

Abstract
Metastasis-associated protein 1 (MTA1), a master chromatin modifier, has been shown to regulate cancer progression and is widely upregulated in human cancer including, hepatitis B virus-associated hepatocellular carcinomas (HCC). Here we provide evidence that hepatitis B virus transactivator protein HBx stimulates the expression of MTA1 but not MTA2 or MTA3. The underlying mechanism of HBx stimulation of MTA1 involves HBx targeting of transcription factor NF-κB and the recruitment of HBx/p65 complex to the NF-κB -consensus motif on the relaxed MTA1 gene chromatin. We also discovered that MTA1 depletion in HBx-expressing cells severely impairs the ability of HBx to stimulate NF-κB signaling and the expression of target pro-inflammatory molecules. Furthermore the presence of HBx in HBx-infected hepatocellular carcinomas correlated well with increased MTA1 and NF-κB-p65. Collectively, these findings revealed a previously unrecognized integral role of MTA1 in HBx stimulation of NF-κB signaling and consequently, the expression of NF-κB targets gene products with functions in inflammation and tumorigenesis.
Keywords: MTA1, hepatitis B virus, HBx, signaling, MTA coregulator, liver cancer
INTRODUCTION
Hepatocellular carcinoma (HCC) is the most common form of liver cancer in adult that accounted for 1 million deaths every year worldwide (Dvorchik et al., 2007). Of those, high morbidity and mortality rate are found in Asia and Africa. In the United States, according to the American Cancer Society, there will be 22,000 new cases of HCC, up to 19,000 of them fatal, during the year of 2008. Over the years, a large number of epidemiological studies suggest that more than 75% of HCC cases are associated with chronic hepatitis B (HBV) and/or C viruses (HVC). The infection of HBV increases the risk of a wide spectrum of clinical manifestations ranging from self-limited acute or fulminant hepatitis, asymptomatic infection, or chronic hepatitis with progression to liver cirrhosis that can lead to hepatocellular carcinoma (HCC). Indeed, elevated serum HBV DNA level is a strong risk predictor of hepatocellular carcinoma (Chen et al., 2006; Lupberger & Hildt, 2007). The circular virion genome of HBV has four overlapping reading frames that are responsible for the generation of seven different hepatitis B proteins. Being encoded by the X gene, the smallest reading frame, HBx has been implicated in the regulation and transactivation of a variety of cellular genes in hepatocytes. HBx targets a gamut of cytoplasmic and nucleus gene products with diverse functions including modulating NF-κB signaling, regulating cell survival, and producing inflammatory cytokines, contributing to HBV infection, replication, pathogenesis, and putatively carcinogenesis (Bouchard & Schneider, 2004).
Among the Rel/NF-κB family of proteins, the RelA (p65) and NF-κB1 (p50; p105) play crucial roles in immune response, cell survival, and cellular transformation (Li & Verma, 2002; Pahl, 1999). While a notable body of literature suggest that HBx interacts with all major forms of NF-κB (Zhang et al., 2006) and activates target promoters bearing NF-κB, cAMP-response element (CRE), and AP-1 consensus motifs (Bergametti et al., 1999; Chirillo et al., 1996; Hoffmann & Baltimore, 2006); induces the degradation of p105 NF-κB1 and IκBα (Su & Schneider, 1996; Zhang et al., 2006); or sequesters IκBα to sustain NF-κB activation (Robert Weil Direct Association and nuclear import of the HBx protein with the NF-KB inhibitor)(Lucito & Schneider, 1992; Yun et al., 2002) where its activity is enhance by VHL binding protein (VBP1) in a cooperative manner (Kim et al., 2008); the mechanism for HBx activation of NF-κB remains undefined.
In addition to HBV, the process of liver carcinogenesis has been also shown to be intimately linked with the upregulation of metastasis-associated protein 1 (MTA1), a component of nuclear remodeling histone deacetylate (NURD) complex (Yoo et al., 2008). The MTA family has emerged not only as a crucial modifier of chromatin remodeling but also a potential prognostic indicator after hepatectomy for HCC (Hamatsu et al., 2003; Manavathi & Kumar, 2007). MTA1 overexpression has been shown to associate with carcinogenesis and angiogenesis in animal tumor model systems and in human cohort studies (Manavathi et al., 2007b). MTA1 overexpression has been shown to be consistently correlated with a higher tumor grade and increased tumor angiogenesis in gastrointestinal carcinoma tumors (Kidd et al., 2006) and in colorectal carcinoma as compared to the matched normal tissues (Giannini & Cavallini, 2005). In postoperative HCC patients, MTA1 expression is higher in HBV-associated HCC than that of HCV-associated HCC; while the median survival-rate of the MTA1-positive HCC patients has been shown to significantly shorter than those with MTA1-negative HCCs (Ryu et al., 2008). Although these studies suggest a strong positive correlation between the HBx and MTA1 in HCC (Yoo et al., 2008), the role of MTA1 in HBx regulation of cellular pathways remains poorly understood. Here we elucidated the molecular basis of MTA1 upregulation in HBx-transfected cells and unexpectedly discovered a mandatory role of MTA1 in HBx regulation of NF-κB signaling and consequently, the expression of NF-κB target genes which collectively play a vital role in HBV-mediated hepatocarcinogenesis.
MATERIALS AND METHODS
Materials
Antibodies against IκBα (sc-371), NF-κB p65 (sc-372), phospho-NF-κB p65 (sc-33020), and NF-κB p65 (286-H) X (sc-7151 X) were from Santa Cruz Biotechnology (Santa Cruz, CA). Cox2 antibody (4842) was from Cell Signaling (Beverly, MA). NF-κB p65 was from Abcam (Cambridge, MA). Flag-M2 monoclonal antibody (F3165), β-actin and vinculin monoclonal antibodies, normal mouse IgG, rabbit IgG were from Sigma Chemical Co. (St. Louis, MO). MTA1 polyclonal antibody (1805) was from Bethyl Laboratories (Montgomery, TX). HBx antibody (MAB8419) was from Millipore/Chemicon (Danver, MA). Rabbit polyclonal anti-HBx was used as previously described (Lee, Elledge, and Butel, 1995). Plasmids pSI-HBx and pSI-GFP plasmids were cloned as described (Keasler, et al, 2007). pCMVXF and pCMV-Core are kind gifts from Dr. A. Siddiqui, University of Colorado Health Sciences, Denver, Colorado. pSG5-HBx full length and pSG5-HBx mutant constructs are from Dr. V. Kumar, International Centre for Genetic Engineering and Biotechnology, India. Cells used were obtained from the American Type Culture Collection. HepG2-X is a gift from Dr. Mien-Chie Hung, MD Anderson Cancer Center. All cell types were cultured as described previously or otherwise stated (Bergametti et al., 1999; Manavathi et al., 2007a).
Cells and tissue
HepG2 liver cells, human embryonic kidney 293 cells, NIH-3T3 cells were obtained from the American Type Culture Collection. HepG2X is a gift from Dr. Mien-Chie Hung, Molecular & Cellular Oncology, M.D. Anderson Cancer Center. All cell types were cultured as described previously or otherwise stated (Bergametti et al., 1999; Manavathi et al., 2007a). Hhuman HCC and nontumor tissues were obtained from the Qidong Liver Cancer Institute in Shanhair, People’s Republic of China, as described in Slagle et al (1991)-see Canc. Res. 61:49–54, 1991. There was evidence of HBV infection (circulating antibody to HBV surface antigen, antibody to HBV core antigen, or HBV DNA integrated into tumor DNA) for all patients. Samples were collected according to institutional IRB approvals. Tissue microarray sections (LVC961) was obtained from Pantomics Inc. (Richmond, CA).
Quantiative RT-PCR
Total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA) and first-strand cDNA synthesis was carried out with SuperScript II reverse transcriptase (Invitrogen) using 2 μg of total RNA and poly (dT) primer.
ChIP and reporter assays
HepG2 cells were transiently transfected with HBx expression vector prior to ChIP analysis and reporter assays. Chromatin immunoprecipitation assays were done by using Flag antibody, NF-κB, MTA1, and H4 Acetylation antibodies as described previously(Gururaj et al., 2006; Manavathi et al., 2007a). The primers used for ChIP are listed in Supplementary Tables I. MTA1-luciferase and NF-κB-luciferase assay were performed according to the manufacturer’s instructions (Promega, Madison, WI).
siRNA transfection
siRNA against MTA1 and negative control siRNA were purchased from Dharmacon (Chicago, IL). Cells were seeded at 40% density 24 hours prior to transfection in 6-well plates. Transfection was performed using Oligofectamine (Invitrogen) according to the manufacturer’s instructions. Cells were subsequently subjected to further studies after 36 hours of transfection. Western blot analyses were performed as described previously (Manavathi et al., 2007a).
EMSA
Nuclear extracts were prepared using a Nonidet P-40 lysis method(Schreiber et al., 1989). EMSA for DNA binding was performed using the annealed and [γ-32P] ATP end-labeled NF-κB Consensus Oligonucleotide (Promega) or MTA1 in a 20μl reaction mixture for 15 min at 20°C. The reactions were then terminated and run on a nondenaturing 5% polyacrylamide gel and imaged by autoradiography. Supershift complex was detected by the addition of 1 μg of antibodies
Immunohistochemical Analysis
The immuno-peroxidase staining method used in these studies was a modification of the avidin-biotin complex technique as described previously. The modifications from the standard method were incorporated to ensure high sensitivity and specificity. Tissue microarray sections (LVC961, Pantomics Inc., Richmond, CA) were deparaffinized, dehydrated, and subjected to antigen retrieval using PT Module (Thermo Fisher Scientific, Fremont, CA) for 30 min at room temperature. The endogenous peroxidase activity was blocked by incubation in 0.3% hydrogen peroxide for 10 min and the slides were then treated with 10% normal horse or goat serum for 30 minutes. Incubation with primary antibodies was performed at 4°C overnight. Following washes with PBS, the slides were incubated with biotinylated secondary antibodies and incubated with avidin-horseradish peroxidase complex (Vector Laboratories, Burlingame, CA). Detection was performed with the 0.125% aminoethylcarbazole chromogen substrate solution (AEC, Sigma). In the study for the correlation of NFκB expression, a polyclonal antibody (Abcam Inc.) was used to detect NFκB in 25 hepatitis cancer (HCC) tumor specimens and 14 non-HCC cases. Concentrations of the antibodies used were as follows: NFκB, 0.200 mg/mL (diluted 1:800), a polyclonal antibody of p-NFκB p65 (Ser 536, SC-33020) from Santa Cruz Biotechnology, Inc. 0.2 mg/mL (diluted 1:100), a polyclonal antibody of MTA1, (IHC-00026) (diluted 1: 200, from Bethyl Laboratories, Inc.) and monoclonal antibody of HBx, 1mg/ml (diluted 1: 50, from Chemicon International a Serologicals company).
Imaging Analysis
To ensure absolute objectivity of the immunohistochemical studies, these experiments used the ACIS III Automated Cellular Imaging System (from DAKO company) to analyze tissues scoring and quantification (nuclear or membrane and cytoplasm applications based on percent and intensity). The percentage of positive tumor cells was used for statistical analysis.
Statistical analysis and reproducibility
The results are given as the mean ± standard error. Statistical analysis of the data was performed by using Student’s t-test, Fisher’s Exact test, or otherwise described.
RESULTS
HBx induces MTA1 expression at the transcription level
To investigate the potential mechanistic basis between the HBx and MTA1, we first tested the effect of increasing amount of HBx upon the levels of MTA1 expression in a human hepatocarcinoma cell line HepG2. We found that HBx expression in HepG2 is accompanied by increased expression of MTA1 (Figure 1A) but not MTA2 or MTA3 (Figure 1B). Similarly, we also noticed a substantial upregulation of MTA1 in HepG2X, a HBx-stable transfectant of HepG2 as compared to vector transfected cells (Ding et al., 2005) or in non-hepatic cell line HEK 293 (Figure 1C). Since HCC has been also linked with hepatitis C virus (HCV)(Ray & Ray, 2001), we examined whether HCV-Core protein also influence MTA1 expression. However, we found only albeit effect of HCV overexpression on the level of MTA1 in HepG2 cells (Figure 1D) suggesting that MTA1 is preferably induced by HBx and not by HCV core protein.
Figure 1.
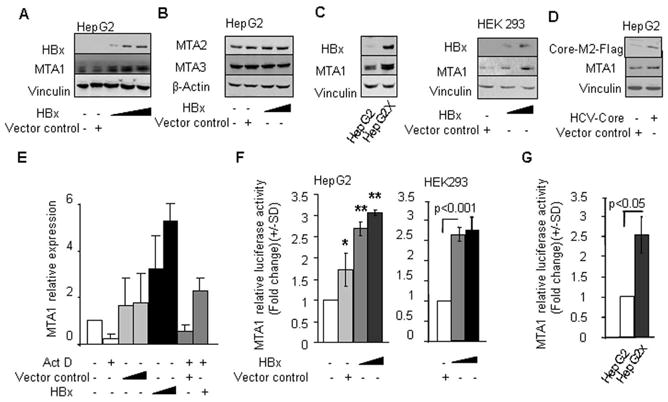
HBx induces MTA1 expression at the transcriptional level. (A) Western blot analysis of HBx and MTA1 in HepG2 cells transfected with either control vector (250 ng/reaction) or HBx expression vector (50, 100, and 250 ng/reaction). (B) Western blot analysis of MTA2 and MTA3 proteins in HepG2 cells transfected with control vector (250 ng/reaction) or HBx expression vector (50 and 250 ng/reaction). (C) Western blot analysis of MTA1 protein in HepG2X, a HBx-stable transfectant of HepG2, and HEK 293 cells transfected with either control vector (250 ng/reaction) or HBx (50 and 250 ng/reaction). (D) Western blot analysis of MTA1 protein in HepG2 cells transfected with either control vector or HCV-core (250 ng/reaction). (E) q-PCR analysis of MTA1 in HepG2 cells treated with actinomycin D (Act D) (5 μg/ml) after being transfected with either control vector or HBx (50–200 ng/reaction). Expression levels of MTA1 were normalized with β-Actin. F, MTA1 promoter activity in HepG2 cells and HEK 293 cells transfected with either control vector (250 ng/reaction) or HBx (100–250 ng/reaction)(*P<0.05,**P<0.001). G, MTA1 promoter activity in HepG2X.
The observed upregulation of MTA1 protein by HBx was at transcription level as HBx-mediated increases the levels of MTA1 mRNA could be effectively blocked by the inclusion of Actinomycin D, a DNA replication inhibitor (Figure 1E). To further validate these findings, we next tested the effect of HBx on a murine MTA1 promoter cloned in a TATA-less-pGL3-luciferase reporter system. We found that HBx expression in HepG2, HEK293 (Figure 1F), and HepG2X cells (Figure 1G) was accompanied by stimulation of the MTA1 transcription. In brief, HBx induces MTA1 expression at the transcription level.
HBx utilizes NF-κB pathway to stimulate MTA1 transcription
The activation of NF-κB and resulting stimulation of the NF-κB transactivation activity confers cell-survival phenotype in HBx-expressing cells (Su & Schneider, 1996; Yun et al., 2002). Recent data from our laboratory suggest that MTA1 promoter contains five NF-κB consensus sites and that p65/RelA directly bind and stimulates MTA1 transcription (Suresh B. Pakala and Rakesh Kumar unpublished findings, manuscript in review elsewhere). We examined the mechanism by which HBx stimulates MTA1 transcription with a particular focus on the NF-κB pathway. To examine the utilization of the NF-κB pathway by HBx to induce MTA1 expression, we showed that HBx-mediated stimulation of the MTA1 promoter activity in HepG2 cells could be effectively blocked by the inclusion of NF-κB-inhibitor parthenolide (Figure 2A). Since HBx uses p65 pathway to stimulate MTA1 transcription (above), there was no additional potentiating effect of co-expressing HBx and p65 on the MTA1 promoter activity (Figure 2B). To define the molecular insights of HBx regulation of MTA1 chromatin, we next determined the region of the MTA1 promoter that is being targeted by HBx by performing a ChIP-based MTA1 promoter-walk in HepG2-transiently expressing HBx. Recruitment of HBx or p65 to all four potential NF-κB consensus sites was analyzed. Flag-tagged antibody- and p65 antibody-based ChIP analysis showed that HBx as well as p65 were recruited onto two regions of the MTA1 promoter (−3814 to −4152 and −2874 to −3207). Since HBx induces the expression of MTA1, we also examined whether acetylated histone H4, a marker of relaxed activated chromatin, is also recruited to the same sites in the MTA1 promoter in the HBx-expressing cells. Indeed, Acetylated H4 antibody-based ChIP analysis again demonstrated that acetylated H4 is recruited to the regions of MTA1 promoter targeted by HBx (Figure 2C). Using sequential ChIPs, we further showed that HBx/p65 complex is recruited to the MTA1 promoter and such recruitment could be substantially reduced by NF-κB-inhibitor parthenolide (Figure 2D). In brief, these observations suggest that HBx/p65 binding to the MTA1 promoter leads to transcriptional stimulation of MTA1 chromatin. Since HBx is not known to bind directly to DNA, this interaction is presumed to be indirect.
Figure 2.
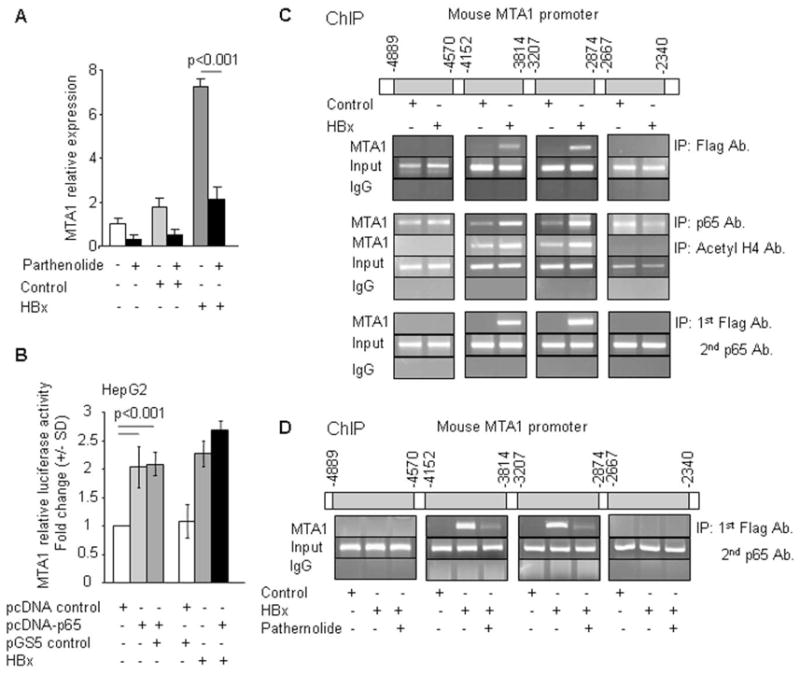
HBx activates MTA1 transcription through NF-κB. (A) qPCR analysis of MTA1 in HepG2 cells treated with parthenolide (5 μM) after being transfected with either control vector or HBx expression vector. Expression levels of MTA1 were normalized with β-Actin. (B) Effect of HBx or NF-κB-p65 on MTA1 promoter activity in HepG2 cells. (C) Recruitment of HBx or NF-κB-p65 or Acetyl H4 to MTA1-chromatin (−3814 to −4152 and −2874 to −3207) by ChIP assay in NIH 3T3 cells using Flag-tagged antibody (Flag Ab.), NF-κB-p65 antibody (p65 Ab.) or Acetyl H4 antibody (Acetyl H4 Ab.) after cotransfecting with pCMV vector control or pCMV-HBx. Row 3: Sequential ChIP assay showing the recruitment of HBx followed by NF-κB-p65 to MTA1-chromatin (−3814 to −4152 and −2874 to −3207). (D) Recruitment of HBx followed by NF-κB-p65 to MTA1-chromatin (−3814 to −4152 and −2874 to −3207) by sequential double ChIP assay in NIH 3T3 cells treated with parthenolide after being transfected with either pCMV vector control or pCMV-HBx.
Essential role of the YFKD motif in HBx regulation of MTA1 transcription
To define the minimal region of HBx required for stimulation of MTA1 transcription, we used a series of well-characterized truncated mutant of HBx. We found that deletions of the N-terminal regions (aa1-84) or the non-conserved region (aa141-154) had marginal effect on the HBx-mediated MTA1 transactivation; while deletions in the region between residue 58 and residue 140 of HBx resulted in significant decrease in the level of HBx-mediated stimulation of MTA1 transcription. Interestingly, there was a good correlation between the effects of HBx-deletion constructs on the MTA1 promoter activity and MTA1 protein expression in HepG2 cells (Figure 3A), providing clue about the biologic significance of the region of HBx encompassing aa58-aa120. Since this region contains the binding motif for p65 (Vijay Kumar, personal communication) and because HBx stimulates MTA1 transcription via NF-κB pathway (this study), we wanted to define the minimal region on HBx that may impact MTA1 expression. We mutated the YFKD motif to FAEN in the full-length HBx protein (designated as mut-HBx) and examined the impact of mut-HBx upon MTA1 promoter activity as well as on MTA1 upregulation. We found that as compared to the wild-type HBx, mut-HBx is unable to efficiently induce the MTA1 promoter activity as well as MTA1 protein in HepG2, NIH3T3, and murine embryonic fibroblasts (MEFs) (Figure 3B).
Figure 3.
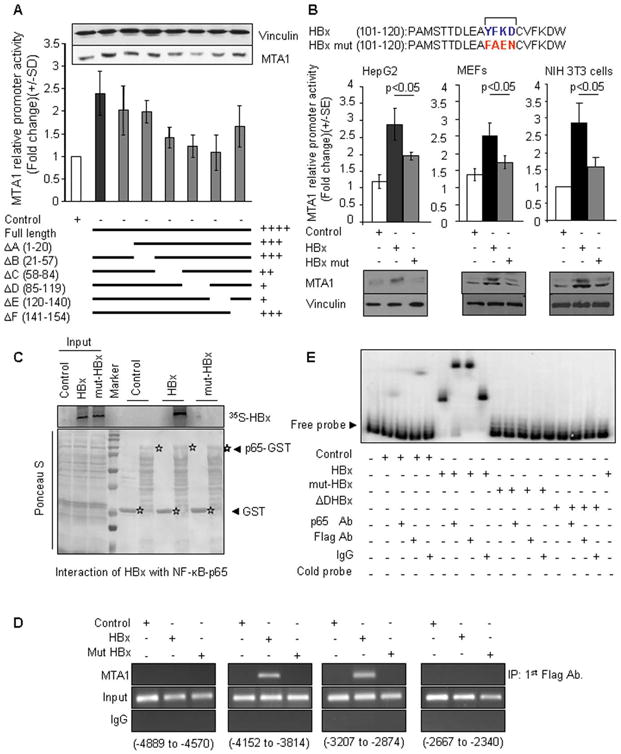
Role of YFKD motif of HBX in regulation of MTA1 transcription. (A) MTA1 promoter activity in HepG2 cells transfected with either control vector or HBx expression vector or HBx-deletion constructs. Inset: Western blot analysis of MTA1 protein in HepG2 cells after transfecting with vector or HBx or HBx- deletion constructs. (B) MTA1 promoter activity in HepG2 or NIH 3T3 or MEF cells after being transfected with either control vector or HBx expression vector or mut-HBx (YFKD was mutated to FAEN) expression vector. Lower panels are Western blot analysis of MTA1 protein in HepG2 or NIH 3T3 or MEF cells after being transfected with either control vector or HBx expression vector or mut-HBx expression vector. (C) GST pulldown assays with 35S-labeled in vitro-translated HBx and mut-HBx and GST- NF-κB-p65 protein. (D) Recruitment of HBx to MTA1-chromatin (−3814 to −4152 and −2874 to −3207) by ChIP assay in the NIH 3T3 cells. (E) EMSA analysis of NF-κB-p65 binding to the mouse MTA1 promoter using PCR product encompassing functional NF-κB consensus sequence in transfected cells and controls. Nuclear extract of HepG2 cell transfected with either Flag-tagged HBx, or Flag-tagged mut-HBx, or Flag-tagged ΔD-deletion HBx (2000 ng/lane), probe control (0.3 ng/lane), NF-κB-p65 antibody (Ab.), anti-Flag antibody, and IgG control (1000 ng/lane), cold probe (15 ng/lane) were used.
While a large body of previous HBx studies have suggested both direct and indirect involvement of NF-κB to HBx transactivation, evidence demonstrating direct interaction between HBx and NF-κB-p65 remains elusive. Hence, we next expressed the wild-type HBx and mut-HBx constructs as 35S-labeled proteins and performed a GST pulldown assay using the GST-NF-κB-p65 fusion protein. Results in Figure 3C showed a direct interaction of the NF-κB-p65 with HBx but not with mut-HBx. These findings also confirmed that the YFKD motif plays an essential role in the noted HBx-NF-κB-p65 interaction. To further validate these results and to establish the significance of the YFKD motif, we next examined the recruitment of the Flag-HBx/p65 complex to the native MTA1 chromatin by a sequential ChIPs involving antibodies against the flag- and p65 in the first and second ChIPs, respectively. We found that the wild-type Flag-HBx but not Flag-mut-HBx was recruited onto the putative binding regions of the MTA1 promoter (−3814 to −4152 and − 2874 to −3207) (Fig. 3D). To establish a direct interact of the HBx protein to the NF-κB consensus motif in the MTA1 promoter DNA and the significance of the YFKD motif, we next performed EMSA. To this end, we subjected the nuclear extracts from the HepG2 cells transfected with HBx or mut-HBx to EMSA analysis using a 300 bp DNA fragment containing a single NF-κB consensus motif in the MTA1 promoter. We found that extracts from the HBx, but not mut-HBx, expressing cells resulted in the formation of a distinct protein/DNA complex (Figure 3E, compare lane 5 with lane 2) which could be competed by the cold DNA probe (lane 8). Interestingly, the observed HBx protein/DNA complex could be effectively super-shifted by anti-p65 antibodies but not by IgG (compare lane 6 with lane 8). These results suggest that HBx presumably uses NF-κB-p65 to interact with the MTA1 DNA as mut-HBx which can not interact with p65, was also unable to bind to MTA1 DNA fragment in the EMSA assay (lanes 9–14).
MTA1 is required for HBx transactivation function
Recent studies suggest that MTA1 is not only a target of p65 but also a component of NF-κB signaling network (Suresh B. Pakala, Tri M. Bui-Nguyen and Rakesh Kumar, unpublished findings). To understand the impact of endogenous MTA1 on HBx transactivation activity, we examined the ability of HBx to induce MTA1-luc activity in the wild-type MEF and MEF-MTA1-KO MEFs. Unexpectedly, we found that HBx is unable to induce MTA1-luc activity in MTA1-deficinent MEFs while it did so in the Wt-MEFs (Figure 4A). To independently validate these results, we next showed that selective siRNA-mediated knockdown of MTA1 in HepG2 cells also impaired both the basal as well as HBx-induced MTA1 promoter activity (Figure 4B). Taken together, these results revealed an integrated role of MTA1 in the transactivation activity of HBx.
Figure 4.
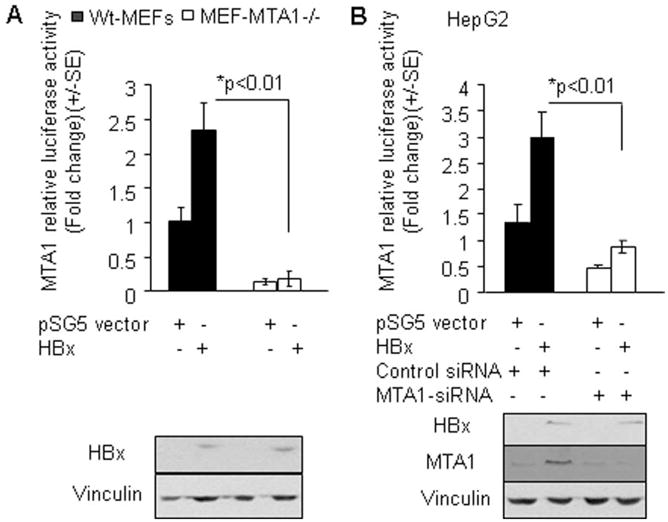
MTA1 is required for HBx transactivation function. (A) MTA1 promoter activity in wild type (WT) and MTA1−/ − MEFs after being transfected with either either control vector or HBx. (B) MTA1-promoter activity in HepG2 cells with or without MTA1 knockdown by siRNA after being transfected with either control vector or HBx.
An essential role of MTA1 in HBx stimulation of NF-κB signaling
As MTA1-depletion compromises HBx transactivation activity, we next examined whether HBx activation of NF-κB requires MTA1. To examine the above hypothesis, we examined the effect of MTA1-siRNA on the ability of HBx to induced NF-κB signaling pathway in HepG2 cells. As expected from the previous studies, HBx expression was accompanied by increased levels of phosphorylated IκB-α and phosphorylated p65 as well as MTA1 expression. However, comparable levels of HBx in HepG2 cells were unable to induce p65RelA activation in the absence of MTA1 (Figure 5A). Consistent with these results, the levels of HBx-mediated stimulation of the NF-κB DNA-binding activity was substantially reduced in MTA1-deficient MEFs as compared to Wt-MEFs (data not shown). We next determined whether the noticed defect in NF-κB signaling pathway by HBx under condition of MTA1-deficiency is also accompanied by a defective NF-κB -target gene expression. To explore this notion, we examined the effect of MTA1-siRNA on the ability of HBx to induce transcription from the NF-κB -luc reporter in HepG2 and MEFs. We found that MTA1 downregulation impairs the ability of HBx to induce NF-κB -luc activity (Figure 5B–C) and to induce the formation of the protein/DNA sequence harboring NF-κB consensus motif complex (Figure 5D). Accordingly, MTA1-depletion in HepG2 by siRNA (Figure 5E, inset) also impairs the ability of HBx to induce the expression of NF-κB target genes such as COX2 and TNF-α (Figure 5E). Since previous studies have shown a strong positive correlation of COX2 upregulation with HBx expression and in hepatitis B virus-associated chronic liver diseases (Cheng et al., 2004), these studies suggest that HBx-dependent stimulation of NF-κB signaling and resulting inflammatory targets such as COX2 and TNF-α are at-least, in-part, are dependent on the presence of MTA1. Together, these findings imply that HBx-induced inflammation, which overtime could contribute to transformation, may be dependent on the cellular status of MTA1. Together, these findings suggest that MTA1 is essential mechanistic role during HBx-mediated stimulation of NF-κB signaling and resulting expression of target genes and functions.
Figure 5.
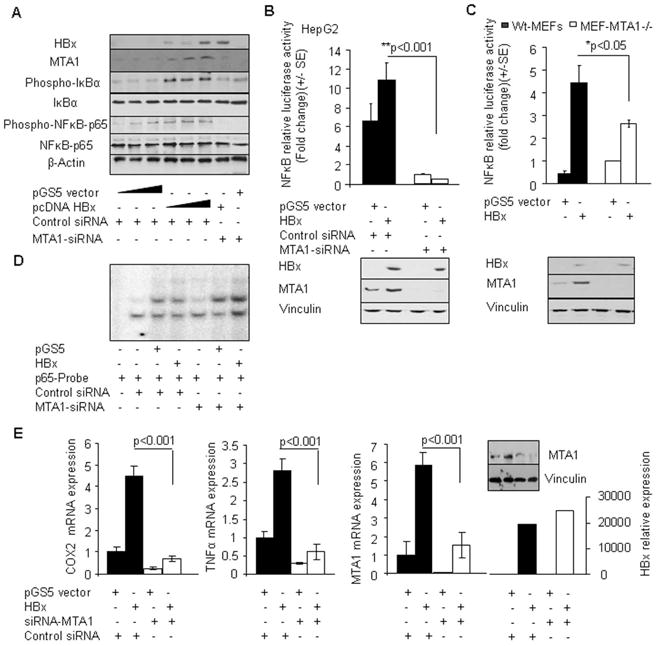
MTA1 is needed for HBx stimulated NF-κB signalling. (A) Effect of selective knockdown of MTA1 on the activation status of the NF-κB signaling components in HepG2 cells by Western blot analysis after being transfected with either control vector or HBx (250 ng per reaction in a 6-well plate). HepG2 cells transfected with increased amount of control vector and HBx (50 ng, 250 ng, and 600 ng per reaction) were used as controls. (B) NF-κB-promoter activity in HepG2 cells with or without MTA1 knockdown by siRNA-MTA1 after being transfected with either vector or HBx. Lower panel is the control Western blot analysis for the aforementioned experiments. Vinculin was used as a control. (C) NF-κB-promoter activity in MEF cells after being transfected with either vector or HBx. Lower panel is the control Western blot analysis for the aforementioned experiments. Vinculin was used as a control. (D) Nucleus extracts from HepG2 cells transfected with either vector control or HBx expression vector after MTA1 knockdown by siRNA-MTA1 were subjected to EMSA analysis using a NF-κB-consensus sequence. Extracts from wild type HepG2 transiently transfected with HBx were used as controls. (E) qPCR analysis of COX2, TNF-α, MTA1, and HBx mRNAs in HepG2 cells with or without MTA1 knockdown by siRNA-MTA1 after being transfected with control vector or vector expressing HBx. Control siRNA was used in indicated experiments. Expression levels of COX2, TNF-α, MTA1, and HBx were normalized with β-Actin. Inset: Western blot analysis for MTA1 in HepG2 cells after being co-transfected with siRNA-MTA1 and HBx or control vector. Vinculin was used as a control.
Levels of MTA1 and p65 in HBV-infected hepatocellular carcinoma
To evaluate the significance of noted HBx-regulation of MTA1 and in-turn, a mechanistic role of MTA1 in HBx-induced NF-κB pathway in human HCC, we examined the expression characteristics of HBx, MTA1, and p65 in HCC specimens and matched non-tumor tissue from the same patient. Western blot analysis of 22 matched sets of specimens indicated that there were 9 HCC-HBx-positive specimens out of 44 analyzed here; while MTA1 and p65 were upregulated in 20 and 19 samples, respectively, and consistently upregulated in HCC as compared to the matching non-tumor tissues (Figure 6A). Overall, there were 7 out of 22 HCC samples wherein HBx presence correlated with increased MTA1 and p65. Interestingly, high levels of MTA1 expression were often ensued by high levels of NF-κB-p65 expression in both tumor and non-HCC samples. To further support these observations, we performed immunohistochemical analysis of 39 additional samples in micro-tissue array format (Figure 6B). 25 HCC and 14 non-HCC samples were examined for the expression of HBx, MTA1, and p65 using immunohistochemical staining (IHC). There was a significant correlation between HBx expression and the detected levels of MTA1 (P=0.0001) in a mixed population of HCC and non-HCC samples. Strong correlation between the intensity of MTA1 and p65 however were only found in HCC samples (P<0.0001) but not in non-HCC samples (P=0.46) (Figure 6C). Together, our data suggest that high expression levels of HBx are accompanied by co-increased expression of MTA1 and p65RelA and that MTA1 plays a pivotal role in HBx transactivation in HCC.
Figure 6.
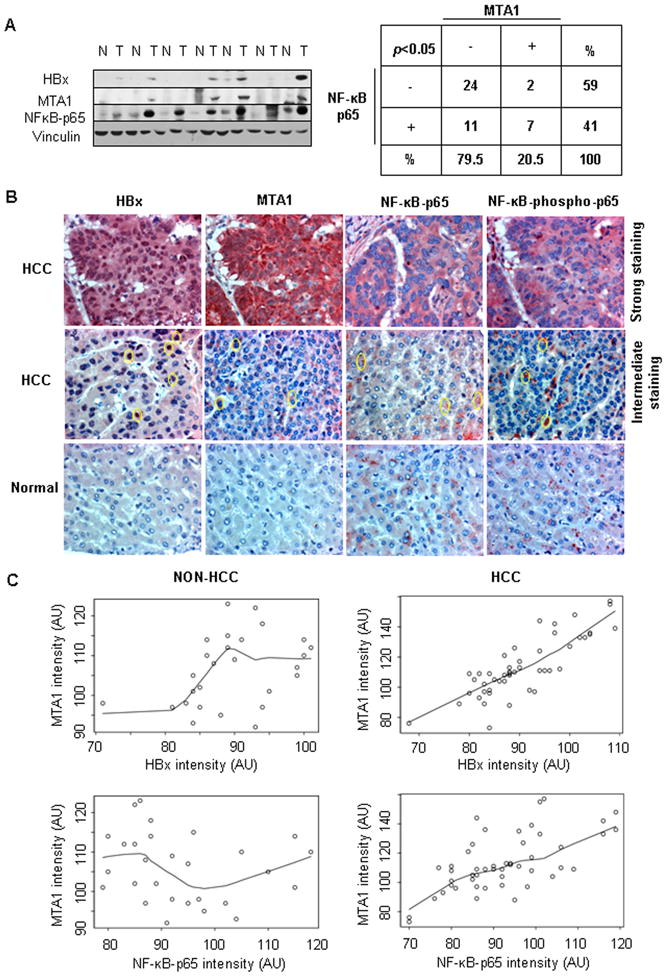
Prevalence of HBx, MTA1, and NF-κB-p65 in hepatocellular carcinoma. (A) Western blot analysis for MTA1, NF-κB-p65, and HBx proteins in human HCC specimens and matched non-tumor tissue from the same patient (n=44). Statistical analysis of the data was performed using Fisher’s Exact test. (B) Representative images of immunohistochemical analysis for HBx, MTA1 NF-κB-p65, and NF-κB-phospho-p65 in HCC and non-HCC samples in microtissue array. Circular marks denoted positive staining regions. (C) A multiple linear mixed model for the MTA1 intensity (arbitrary unit, AU) with those of HBx or NF-κB-p65 and the interaction between these markers and disease group fitted as covariates was assembled. Positive association between MTA1 and HBx and MTA1 and NF-κB-p65 intensity were observed in the HCC patients but non-HCC patients. Particularly, in HBx positive HCC patients, there was a significant positive association between NF-κB-p65 intensity and MTA1 intensity (P< 0.0001).
DISCUSSION
The expression of HBx, a multifunctional HBV viral transactivator, targets a variety of cellular proteins, including NF-κB with putative functions ranging from transcription, inflammation, survival, and cancerous phenotypes. However, at the moment, cellular targets of HBx have not been exploited to develop strategies to inhibit or slow-down the process of HBV-mediated HCC carcinogenesis. Whereas, increased expression of MTA1, a metastasis associated coregulatory protein has been also linked with the degree of intrahepatic invasion and metastasis in HCC patients (Hamatsu et al., 2003; Ryu et al., 2008). While this study was in process, Yoo et al showed that HBx induces MTA1 but that study did not address possible mechanistic insights (Moon et al., 2004). Here we delineated the molecular mechanism underlying the induction of MTA1 by HBx and demonstrated the integral role of MTA1 during HBx-mediated activation of NF-κB signaling and its targets. We further established that although both HBV and HCV have been linked with HCC, MTA1 is targeted by HBV-HBx protein and not by HCV core protein. We further demonstrated that HBx selectively induces MTA1 but not MTA2 and MTA3. On-going cloning of a murine MTA1 promoter allowed us to provide a detailed mechanistic insight of HBx regulation of MTA1 transcription. We showed that HBx targets p65 to physically interact with the MTA1 promoter containing a functional p65 motif, and such interactions leads to a productive MTA1 transcription in HBx-expressing cells. Indeed, we demonstrated that HBx uses the YFKD motif to interact with the p65 subunit and consequently, the HBx-p65 complex interacts with the MTA1 promoter element through the NF-κB response elements. Nevertheless, the inability of both ΔC-HBx (HBx with intact 101-120 amino acids) and ΔE-HBx (HBx with intact 101-120 amino acids) to stimulate the p65 luc-promoter activity (supplementary Fig. S1D) suggests that individual domains of HBx may not mimic the activity conferred by the intact HBx.
The HBx regulatory protein has been established by many laboratories to activate NF-κB activities in transfected cells, and this is presumed to play a role in HCC (Kim et al., 2008; Su & Schneider, 1996). As shown in Fig. 5A, we did observe a transient induction of IKBα phosphorylation, which is a direct indicator of functional upstream signaling feeding into the IKBα-p65 linearity. For reasons not fully understood at the moment, we did not observe any down-regulation of IκBα protein (Fig. 5A, 4th panel) as was the case in Chirillo et al., 1996. Several possibilities may be attributable to this discrepancy such as the use of the HeLa cells by Chirillo et al. as oppose to the relevant use of HepG2 cells in our study; possible HBx interaction with papilloma virus in the HeLa cells, leading to the observed effects reported by Chirillo et al. While it is clear that MTA1 is essential for HBx-transactivation of NF-κB, the manner by which MTA1 could feed into HBx-NF-κB pathway remains to be an important question for future investigation. Since MTA1 now affects the stability of proteins (Li et al., 2009), it is possible that HBx-induced MTA1 in-turn, stabilizes specific component or components of proximal NF-κB signaling. If so, such changes may translate into an amplified NF-κB signaling. Furthermore, because MTA1 is a chromatin modifier, it is conceivable that MTA1 may affect the net transcription of the components of NF-κB pathway, and consequently, affect the signaling.
Both inflammation and MTA1 have been linked to cancer, particularly, to HCC. Chronic inflammation in liver, depending on the magnitude of NF-κB activity, promotes hepatocarcinogenesis while MTA1 overexpression is known to associate with tumor growth and metastasis (Bagheri-Yarmand et al., 2004; Giannini & Cavallini, 2005; Hamatsu et al., 2003; Jang et al., 2006; Kidd et al., 2006; Maeda et al., 2005; Manavathi & Kumar, 2007; Manavathi et al., 2007b; Toh et al., 1995). Nevertheless, the role of the nucleosome remodeling deacetylase complex (NuRD) in NF-κB signaling remains unknown until our recent discovery. Our recent study showed that MTA1, the founding member of the NuRD family of coregulators, is a part of NF-κB pathway and regulates host inflammatory response. Here we took this observation further and established regulatory contribution of MTA1-NF-κB cross-talk in the transactivation activity of HBx. Results from ChIP studies suggested that HBx may utilize p65 as a coactivator and that HBx/p65 recruitment to the MTA1 promoter is correlated with the recruitment of H4-specific acetyltransferase complexes. The overexpression of p65 can also mimic the effect of HBx-mediated stimulation of the MTA1 transcription. However NF-κB, which is known to regulate the transcription of many acute-phase proteins (Pahl, 1999), requires other transcription factor and coregulator such as SRC-3 (Anderson & Kedersha, 2007) for its maximum activity. In this context, it is worth mentioning that the removal of MTA1 had a profound impairment of HBx-transactivation and expression of the NF-κB’s targets, e.g. Cox-2, and TNF-α. These findings suggest an integral role of MTA1 in the previously shown regulation of NF-κB activity by HBx and support the notion that MTA1 is required efficient transactivation of HBx in hepatocytes. Although HBx has been shown to influence a large number of molecules in the cell lines, only a few have been shown to be involved in the physiologically relevant human specimens as well. In this context, the present study has utilized both the model systems and point to the fact that the newly described HBx-MTA1-NFκB pathway may be physiologically relevant in the pathogenesis of HCC. Since TNF-α is a target as well as activator of NF-κB, the status of MTA1 in HBx-expressing cells is likely to have an important effect upon carcinogenesis process. These findings suggest that MTA1 may not only represent as a novel HCC prognostic indicator (Hamatsu et al., 2003), but also can be a therapeutic target.
Supplementary Material
MTA1 promoter activity in HepG2 cells (A) or HEK 293 (B) transfected with either control vector (250 ng/reaction) or HBx expression vector (250 ng/reaction) from different expression systems (pcDNA 3.1, pSI-GFP, pSG5) was analyzed. Full-length murine MTA1 promoter (■)(−140 to −5200) or its minimal promoter (−2872−5200)(□) were cloned into pGCL3 luciferase reporter vector (Promega, Madison, WI). Luciferase assay were performed according to the manufacturer’s instructions (Promega, Madison, WI), and the results were normalized against the β-galactosidase activity, an internal control. C, Western blot analysis for transfection efficiency in HepG2 after being cotransfected with pcDNA-p65 expression vector and HBx expression vector. Transfected cells were analyzed for HBx, NF-κB-p65 48 hours after transfection. Vinculin was used as a control. D. NF-κB promoter activity in HepG2 cells transfected with either control vector or HBx expression vector or HBx-deletion constructs (ΔC-HBx-HBx with intact 101-120 amino acids and ΔE-HBx-HBx with intact 101-120 amino acids).
qPCR analysis of MTA1 and HBx (WT and mutant) mRNAs in HepG2 cells transfected with either control vector or HBx or mut-HBx expression vector. Expression levels of analyzed mRNAs were normalized by β-Actin.
Acknowledgments
We would like to thank Aleem Siddiqui for the pCMVXF and pCMV-Core constructs, Ranjit Banerjee for the HVC core plasmid, Amanda J. Hodgson for technical assistance, and to Xuemei Wang for statistical analysis. The work was supported by NIH Grant CA98823 (RK). Work in Dr. Betty L. Slagle laboratory is supported by the NIH Grant 95388.
References
- Anderson P, Kedersha N. Mol Cell. 2007;25:796–7. doi: 10.1016/j.molcel.2007.03.005. [DOI] [PubMed] [Google Scholar]
- Bagheri-Yarmand R, Talukder AH, Wang RA, Vadlamudi RK, Kumar R. Development. 2004;131:3469–79. doi: 10.1242/dev.01213. [DOI] [PubMed] [Google Scholar]
- Bergametti F, Prigent S, Luber B, Benoit A, Tiollais P, Sarasin A, Transy C. Oncogene. 1999;18:2860–71. doi: 10.1038/sj.onc.1202643. [DOI] [PubMed] [Google Scholar]
- Bouchard MJ, Schneider RJ. J Virol. 2004;78:12725–34. doi: 10.1128/JVI.78.23.12725-12734.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CJ, Yang HI, Su J, Jen CL, You SL, Lu SN, Huang GT, Iloeje UH. JAMA. 2006;295:65–73. doi: 10.1001/jama.295.1.65. [DOI] [PubMed] [Google Scholar]
- Cheng AS, Chan HL, Leung WK, To KF, Go MY, Chan JY, Liew CT, Sung JJ. Mod Pathol. 2004;17:1169–79. doi: 10.1038/modpathol.3800196. [DOI] [PubMed] [Google Scholar]
- Chirillo P, Falco M, Puri PL, Artini M, Balsano C, Levrero M, Natoli G. J Virol. 1996;70:641–6. doi: 10.1128/jvi.70.1.641-646.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Xia W, Liu JC, Yang JY, Lee DF, Xia J, Bartholomeusz G, Li Y, Pan Y, Li Z, Bargou RC, Qin J, Lai CC, Tsai FJ, Tsai CH, Hung MC. Mol Cell. 2005;19:159–70. doi: 10.1016/j.molcel.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Dvorchik I, Demetris AJ, Geller DA, Carr BI, Fontes P, Finkelstein SD, Cappella NK, Marsh JW. Curr Pharm Des. 2007;13:1527–32. doi: 10.2174/138161207780765846. [DOI] [PubMed] [Google Scholar]
- Giannini R, Cavallini A. Anticancer Res. 2005;25:4287–92. [PubMed] [Google Scholar]
- Gururaj AE, Singh RR, Rayala SK, Holm C, den Hollander P, Zhang H, Balasenthil S, Talukder AH, Landberg G, Kumar R. Proc Natl Acad Sci U S A. 2006;103:6670–5. doi: 10.1073/pnas.0601989103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamatsu T, Rikimaru T, Yamashita Y, Aishima S, Tanaka S, Shirabe K, Shimada M, Toh Y, Sugimachi K. Oncol Rep. 2003;10:599–604. [PubMed] [Google Scholar]
- Hoffmann A, Baltimore D. Immunol Rev. 2006;210:171–86. doi: 10.1111/j.0105-2896.2006.00375.x. [DOI] [PubMed] [Google Scholar]
- Jang KS, Paik SS, Chung H, Oh YH, Kong G. Cancer Sci. 2006;97:374–9. doi: 10.1111/j.1349-7006.2006.00186.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd M, Modlin IM, Mane SM, Camp RL, Eick G, Latich I. Ann Surg Oncol. 2006;13:253–62. doi: 10.1245/ASO.2006.12.011. [DOI] [PubMed] [Google Scholar]
- Kim SY, Kim JC, Kim JK, Kim HJ, Lee HM, Choi MS, Maeng PJ, Ahn JK. BMB Rep. 2008;41:158–63. doi: 10.5483/bmbrep.2008.41.2.158. [DOI] [PubMed] [Google Scholar]
- Li Q, Verma IM. Nat Rev Immunol. 2002;2:725–34. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- Li Da-Q, Ohshiro K, Reddy SDN, Pakala SB, Lee MH, Zhang Y, Rayala SK, Kumar R. Proc Natl Acad Sci USA. 2009 doi: 10.1073/pnas.0908027106. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucito R, Schneider RJ. J Virol. 1992;66:983–91. doi: 10.1128/jvi.66.2.983-991.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupberger J, Hildt E. World J Gastroenterol. 2007;13:74–81. doi: 10.3748/wjg.v13.i1.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Kamata H, Luo JL, Leffert H, Karin M. Cell. 2005;121:977–90. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Manavathi B, Kumar R. J Biol Chem. 2007;282:1529–33. doi: 10.1074/jbc.R600029200. [DOI] [PubMed] [Google Scholar]
- Manavathi B, Peng S, Rayala SK, Talukder AH, Wang MH, Wang RA, Balasenthil S, Agarwal N, Frishman LJ, Kumar R. Proc Natl Acad Sci U S A. 2007a;104:13128–33. doi: 10.1073/pnas.0705878104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manavathi B, Singh K, Kumar R. Nucl Recept Signal. 2007b;5:e010. doi: 10.1621/nrs.05010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon EJ, Jeong CH, Jeong JW, Kim KR, Yu DY, Murakami S, Kim CW, Kim KW. FASEB J. 2004;18:382–4. doi: 10.1096/fj.03-0153fje. [DOI] [PubMed] [Google Scholar]
- Pahl HL. Oncogene. 1999;18:6853–66. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- Ray RB, Ray R. FEMS Microbiol Lett. 2001;202:149–56. doi: 10.1111/j.1574-6968.2001.tb10796.x. [DOI] [PubMed] [Google Scholar]
- Ryu SH, Chung YH, Lee H, Kim JA, Shin HD, Min HJ, Seo DD, Jang MK, Yu E, Kim KW. Hepatology. 2008;47:929–36. doi: 10.1002/hep.22124. [DOI] [PubMed] [Google Scholar]
- Schreiber E, Matthias P, Muller MM, Schaffner W. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su F, Schneider RJ. J Virol. 1996;70:4558–66. doi: 10.1128/jvi.70.7.4558-4566.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toh Y, Pencil SD, Nicolson GL. Gene. 1995;159:97–104. doi: 10.1016/0378-1119(94)00410-t. [DOI] [PubMed] [Google Scholar]
- Yoo YG, Na TY, Seo HW, Seong JK, Park CK, Shin YK, Lee MO. Oncogene. 2008;27:3405–13. doi: 10.1038/sj.onc.1211000. [DOI] [PubMed] [Google Scholar]
- Yun C, Um HR, Jin YH, Wang JH, Lee MO, Park S, Lee JH, Cho H. Cancer Lett. 2002;184:97–104. doi: 10.1016/s0304-3835(02)00187-8. [DOI] [PubMed] [Google Scholar]
- Zhang X, Zhang H, Ye L. J Lab Clin Med. 2006;147:58–66. doi: 10.1016/j.lab.2005.10.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
MTA1 promoter activity in HepG2 cells (A) or HEK 293 (B) transfected with either control vector (250 ng/reaction) or HBx expression vector (250 ng/reaction) from different expression systems (pcDNA 3.1, pSI-GFP, pSG5) was analyzed. Full-length murine MTA1 promoter (■)(−140 to −5200) or its minimal promoter (−2872−5200)(□) were cloned into pGCL3 luciferase reporter vector (Promega, Madison, WI). Luciferase assay were performed according to the manufacturer’s instructions (Promega, Madison, WI), and the results were normalized against the β-galactosidase activity, an internal control. C, Western blot analysis for transfection efficiency in HepG2 after being cotransfected with pcDNA-p65 expression vector and HBx expression vector. Transfected cells were analyzed for HBx, NF-κB-p65 48 hours after transfection. Vinculin was used as a control. D. NF-κB promoter activity in HepG2 cells transfected with either control vector or HBx expression vector or HBx-deletion constructs (ΔC-HBx-HBx with intact 101-120 amino acids and ΔE-HBx-HBx with intact 101-120 amino acids).
qPCR analysis of MTA1 and HBx (WT and mutant) mRNAs in HepG2 cells transfected with either control vector or HBx or mut-HBx expression vector. Expression levels of analyzed mRNAs were normalized by β-Actin.