as active enzymes.8−10 A recent advance in kinase inhibitor profiling uses a chemical proteomic methodology that captures a substantial portion of the expressed kinome (and related proteins) contained in cell lysates on a mixed kinase-inhibitor matrix known as kinobeads.11,12 Addition of a kinase inhibitor to the cell lysates enables it to bind to its specific target(s), occupying the binding sites and preventing binding to the kinobeads, whereas the binding of nontargeted kinases and other proteins are unaffected. Incubation of the lysate with varying concentrations of the inhibitor and subsequent analysis of the kinobead-bound subproteome by quantitative mass spectrometry allows inhibition curves to be generated for each protein observed (Figure 1). We reasoned that this methodology should be species independent, provided that the kinobeads are sufficiently promiscuous to capture a sizable portion of the parasite kinome.
Figure 1.
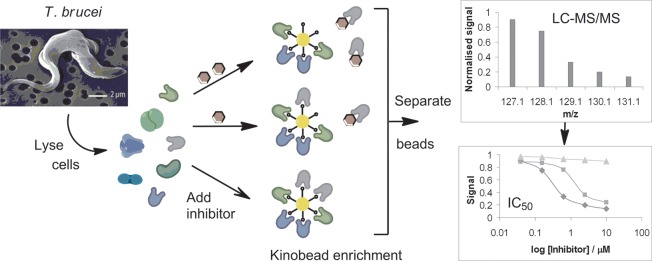
Chemical proteomics approach to profiling the targets of kinase inhibitors. T. brucei cell lysates are incubated in the presence or absence of the test inhibitor prior to the addition of mixed kinase-inhibitor beads (kinobeads) to enrich kinases and related proteins. The presence of the test kinase inhibitor prevents the binding of its target(s) to the kinobeads. Analysis of the kinobead-bound subproteome by quantitative tandem mass spectrometry using isobaric tags allows inhibition curves to be calculated for each protein observed.
Here, we present the results of our efforts to establish kinobead chemoproteomics profiling in T. brucei and estimate the coverage of the parasite kinome. Our strategy enabled us to access more than 50 parasite kinases for inhibitor profiling, which by far exceeds any currently available enzyme panels. We report the profile of the mammalian kinase inhibitors staurosporine and BMS-387032 and several early hit compounds identified as parasite protein kinase inhibitors by the Drug Discovery Unit at the University of Dundee.
Results and Discussion
Coverage of the T. brucei Kinome
Kinobeads consist of immobilized analogues of a variety of ATP competitive kinase inhibitors that show relatively promiscuous binding to the mammalian kinases, but their ability to bind kinases from more divergent organisms has not been examined. To establish to what extent T. brucei protein kinases were able to bind to the kinobeads, we examined the subproteome enriched from cell lysates of the clinically relevant bloodstream form of T. brucei. Initially, we compared four versions of kinobeads that differ in the identity of the immobilized inhibitors (see Methods). In these experiments, we observed the enrichment of a total of a 57 protein kinases (Supplementary Table S2). The enrichment of a significant number of trypanosome protein kinases by these promiscuous mammalian kinase inhibitors suggests that the ATP binding pocket architecture is broadly conserved between the two species.
Estimating the portion of the trypanosome kinome captured is not straightforward, as the bloodstream form represents just one of the multiple lifecycle stages of T. brucei and may not express every kinase encoded by the genome. Before we could determine the proportion of the bloodstream form kinome that was captured by kinobeads, we needed to estimate how many protein kinases were present in the cell lysates. To achieve this, we took advantage of the differing bias of two orthogonal proteomic techniques. By analyzing the total proteome contained in the non-enriched cell lysate we identified the most abundant 3248 proteins, which included 90 protein kinases (Supplementary Table S3). This data shows good overlap with a recent comparative SILAC proteomic study of the bloodstream and procyclic form T. brucei, which identified 65 protein kinases,13 including 18 not seen in this study. A separate phosphoproteomic study by Nett et al.,5 which used strong cation exchange and TiO2 chromatography to enrich for phosphopeptides, identified 43 phosphorylated protein kinases in bloodstream form T. brucei. Comparison of the protein kinases identified by these three orthogonal proteomics techniques revealed overlapping and complementary coverage of the bloodstream form kinome (Figure 2 and Supplementary Table S4), providing experimental observation of a total of 124 protein kinases out of the predicted 182 (68%). The kinases that bind to kinobeads are not significantly biased toward abundant (Supplementary Figure S1) or phosphorylated protein kinases or any particular kinase group. Mapping the kinases that bind to kinobeads onto the phylogeny of the T. brucei kinome shows that kinobead enrichment appears to be independent of the degree of sequence homology (Figure 2). While it is likely that the coverage of this observable bloodstream form kinome is not complete, it is in reasonable agreement with transcriptome studies that suggest 25% of the genome is differentially expressed between bloodstream and procyclic form T. brucei cells.14 The kinobead-enriched subproteome contains 46% of the observed bloodstream form kinome (31% of the predicted genome), comparable to the coverage obtained from analysis of human cell lysates (52% of the predicted genome).
Figure 2.

Profiling the kinome expressed in bloodstream form T. brucei using complementary mass spectrometry-based observations. (a) Venn diagram summarizing overlapping protein kinase observations. (b) Details of protein kinases observed (black square), divided by kinase group classification according to the similarity of their catalytic domains. Proteome: detection at natural abundance. Phosphorylated: enrichment of phosphorylated peptides.5 Kinobeads: enriched by immobilized mixed kinase-inhibitors.
Profiling of Known Kinase Inhibitors
Kinobead-based profiling enables access to a sizable fraction of the expressed trypanosome kinome, which can be used to determine the potency and selectivity of kinase inhibitors in cell extracts by means of a multiplexed competition binding assay. The kinobeads version producing the best coverage of the T. brucei kinome was used to determine the kinase inhibition profile of two well-studied kinase inhibitors: Staurosporine and BMS-387032. The binding of these inhibitors to their cellular targets was quantified by mass spectrometry using isobaric tags for relative and absolute quantification (iTRAQ)15,16 for 51 protein kinases and 67 other kinobead-binding proteins (Figure 3 and Supplementary Table S5). Staurosporine, a natural product, is a prototypical ATP-competitive pan-kinase inhibitor that binds to many protein kinases with high affinity and little selectivity.11 In a previous study we reported kinobead profiling of staurosporine in primary chronic lymphocytic leukemia cells17 and demonstrated that more than a third of the observed human kinome (41/112) displayed submicromolar IC50 values (Figure 3). This pan-kinase activity was retained against the T. brucei kinome, with more than a third of the observed trypanosome kinome (18/44) displaying IC50 values <1 μM, including 10 with IC50 values below 100 nM (Figure 3).
Figure 3.

Chemical proteomics profiling of Staurosporine and BMS-387032 against the trypanosome and human kinome. Horizontal bars represent IC50 value calculated from the isobaric reporter signals, gray bars indicate where binding was not quantified. Lysates were incubated with varying concentrations of compounds prior to incubation with kinobeads, and the bound fraction was quantified by tandem mass spectrometry. Data for TK, TKL, atypical, and lipid kinase are not shown for clarity; the full data for the trypanosome profile can be found in Supplementary Table S5, and the CLL cell data has been reported previously.17
The second kinase inhibitor to be profiled, BMS-387032, an established pan-cyclin-dependent kinase (CDK) inhibitor, was selected because T. brucei have a relatively expanded CMGC group including many putative CDK or CDK-like (CDKL) family members.4,5 Kinobead profiling of BMS-387032 against primary chronic lymphocytic leukemia cells showed that all of the seven observed CDKs were inhibited, with CDK2, CDK9, CRK7, and PCSTAIRE2 displaying submicromolar IC50 values (Figure 3).17 BMS-387032 retained the ability to inhibit the majority of the observed trypanosome CDKs, including targeting the CDK2-related kinases CRK2 and CRK3 with submicromolar potency (IC50 of 148 and 57 nM, respectively), although no inhibition of CRK1 was observed (Figure 3). In addition, the compound selectivity was slightly broader than just the CDKs, with two additional kinases (CMGC and CAMK) also inhibited with IC50 values below 500 nM, which may reflect the divergence of the trypanosome kinome. These data provide the first molecular evidence that the trypanosome kinome is sensitive to typical mammalian kinases inhibitors with nanomolar potencies and suggest that other standard kinase inhibitor scaffolds may retain substantial activity.
Profiling Trypanosome Kinase Inhibitors
The Drug Discovery Unit at the University of Dundee has conducted a number of screening campaigns to help identify preclinical candidates for the treatment of African sleeping sickness, including target-based screens against the T. brucei protein kinases Glycogen Synthase Kinase 3 (GSK3, Tb427.10.13780) and the Nuclear DBF-2-related (NDR) kinases PK50 (Tb427.10.4940) and PK53 (Tb427.07.5770).8 Details of the compound screening and optimization will be reported elsewhere. To further probe the chemical space that mammalian and trypanosome kinase inhibitors occupy and demonstrate the utility of chemical proteomics in antiparasitic drug discovery, we profiled hits selected from these screens (Supplementary Tables S6 and S7). The compounds selected all show nanomolar potency against their respective molecular target in vitro and variable efficacy against cultured trypanosome and human hepatocyte (MRC5) cells (Table 1).
Table 1. Trypanosome Kinase Inhibitors.
| GSK3 | PK50 | PK53 | |
|---|---|---|---|
| compound ID | DDD85893 | DDD34425 | DDD88213 |
| T. brucei enzyme IC50 (μM)a | <0.002 | 0.013 ± 0.006 | 0.73 ± 0.14 |
| T. brucei kinobead IC50 (μM)b | <0.039 | not observed | 5.7 |
| T. brucei EC50 (μM)c | 1.3 ± 1.2 | 0.86 ± 0.52 | 45 ± 4 |
| H. sapiens MRC5 EC50 (μM)c | 28 ± 9.7 | >50 | >50 |
| H. sapiens kinobead IC50 (μM)b | <0.06 | not observed | not observed |
The compound DDD85893 was identified as a potent inhibitor of T. brucei GSK3 (TbGSK3) in vitro, with good efficacy against cultured T. brucei and good selectivity against cultured human cells. The kinobead profiling of DDD85893 against T. brucei cell lysates confirmed that TbGSK3 was inhibited with nanomolar potency, with three other CMGC kinases inhibited at micromolar level (Figure 4). The compound also showed a very clean profile against human MRC5 cell lysates, with only human GSK3α, GSK3β, and CDK9 inhibited with nanomolar potency. These data show that the compound DDD85893 has excellent selectivity for GSK3 and limited other CMGC members but does not display any species specificity.
Figure 4.

Chemical proteomics profiling of trypanosome kinase inhibitors against the kinome of bloodstream form T. brucei. Horizontal bars represent IC50 value calculated from the isobaric reporter signals; gray bars indicate where binding was not quantified. Lysates were incubated with varying concentrations of compounds prior to incubation with kinobeads, and the bound fraction was quantified by tandem mass spectrometry. Data for TK, TKL, atypical, and lipid kinase are not shown for clarity; the full data can be found in Supplementary Tables S6 and S7.
The second compound to be profiled (DDD34425) was identified as a potent inhibitor of T. brucei PK50 in vitro, with good efficacy against cultured T. brucei and good selectivity against cultured human cells. Unfortunately, the expected target PK50 was not among the kinases that bound to the kinobeads, suggesting that the ATP binding site contains features that are not recognized by the set of standard ligands immobilized on the beads. Indeed, none of the four human NDR kinases (NDR1, NDR2, LATS1, LATS2) were among the kinases in MRC5 lysates that bound to the kinobeads. However, the kinobead profile of DDD34425 against T. brucei cell lysates revealed that MAPK10 and MAPK5 were inhibited with nanomolar potency, with three other kinases inhibited at micromolar level (Figure 4). As T. brucei bloodstream form MAPK5 null mutants grow normally in vitro(18) and little is known about MAPK10,19 it is unclear what effect, if any, their inhibition may contribute to the observed trypanocidal effect of DDD34425. The kinobead profile against MRC5 cell lysates showed a broader specificity, with the MAPK NKL and TKLs RIPK2 and ALK2 inhibited with nanomolar potency. Six additional kinases from four different groups were inhibited at the micromolar level. These data revealed that the compound DDD34425 lacks specificity against the observable trypanosome kinome and significant poly pharmacology against the observable human kinome, including a number of tyrosine-specific kinases that are absent from T. brucei.
The compound DDD88213 was identified as a submicromolar inhibitor of T. brucei PK53 in vitro but lacked efficacy against both cultured T. brucei and human cells. The kinobeads profile of DDD88213 against T. brucei cell lysates revealed that PK53 was inhibited with an IC50 of 5.7 μM, with CK2α2 also inhibited with similar potency (Figure 4). The 10-fold drop in potency between the enzyme IC50 and the kinobeads binding assay is not significantly different from that seen with other inhibitors and may reflect differences between purified proteins and cell extracts. Moreover, the potency determined with kinobeads is in line with the weak activity of the compound in the Alamar blue assay. The kinobead profile against MRC5 cell lysates showed that none of the observable human kinome was significantly inhibited by DDD88213. These data show that while this compound does appear to be specifically targeting the desired trypanosome kinase and appears to have little effect on the human kinome, it lacks sufficient potency to achieve the desired trypanocidal effect and would require further optimization.
The final compound to be profiled was recently identified as a potent inhibitor of the Leishmania major cyclin dependent kinase 2-related kinase 3 (LmCRK3) (compound 33, Cleghorn et al.(20)) but lacked efficacy against cultured L. major or the related parasite T. brucei.20 As active recombinant T. brucei CRK3 (TbCRK3) is not available for in vitro screening, we attempted to use kinobead profiling to investigate whether the lack of cellular potency against T. brucei was due to lack of potency against TbCRK3. The kinobead profile of the compound revealed that none of the observed T. brucei kinases were significantly inhibited, including TbCRK3, and neither were any mammalian kinase (Supplementary Tables S6 and S7). The complete lack of inhibition suggests that the native state of parasite CRK3s is distinct from the recombinant form used for in vitro screening. While these differences may be due to the absence of the associated cyclin, a recent screen against LmCRK3-cyclin 6 also resulted in compounds that lacked efficacy against cultured L. major.21
Summary and Conclusions
We set out to examine the similarity of the chemical space that parasite and mammalian protein kinase inhibitors occupy using a recently developed chemical proteomics approach to profile kinase inhibitors. The data presented here represent the first molecular evidence that typical ATP-competitive inhibitors can retain low nanomolar potency against T. brucei protein kinases. The inhibition profile of the compounds does not seem to map directly between the two species, suggesting that it may be possible to exploit these differences to obtain host/parasite specificity. It is also possible that the differences observed are an artifact due to the limited coverage of the kinome achieved by kinobead enrichment. However, the lack of bias in the enriched kinome suggested that this is unlikely.
Our data suggest that phenotypic screening of known kinase inhibitors against T. brucei is likely to identify potent compounds but also show that it is inappropriate to infer the molecular target on the basis of the inhibition profile established in mammalian systems. The chemoproteomics approach presented to profile potential kinase inhibitors simultaneously covers as much as half the observed bloodstream form kinome, representing a 10-fold increase in the selection of active parasite kinases currently available for drug discovery. Development of kinobeads tailored to the trypanosome kinome, for instance, by immobilizing novel inhibitors identified through phenotypic screening, is an attractive approach to extend the coverage of the parasite kinome. Importantly, the approach presented here is species-independent and can be applied to any clinically relevant pathogen for which a genome sequence is available.
Methods
Reagents and Drugs
All reagents were purchased from Sigma unless otherwise noted. Staurosporine was purchased from IRIS Biotech, and BMS-387032 was custom synthesized by Park Place Research.
Preparation of Bloodstream Form T. brucei Cell Lysates
Bloodstream form T. brucei brucei variant 117 (MITat1.4) was purified from infected rodent blood over DE52 cellulose as described previously.22 The cells were centrifuged at 800 × g for 10 min at 4 °C and resuspended at 1 × 109 cells/mL in ice-cold buffer 1 containing protease and phosphatase inhibitors (0.1 mM TLCK, 1 μg/mL Leupeptin, 1 μg/mL aprotinin, 1 mM PMSF, 1 mM benzamidine, Phosphatase Inhibitor Cocktail II (Roche)), and hypotonic lysis was allowed to proceed for 10 min on ice. An equal volume of ice-cold buffer 2 (100 mM Tris pH 7.5, 10% glycerol, 300 mM NaCl, 50 mM NaF, 3 mM MgCl2, 0.2 mM Na3VO4, 1.6% Igepal-CA630, 2 mM DTT, 0.1 mM TLCK) was added, and the lysate was centrifuged at 145,000 × g for 1 h at 4 °C. The BCA assay (Pierce) was used to determine the total protein content in the supernatant, and the concentration was adjusted to 5 mg/mL. Aliquots were frozen in liquid nitrogen and stored at −80 °C prior to use.
Preparation of H. sapiens MRC5 Cell Lysates
MRC5 cells were grown at 37 °C, 5% CO2 in MEM supplemented with 10% FCS. Cell lysates were prepared washing the cells briefly in PBS, incubating with an equal volume of buffers 1 and 2 for 15 min, and then processing the crude lysate as described above.
Proteomic Analysis
The total proteome was determined in duplicate by fractionating 25 μg of the bloodstream form T. brucei cell lysate by SDS-PAGE and pixilation into 24 bands, followed by in-gel reductive alkylation and tryptic digest. Samples were analyzed by liquid chromatography – tandem mass spectrometry on a Eksigent 1D+ HPLC system coupled to a LTQ-Orbitrap mass spectrometer (Thermo scientific). MS spectra were searched using Mascot (Matrix Science) against a nonredundant, in-house compiled database of Trypanosoma brucei 927 and 427 strains obtained from TriTrypDB 3.0 (23) with additional protein sequences from SwissProt and RefSeq databases, as well as known contaminant sequences such as keratins and trypsin. To assess the false discovery rate (FDR) “decoy” proteins (reverse of the protein sequence) were added to the database. Protein identifications were accepted as follows: (i) For single spectrum to sequence assignments, we required the assignment to be the best match and a minimum Mascot score of 37 and a 10× difference of the assignment over the next best assignment. On the basis of these criteria, the decoy search results indicated <1% false discovery rate (FDR). (ii) For multiple spectrum to sequence assignments and using the same parameters, the decoy search results indicate <0.1% FDR. To make our data accessible to the scientific community, we have uploaded the results of this study to TriTrypDB (http://www.tritrypdb.org).23
Kinobead Profiling
Procedures are essentially as described previously.11,17,24 Kinobeads were prepared by immobilization of ATP-mimetics on sepharose beads, with the four versions differing in the identity of the immobilized kinase ligands, as described in Supplementary Table S8.
For kinobead profiling, compounds were dissolved in DMSO, added at various concentrations (0, 0.039, 0.156, 0.625, 2.5, and 10 μM) to 1-mL cell lysate samples, and incubated for 45 min at 4 °C. Subsequently, kinobeads were added to each sample and incubated for a further 60 min at 4 °C. The kinobeads were collected by centrifugation and washed with lysis buffer containing 0.2% Igepal-CA630, and bead-bound proteins were eluted with NuPAGE LDS buffer (Invitrogen) containing 50 mM DTT for 30 min at 50 °C followed by alkylation with 20 mg/mL iodoacetamide for 30 min. Samples were purified on 4–12% NuPAGE gels, stained with colloidal Coomassie blue, digested with trypsin, and subsequently labeled with TMT isobaric tagging reagents (ThermoFisher Scientific).15 Tryptic peptides were separated over 4 h using nanoflow reversed-phase chromatography online coupled to an Orbitrap mass spectrometer. Peptide fragmentation was performed using PQD, and peptides were identified with Mascot and quantified as described.25
Identification of the T. brucei 427 Strain Kinome
The annotated proteins from T. brucei brucei 427 strain was obtained from TriTrypDB 3.023 and scanned through a highly sensitive and specific multilevel HMM library of the protein kinase superfamily,26 followed by expert curation. Assignment of putative protein kinases to the main ePK and aPK groups was done by using the E-value cutoffs specific for each group as described previously.26,27 This procedure identified and assigned 187 protein kinases (Supplementary Table S1).
Phylogenetic Analysis
The phylogeny of the kinase groups identified in T. brucei 427 strain was determined using the Phylogeny.fr platform28 and comprised the following steps: Sequences were aligned with T-Coffee (v6.85) using pairwise alignment methods,29 and ambiguous regions (i.e., containing gaps and/or poorly aligned) were removed with Gblocks (v0.91b)30 with low stringency (Min. seq. for flank pos.: 55%, Max. contig. nonconserved pos.: 8, Min. block length: 5, Gaps in final blocks: half). The phylogenetic tree was reconstructed using the maximum likelihood method implemented in the PhyML program (v3.0 aLRT)31 with reliability for internal branch assessed using the aLRT test (minimum of SH-like and Chi2-based parametric).32 Graphical representation and editing of the phylogenetic tree was performed with TreeDyn (v198.3).33
Acknowledgments
This work was funded by the Welcome Trust (Grants 085622 and 077705, and Strategic award 083481). We thank F. Simeons, L. Stojanovski, and K. Read of the University of Dundee Drug Discovery Unit for assistance in the culture of T. brucei in rodents and M. Boesche (Cellzome) for performing mass spectrometry.
Supporting Information Available
This material is free via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): The authors T.M., D.E., M.B., and G.D. are employees of Cellzome AG, which contributed to the funding of this work by payment-in-kind.
Supplementary Material
References
- Simarro P.; Diarra A.; Ruiz Postigo J.; Franco J.; Jannin J. (2011) The Human African Trypanosomiasis control and Surveillance Programme of the WHO 2000–2009: The Way Forward. PLoS Negl. Trop. Dis. 5, e1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frearson J. A.; Wyatt P. G.; Gilbert I. H.; Fairlamb A. H. (2007) Target assessment for antiparasitic drug discovery. Trends Parasitol. 23, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naula C.; Parsons M.; Mottram J. C. (2005) Protein kinases as drug targets in Trypanosomes and Leishmania. Biochem. Biophys. Acta 1754, 151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons M.; Worthey E. A.; Ward P. N.; Mottram J. C. (2005) Comparative analysis of the kinomes of three pathogenic trypanosomatids: Leishmania major, Trypanosoma brucei and Trypanosoma cruzi. BMC Genomics 6, 127–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nett I. R. E.; Martin D. M. A.; Miranda-Saavedra D.; Lamont D.; Barber J. D.; Mehlert A.; Ferguson M. A. J. (2009) The phopshoproteome of bloodstream form Trypanosoma brucei, causative agent of African sleeping sickness. Mol. Cell. Proteomics 8, 1527–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsford S.; Turner D. J.; Obado S. O.; Sanchez-Flores A.; Glover L.; Berriman M.; Hertz-Fowler C.; Horn D. (2011) High-Throughput phenotyping using parallel sequencing of RNA interference targets in the African Trypanosome. Genome Res. 21, 915–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies S. P.; Reddy H.; Caivano M.; Cohen P. (2000) Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 351, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J.; Benz C.; Grimaldi R.; Stockdale C.; Wyatt P.; Frearson J.; Hammarton T. C. (2010) Nuclear DBF-2-related kinases are essential regulators of cytokinesis in bloodstream stage Trypanosoma brucei. J. Biol. Chem. 285, 15356–15368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbaniak M. D. (2009) Casein kinase 1 isoform 2 is essential for bloodstream form Trypanosoma brucei. Mol. Biochem. Parasitol. 166, 183–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojo K. K.; Gillespie J. R.; Reichers A. J.; Napuli A. J.; Verlinde C. L.; Buckner F. S.; Gelb M. H.; Domostoj M. M.; Wells S. J.; Scheer A.; Wells T. N.; Van Voorhis W. C. (2008) Glycogen synthase kinase 3 is a potential drug target for African trypanosomiasis therapy. Antimicrob. Agents Chemother. 52, 3710–3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bantscheff M.; Eberhard D.; Abraham Y.; Bastuck S.; Boesche M.; Hobson S.; Mathieson T.; Perrin J.; Raida M.; Rau C.; Reader V.; Sweetman G.; Bauer A.; Bouwmeester T.; Hopf C.; Kruse U.; Neubauer G.; Ramsden N.; Rick J.; Kuster B.; Drewes G. (2007) Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat. Biotechnol. 25, 1035–1044. [DOI] [PubMed] [Google Scholar]
- Bantscheff M.; Drewes G. (2012) Chemoproteomic approaches to drug target identification and drug profiling. Bioorg. Med. Chem. 20, 1973–1978. [DOI] [PubMed] [Google Scholar]
- Urbaniak M. D.; Guther M. L. S.; Ferguson M. A. J. (2012) Comparative SILAC proteomic analysis of Trypanosoma brucei bloodstream and procyclic lifecycle stages. PLoS One 7, e36619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen B. C.; Sivam D.; Kifer C. T.; Myler P. J.; Parsons M. (2009) Widespread variation in transcript abundance within and across developmental stages of Trypanosoma brucei. BMC Genomics 10, 482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson A.; Schafer J.; Kuhn K.; Kienle S.; Schwarz J.; Schmidt G.; Naeumann T.; Johnstone R.; Mohammed A. K.; Hamon C. (2003) Tandem mass tags: a novel quantification stratergy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 75, 1895–1904. [DOI] [PubMed] [Google Scholar]
- Ross P.; Huang Y.; Marchese J.; Williamson B.; Parker K.; Hattan S.; Khainovski N.; Pillai S.; Dey S.; Daniels S.; Purkayastha S.; Junhasz P.; Martin S.; Bartlet-Jones M.; He F.; Jacobson A.; Pappin D. (2004) Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics 3, 1154–1169. [DOI] [PubMed] [Google Scholar]
- Kruse U.; Pallasch C. P.; Bantscheff M.; Eberhard D.; Frenzel L.; Ghidelli S.; Maier S. K.; Werner T.; Wendtner C. M. (2011) Chemoproteomics-based kinome profling and target deconvolution of clinical multi-kinase inhibitors in primary lymphocytic leukemia cells. Leukemia 25, 89–100. [DOI] [PubMed] [Google Scholar]
- Pfister D. D.; Burklard G.; Morand S.; Renggli C. K.; Roditi I.; Vassella E. (2006) A mitogen-activated protein kinase controls differentiation of bloodstream forms of Trypanosoma brucei. Eukaryotic Cell 5, 1126–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotureau B.; Morales M. A.; Bastin P.; Spath G. F. (2009) The flagellum-mitogen-activated protein kinase connection in Trypanosomatids: a key sensory role in parasite signalling and development?. Cell. Microbiol. 11, 710–718. [DOI] [PubMed] [Google Scholar]
- Cleghorn L. A.; Woodland A.; Collie I. T.; Torrie L. S.; Norcross N.; Luksch T.; Mpamhanga C.; Walker R. G.; Mottram J. C.; Brenk R.; Frearson J. A.; Gilbert I. H.; Wyatt P. G. (2011) Identification of inhibitors of the Leishmania cdc2-related protein kinase CRK3. ChemMedChem 6, 2214–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker R. G.; Thomson G.; Malone K.; Nowicki M. W.; Brown E.; Blake D. G.; Turner N. J.; Walkinshaw M. D.; Grant K. M.; Mottram J. C. (2011) High throughput screening yield small molecule inhibitors of Leishmania CRK3:CYC6 cyclin-dependant kinase. PLoS Negl. Trop. Dis. 5, e1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross G. A. M. (1984) Release and purification of Trypanosoma brucei variant surface glycoprotein. J. Cell. Biochem. 24, 79–90. [DOI] [PubMed] [Google Scholar]
- Aslett M.; Aurrecoechea C.; Berriman M.; Brestelli J.; Brunk B. P.; Carrington M.; Depledge D. P.; Fischer S.; Gajria B.; Gao X.; Gardner M. J.; Gingle A.; Grant G.; Harb O. S.; Heiges M.; Hertz-Fowler C.; Houston R.; Innamorato F.; Iodice J.; Kissinger J. C.; Kraemer E.; Li W.; Logan F. J.; Miller J. A.; Mitra S.; Myler P. J.; Nayak V.; Pennington C.; Phan I.; Pinney D. F.; Ramasamy G.; Rogers M. B.; Roos D. S.; Ross C.; Sivam D.; Smith D. F.; Srinivasamoorthy G.; Stoeckert C. J. Jr.; Subramanian S.; Thibodeau R.; Tivey A.; Treatman C.; Velarde G.; Wang H. (2010) TriTrypDB: a functional genomic resource for the Trypanosomatidae. Nucleic Acid Res. 38, D457–D462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergamini G.; Bell K.; Shimamura S.; Werner T.; Cansfield A.; Mueller K.; Perrin J.; Rau C.; Ellard K.; Hopf C.; Doce C.; Leggate D.; Mangano R.; Mathieson T.; O’Mahony A.; Plavec I.; Rharbaoui F.; Reinhard F.; Savitski M. M.; Ramsden N.; Hirsch E.; Drewes G.; Rausch O.; Bantscheff M.; Neubauer G. (2012) A selective inhibitor reveals PI3Kγ dependence of TH17 cell differentiation. Nat. Chem. Biol. 8, 576–582. [DOI] [PubMed] [Google Scholar]
- Bantscheff M.; Boesche M.; Eberhard D.; Matthieson T.; Sweetman G.; B. K. (2008) Robust and sensitive iTRAQ quantification on an LTQ Orbitrap mass spectrometer. Mol. Cell. Proteomics 7, 1702–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda-Saavedra D.; Barton G. J. (2007) Classification and functional annotation of eukaryotic protein kinases. Proteins 68, 893–914. [DOI] [PubMed] [Google Scholar]
- Martin D. M.; Miranda-Saavedra D.; Barton G. J. (2009) Kinomer v.1.0: a database of systematically classified eukaryotic protein kinases. Nucleic Acid Res. 37, D244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dereeper A.; Guignon V.; Blanc G.; Audic S.; Buffet S.; Chevenet F.; Dufayard J. F.; Guindon S.; Lefort V.; Lescot M.; Claverie J. M.; Gascuel O. (2008) Phylogeny.fr: robust phylogentic analysis for the non-specialist. Nucleic Acid Res. 36, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notredame C.; Higgins D.; Heringa J. (2000) T-Coffee: A novel method for multiple sequence alignment. J. Mol. Biol. 302, 205–217. [DOI] [PubMed] [Google Scholar]
- Castresana J. (2000) Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. [DOI] [PubMed] [Google Scholar]
- Guindon S.; Gascuel O. (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704. [DOI] [PubMed] [Google Scholar]
- Anisimova M.; Gascuel O. (2006) Approximate likelihood ration test for branches: a fast, accurate and powerful alternative. Syst. Biol. 55, 539–552. [DOI] [PubMed] [Google Scholar]
- Chevenet F.; Brun C.; Banuls A. L.; Jacq B.; Chisten R. (2006) TreeDyn: towards dynamic graphic and annotation analyses of trees. BMC Bioinfomatics 7, 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant J. D.; Urbaniak M. D.; Ferguson M. A. J.; McCammon J. A. (2010) Computer-aided identification of Trypanosoma brucei uridine diphosphatase galactose 4′-epimerase inhibitors: towards the development of novel therapies for African sleeping sickness. J. Med. Chem. 53, 5025–5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.