

Abstract
NRIF3 is an estrogen-inducible nuclear receptor coregulator that stimulates estrogen receptor-alpha (ERα) transactivation functions and associates with the endogenous ER and its target gene promoter. P21-activated protein kinase 1 (Pak1) phosphorylates ERα at Ser305 and this modification plays an important role in ERα transactivation function. Although ERα transactivation functions are regulated by coactivator activity of NRIF3(Talukder et al., 2004), it remains unclear whether Pak1 could impact ER functions via a posttranslational modification of NRIF3. Here, we report that Pak1 phosphorylates NRIF3 at Serine28 and that NRIF3 binds to Pak1 in vitro and in vivo. We found that NRIF3 phosphorylation, coactivator activity, and association with ERα increased following Pak1 phosphorylation of NRIF3’s Ser28 and that activated ERα-Ser305 and NRIF3-Ser28 cooperatively support transactivation of ERα. NRIF3 expression increased significantly in cells with inducible Pak1 expression. We found that NRIF3 and ERα interaction, sub-cellular localization, and ERα transactivation activity all increased in cells expressing the Pak1 phosphorylation mimicking mutant NRIF3-Ser28Glu. Consistently, the NRIF3-Ser28Glu mutant exhibited an enhanced recruitment to the endogenous ER target genes and increased expression following estrogen stimulation. Finally, breast cancer cells with stable over-expression of NRIF3 showed increased proliferation and enhanced anchorage-independent growth. These findings suggest that NRIF3-Ser28 is a physiologic target of Pak1 signaling and contributes to the enhanced NRIF3 coactivator activity, leading to coordinated potentiation of ERα transactivation, its target gene expression, and estrogen responsiveness of breast cancer cells.
Keywords: NRIF3, Pak1, ERα, phosphorylation
Introduction
Coactivators are a class of proteins that interact with sequence-specific transcription factors to enhance the transcription of target genes. Recent studies have identified a number of coactivators that associate with nuclear receptors (NR) in a ligand-dependent manner to stimulate the transactivation functions of NR. A partial list of these coactivators includes SRC-1/NCoA-1, TIF2/GRIP1/NCoA-2, pCIP/ACTR/AIB1, the TRAP/DRIP complex (Kamei et al., 1995; Onate et al., 1995), and NRIF3, an estrogen-inducible nuclear receptor coactivator (Talukder et al., 2004). Interaction between the ligand-activated NR and these coactivators is mediated by a small α-helical motif containing the sequence LXXLL (where L denotes leucine and X is any amino acid) in the coactivator proteins. The LXXLL motif is crucial but not sufficient for the interaction with the NR, since different nuclear receptors show overlapping but distinct preferences for individual LXXLL motifs, and sequences outside this motif are also important. (Freedman 1999; Glass and Rosenfeld, 2000).
The activities of transcription factors are also regulated by phosphorylation. The importance of phosphorylation for ERα function is exemplified by growth factors such as epidermal growth factor, heregulin, insulin, and the insulin-like growth factor TGFα, as well as cyclic AMP (cAMP) and phorbol esters, can also activate ERα (Aronica and Katzenellenbogen, 1993; Bunone et al., 1996; Ignar et al., 1993; Ma et al., 1994; Metzger et al., 1995; Patrone et al., 1996; Pietras et al., 1995). Activated ERα, in-turn, stimulates transcription of its target genes. Phosphorylation of ERα on Ser104 and/or Ser106, Ser118 and Ser167, and Tyr537 has been shown to support it transactivation function (Ali et al., 1993; Castoria et al., 1993; Joel et al., 1995; Le Goff et al., 1994). Phosphorylation of human ERα at serine 118 is mediated by the RAS/MAPK pathway(Bunone et al., 1996; Kato et al., 1995); activation of the MAPK pathway enables ligand-independent transactivation of human ERα by MAPK or its upstream activator (Bunone et al., 1996). Mutation of Ser118 to Ser118-Ala, which impairs the phosphorylation on Ser118, reduces its transcription ability (Ali et al., 1993; Le Goff et al., 1994). Similarly, Pak1 directly phosphorylates ERα at the N-terminal residue Ser305, and mutation of this residue to Ala (Ser305-Ala) abolishes the Pak1-mediated phosphorylation and transactivation functions of the ERα, while its substitution by glutamic acid (Ser305-Glu) which mimic phosphorylation, promotes transactivation activity of ERα (Balasaenthil et al., 2004; Wang et al., 2002). Intriguingly, several lines of evidence show that polypeptide factor signaling can also modulate the functional activity of NR coregulatory factors via posttranslational phosphorylation events (reviewed by Hermanson et al., 2002; Spiegelman and Heinrich, 2004; Wu et al., 2005). For example, phosphorylation of specific members of the steroid receptor coactivator (SRC)/p160 family of proteins via different signal transduction pathways can enhance nuclear localization of SRC(Wang et al., 2000; Wu et al., 2002), inhibit interactions with non-NR activators (Lazaro et al., 2002), or stimulate intrinsic SRC/p160 coactivator activity (Font de Mora et al., 2000; Rowan et al., 2000; Wu et al., 2004). Similarly, phosphorylation of the NR coactivator PGC-1 via theP38-MAPK significantly stabilizes protein expression, leading to increased levels of coactivator activity (Pierce et al., 2001).
Earlier studies have shown a role of NRIF3 in potentiation of transactivation functions of the NR (Li et al., 1999; Talukder et al., 2004). Further NRIF3 itself is an estrogen-inducible gene with an ER responsive motif in its gene promoter sequence (Talukder et al., 2004). It was also shown that the N-terminal LXXLL (RID2) motif of NRIF3 is necessary for binding with ERα, that NRIF3 association with estrogen-responsive gene-chromatin is estrogen sensitive, and that this association could be inhibited by the transcriptional corepressor MTA1. Deletion of the first 28 amino acids of NRIF3 (which contains the RID2 domain and LXXLL motif) mimics NRIF3’s ERα transactivation functions (Talukder et al., 2004). Although the deletion of the first 28 amino acids in NRIF3 impairs its binding to ERα (Talukder et al., 2004), it remains unclear whether the impairment is due to inefficient binding or another mechanism that modifies ER-transactivation function of NRIF3 in a physiological setting. The purpose of our study was to explore the existence of mechanistic linkage between NRIF3 and ER. We found that NRIF3 is a phosphoprotein that has a consensus Pak1 phosphorylation site on Ser28 [(K/R) (R/X) (X) (S/T)] and that NRIF3 is phosphorylated on Ser28 by Pak1. We show that this phosphorylation of NRIF3 on Ser28 promotes the ability of NRIF3 to transactivate ERα by enhancing recruitment of NRIF3 to the ERα target gene chromatin, leading to enhanced anchorage-independent growth of breast cancer cells.
Results and Discussion
Pak1 phosphorylates and interacts with NRIF3
To identify the potential posttranslational modifications that might affect NRIF3’s coactivation of ERα, we searched the NetPhos 2.0 server (www.cbs.dtu.dk/services/Netphos ) for phosphorylation sites in NRIF3. We found several phosphorylation sites. Among these, we identified Ser28 (RKKS) as a putative Pak1 phosphorylation site [(K/R) (R/X) (X) (S/T)]. Using GST-purified Pak1 enzyme, we performed an in vitro kinase assay using GST-NRIF3 and GST-NRIF3-29del (Talukder et al., 2004). We found that Pak1 phosphorylates NRIF3 profoundly but not GST alone or the mutant NRIF3 lacking the first 28 amino acids (Fig. 1A). We next generated NRIF3 mutant wherein Ala was substituted for Ser28: Pak1 was unable to phosphorylate this mutant NRIF3 (Fig. 1B). These results indicate that Pak1 phosphorylated NRIF3 specifically at Ser28 in an in vitro system.
Figure 1. Pak1 phosphorylates NRIF3.
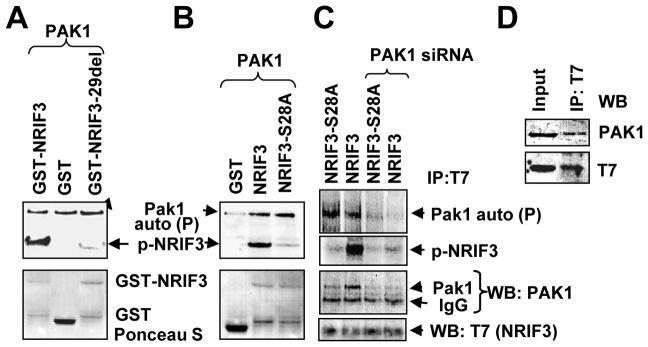
(a–b) In vitro kinase assays were performed using purified Pak1 enzyme either with GST-NRIF3-29del (a) or GST-NRIF3-S28A (b) or GST alone as a negative control and GST-NRIF3 as a positive control. On the upper panels (autoradiogram), the upper band indicates Pak1 autophosphorylation and the lower band indicates NRIF3 phosphorylation (p-NRIF3). The lower panels (ponceau-S staining) indicate the amount of transferred GST-NRIF3 or GST on the blot. (c) Phosphorylation and binding of transiently expressed T7-NRIF3 (or T7-NRIF3-S28A) with endogenous Pak1 in MCF-7 cells in the presence of 32P-orthophosphate. The cell lysates were immonoprecipitated with anti-T7-antibody and developed by autoradiography or by Western blotting. The upper two panels show Pak1 auto-phosphorylation and phosphorylation of NRIF3; the lower two panels show the same blots developed against anti-Pak1 antibody and anti-T7-antibody (NRIF3). (d) Transiently expressed T7-NRIF3 in MCF-7 cells binds with endogenous Pak1. Cell lysate was immunopecipitated with anti-T7 monoclonal antibody and Western blot with anti-Pak1 antibody and anti-T7 antibody.
To identify the mechanism of NRIF3 phosphorylation in a mammalian system, we generated T7-tagged NRIF3-S28A and transfected MCF-7 cells with T7-NRIF3 or T7-NRIF3-S28A in the presence of 32P-orthophosphoric acid and in the presence or absence of control or Pak1-specific siRNAs. Cell lysates were immunoprecipitated with anti-T7 monoclonal antibody, resolved by SDS-PAGE, transferred to a blotting membrane, and auto-radiographed. The same blot was probed against either anti-Pak1 or anti-T7 antibody. As shown in Fig. 1C, transiently expressed T7-NRIF3 was substantial phosphorylation in MCF-7 cells (lane 2) and such phosphorylation was inhibited in NRIF3-S28A (lane 1) or by Pak1-siRNA (lanes 3, 4). The lower two panels in Fig. 1C show that T7-NRIF3 also interacted with endogenous Pak1. Transiently expressed T7-NRIF3 in MCF-7 cells also bound with endogenous Pak1 (Fig. 1D). These findings indicate that NRIF3 is a phosphoprotein and that Pak1 interacts with and phosphorylates NRIF3 at Ser28.
NRIF3 binds with Pak1 and its expression and phosphorylation is Pak1 sensitive
Since Pak1 interacted with and phosphorylated NRIF3, we next explored whether NRIF3 would also interact with endogenous Pak1. Total cell lysates from MCF-7 cells were immunoprecipitated with Pak1 antibody or NRIF3 antibody and blotted with antibodies against NRIF3 or Pak1. Results showed that Pak1 weakly bound to NRIF3 (Fig. 2A) and that NRIF3 binding to Pak1 increased when the cells were stimulated with EGF (Fig. 2B). The self-immunoprecipitation of NRIF3 was shown in Fig. 2C. We also examined the phosphorylation status of endogenous NRIF3. As shown in Fig. 2D, endogenous NRIF3 was phosphorylated in the presence of 32P-orthophosphoric acid in a Pak1-sensitive manner: Pak1 overexpression and Pak1 depletion in MCF-7 cells increased and decreased the phosphorylation of NRIF3, respectively (Fig. 2E).
Figure 2. NRIF3 is a Pak1-interacting substrate in a Pak1-sensitive manner.

(a–c) NRIF3 binds with Pak1 in vivo. Total lysates were made from MCF-7 cells treated with or without EGF overnight, immunoprecipitated with IgG, Pak1, or NRIF3 antibodies, and resolved on SDS-PAGE. Western blot analysis was performed using antibody against Pak1 or NRIF3. (a) Pak1 binds with NRIF3 (4th lane from left; 1st and 3rd lane as positive control). (b) NRIF3 binds strongly with Pak1 in presence of EGF (3rd lane, lower band). (c) NRIF3 can be detected by self-immunoprecipitation. (d) NRIF3 is phosphorylated in vivo. MCF-7 cells were labeled with 32P orthophosphate and immunoprecipitated with NRIF3 antibody or IgG, resolved on SDS-PAGE, transferred to a blot, and examined by autoradiography or Western blot analysis using anti-NRIF3 antibody. Upper panel is the autoradiogram and lower panel is the Western blot analysis. (e) Over-expressing Pak1 clones (left panel), MCF-7 cells with or without serum (middle panel), Pak1 silencing SH-RNA Pak1 or vector clones were labeled with 32P orthophosphate and immunoprecipitated with NRIF3 antibody. Upper panels show the change in NRIF3 phosphorylation as Pak1 is included or depleted.
NRIF3 promotes ER transactivation function in a Pak1 sensitive manner
Since Pak1 phosphorylated NRIF3 and bound to the endogenous NRIF3, we hypothesized that Pak1-mediated phosphorylation of NRIF3 influences its transactivation activity. Since NRIF3 is an estrogen response gene (Talukder et al., 2004) and estrogen induced the expression of NRIF3 in an anti-estrogen dependent manner (Fig. 3A), we investigated whether phosphorylation of NRIF3 itself had any coactivator activity upon ER transactivation function. To determine whether Ser28 phosphorylation affects the ability of NRIF3 to coactivate the ERα transactivation function, we transfected MCF-7 cells with ERE-luc along with T7-NRIF3 or NRIF3-S28A or NRIF3-specific or control siRNA. NRIF3 proved to be a potent coactivator of ER-transactivation activity (Fig. 3B), and its coactivator function severely impaired when the Pak1 phosphorylation site in NRIF3 was inactivated (i.e. NRIF3-S28A) or when endogenous NRIF3 was depleted by siRNA (Fig. 3B). The results suggest that NRIF3 has a Ser28 phosphorylation-dependent ability to coactivate ER-transactivation activity. This finding provides a new perspective to the earlier report of Pak1 phosphorylation of ERα-Ser305 and transactivation function (Wang et al., 2002) and raises the possibility that Pak1 phosphorylation of NRIF3 contributes to the coactivator activity of NRIF3. Consistent with this notion, NRIF3 and Pak1 co-stimulated ERE-transactivation, whereas Pak1 knockdown (Fig. 3C) or dominant-negative Pak1 (DN-Pak1) (Fig. 3D) reduced the abililty of NRIF3 to coactivate ERE transactivation.
Figure 3. Pak1 stimulates NRIF3’s ER transactivation functions.

(a) Western blots showing estrogen responsiveness by NRIF3. NRIF3 is increased by estrogen but decreased by the estrogen antagonist ICI. (b–d) Transactivation function of ERα was assessed by transfecting an ERE-luciferase construct (200 ng per well in six-well plate) in presence or in absence of Pak1 or NRIF3 in MCF-7 cells. All the experiments were carried out at 3% DCC and cells were treated for last 15 h with or without estrogen (10−9 M). Luciferase activity was measured 60 h after transfection. (b) ERE luciferase activities measured in presence of NRIF3 or NRIF3-S28A (200 ng) or NRIF3-siRNA (50nM final concentration). (c) In presence of NRIF3, Pak1, or both, and in some cases Pak1 siRNA. (d) In presence of pcDNA3.1A, NRIF3, Pak1, and dominant negative Pak1 (DN-Pak1). All the data are the mean value of three independent experiments.
NRIF3 Ser28 phosphorylation affects its interactions with ERα
We next examined the binding of ER to NRIF3 and to the phosphorylation mimic mutant of NRIF3 (NRIF3-Ser28 to glutamic acid modification) (ER-S305E) and an inactive mutant (ER-S305A). We found that negatively charged phosphorylation mimic mutant of NRIF3 bound more strongly with ERα than NIRIF3 (Fig. 4A). Like-wise, negatively charged phosphorylation mimic mutant of ERα bound more strongly to NRIF3 than the phosphorylation inactive mutant of ERα (Fig. 4B). These findings suggest that negative charge on these serine residues promotes interaction between the two molecules. This constitutive negative charge might also promote colocalization of NRIF3 with ERα. To examine this possibility, we first determined the colocalization of T7-NRIF3 with endogenous ERα. Results in Fig. 4C show that T7-NRIF3 colocalized with the endogenous ERα. We also found that endogenous NRIF3 bound strongly with endogenous ERα in an E2-sensitive manner (Fig. 4D).
Figure 4. Binding and colocalization of NRIF3 with ERα.
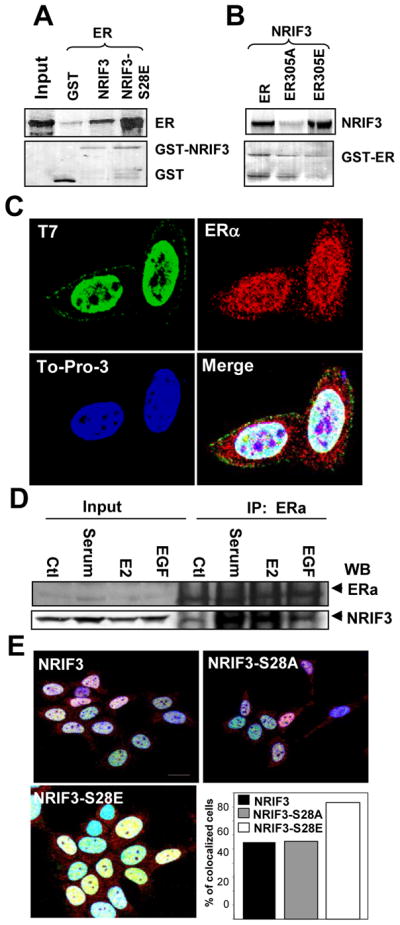
(a–b)GST-pull down assay was carried out either with GST, GST-NRIF3 or GST-NRIF3-S28E with 35S ERα (a) and GST-ERs with 35S NRIF3 (b). Upper panels are the autoradiogram showing bound ERα (a) and bound NRIF3 (b). Lower panels are ponceau-S staining of the blots showing the amount of protein transferred to the blots. (c) Immunofluorescent localization of transfected T7-NRIF3 (green) and endogenous ERα (red) in MCF-7 cells grown in regular medium for 24 h. Blue, DNA dye TO-PRO-3; yellow indicates colocalization of green and red fluorescence. Bar=10 μm. (d) NRIF3 binds with ERα in vivo. MCF-7 cells were treated with 10% serum, E2, or EGF and IP with ERα antibody. Western blots analysis was performed with ERα or with NRIF3 antibody. (e) Colocalization of NRIF3 and ERα is affected by ser28 of NRIF3. Cells were grown in DCC medium for 48 h. Blue, DNA dye TO-PRO-3; yellow, colocalization of green and red fluorescence. For quantification, cells demonstrating nuclear colocalization (yellow) of transfected NRIF3 (green) and endogenous ERα (red) were counted in at least 150 cells per treatment group and graphed as a percentage of the total cells counted. Bar=10 μm.
Next we determined the localization of wild-type NRIF3, NRIF3-S28A, and NRIF3-S28E with ERα in MCF-7 cells. Cells were transfected as described in the Methods section. As shown in Figure 4E, consistent weak colocalization of ERα with NRIF3 or NRIF3-S28A was seen in 54% and 55%, respectively, of the cells in DCC media. However, NRIF3-S28E was colocalized with ERα in 83% of transfected cells, suggesting that NRIF3 phosphorylation mimicking modification displayed better colocalization with ERα. Together these studies suggest that the phosphorylation of NRIF3-Ser28 plays a role in promoting interaction with ERα in breast cancer cells.
NRIF3-S28E and ER-S305E stimulate ER’s transactivation in a cooperative manner
Since the interaction of phosphorylation mimic mutants of NRIF3 with ER was enhanced, we examined the possibility that NRIF3-S28E would further promote the transactivation function of ER-S305E. Indeed, NRIF3 promoted the transactivation activity of ER-S305E but not ER-S305A in ER-negative HeLa cells (Fig. 5A), and the contribution of NRIF3 to ERE-transactivation was much more significant with ER-S305E than with ER (Fig. 5B). To directly delineate the possible role of Ser28 of NRIF3 on the ERE-transactivation, we co-expressed NRIF3-S28E and ER-S305E and measured ERE-transactivation activity: the increase in transactivation activity was robust (Fig. 5C), suggesting that phosphorylated NRIF3 is a very strong coactivator of ER’s transactivation function.
Figure 5. NRIF3-S28E stimulates ERα’s transactivation function.

(a) HeLa cells were transfected with ERα, ERα-S305E, or ERα-S305A with or without NRIF3 in presence of ERE-luciferase construct. (b) NRIF3 was transfected into HeLa cells along with ERα or ERα-S305E, treated with or without estrogen. (c) ERα and ERα-S305E were transfected with NRIF3, NRIF3-S28A, or NRIF3-S28E in six different combinations treated with or without estrogen. All the luciferase assays were done 48 h after transfection, which included last 8 h of estrogen treatment. All data are the mean value of three independent experiments.
A role of Ser28 of NRIF3 in the stimulation of ER target genes
Earlier we showed that NRIF3 promotes the expression of PS2 and cyclinD1 (Talukder et al., 2004). To examine the role of NRIF3’s Ser28 phosphorylation in the stimulation ER target genes such as pS2 and cyclin D1, we generated stable clones of T7-tagged vector (pcDNA), NRIF3, NRIF3-S28A, and NRIF3-S28E (Supplementary Fig. 1) and measured levels of pS2 and cyclin D1. We found increased levels of estrogen-inducible pS2 and cyclin D1 mRNAs in cells overexpressing NRIF3 or NRIF3-S28E but not NRIF3-S28A (Fig. 6A). The levels of cyclin D1 protein (Fig. 6B) and cyclin D1 promoter activity (Fig. 6C) were also increased in MCF7/NRIF3 and in MCF-7/NRIF3-S28E clones. Consistent with these results, chromatin-immunoprecipitation (ChIP) assays showed that NRIF3 and NRIF3-S28E (as compared to NRIF3-S28A) were effectively recruited to the promoters of pS2 and cyclin D1 in estrogen-stimulated cells (Figs. 6D, 6E). In brief, these findings suggest that Ser28 phosphorylation of NRIF3 plays an important role in stimulating ER-responsive genes, presumably due to increased NRIF3-ER interaction and recruitment to the target gene chromatins.
Figure 6. NRIF3 enhances expression and promoter activity of other ER target genes and associates with their gene promoters.

(a) Expression of PS2 and cyclinD1 in control pcDNA3.1 and other NRIF3 stable clones were analyzed by RT-PCR. Actin was used as an internal control. (b) Expression of cyclinD1 was measured by Western blot analysis; vinculin was used as internal control. (c) CyclinD1 luciferase activity was measured in the overexpressing NRIF3 clones; in some cases, estrogen was added for the last 8 hours. All data are the mean value of three independent experiments. (d–e) The association of NRIF3 with E2-responsive genes as analyzed by ChIP assay. Stable transfectants of pcDNA-, NRIF3-, NRIF3-S28A, or NRIF3-S28E-MCF-7 cells were treated with estrogen (10 nM for 45 min), and chromatin lysates were immunoprecipitated with anti-T7 antibody or control IgG; samples were processed as described in Methods. Upper panels show PCR analysis of pS2 (d) and cycD1(e) promoter fragments associated with T7–NRIF3.
Ser28 phosphorylation of NRIF3 stimulates proliferation of breast cancer cells
To understand the significance of Ser28 phosphorylation of NRIF3 upon the biology of breast cancer cells, we next examined the effect of estrogen on the proliferation of the pooled clones of MCF-7 cells stably expressing NRIF3, NRIF3-S28A, NRIF3-S28E or pcDNA. Results indicated that overexpression of NRIF3 or NRIF3-S28E promoted the baseline as well as estrogen-induced growth of MCF-7 cells which could be further enhanced by estrogen (Fig. 7A), implicating NRIF3-Ser28 in the estrogen-responsiveness of breast cancer cells. Consistent with these results, expression of NRIF3 or NRIF3-S28E but not NRIF3-S28A promoted both basal and estrogen-inducible growth of MCF-7 in an anchorage-independent manner (Fig. 7B). In brief, Ser28 phosphorylation of NRIF3 leads to cellular consequences.
Figure 7. Ser28 plays a role in NRIF3’s stimulation of cell growth.
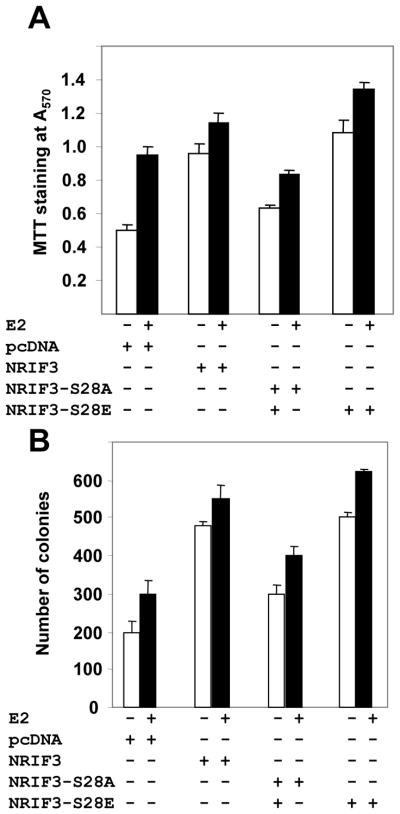
(a) NRIF3, NRIF3-S28A, NRIF3-S28E or pcDNA3.1 over expressing stable clones in MCF-7 cells were grown in regular medium. After 24 h regular medium was changed to white medium containing 3% DCC for estrogen treatment. At 72 h, estrogen was added to some cells for and additional 15 h and an MTT assay for cell proliferation performed. (b) Colony growth assays were performed with the same clones; they were seeded in soft agar in triplicate and maintained for 28 days in absence or presence of E2 (10−9M); total number of colonies were recorded.
Conclusions
In our study described here, we identified Serine 28 (RKKS) of NRIF3 as a Pak1 phosphorylation site. In addition, we found that NRIF3 but not its Ser28-phosphorylation mutant confers coactivator function upon ER-target genes. Phosphorylation of co-activators modifies 3D-conformation of coregulators in favor of a stabilized coactivator, leading to increased coactivator activity. Consistent with this notion, NRIF3-Ser28 phosphylation potentiated ERα transactivation function, presumably due to an enhanced NRIF3-ERα interaction and its resulting recruitment to the ER-target gene chromatin. Since Pak1 signaling is known to phosphorylate ERα on Ser305 and we have now established that Pak1 also phosphorylates NRIF3-Ser28, we also established a positive co-operative role of NRIF3-Ser28 and ERα-Ser305 in stimulating ER-transactivation function and expression of the target genes pS2 and Cyclin D1. Our results implicated activated NRIF3 (i.e. NRIF3-S28E) in the ER-induced stimulation of proliferation and anchorage-independent growth in breast cancer cells, thus demonstrating the functional significance of these molecular interactions. Collectively, the results presented here provide novel insights into NRIF3 function, leading to recognization of its phosphorylation-dependent coactivator activity upon ERα transcriptional action, and consequently, growth stimulation of breast cancer cells.
Materials and Methods
Cell culture and antibodies
Human breast cancer cells were cultured in DMEM/F12 medium supplemented with 10% fetal bovine serum. For estrogen treatment, regular medium was replaced by medium containing 3% DCC (charcoal-stripped serum). Antibody against NRIF3 (Cat# 10743-1-AP) was purchased from Proteintech Group Inc., Chicago, IL; anti-T7-tag (Cat# 69522) from EMD Chemicals, Inc., San Diego, CA; anti-HA-tag (Cat# sc-7392), anti-ERα (Cat# sc-7207), and anti-cyclin D1 from the Santa Cruz Biotechnology, Santa Cruz, CA; anti-actin (Cat# a-4700) and anti-vinculin (Cat# v-9131) from Sigma-Aldrich, St. Louis, MO; anti-Pak1 (Cat# 2602) from Cell Signaling Technology, Boston, MA. Anti-mouse- and anti-rabbit-horseradish peroxidase-conjugates were purchased from Amersham, Piscataway, NJ.
Pak1 kinase assay
In vitro kinase assays using GST-NRIF3 fusion protein were performed in HEPES buffer (50 mM HEPES, 10 mM MgCl2, 2 mM MnCl2, 1 mM DTT) containing 1 μg of purified bacterially expressed GST-Pak1 enzyme, 10 μCi of [γ-32P]ATP, and 25 μM unlabeled ATP. The reaction was carried out in a 30 μl volume for 30 min at 30°C and then stopped by addition of 10 μl of 4X SDS buffer. Reaction products were then analyzed on a SDS-PAGE, transferred to a blot, and developed by autoradiography.
Plasmid Construction and Point Mutations at Serine28 of NRIF3
A PCR based approach was used for site-directed mutagenesis of the putative Pak1 phosphorylation site at ser28 of full-length GST-NRIF3. We used the following primers: for Ser28Ala, the forward primer was 5′AGGAAGAAAGCTGTTATAACTTAT 3′ and the reverse primer was 5′ATAAGTTATAACAGCTTTCTTCCT 3′; for Ser28Glu, the forward primer was 5′AGGAAGAAAGAGGTTATAACTTAT 3′ and the reverse primer was 5′ATAAGTTATAACCTCTTTCTTCCT 3′ using Stratagene’s Quick Change site-directed mutagenesis kit. To make T7-tagged S28A and S28E-mutated full-length NRIF3 for mammalian expression, we digested the GST-constructs with BamHI and NotI and ligated them into pcDNA3.1A (Invitrogen) at the same sites.
In vitro transcription, translation, and GST pull-down assays
In vitro transcription and translation of the test proteins were performed using the TNT-transcription-translation system (Promega). One microgram of T7-tagged plasmid DNA was translated in the presence of 35S-labeled methionine in a 50-μl reaction volume by using the T7-TNT reaction mixture. The reaction mixture was diluted to 1 ml with the NP40 lysis buffer, and an aliquot (250 μl) was used for each GST pull-down assay. The identity of of the translated reaction mixture was verified by subjecting 2 μl to SDS-PAGE and autoradiography. The GST pull-down assays were performed by incubating equal amounts of GST or GST-fusion protein immobilized to glutathione-Sepharose beads (Amersham) with in vitro translated 35S-labeled test protein. The mixtures were incubated for 2 h at 4°C and washed six times with NP40 lysis buffer. Bound proteins were eluted with 2xSDS buffer, separated by SDS-PAGE, and visualized by autoradiography. The transferred protein on the blot was visualized using Ponceau S stain.
Interference of NRIF3 and Pak1 expression by siRNA
We used small interfering RNA (siRNA) designed by Qiagen to target human NRIF3 (Talukder et al., 2004). Human Pak1 siRNA was bought from Cell Signaling (Beverly, MA). As a control, we used a commercially available nonspecific random siRNA. MCF-7 cells were seeded at 30% density the day before transfection in 6-well plates. On the day of transfection, cells were changed to antibiotic-free 10% serum or 3% DCC medium (for estrogen treatment). siRNA transfections were performed by using Oligofectamin reagent (Invitrogen) according to the manufacturer’s instructions with 12 μl of 20 μM siRNA and 4 μl of oligofectamin reagent/well in 6-well plates (50 nM final concentration). For estrogen treatment, cells were transfected with siRNA in 3% DCC serum. For luciferase assay, cells were re-transfected after 24 h of siRNA transfection with ERE-luciferase plus other plasmids and incubated in 3% DCC medium for another 48 h; estrogen (10−9M) was added during the last 8 h before harvest.
Transient transfection, immunoprecipitation, in vivo phosphorylation, and Western blot
MCF-7 cells at 70% confluence in 60-mm plates were transiently transfected with 4 μg of T7-tagged wtNRIF3 or S28A-NRIF3 using Fugene method (Roche). Some plates were also transfected with Pak1 siRNA (50 nM final concentration) 24 h before plasmid transfection. For in vivo phosphorylation, cells were washed with phosphate-free medium containing 1X Na-pyruvate, 1X L-glutamine, and 2% dialyzed FCS and incubated for 1 h in the same medium. Then 32P-labeled orthophosphate (20 μl/ml; specific activity, 20 μCi/ml) was added and incubated overnight at 37°C. Cells were washed with 1XPBS and lysed in TritonX-100 lysis buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 1.5 mM MgCl2, 1mM EGTA, 1 mM DTT, 1% Triton X-100, 10% glycerol containing 1 mM NaVO5, protease inhibitor cocktail (Roche) for 15 min on ice and immunoprecipitated with monoclonal anti-T7-agarose beads or NRIF3-antibody for 4 h at 4°C, washed three times with washing buffer, boiled for 5 min in 2X SDS-loading buffer and run into 12 % SDS-PAGE, and finally transferred to nitrocellulose blot overnight at 4°C. For in vivo phosphorylation, the blot was developed by autoradiogram, and for Western blot the same membrane was blocked with 5% fat-free milk and developed against monoclonal T7-antibody or anti-Pak1 antibody using ECL method.
Stable cell lines
MCF-7 cells were transfected with three different plasmids of T7-NRIF3 constructs (wtNRIF3, S28A-NRIF3, and S28E-NRIF3). After 48 h, G418 (500 μg/ml final concentration) was added and selection pressure maintained for 3 weeks. Clones from each plate were pooled and maintained as individual clones. Total RNA was isolated from cell lines stably transfected with pcDNA and NRIF3 using Trizol (Life Technologies, Inc., Rockville, MD). Expression of T7-NRIF3 was verified by RT-PCR using T7-tagged forward primer is pcDNA982For 5′ CAGCAAATGGGTCGGGATC 3′ and the reverse primer was NRIF3-373Rev 5′ GCTCTCTACTGCCCTCCAAA 3′ and Western blot using anti-T7 mAb and NRIF3 antibody (PTG Inc. Chicago, IL)
Immunofluorescent labeling and confocal microscopy
Indirect immunofluorescence was used to establish the cellular localization of the T7 tag of transfected wild-type NRIF3 and endogenous ERα. Briefly, MCF-7 cells were plated on sterile glass cover slips in 6-well plates and allowed to attach overnight. Cells were then co-transfected with wild type T7-NRIF3. After 24 h, cells were rinsed twice in PBS, fixed in 4% phosphate-buffered paraformaldehyde for 15 min, and permeabilized in acetone at −20°C for 4 min. Cells were then blocked in 5% normal goat serum-PBS for 30 min, incubated with primary antibodies raised against T7 (Bethyl, Montgomery, TX, USA) or ERα (sc-7207, Santa Cruz Biotechnology, Santa Cruz, CA) diluted 1:1000 and 1:100, respectively, in 1% NGS-PBS for 1 h at room temperature. Following incubation, cells were washed three times in PBS and then incubated with goat-anti-mouse or goat-anti-rabbit secondary antibodies conjugated with 546-Alexa (red) or 488-Alexa (green) from Molecular Probes (Eugene, OR, USA). The DNA dye TO-PRO-3 (Molecular Probes) was used for nuclear localization (blue). Microscopic analyses were performed using an Olympus FV300 laser scanning confocal microscope in accordance with established methods, utilizing sequential laser excitation to minimize the possibility of fluorescence emission bleed through. Each image consisted of a single Z section captured at the same cellular level and magnification.
For the effect of estrogen on localization of NRIF3, MCF-7 cells were transfected with either wild-type or mutant T7-NRIF3. After 24 h, cells were incubated in DCC media without serum for 48 hand then treated with 10% fetal calf serum with and without pretreatment with the pure antiestrogen ICI (10−8M for 60 min), or with 17β-estradiol (10−7M for 30 min). Following treatment, cells were fixed and stained with primary antibodies raised against T7 (Novegen, Madison, WI USA) or ERα. Cells demonstrating nuclear colocalization (yellow) of transfected NRIF3 (green) and endogenous ERα (red) in at least 150 cells per treatment group were counted and graphed as a percentage of the total cells counted.
Transient transfection and promoter assays
Cells were maintained in DMEM/ F12 (1:1) supplemented with 10% fetal calf serum. For reporter-gene transient transfections, cells were cultured in medium without phenol red and containing 3% charcoal-stripped (DCC) serum for 24–36 h. Transient transfection with the appropriate plasmids was performed using Fugene-6 (Roche Biochemical, New Jersey) according to the manufacturer’s instructions, and promoter assays were performed 48 hafter transfection.
Chromatin immunoprecipitation (ChIP) assay
T7-NRIF3 stable clones and pcDNA control vector were used for the ChIP assay. The cells were kept in a medium without phenol red and containing 3% charcoal-stripped (DCC) serum 48 h before estrogen or ICI treatment or both; when both were used, ICI was added 1 h before estrogen. ChIP assay was performed as described (Talukder et al., 2004)). Approximately 1x106 cells were treated with 1% formaldehyde (final concentration, v/v) for 10 min at 37°C to cross-link histones to DNA and washed twice with PBS containing protease inhibitor cocktail. Cells were lysed by sonication and immunoprecipitated with T7 monoclonal antibody (Novagen). The immunoprecipitates were washed, and the DNA was eluted off the beads; purified DNA (phenol-chloroform extraction) was subjected to PCR. The sequence of the forward and reverse primers for pS2 promoter used in this study was 5′GAATTAGCTTAGGCCTAGACGGAATG-3′ and 5′AGGATTTGCTGATAGACAGAGACGAC-3′ respectively. 17-β-Estradiol, ICI-182780, and stock chemicals were obtained from Sigma. The sequences of the forward and reverse primers for NRIF3 promoter were 5′ACTACCTTTCTCAGCCTCTGGTAACC-3′ (spanning region -5193 to -5218 bp) and 5′GGGAACCCTCTTACACTGTTGGTAG-3′ (spanning region -4730 to -755 bp), respectively.
MTT cell viability assay
The cell viability assay is based on the ability of viable mitochondria to convert 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), a soluble tetrazolium salt, into an insoluble formazan precipitate, which can be quantitated by spectrophotometry. NRIF3 and pcDNA clones were seeded in a 96-well plate (2000 cells/plate). After 24 h, white medium containing 3%DCC was exchanged for regular medium. At 72 h, cells were treated with or without estrogen for 15 h. Then cells were rinsed with 1XPBS and 0.1 ml MTT solution (5mg/ml in PBS) was added. Cells were incubated at 37°C for cleavage of MTT for 4 h (at the end of this time, the MTT formazan produced in wells containing live cells appears as black, fuzzy crystals on the bottom of the well), and 0.1 ml isopropanol/HCl (0.4 N) was added to each well and mixed thoroughly by repeated pipetting with a multichannel pipettor. (Isopropanol dissolves the formazan to give a homogeneous blue solution suitable for absorbance measurement.) The absorbance was measured within an hour on an ELISA plate reader at 570 nm.
Cell growth and soft agar assays
Colony growth assays were performed as described previously (14). Briefly. a 1 ml of solution of 0.6% DIFCO agar in DMEM supplemented with 10% FBS with insulin was layered onto 35x15 mm graded tissue culture plates. Stable clones of pcDNA and NRIF3 (10,000 cells/plate) were mixed with 1 ml of 0.36% Bactoagar solution in DMEM prepared in a similar manner and layered on top of the 0.6% Bactoagar layer. Plates were incubated for 28 days. For treatment with estrogen, cells were replaced with DCC serum and then added 10−9M estrogen.
Supplementary Material
NRIF3, NRIF3-S28A, NRIF3-S28E or pcDNA3.1 (control vector) overexpressing stable clones in MCF-7 cells. Western blot was performed using 100 μg protein and probed against NRIF3 or T7 antibody. RT-PCR performed using total RNA from the clones. Forward primer is vector specific and reverse primer is NRIF3 specific.
Acknowledgments
We thank Christopher J. Barnes and Bramanandam Manavathi for the confocal microscopy and ChIP analysis studies. This study was supported by the NIH grant CA098823 to R.K.
The abbreviations used are
- ER
estrogen receptor-α
- ERE
estrogen response element
- NR
nuclear receptor
- MTA1
metastasis tumor antigen1
- E2
17-beta-estrodiol
- luc
luciferase
References
- Ali S, Metzger D, Bornert JM, Chambon P. Phosphorylation of the human oestrogen receptor: identification of a phosphorylation site required for trans-activation. EMBO J. 1993;12:1153–1160. doi: 10.1002/j.1460-2075.1993.tb05756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold SF, Obourn JD, Jaffe H, Notides AC. Serine 167 is the major estradiol-induced phosphorylation site on the human estrogen receptor. Mol Endocrinol. 1994;8:1208–1214. doi: 10.1210/mend.8.9.7838153. [DOI] [PubMed] [Google Scholar]
- Arnold SF, Obourn JD, Jaffe H, Notides AC. Phosphorylation of the human estrogen receptor by mitogen-activated protein kinase and casein kinase II: consequence on DNA binding. J Steroid Biochem Mol Biol. 1995;55:163–172. doi: 10.1016/0960-0760(95)00177-2. [DOI] [PubMed] [Google Scholar]
- Arnold SF, Obourn JD, Jaffe H, Notides AC. Phosphorylation of the human estrogen receptor on tyrosine 537 in vivo and by src family tyrosine kinases in vitro. Mol Endocrinol. 1995;9:24–33. doi: 10.1210/mend.9.1.7539106. [DOI] [PubMed] [Google Scholar]
- Arnold SF, Obourn JD, Yudt MR, Carter TH, Notides AC. In vivo and in vitro phosphorylation of the human estrogen receptor. J Steroid Biochem Mol Biol. 1995;52:159–171. doi: 10.1016/0960-0760(94)00166-j. [DOI] [PubMed] [Google Scholar]
- Aronica SM, Katzenellenbogen BS. Stimulation of estrogen receptor-mediated transcription and alteration in the phosphorylation state of the rat uterine estrogen receptor by estrogen, cyclin adenosine monophosphate and insulin-like growth factor-1. Mol Endocrinol. 1993;7:743–752. doi: 10.1210/mend.7.6.7689695. [DOI] [PubMed] [Google Scholar]
- Balasenthil S, Sahin AA, Barnes CJ, Wang RA, Pestell RG, Vadlamudi RK, et al. p21-activated Kinase-1 Signaling Mediates Cyclin D1 Expression in Mammary Epithelial and Cancer Cells. J Biol Chem. 2004;279:1422–1428. doi: 10.1074/jbc.M309937200. [DOI] [PubMed] [Google Scholar]
- Bunone G, Briand PA, Miksicek RJ, Picard D. Activation of the unliganded estrogen receptor by EGF involves the MAP kinase pathway and direct phosphorylation. EMBO J. 1996;15:2174–2183. [PMC free article] [PubMed] [Google Scholar]
- Castoria G, Migliaccio A, Green S, Di-Domenico M, Chambon P, Aurrichio F. Properties of a purified estradiol-dependent calf uterus tyrosine kinase. Biochemistry. 1993;32:1740–1750. doi: 10.1021/bi00058a007. [DOI] [PubMed] [Google Scholar]
- Font de Mora J, Brown M. AIB1 is a conduit for kinase-mediated growth factor signaling to the estrogen receptor. Mol Cell Biol. 2000;20:5041–5047. doi: 10.1128/mcb.20.14.5041-5047.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman LP. Increasing the complexity of coactivation in nuclear receptor signaling. Cell. 1999;97:5–8. doi: 10.1016/s0092-8674(00)80708-4. [DOI] [PubMed] [Google Scholar]
- Glass CK, Rosenfeld MG. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000;14:121–141. [PubMed] [Google Scholar]
- Hermanson O, Glass CK, Rosenfeld MG. Nuclear receptor coregulators: multiple modes of modification. Trends Endocrinol Metab. 2002;13:55–60. doi: 10.1016/s1043-2760(01)00527-6. [DOI] [PubMed] [Google Scholar]
- Ignar-Trowbridge D, Teng CT, Ross KA, Parker MG, Korach KS, McLachlan JA. Peptide growth factors elicit estrogen receptor-dependent transcriptional activation of an estrogen-responsive element. Mol Endocrinol. 1993;7:992–998. doi: 10.1210/mend.7.8.8232319. [DOI] [PubMed] [Google Scholar]
- Joel PB, Traish AM, Lannigan DA. Estradiol and phorbol ester cause phosphorylation of serine 118 in the human estrogen receptor. Mol Endocrinol. 1995;9:1041–1052. doi: 10.1210/mend.9.8.7476978. [DOI] [PubMed] [Google Scholar]
- Kamei Y, Xu L, Heinzel T, Torchia J, Kurokawa R, Gloss B, et al. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell. 1996;85:403–414. doi: 10.1016/s0092-8674(00)81118-6. [DOI] [PubMed] [Google Scholar]
- Kato S, Endoh E, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. 1995;270:1491–14941. doi: 10.1126/science.270.5241.1491. [DOI] [PubMed] [Google Scholar]
- Kumar R, Vadlamudi RK. Emerging functions of p21-activated kinases in human cancer cells. J Cell Physiol. 2002;193:133–144. doi: 10.1002/jcp.10167. [DOI] [PubMed] [Google Scholar]
- Lazaro JB, Bailey PJ, Lassar AB. Cyclin D-cdk4 activity modulates the subnuclear localization and interaction of MEF2 with SRC-family coactivators during skeletal muscle differentiation. Genes Dev. 2002;16:1792–1805. doi: 10.1101/gad.U-9988R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Goff P, Montano MM, Schodin DJ, Katzenellenbogen BS. Phosphorylation of the human estrogen receptor. Identification of hormone-regulated sites and examination of their influence on transcriptional activity. J Biol Chem. 1994;269:4458–4466. [PubMed] [Google Scholar]
- Li D, Desai-Yajnik V, Lo E, Schapira M, Abagyan R, Samuels HH. NRIF3 is a novel coactivator mediating functional specificity of nuclear hormone receptors. Mol Cell Biol. 1999;19:7191–7202. doi: 10.1128/mcb.19.10.7191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Wang F, Samuels HH. Domain structure of the NRIF3 family of coregulators suggests potential dual roles in transcriptional regulation. Mol Cell Biol. 2001;21:8371–8384. doi: 10.1128/MCB.21.24.8371-8384.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma ZQ, Santagati S, Patrone C, Pollio G, Vegeto E, Maggi A. Insulin-like growth factors activate estrogen receptor to control the growth and differentiation of the human neuroblastorna cell line SK-ER3. Mo Endocrinol. 1994;8:910–8. doi: 10.1210/mend.8.7.7984152. [DOI] [PubMed] [Google Scholar]
- Metzger D, Berry M, Ali S, Chambon P. Effect of antagonists on DNA binding properties of the human oestrogen receptor in vitro and in vivo. Mo Endocrinol. 1995;9:579–591. doi: 10.1210/mend.9.5.7565805. [DOI] [PubMed] [Google Scholar]
- Onate SA, Tsai SY, Tsai MJ, O’Malley BW. Sequence and Characterization of a Coactivator for the Steroid Hormone Receptor Superfamily. Science. 1995;270:1354–1357. doi: 10.1126/science.270.5240.1354. [DOI] [PubMed] [Google Scholar]
- Patrone C, Ma ZQ, Pollio G, Agrati P, Parker MG, Maggi A. Cross-coupling between insulin and estrogen receptor in human neuroblastoma cells. Mol Endocrinol. 1996;10:499–507. doi: 10.1210/mend.10.5.8732681. [DOI] [PubMed] [Google Scholar]
- Pierce KL, Luttrell LM, Lefkowitz RJ. New mechanisms in heptahelical receptor signaling to mitogen activated protein kinase cascades. Oncogene. 2001;20:1532–1539. doi: 10.1038/sj.onc.1204184. [DOI] [PubMed] [Google Scholar]
- Pietras RJ, Arboleda J, Reese DM, Wongvipat N, Pelgram MD, Ramos L, et al. HER-2 tyrosine kinase pathway targets estrogen receptor and promotes hormone-independent growth in human breast cancer cells. Oncogene. 1995;10:2435–2446. [PubMed] [Google Scholar]
- Power RF, Mani SK, Codina J, Conneely OM, O’Malley BW. Dopaminergic and ligand-independent activation of steroid hormone receptors. Science. 1991;254:1636–1639. doi: 10.1126/science.1749936. [DOI] [PubMed] [Google Scholar]
- Rowan BG, Garrison N, Weigel NL, O’Malley BW. 8-Bromo-cyclic AMP induces phosphorylation of two sites in SRC-1 that facilitate ligand-independent activation of the chicken progesterone receptor and are critical for functional cooperation between SRC-1 and CREB binding protein. Mol Cell Biol. 2000;20:8720–8730. doi: 10.1128/mcb.20.23.8720-8730.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegelman BM, Heinrich R. Biological control through regulated transcriptional coactivators. Cell. 2004;119:157–167. doi: 10.1016/j.cell.2004.09.037. [DOI] [PubMed] [Google Scholar]
- Talukder AH, Gururaj A, Mishra SK, Vadlamudi RK, Kumar R. Metastasis-associated protein 1 interacts with NRIF3, an estrogen-inducible nuclear receptor coregulator. Mol Cell Biol. 2004;24:6581–6591. doi: 10.1128/MCB.24.15.6581-6591.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RA, Mazumdar A, Vadlamudi RK, Kumar R. P21-activated kinase-1 phosphorylates and transactivates estrogen receptor-alpha and promotes hyperplasia in mammary epithelium. EMBO J. 2002;21:5437–5447. doi: 10.1093/emboj/cdf543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Rose DW, Hermanson O, Liu F, Herman T, Wu W, et al. Regulation of somatic growth by the p160 coactivator p/CIP. Proc Natl Acad Sci USA. 2000;97:13549–13554. doi: 10.1073/pnas.260463097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu RC, Qin J, Hashimoto Y, Wong J, Xu J, Tsai SY, et al. Regulation of SRC-3 (pCIP/ACTR/AIB-1/RAC-3/TRAM-1) coactivator activity by I B kinase. Mol Cell Biol. 2002;22:3549–3561. doi: 10.1128/MCB.22.10.3549-3561.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu RC, Qin J, Yi P, Wong J, Tsai SY, Tsai MJ, et al. Selective phosphorylations of the SRC-3/AIB1 coactivator integrate genomic responses to multiple cellular signaling pathways. Mol Cell. 2004;15:937–949. doi: 10.1016/j.molcel.2004.08.019. [DOI] [PubMed] [Google Scholar]
- Wu RC, Smith CL, O’Malley BW. Transcriptional regulation by steroid receptor coactivator phosphorylation. Endocr Rev. 2005;26:393–399. doi: 10.1210/er.2004-0018. [DOI] [PubMed] [Google Scholar]
- Vadlamudi RK, Adam L, Wang RA, Mandal M, Nguyen D, Sahin A, et al. Regulatable expression of p21-activated kinase-1 promotes anchorage-independent growth and abnormal organization of mitotic spindles in human epithelial breast cancer cells. J Biol Chem. 2000;275:36238–36244. doi: 10.1074/jbc.M002138200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
NRIF3, NRIF3-S28A, NRIF3-S28E or pcDNA3.1 (control vector) overexpressing stable clones in MCF-7 cells. Western blot was performed using 100 μg protein and probed against NRIF3 or T7 antibody. RT-PCR performed using total RNA from the clones. Forward primer is vector specific and reverse primer is NRIF3 specific.