

Abstract
Each time a cell divides its chromosome content must be equally segregated into the two daughter cells. This critical process is mediated by a complex microtubule based apparatus, the mitotic spindle. In most animal cells the centrosomes contribute to the formation and the proper function of the mitotic spindle by anchoring and nucleating microtubules and by establishing its functional bipolar organization. Aberrant expression of proteins involved in centrosome biogenesis can drive centrosome dysfunction or abnormal centrosome number, leading ultimately to improper mitotic spindle formation and chromosome missegregation. Here we review recent work focusing on the importance of the centrosome for mitotic spindle formation and the relation between the centrosome status and the mechanisms controlling faithful chromosome inheritance.
Introduction
The main function of mitosis is to achieve the faithful segregation of one copy of each chromosome into each daughter cell. The physical apparatus responsible for this fundamental cellular process, the mitotic spindle, is assembled of microtubules and hundreds of proteins that are involved in its morphogenesis, the regulation of its dynamic properties, and the assembly of the microtubule attachment site on the chromosome, the kinetochore [1]. In most animal cells the poles of the spindle contain centrosomes which function to mediate microtubule nucleation and anchoring and to facilitate the organization of the overall structure. Those complex organelles are made of two barreled shaped, orthogonally-assembled structures, the centrioles, embedded in an amorphous proteinacious material called the pericentriolar material (PCM). The PCM contains proteins which are directly involved in microtubule nucleation, including the γ-tubulin ring complex and pericentrin [2,3].
Similar to the chromosomes, the centrosome at each spindle pole is delivered into the corresponding daughter cell at cell division and then is duplicated in the following S phase. Each centriole pair so produced is used to nucleate microtubules to form a spindle pole in the next mitosis. Despite their similar overall organization, the two centrioles in each centrosome are not equivalent, with a mother centriole that is one cell cycle older (and characterized, in mammals, by the presence of distal and sub-distal appendages involved in microtubule anchoring) and a more newly born daughter centriole [4].
The centrosome duplication cycle and the cell cycle share common key regulators. Active Cdks (Cyclin-dependent kinases) have been shown to be essential to initiate centriole duplication in S phase [5,6] and to allow centrosome maturation and separation, together with Plk1 and Aurora A in late S/G2, G2 and M phases [7-10]. Although the sequence of events is not entirely understood, it has been proposed that CDK2 activates the proteins NPM, MPS1 and CP110 at the onset of S phase. Those proteins in turn trigger the sequential recruitment of the five core centriolar components SPD2/CEP192, ZYG1/Plk4, SAS5/STIL, SAS6/hSAS6 and SAS4/CPAP in the vicinity of the mother centriole so as to enable daughter centriole formation [4,11,12]. Both cell cycle regulator and centriolar protein levels are tightly controlled and disruption of their expression levels or their enzymatic activities results in abnormal centrosome number or microtubule nucleation, conditions which are often associated with abnormal mitotic spindle activity.
In this review, we describe the importance of centrosome homeostasis for the accuracy of chromosome segregation and the maintenance of cell ploidy and we discuss the cellular pathways that prevent cell division in the presence of centrosome anomalies.
Chromosome instability (CIN) and centrosome abnormalities
Genome stability is dependent on the accuracy of chromosome segregation by the mitotic spindle. Errors produce aneuploidy, a state in which a cell possesses a chromosome content other than a multiple of the haploid number. More than 90% of human solid tumors (and the majority of haemotopoietic cancers) are aneuploid [13]. In contrast to simple aneuploidy – from missegregation of one or a handful of chromosomes – many tumor cells acquire chromosomal instability (CIN), a condition in which the cells have a continuously changing aneuploidy arising from continuing segregation errors. Various defects in mitotic processes can be responsible for CIN and the first suspected one was a defective mitotic checkpoint (also known as the spindle assembly checkpoint). This checkpoint is the major cell cycle control mechanism governing advance from metaphase to anaphase. A “wait anaphase” inhibitor is produced by unattached kinetochores so that anaphase onset is delayed until the centromere of each chromosome has made stable attachment to the mitotic spindle. The linkage of mitotic checkpoint components to CIN was initially proposed from an apparent high rate of mutation in a core component of the checkpoint (Bub1) in CIN cancer cells [14]. However, most CIN cells subsequently reported have an intact mitotic checkpoint despite an elevated rate of chromosome missegregation. This has been described for a variety of chromosomally unstable cancer cell lines harboring an active checkpoint in presence of unaligned chromosomes [15] or in presence of defective spindle activity following treatment with spindle poisons [16•].
It has become increasingly recognized that perturbation of other mitotic elements can be responsible for CIN. One of these is defective cohesion between sister chromatids, which has been recently demonstrated to be responsible for CIN in various cancer cell lines [17••]. Here, the authors clearly demonstrated the necessity of intact cohesion: normally stable cell lines were converted to unstable ones after depletion of the cohesin subunit STAG2. Even more remarkably, chromosomal stability was rescued in highly unstable aneuploid glioblastoma cell lines by substituting a non-mutated form of STAG2 in place of the previously mutated one.
But the most prominent cause of CIN is probably the presence of abnormally high numbers of centrosomes. Indeed, this state - called centrosome amplification - is repeatedly observed in cancer cells lines which also harbor a CIN phenotype [18-20]. Cells can acquire extra centrosomes by diverse mechanisms including cell fusion, failure in completion of cytokinesis, and the deregulation of centrosome biogenesis, i.e., the overduplication of centrosomes or the de novo assembly of extra centrosomes. Production of multiple centrosomes by cell fusion is a mechanism used during development, especially by skeletal muscle as a means to produce multi-nucleated, multi-centrosome containing cells. Such fusion events, of course, are unlikely drivers of chromosome missegregation since terminally differentiated muscle cells do not continue to divide. It should be noted as well that the multiple centrosomes in these cells do not nucleate microtubules [21], suggesting the presence of mechanisms to suppress nucleation activity as a means to prevent any potential deleterious effects from the extra centrosomes.
Skipping mitosis altogether [22] and failure to complete cytokinesis [23-25] have both been shown to induce centrosome amplification in transformed cells. However, the potential of cytokinesis failure to generate cells with stable centrosome amplification has been questioned [26]. In this work, Krzywicka-Racka and Sluder tested if multiple rounds of chemically-induced cytokinesis failure in transformed and non-transformed cells can drive centrosome amplification. In two of the cell types tested (RPE1 and HCT116 cells), cells failing cytokinesis did not accumulate extra centrosomes. In the third cell type (CHO cells), cells did accumulate modest levels of extra centrosomes, but the cells did not continue to proliferate as a consequence of a p53-p21 dependent cell cycle arrest in the subsequent interphase [26].
Cells can also acquire multiple centrosomes by amplification through perturbation in centrosome biogenesis when the levels, timing, or localization of proteins involved in centriole formation are missregulated. Recent studies have described how Plk4, a kinase essential for centrosome duplication, is very tightly autoregulated: its self-phosphorylation within a 24 amino acid phospho-degron makes it a substrate for ubiquitination by the SKP1-CUL1-F-Box (SCF) complex in order to regulate its cellular levels (and kinase activity) through subsequent proteosomal degradation [27••]. Furthermore, in Drosophilla, stabilization of Plk4 can be achieved during mitosis through the dephosphorylation of Plk4 by PP2A, an activity necessary to promote normal centriole duplication [28•]. Other work in Drosophilla embryos and in human cells has revealed the importance of the recruitment of core centriolar proteins CPAP and Plk4 to the centrosome by CEP152 to support normal centrosome biogenesis [29]. Finally, recent work has described another level of complexity in centrosome biogenesis. Using an RNA interference approach in human cells, an indispensable recruitment of STIL by CPAP at the G1/S transition has been reported to enable the initiation of procentriole formation [12,30-32]. Altogether, these studies underlined the importance of tight control of synthesis and accumulation of centriolar components in order to maintain normal centrosome homeostasis.
Mechanisms of chromosomal instability induced by centrosome amplification
The correlation between centrosome amplification and aneuploidy was described more than a century ago by Theodor Boveri, but only recently were detailed mechanisms proposed to explain this relationship. Centrosome amplification can predispose cells to CIN in a two-step mechanism: 1) the formation of a multipolar spindle and 2) the resolution of this aberrant mitotic configuration into a bipolar spindle (Figure 1). This transition from a multipolar to a bipolar spindle promotes the formation of incorrect kinetochore microtubule attachment that then provokes subsequent chromosome segregation errors during anaphase and that are maintained at cytokinesis. Cells arising from multipolar divisions are often inviable [33•,34]. This was especially highlighted by use of live cell imaging to demonstrate that multipolar division is lethal and that transient multipolar spindles in the presence of extra centrosomes do promote segregation errors [33•]. Despite this, there are notable exceptions, such as liver, where multipolarity has been proposed [35] to be beneficial by generating genetic diversity in hepatocytes.
Figure 1. Chromosomes segregation in presence of extra centrosomes.
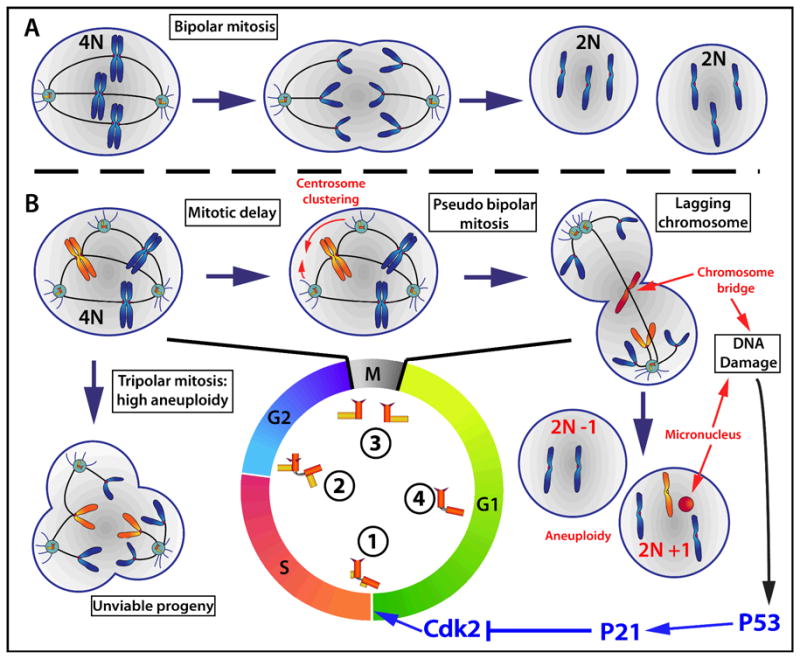
Centrosome duplication starts at the G1/S transition with procentriole formation (1). Centrosomes are matured throughout the G2 phase (2) and they are separated to form the poles of the mitotic spindle in M phase (3). In G1, the centrosomes lose their orthogonal configuration in preparation for their duplication (4). A. Cells with two centrosomes form a bipolar spindle and chromosomes are usually accurately segregated. Cellular ploidy is maintained and cell proliferation is sustained. B. Cells entering mitosis with more than two centrosomes can either go through a multipolar mitosis or cluster their centrosomes to form a bipolar spindle. Progeny of multipolar divisions are highly aneuploid and are usually inviable. Centrosome clustering favors the formation of merotelic kinetochore/microtubule attachments resulting in lagging chromosomes. Lagging chromosomes can be included in DNA bridges or micronuclei during cell cleavage to produce daughter cells. These phenomena induce p53- dependent activation of p21, which inhibits Cdk2 and produces cell cycle arrest at the G1/S transition.
Coalescence of centrosomes nucleating a multipolar spindle into a bipolar spindle requires the minus end directed motor dynein, as demonstrated by Quintyne and coauthors [36]. Studies using genome wide RNA interference approaches (in squamous cell carcinoma cells [34] and in Drosophilla S2 cells [37]) subsequently highlighted the importance of the actin cytoskeleton and functional kinetochore-microtubule interface in mediating proper centrosome clustering in cells with a multipolar spindle. In parallel with the mechanical forces involved, work in cells [37,38] and fly [39] models has demonstrated that the delay in kinetochore attachment or production of aberrant attachments leads to failure to silence the mitotic checkpoint with normal timing, thus delaying advance to anaphase in cells with extra centrosomes. This delay facilitatescentrosome clustering that can mitigate the deleterious consequences of multipolar division.
The transition from a multipolar spindle to a bipolar spindle also enables an increased frequency of abnormal kinetochore/microtubule configurations. In cells initially forming bipolar mitotic spindles, the kinetochores of each duplicated sister chromatid normally bi-orient, with the two kinetochores of each pair attaching to microtubules nucleated by opposite spindle poles, thereby generating a configuration called amphitelic attachment. In cells with an initial multipolar spindle, however, each kinetochore has an elevated chance of capturing microtubules emanating from more than one pole. If in subsequent centrosome clustering those poles end up coalescing into different composite poles, the kinetochore will now be attached to microtubules from both spindle poles, a configuration called merotely. As the both mitotic and meiotic checkpoints in vertebrate mitosis [40,41] and female meiosis [42••], respectively, are silenced by microtubule attachment, merotelic attachments go unrecognized as attachment errors. At anaphase entry, the merotelically attached chromosome is pulled toward both spindle poles, thereby producing a lagging chromosome with a high potential for missegregation.
This sequence of events has been clearly described to be responsible for CIN in a human cancer cell line (U2OS) where centrosome amplification - induced by Plk4 overexpression - was shown to be directly linked to chromosome missegragation [33•]. However, in a further study where the fate of lagging chromosomes was followed using a LacI-GFP to target a LacO array integrated at a specific chromosomal locus, it was reported that the merotelically attached chromosomes were actually segregating toward the correct daughter cell in a majority of the anaphases [43] suggesting that lagging anaphase chromosome may not be the sole mechanism responsible for CIN. In line with this idea, recent work using live imaging of a cancer cell line (a Wilm's tumor kidney cancer cell line) along with analysis of tumor chromosome content has offered support for the combination of an initial tripolar mitosis followed - without centrosome clustering - by cytokinesis failure between two of three nascent daughter cells as a mechanism for inducing the missegregation of multiple chromosomes in a single mitotic event [44••].
Centrosomes, p53, and the cell cycle: a continuing controversy
The contribution of centrosome amplification to CIN and to the establishment of ananeuploid chromosome content raises the question of the existence of mechanisms controlling centrosome number. Can cells monitor or detect the presence of extra centrosomes? Evidence emerging over the past two decades has provided contradictory answers to this question.
On the one hand, work a decade ago originally proposed that specific cell types depleted of centrosomes can go through mitosis normally but then arrest in the following interphase without replicating their DNA, thus implicating a requirement of centrosomes in cell cycle advance in interphase [45,46]. However, a following study opposed this proposition, reporting that non-transformed human cells were able to sustained normal cell cycle progression after laser ablation of their centrosomes [47]. Furthermore, the evidence of Fukasawa et al lead to the initial proposal that mouse embryonic fibroblasts lacking p53 accumulate an abnormal number of centrosomes [48]. Hence, this work suggested that p53 opposes cell cycle progression in cells with extra centrosomes.
Additional work following cells with too many centrosomes lead to an even more direct proposal of a p53-dependent G1/S arrest in response to centrosome amplification and has been referred to as a tetraploidy checkpoint [49]. The existence of such a checkpoint responsible for arresting cell cycle progression following cytokinesis failure and doubling of the DNA and centrosome content remains highly controversial. The notion of a tetraploidy checkpoint was quickly challenged, with the observed p53-dependent G1/S arrest after cytokinesis failure linked to direct DNA damage [50,51] or consequences of damage to the spindle machinery arising from the drug treatment used to generate the tetraploid cells [52].
Another possible linkage of p53 activation to centrosome amplification has recently emerged from efforts proposing that in otherwise unperturbed U2OS cells in which centrosome amplification was induced - by overexpressing a CDK6 activator (a D-type cyclin encoded by a Kaposis's sarcoma herpes virus) - a proapoptotic signal dependent on p53 was triggered [53]. Additionally, depletion of p53 by shRNA in an unperturbed immortalized diploid human cell line (RPE1) was reported to lead to a mild level of centrosome amplification [54•], an observation which can either support the idea of a direct role of p53 in centrosome duplication or the possibility that the p53 arrests cell cycle progression in response to spontaneously acquired extra centrosomes.
Support for the proposal that the absence of p53 might be directly responsible for driving centrosome amplification has come from two lines of evidence. First, the absence of activation of the CDK inhibitor p21 by p53 may trigger high CDK activity to drive multiple rounds of centrosome duplication in G1/S arrested cells [55]. Alternatively, p53 may be directly required at the centrosome to mediate its duplication at the G1/S transition [56].
Furthermore, evidence for the recruitment of cyclinE/Cdk2 to the centrosome led to the proposal that this was necessary to allow the initiation of DNA replication in S phase [57]. This study, along with another describing the activation of a p38-p53-p21 dependent G1/S arrest in RPE1 cells depleted of centriolar components [58], provided additional evidence to support the idea that cell cycle progression was sensitive to centrosome integrity.
Consequences of centrosome amplification on aneuploidy and cell proliferation
In light of the contradictory evidence linking p53 and cell cycle advance to centrosome number, our view is that the most plausible explanation for a G1/S arrest observed in cells with extra centrosomes is the result of DNA damage caused by an aberrant event in the previous mitosis (Figure 1). For cells with extra centrosomes and the corresponding erroneous kinetochore/microtubule attachments, lagging chromosomes (as described above) are frequent and those chromosomes are likely to be damaged. This can happen because of trapping the lagging chromosome into DNA bridges during cytokinesis (as first suggested 30 years ago by the evidence of Mullins and Biesele [59]) with the direct result of DNA damage and activation of a DNA damage response involving p53 [60••].
Lagging chromosomes arising from aberrant spindles can also be included into micronuclei, and work from Pellman and colleagues [61••] has recently shown that the micronuclear environment so produced is partially dissociated from the cell cycle (at least in part because of a paucity of nuclear pores). As a consequence, in the subsequent interphase, DNA replication in a micronucleus can be delayed, but not recognized, and mitotic entry ensues before DNA replication has been completed within the micronucleus. This situation produces massive DNA damage and chromosome rearrangements (known as chromothripsis) that are restricted to the chromosome(s) encapsulated within the micronucleus. A final possible cause for acquiring DNA damage in cells with extra centrosomes is the long mitotic delay required for clustering centrosomes, as prolonged mitotic delays have been shown recently to induce telomere deprotection, telomere damage and a p53-dependent arrest at the next G1 to S phase transition [62•,63].
Conclusion
Aneuploidy is a common feature of cancer cells, and it is now well established that centrosome amplification can strongly contribute to the establishment of this state by favoring chromosome segregation errors during mitosis. Although the mechanisms linking centrosome amplification to chromosome instability are well understood, it is uncertain if centrosome amplification by itself can be sufficient to drive tumor formation. It is possible that to be able to drive cell transformation and tumor formation centrosome amplification may need to be added to other defects, such as mutation or loss of a tumor suppressor. To establish the sequence of events leading to tumor formation in presence of extra centrosome and to understand the importance of centrosome amplification in this process, it is now essential to develop mouse models where this state can be induced independently of any other factors as a means to test how it contributes to neoplasia.
Acknowledgments
The authors would like to thank Andrew Holland for helpful comments on this manuscript. We apologize to all whose work cannot be cited because of space restrictions. This work was supported by a grant (GM29513) from the National Institutes of Health to D.W.C, who receives salary support from the Ludwig Institute for Cancer Research. B.D.V. is supported by a Human Frontier Science Program Long Term Fellowship (LT000855/2010).
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Bonner MK, Poole DS, Xu T, Sarkeshik A, Yates JR, 3rd, Skop AR. Mitotic spindle proteomics in Chinese hamster ovary cells. PLoS One. 2011;6:e20489. doi: 10.1371/journal.pone.0020489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doxsey SJ, Stein P, Evans L, Calarco PD, Kirschner M. Pericentrin, a highly conserved centrosome protein involved in microtubule organization. Cell. 1994;76:639–650. doi: 10.1016/0092-8674(94)90504-5. [DOI] [PubMed] [Google Scholar]
- 3.Stearns T, Kirschner M. In vitro reconstitution of centrosome assembly and function: the central role of gamma-tubulin. Cell. 1994;76:623–637. doi: 10.1016/0092-8674(94)90503-7. [DOI] [PubMed] [Google Scholar]
- 4.Strnad P, Gonczy P. Mechanisms of procentriole formation. Trends Cell Biol. 2008;18:389–396. doi: 10.1016/j.tcb.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 5.Hinchcliffe EH, Li C, Thompson EA, Maller JL, Sluder G. Requirement of Cdk2-cyclin E activity for repeated centrosome reproduction in Xenopus egg extracts. Science. 1999;283:851–854. doi: 10.1126/science.283.5403.851. [DOI] [PubMed] [Google Scholar]
- 6.Matsumoto Y, Hayashi K, Nishida E. Cyclin-dependent kinase 2 (Cdk2) is required for centrosome duplication in mammalian cells. Current Biology. 1999;9:429–432. doi: 10.1016/s0960-9822(99)80191-2. [DOI] [PubMed] [Google Scholar]
- 7.Hannak E, Kirkham M, Hyman AA, Oegema K. Aurora-A kinase is required for centrosome maturation in Caenorhabditis elegans. J Cell Biol. 2001;155:1109–1116. doi: 10.1083/jcb.200108051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee K, Rhee K. PLK1 phosphorylation of pericentrin initiates centrosome maturation at the onset of mitosis. J Cell Biol. 2011;195:1093–1101. doi: 10.1083/jcb.201106093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loncarek J, Hergert P, Khodjakov A. Centriole reduplication during prolonged interphase requires procentriole maturation governed by Plk1. Curr Biol. 2010;20:1277–1282. doi: 10.1016/j.cub.2010.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith E, Hegarat N, Vesely C, Roseboom I, Larch C, Streicher H, Straatman K, Flynn H, Skehel M, Hirota T, et al. Differential control of Eg5-dependent centrosome separation by Plk1 and Cdk1. EMBO J. 2011;30:2233–2245. doi: 10.1038/emboj.2011.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tang CJ, Fu RH, Wu KS, Hsu WB, Tang TK. CPAP is a cell-cycle regulated protein that controls centriole length. Nat Cell Biol. 2009;11:825–831. doi: 10.1038/ncb1889. [DOI] [PubMed] [Google Scholar]
- 12.Tang CJ, Lin SY, Hsu WB, Lin YN, Wu CT, Lin YC, Chang CW, Wu KS, Tang TK. The human microcephaly protein STIL interacts with CPAP and is required for procentriole formation. EMBO J. 2011;30:4790–4804. doi: 10.1038/emboj.2011.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mitelman F, Johansson B, Mertens F, editors. Mitelman Database of Chromosome Aberrations and Genes Fusions in Cancer. 2012 http://cgap.nci.nih.gov/Chromosomes/Mitelman.
- 14.Cahill DP, Lengauer C, Yu J, Riggins GJ, Willson JK, Markowitz SD, Kinzler KW, Vogelstein B. Mutations of mitotic checkpoint genes in human cancers. Nature. 1998;392:300–303. doi: 10.1038/32688. [DOI] [PubMed] [Google Scholar]
- 15.Thompson SL, Compton DA. Examining the link between chromosomal instability and aneuploidy in human cells. J Cell Biol. 2008;180:665–672. doi: 10.1083/jcb.200712029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16•.Gascoigne KE, Taylor SS. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell. 2008;14:111–122. doi: 10.1016/j.ccr.2008.07.002. This article describes the fate of multiple cell lines cells treated with a wide range of mitotic drugs. This study also shows that CIN is independent of mitotic checkpoint functionality. [DOI] [PubMed] [Google Scholar]
- 17••.Solomon DA, Kim T, Diaz-Martinez LA, Fair J, Elkahloun AG, Harris BT, Toretsky JA, Rosenberg SA, Shukla N, Ladanyi M, et al. Mutational inactivation of STAG2 causes aneuploidy in human cancer. Science. 2011;333:1039–1043. doi: 10.1126/science.1203619. This article demonstrates the importance of the cohesin component STAG2 in accurate chromosome segregation by depleting this component and making chromosome segregation unstable in a nearly diploid cell line. The effort also confirmed this role by rescuing deficient STAG2 and restoring chromosomal stability in otherwise highly unstable aneuploid glioblastoma cell lines. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giehl M, Fabarius A, Frank O, Hochhaus A, Hafner M, Hehlmann R, Seifarth W. Centrosome aberrations in chronic myeloid leukemia correlate with stage of disease and chromosomal instability. Leukemia. 2005;19:1192–1197. doi: 10.1038/sj.leu.2403779. [DOI] [PubMed] [Google Scholar]
- 19.Lingle WL, Barrett SL, Negron VC, D'Assoro AB, Boeneman K, Liu W, Whitehead CM, Reynolds C, Salisbury JL. Centrosome amplification drives chromosomal instability in breast tumor development. Proc Natl Acad Sci U S A. 2002;99:1978–1983. doi: 10.1073/pnas.032479999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nam HJ, Chae S, Jang SH, Cho H, Lee JH. The PI3K-Akt mediates oncogenic Met-induced centrosome amplification and chromosome instability. Carcinogenesis. 2010;31:1531–1540. doi: 10.1093/carcin/bgq133. [DOI] [PubMed] [Google Scholar]
- 21.Fant X, Srsen V, Espigat-Georger A, Merdes A. Nuclei of non-muscle cells bind centrosome proteins upon fusion with differentiating myoblasts. PLoS One. 2009;4:e8303. doi: 10.1371/journal.pone.0008303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laronne A, Rotkopf S, Hellman A, Gruenbaum Y, Porter AC, Brandeis M. Synchronization of interphase events depends neither on mitosis nor on cdk1. Mol Biol Cell. 2003;14:3730–3740. doi: 10.1091/mbc.E02-12-0850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature. 2005;437:1043–1047. doi: 10.1038/nature04217. [DOI] [PubMed] [Google Scholar]
- 24.Meraldi P, Honda R, Nigg EA. Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53-/- cells. EMBO J. 2002;21:483–492. doi: 10.1093/emboj/21.4.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu Q, Sahasrabudhe RM, Luo LZ, Lewis DW, Gollin SM, Saunders WS. Deficiency in myosin light-chain phosphorylation causes cytokinesis failure and multipolarity in cancer cells. Oncogene. 2010;29:4183–4193. doi: 10.1038/onc.2010.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krzywicka-Racka A, Sluder G. Repeated cleavage failure does not establish centrosome amplification in untransformed human cells. J Cell Biol. 2011;194:199–207. doi: 10.1083/jcb.201101073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27•.Holland AJ, Lan W, Niessen S, Hoover H, Cleveland DW. Polo-like kinase 4 kinase activity limits centrosome overduplication by autoregulating its own stability. J Cell Biol. 2010;188:191–198. doi: 10.1083/jcb.200911102. This study describes how Plk4 self phosphorylations tightly regulate its cellular levels and implicates this mechanism in preventing cells from acquiring an abnormal number of centrosomes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28•.Brownlee CW, Klebba JE, Buster DW, Rogers GC. The Protein Phosphatase 2A regulatory subunit Twins stabilizes Plk4 to induce centriole amplification. J Cell Biol. 2011;195:231–243. doi: 10.1083/jcb.201107086. Using RNA interference and ectopic expression of PP2A in Drosophila cells, this article describes how PP2A phosphatase activity is required to counteract Plk4 self phosphorylation-mediated degradation, thereby stabilizing Plk4 in mitosis and allowing normal centriole duplication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dzhindzhev NS, Yu QD, Weiskopf K, Tzolovsky G, Cunha-Ferreira I, Riparbelli M, Rodrigues-Martins A, Bettencourt-Dias M, Callaini G, Glover DM. Asterless is a scaffold for the onset of centriole assembly. Nature. 2010;467:714–718. doi: 10.1038/nature09445. [DOI] [PubMed] [Google Scholar]
- 30.Arquint C, Sonnen KF, Stierhof YD, Nigg EA. Cell-cycle-regulated expression of STIL controls centriole number in human cells. J Cell Sci. 2012;125:1342–1352. doi: 10.1242/jcs.099887. [DOI] [PubMed] [Google Scholar]
- 31.Kitagawa D, Kohlmaier G, Keller D, Strnad P, Balestra FR, Fluckiger I, Gonczy P. Spindle positioning in human cells relies on proper centriole formation and on the microcephaly proteins CPAP and STIL. J Cell Sci. 2011;124:3884–3893. doi: 10.1242/jcs.089888. [DOI] [PubMed] [Google Scholar]
- 32.Vulprecht J, David A, Tibelius A, Castiel A, Konotop G, Liu F, Bestvater F, Raab MS, Zentgraf H, Izraeli S, et al. STIL is required for centriole duplication in human cells. J Cell Sci. 2012;125:1353–1362. doi: 10.1242/jcs.104109. [DOI] [PubMed] [Google Scholar]
- 33•.Ganem NJ, Godinho SA, Pellman D. A mechanism linking extra centrosomes to chromosomal instability. Nature. 2009;460:278–282. doi: 10.1038/nature08136. Using live cell imaging, this article demonstrates that multipolar division is lethal to cell progeny and describes how the formation of a transient multipolar spindle in the presence of extra centrosomes can promote chromosome segregation errors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leber B, Maier B, Fuchs F, Chi J, Riffel P, Anderhub S, Wagner L, Ho AD, Salisbury JL, Boutros M, et al. Proteins required for centrosome clustering in cancer cells. Sci Transl Med. 2010;2:33ra38. doi: 10.1126/scitranslmed.3000915. [DOI] [PubMed] [Google Scholar]
- 35.Duncan AW, Taylor MH, Hickey RD, Hanlon Newell AE, Lenzi ML, Olson SB, Finegold MJ, Grompe M. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature. 2010;467:707–710. doi: 10.1038/nature09414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Quintyne NJ, Reing JE, Hoffelder DR, Gollin SM, Saunders WS. Spindle multipolarity is prevented by centrosomal clustering. Science. 2005;307:127–129. doi: 10.1126/science.1104905. [DOI] [PubMed] [Google Scholar]
- 37.Kwon M, Godinho SA, Chandhok NS, Ganem NJ, Azioune A, Thery M, Pellman D. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev. 2008;22:2189–2203. doi: 10.1101/gad.1700908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang Z, Loncarek J, Khodjakov A, Rieder CL. Extra centrosomes and/or chromosomes prolong mitosis in human cells. Nat Cell Biol. 2008;10:748–751. doi: 10.1038/ncb1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Basto R, Brunk K, Vinadogrova T, Peel N, Franz A, Khodjakov A, Raff JW. Centrosome amplification can initiate tumorigenesis in flies. Cell. 2008;133:1032–1042. doi: 10.1016/j.cell.2008.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rieder CL, Cole RW, Khodjakov A, Sluder G. The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores. J Cell Biol. 1995;130:941–948. doi: 10.1083/jcb.130.4.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Waters JC, Chen RH, Murray AW, Salmon ED. Localization of Mad2 to kinetochores depends on microtubule attachment, not tension. J Cell Biol. 1998;141:1181–1191. doi: 10.1083/jcb.141.5.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42••.Kolano A, Brunet S, Silk AD, Cleveland DW, Verlhac MH. Error-prone mammalian female meiosis from silencing the spindle assembly checkpoint without normal interkinetochore tension. Proc Natl Acad Sci U S A. 2012;109:E1858–1867. doi: 10.1073/pnas.1204686109. By genetically removing the spindle pole focusing NuMA-dynein complex from the centrosome-free spindles in female meiosis in mice, this study establishes that in mammalian meiosis I the mitotic checkpoint cannot sustain meiotic arrest in the absence of normal interkinetochore tension. This discovery provides an explanation for why female meiosis I in mammals is so error prone. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thompson SL, Compton DA. Chromosome missegregation in human cells arises through specific types of kinetochore-microtubule attachment errors. Proc Natl Acad Sci U S A. 2011;108:17974–17978. doi: 10.1073/pnas.1109720108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44••.Gisselsson D, Jin Y, Lindgren D, Persson J, Gisselsson L, Hanks S, Sehic D, Mengelbier LH, Ora I, Rahman N, et al. Generation of trisomies in cancer cells by multipolar mitosis and incomplete cytokinesis. Proc Natl Acad Sci U S A. 2010;107:20489–20493. doi: 10.1073/pnas.1006829107. This study, performed on cells and tumor tissues, does what? – add more description. From those findings, it proposes an elegant model for how cells can missegregate multiple chromosomes in a single mitotic event. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hinchcliffe EH, Miller FJ, Cham M, Khodjakov A, Sluder G. Requirement of a centrosomal activity for cell cycle progression through G1 into S phase. Science. 2001;291:1547–1550. doi: 10.1126/science.1056866. [DOI] [PubMed] [Google Scholar]
- 46.Khodjakov A, Rieder CL. Centrosomes enhance the fidelity of cytokinesis in vertebrates and are required for cell cycle progression. J Cell Biol. 2001;153:237–242. doi: 10.1083/jcb.153.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Uetake Y, Loncarek J, Nordberg JJ, English CN, La Terra S, Khodjakov A, Sluder G. Cell cycle progression and de novo centriole assembly after centrosomal removal in untransformed human cells. J Cell Biol. 2007;176:173–182. doi: 10.1083/jcb.200607073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fukasawa K, Choi T, Kuriyama R, Rulong S, Vande Woude GF. Abnormal centrosome amplification in the absence of p53. Science. 1996;271:1744–1747. doi: 10.1126/science.271.5256.1744. [DOI] [PubMed] [Google Scholar]
- 49.Andreassen PR, Lohez OD, Lacroix FB, Margolis RL. Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1. Mol Biol Cell. 2001;12:1315–1328. doi: 10.1091/mbc.12.5.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Uetake Y, Sluder G. Cell cycle progression after cleavage failure: mammalian somatic cells do not possess a “tetraploidy checkpoint”. J Cell Biol. 2004;165:609–615. doi: 10.1083/jcb.200403014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wong C, Stearns T. Mammalian cells lack checkpoints for tetraploidy, aberrant centrosome number, and cytokinesis failure. BMC Cell Biol. 2005;6:6. doi: 10.1186/1471-2121-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aylon Y, Michael D, Shmueli A, Yabuta N, Nojima H, Oren M. A positive feedback loop between the p53 and Lats2 tumor suppressors prevents tetraploidization. Genes Dev. 2006;20:2687–2700. doi: 10.1101/gad.1447006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cuomo ME, Knebel A, Morrice N, Paterson H, Cohen P, Mittnacht S. p53-Driven apoptosis limits centrosome amplification and genomic instability downstream of NPM1 phosphorylation. Nat Cell Biol. 2008;10:723–730. doi: 10.1038/ncb1735. [DOI] [PubMed] [Google Scholar]
- 54•.Holland AJ, Fachinetti D, Da Cruz S, Zhu Q, Vitre B, Lince-Faria M, Chen D, Parish N, Verma IM, Bettencourt-Dias M, et al. Polo-like kinase 4 controls centriole duplication but does not directly regulate cytokinesis. Mol Biol Cell. 2012;23:1838–1845. doi: 10.1091/mbc.E11-12-1043. Following from a report that reduction in Plk4 kinase regulates cytokinesis by regulating the Rho A guanine exchange factor, this report demonstrates that Plk4 is localized to centrosomes, but not the cytokinetic furrow. Further, from efforts using targeted inactivation of one Plk4 allele or shRNA to Plk4, it is concluded that Plk4 governs centrosome duplication, but plays no direct role in cytokinesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tarapore P, Horn HF, Tokuyama Y, Fukasawa K. Direct regulation of the centrosome duplication cycle by the p53-p21Waf1/Cip1 pathway. Oncogene. 2001;20:3173–3184. doi: 10.1038/sj.onc.1204424. [DOI] [PubMed] [Google Scholar]
- 56.Shinmura K, Bennett RA, Tarapore P, Fukasawa K. Direct evidence for the role of centrosomally localized p53 in the regulation of centrosome duplication. Oncogene. 2007;26:2939–2944. doi: 10.1038/sj.onc.1210085. [DOI] [PubMed] [Google Scholar]
- 57.Ferguson RL, Maller JL. Centrosomal localization of cyclin E-Cdk2 is required for initiation of DNA synthesis. Curr Biol. 2010;20:856–860. doi: 10.1016/j.cub.2010.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mikule K, Delaval B, Kaldis P, Jurcyzk A, Hergert P, Doxsey S. Loss of centrosome integrity induces p38—p53—p21-dependent G1—S arrest. Nature Cell Biology. 2006;9:160–170. doi: 10.1038/ncb1529. [DOI] [PubMed] [Google Scholar]
- 59.Mullins JM, Biesele JJ. Terminal phase of cytokinesis in D-98s cells. J Cell Biol. 1977;73:672–684. doi: 10.1083/jcb.73.3.672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60••.Janssen A, van der Burg M, Szuhai K, Kops GJ, Medema RH. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science. 2011;333:1895–1898. doi: 10.1126/science.1210214. Using non transformed RPE-1 cells, the authors demonstrate that missegregated chromosomes are often damaged during cytokinesis, producing sustained DNA double strand breaks and structural rearrangements. As a consequence of that damage, an ATM/Chk2 and p53 DNA damage response is activated leading to cell cycle arrest at the next G1/S transition. [DOI] [PubMed] [Google Scholar]
- 61••.Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, Nezi L, Protopopov A, Chowdhury D, Pellman D. DNA breaks and chromosome pulverization from errors in mitosis. Nature. 2012;482:53–58. doi: 10.1038/nature10802. This article describes how missegregated chromosomes are encapsulated into micronuclei which assemble with a surprisingly limited number of nuclear pores. The paucity in pores leads to delayed DNA replication within the micronucleus, but not a delay in cell cycle progression, thereby producing massive DNA damage within the micronuclear DNA following premature mitotic entry. This study proposes that this series of events could be responsible for the high levels of genomic rearrangements observed in cells affected by chromothripsis, an event frequently seen in tumors in which one (or a few) chromosome(s) undergoes tens to hundreds of highly localized rearrangements. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62•.Hayashi MT, Cesare AJ, Fitzpatrick JA, Lazzerini-Denchi E, Karlseder J. A telomere-dependent DNA damage checkpoint induced by prolonged mitotic arrest. Nat Struct Mol Biol. 2012;19:387–394. doi: 10.1038/nsmb.2245. This work proposes a novel mechanism involving telomere deprotection and activation of a p53 DNA damage response to explain why cells stop dividing after a prolonged mitotic arrest. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Uetake Y, Sluder G. Prolonged prometaphase blocks daughter cell proliferation despite normal completion of mitosis. Curr Biol. 2010;20:1666–1671. doi: 10.1016/j.cub.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]