

Abstract
Mitotic spindle-disrupting agents target and alter microtubule dynamics. These agents include clinically important chemotherapies, such as taxanes (paclitaxel [Taxol], docetaxel [Taxotere]) and vinca alkaloids (vincristine [Oncovin], vinblastine). Taxanes are a standard component of treatment for many malignancies, often in conjunction with other cytotoxic agents. However, the optimal sequencing of these treatments and whether efficacy may be influenced by in vitro cellular growth conditions remain incompletely investigated. Yet such preclinical investigations may guide clinical decision making. We therefore studied the effect of cell density on rapid killing by paclitaxel and vincristine. Breast, ovarian and prostate cancer cells were sensitive to rapid killing by either agent when grown at low density, but were markedly resistant when grown at high density, i.e., nearly confluent. The resistance of densely growing cells to rapid killing by these drugs translated to increased clonogenic survival. Pretreatment of densely growing cancer cells with cisplatin followed by paclitaxel, partially reversed the treatment resistance. Gene ontology associations from microarray analyses of cells grown at low and high density, suggested roles for membrane signal transduction and adhesion, but potentially also DNA damage repair and metabolism. Taken together, the treatment resistance at higher cell density may be associated with a lower proportion of active cycling in cells growing at high density as well as transduction of survival signals induced by increased cell-cell adhesion. Collectively these findings suggest mechanisms by which growth conditions may contribute to resistance to rapid killing by microtubule-disrupting drugs.
Keywords: cellular density, paclitaxel, microtubule-targeting agents, microarray, cell cycle
Introduction
Drugs that disrupt microtubule dynamics include those that either accelerate or inhibit the intracellular polymerization of tubulin heterodimers into microtubules. Taxanes, including paclitaxel (Taxol) and docetaxel (Taxotere), are the most prominent class of drugs that accelerate polymerization; this is mediated through drug binding to tubulin subunits which then results in stabilization and persistent polymerization of microtubules.1 In contrast, nocodazole and the vinca alkaloids, vincristine (Oncovin) and vinblastine, inhibit the polymerization of the tubulin subunits, so microtubules do not form.2–5 These microtubule-targeting drugs share the common property of preventing the proper formation and function of the mitotic spindle. The lack of a functional spindle impedes mitotic progression, leading to a mitotic delay in checkpoint-competent cells, or rapid death for cells with an inactivated mitotic checkpoint.6–10
Taxanes are widely used because of their efficacy and favorable toxicity profile in treating cancer patients. More recently this class of drugs was found to surpass doxorubicin’s ability to improve the survival of women with breast cancer,11–13 and is the first chemotherapy to improve the survival of patients with hormone-refractory prostate cancer.14,15 Despite their proven efficacy in treating specific human cancers, the factors determining treatment success or failure with taxanes remain surprisingly unclear. Preclinical studies have identified a number of mechanisms that mediate in vitro resistance to taxanes. These include mutations in the alpha or beta-subunits of tubulin that decrease binding by the drug,16–19 activation of the multidrug drug resistance (MDR) gene,20–22 alterations in selective cytokines/chemokines23 and levels of activation of pro- or anti-apoptotic mechanisms.24
While the clinical relevance of these factors has yet to be extensively studied in clinical trials, a consistent theme in oncology practice is that smaller solid tumors respond better to treatment. In contrast, the probability of a complete response is less likely for highly cellular, locally-advanced tumors. It has been hypothesized that in larger tumors, physiologic factors such as hypoxia, poor perfusion or tumor interstitial fluid pressure may come into play to limit the effectiveness of chemotherapy. There are probable cell-type differences in sensitivity to rapid killing by spindle-disrupting drugs. Cells with robust mitotic spindle checkpoints may be able to tolerate brief exposure to the drugs, while those that are checkpoint-deficient are rapidly killed.25 It has also been demonstrated that cells derived from tumors with high cellularity, enhanced cell-cell interactions and minimal stromal involvement have increased viability and can be serially transplanted preferentially in nude mice.26 Similar studies conducted with cells isolated from primary human breast tumors support the idea that increased cell-cell interactions resulting from higher cell densities may be critical for promoting survival in primary breast tumors and potentially diminish therapeutic response. It is likely these factors ultimately work together to orchestrate chemotherapy resistance or sensitivity in the clinic.
To address the some of the proposed mechanisms of treatment resistance, we studied the effect of cancer cell growth density on sensitivity to rapid killing by the microtubule-targeting drugs paclitaxel and vincristine. We studied previously established mitotic checkpoint deficient human cancer cell lines. As expected, cells grown under sparse conditions with diminished cell-cell interactions were rapidly killed by paclitaxel and vincristine. Conditions permitting exponential growth resulted in rapid nuclear fragmentation in cells treated with microtubule-targeting drugs.25 However, when grown under dense, near-confluent conditions with high cell-cell interaction, cells became considerably more resistant to being killed by these drugs. This finding was consistent among breast, prostate and ovarian cancer cells. Finally, to gain insight into the cellular components, biologic processes and molecular functions underlying this differential sensitivity, we performed a microarray analyses to determine gene ontology associations from cells grown at low and high cell density. Taken together, these findings help identify potential factors that contribute to the therapeutic resistance of densely-growing, locally-advanced tumors.
Results
Rapid killing of SKBR3 cells by microtubule-targeting agents is reduced and clonogenic survival is enhanced when grown at high density
We have previously established that due to the lack of a robust mitotic spindle checkpoint, SKBR3 breast cancer cells maintained in conditions allowing exponential growth are sensitive to being rapidly killed by even brief exposure to paclitaxel.25 To determine whether different growth conditions might influence the susceptibility of SKBR3 cells to rapid killing by paclitaxel, the cells were grown at three different densities, from low (10–30% confluency, 10,000 cells per cm2) to moderate (50–70% confluency, 30,000 cells per cm2) to high density (80–100% confluency, 60,000 cells per cm2).
Paclitaxel was added to all plates simultaneously with fresh media, and groups of cells growing under each condition were harvested sequentially for cell cycle analysis. The cells grown sparsely were killed quicker and more thoroughly by the same exposure to paclitaxel than those grown under denser conditions. A difference in the proportion of cells being killed between the cell growth conditions was evident by 6 h after addition of paclitaxel, and become increasingly evident with passage of time (Fig. 1A–C). After 15 h of paclitaxel exposure very few cells grown at low density were still viable, while approximately 58% of the cells grown under conditions of high density remained intact (Fig. 1B and C). To test whether the effect of cell growth density on rapid killing also applied to other drugs that disrupt microtubule dynamics, such as vincristine or nocodazole, we repeated these experiments using these drugs in place of paclitaxel. A similar effect on viability, dependent on cell density at time of treatment, was confirmed in vincristine- and nocodozole-treated SKBR3 cells at 15 h (Fig. 1D and data not shown, respectively).
Figure 1.

Cell growth density determines proportion of SKBR3 cells that are rapidly killed by paclitaxel and vincristine. (A) Cell death after Taxol of cells grown at three different densities. Parallel plates of SKBR3 cells were grown at the respective densities, and then harvested prior to treatment (“0 hours”) or at the indicated times after treatment with paclitaxel (“Taxol”, 20 nM) for cell cycle analysis via FACS. Low density = 10–30% confluency. Moderate density = 50–70% confluency. High density = 80–100% confluency. Histograms show cell cycle distribution, with the respective phases indicated. By 15 h after treatment, virtually none of the cells grown at low density were still alive, while a substantial proportion of cells grown at high density were viable. The arrow indicates cells in G1 that remain viable and intact, especially prominent in the cells grown under dense conditions. (B) Bar graphs showing the percentage of nonviable cells with sub-G1 DNA content in each of the FACS histograms shown in (A). The bars represent the respective times after the addition of paclitaxel according to the figure legend. (C) SKBR3 cells grown at the three different densities as described in (A), were imaged (x40) before or 12 h after treatment with paclitaxel (Taxol). The area within the dashed box is shown at higher magnification. (D) SKBR3 cells growing at the three densities were prepared as in (A), but treated with vincristine (20 nM) for 15 h. All cells were then harvested for cell cycle analysis via FACS. Bar graphs show the percentages of treated cells at each growth density (indicated in the figure legend) undergoing nuclear fragmentation and resulting in sub-G1 DNA accumulation. Error bars represent standard deviation. Open and filled stars indicate significant differences between the low density and the moderate and high density treatment groups, respectively, with associated p values.
We sought to determine if the rapid cell death of most paclitaxel-treated cells grown at low density was due to the loss of replicative potential. In other words, to determine whether or not cell death was merely delayed, such that all cells exposed to paclitaxel, irrespective of cell growth density, were ultimately programmed for eventual death. Alternatively, we sought to assess if brief exposure to paclitaxel gave rise to cells that proliferated more aggressively (as one might expect if few surviving cells had developed increased aneuploidy). Therefore, we repeated the experiment, but this time treated cells were replated for analysis by clonogenic survival, the standard assay for replicative potential. Cells grown under all three densities were treated with paclitaxel, washed, counted and equal numbers of each treatment group were replated into fresh dishes left to grow undisturbed. Significantly fewer cells grown at low density during paclitaxel exposure survived to form colonies, but almost four times as many cells grown at high density during treatment formed colonies (Fig. 2A and B). Thus, the rapid nuclear fragmentation of paclitaxel-treated cells grown at low density ultimately results in significantly fewer colonies.
Figure 2.
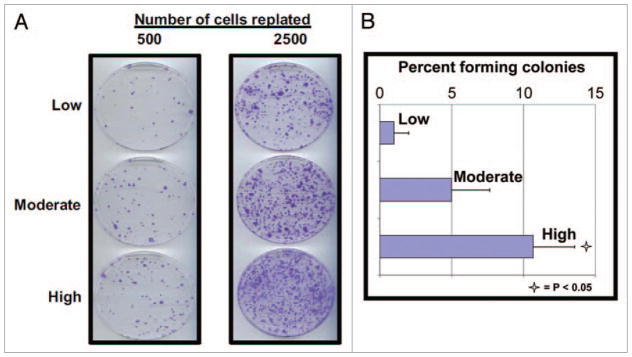
Clonogenic survival after paclitaxel treatment is affected by cell growth density at the time of treatment. (A) SKBR3 cells were grown at the at the three different cell growth densities and then treated with paclitaxel (20 nM) for 12 h. All cells were then washed extensively and the indicated numbers of cells (either 500 or 2,500) of each treatment group were plated into new plates with fresh drug-free media. The cells were then left to grow undisturbed for 10 d. All plates were then fixed, stained, and the colonies counted. Plates containing the same number of cells for each group of cells were prepared in triplicate, fixed, and stained identically, with the resulting numbers of colonies averaged. The experiment was repeated three times, with similar results. Representative plates containing identically treated cells of each original growth density are shown. (B) Bar graphs showing the percentages of cells of each growth density forming colonies after treatment with paclitaxel as described. Each data point and its standard deviation (error bars) represents the results from at least six different plates of cells. Open star indicates a significant difference between the low density and the high density groups with the associated p value.
Sensitivity to microtubule-targeting drugs is consistently reduced in mitotic checkpoint-deficient human cancer cell lines when grown at high density
We wanted to confirm that the reduced killing of cells grown in high density was not specific to SKBR3 cells but rather a general property of paclitaxel-treated cell lines deficient for the mitotic checkpoint. Therefore, we assessed paclitaxel-induced cell death in PC3 (a hormone independent prostate cancer cell line), A2780 and OVCAR-3 (both ovarian cancer cell lines) when these cells were grown in different growth densities. We had previously established these checkpoint-deficient cell lines were efficiently killed by paclitaxel under conditions allowing exponential growth.27 Confirming our results in SKBR3 cells, rapid killing by paclitaxel was dramatically affected by cell growth densities in all three cell lines tested (Fig. 3A). Paclitaxel treated cells grown at low density showed a sub-G1 content of 84–86% at 15 h (Fig. 3B). In contrast, PC3, A2780 and OVCAR cells grown at high densities with identical drug exposure displayed a sub-G1 content of 13–29% (Fig. 3B).
Figure 3.
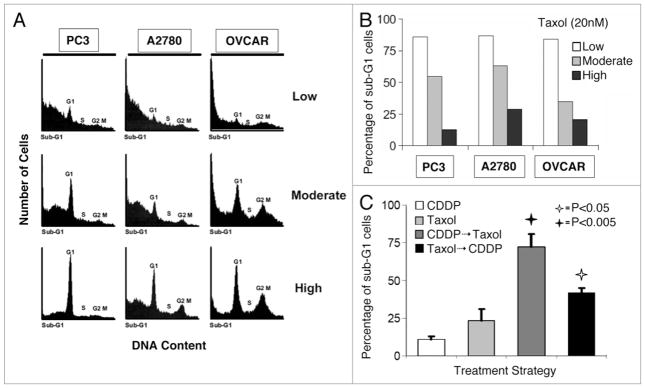
Cell lines susceptible to rapid killing by paclitaxel are rendered more resistant when grown at a high growth density and resistance is abrogated when cells are pretreated with cisplatin. (A) PC3, A2780 and OVCAR-3 cells were grown under the three different cell growth densities as described previously for SKBR3 cells (Fig. 1), treated with paclitaxel (“Taxol”, 20 nM), and were harvested 15 h later for cell cycle analysis via FACS. (B) Bar graphs showing the percentage of cells of each cell line and of each growth density undergoing nuclear fragmentation, resulting in sub-G1 DNA content, after treatment with paclitaxel. Low density = 10–30% confluency. Moderate density = 50–70% confluency. High density = 80–100% confluency, according to figure legend. (C) The resistance to rapid killing by paclitaxel of cancer cells growing at high density is abrogated when cells are pretreated with cisplatin. SKBR3 cells were grown at high density, and treated with cisplatin (5 μg/ml) for 12 h, paclitaxel (20 mM) for 12 h, cisplatin for 6 h followed by paclitaxel for 6 h, or paclitaxel for 6 h followed by cisplatin for 6 h. All cells were then harvested for cell cycle analysis via FACS. Bar graphs show the percentages of treated cells at each growth density undergoing nuclear fragmentation, resulting in sub-G1 DNA content. The experiment was repeated three times. Error bars represent standard deviation. Open and filled stars indicate significant differences between the following treatment strategies: Taxol vs. Taxol → CDDP and CDDP vs. CDDP → Taxol, respectively, with associated p values.
Together, these results suggest that cell growth density may have a substantial effect on the rapid killing of cancer cell lines by microtubule/mitotic spindle-disrupting drugs, potentially through the level of cell-cell interaction. Thus cell lines sensitive to these drugs when grown at low density and without significant cell-cell contact may show increased drug resistance at high growth densities.
Resistance to paclitaxel-induced killing of cells at high density can be partially reversed by cisplatin pretreatment
Cisplatin is a well-known anticancer agent that has been in use for 30 y. It is a chemotherapeutic drug that forms inter- and intra-strand adducts with DNA.28 Cisplatin was recently found to enhance cytotoxic efficacy in cancer cells grown at a high density, a result attributed to enhanced kinase signaling mediated through cell-to-cell gap-junctions not present in cells grown more sparsely.29 We treated SKBR3 cells grown at the highest density with cisplatin alone, paclitaxel alone, cisplatin followed by paclitaxel or paclitaxel followed by cisplatin (Fig. 3C). The cumulative drug exposure time for all four groups was identical, and all cells were harvested at the same time. While cisplatin and paclitaxel alone were comparable in the proportion of cancer cells killed, pretreatment with cisplatin followed by paclitaxel led to markedly increased killing. The administration of paclitaxel followed by cisplatin also led to increased cell killing compared to either drug alone, but was less effective than administering cisplatin first.
Active cycling is decreased in cells growing at high growth density in vitro and in vivo
At drug concentrations that are the most clinically relevant to patients, the killing of cancer cells by microtubule-targeting drugs is cell cycle-dependent, in that death occurs when the cells progress into mitosis.30 Consequently, it may be possible that a reduced proportion of cells actively cycling may influence the therapeutic efficacy of these drugs. To begin investigating whether the resistance of densely grown cells to rapid killing by chemotherapy was related to a smaller proportion of cycling and mitotic cells, we analyzed BrdU (Bromodeoxyuridine) incorporation at low, moderate and high cell growth densities. SKBR3 cells were grown on cover-slips and grown at densities as in the previous experiments. All cells were briefly exposed to BrdU a drug which only replicating cells can incorporate. The cells were then fixed and immunofluorescence for incorporated BrdU was performed. The majority (77 ± 4%) of cells grown sparsely had taken up BrdU, consistent with exponential growth when cells are allowed to grow at low density (Fig. 4A and B). In contrast, BrdU was incorporated by only a minority (12 ± 5%) of the cells grown at high density, and the cells grown at moderate density showed an intermediate percentage (38 ± 6%) of BrdU labeled cells. We found similar results when the same experiment was repeated with PC3 and OVCAR-3 cells (data not shown). These observations together indicate higher density correlated with a smaller proportion of cells actively traversing the cell cycle. A potential consequence of fewer cells entering mitosis at the time of drug administration was less cell death by microtubule-targeting agents.
Figure 4.

The proportion of actively replicating cells is inversely proportional to cell growth density, in vitro as well as in vivo. (A) SKBR3 cells were grown on coverslips at the at the three different cell growth densities, pulse-labeled with BrdU for 10 min, and then fixed and probed for BrdU and total chromatin via DAPI. The proportion of cells showing BrdU uptake indicative of active replication as a function of total cells was counted. Representative images of cells grown at each growth density staining for BrdU and total chromatin (DAPI). (B) Bar graphs showing the percentages of cells of each growth density that are actively replicating. Each data point and its standard deviation (error bars) represents the results from at least three different plates of cells. The experiment was repeated three times, with similar results. Open and filled stars indicate significant differences between the low density and the moderate and high density treatment groups, respectively, with associated p values. (C) Breast cancer cell growth density correlates with increasing tumor stage. Excisional biopsies from three representative patients presenting with AJCC Stages T1a (background of DCIS with microinvasion), T1c, and T2 breast cancer were fixed onto slides. These were subsequently deparaffinized and probed for CENP-F (specific marker of G2/M cells) and counterstained with hematoxylin. Representative image of 1 mm2 of tissue from each patient is shown. The area within the dashed box is shown adjacent at a higher magnification. Tumor cells show abundant cytoplasm, and those in G2/M show high expression of CENP-F (individual CENP-F expressing cells indicated by arrows in images at higher magnification). (D) Bar graphs showing the average number of cells per 1 mm2 areas of tumor tissue. Each data point and its standard deviation (error bars) represents the results from at least three samples. Five separate areas were counted. Stars indicate significant differences between the T1a samples and the T1c and T2 samples with associated lc value. Figure inset: average percentage of tumor cells of each mm2 area strongly expressing CENP-F (% +++ CENP-F staining) according to Stage (T1a, T1c and T2).
The next step was to assess whether a decreased fraction of cycling cells might be detectable in clinically dense tumors. We analyzed paraffin-fixed tissue samples from excisional biopsies of women presenting for treatment of localized breast cancer. These samples were obtained as part of an IRB-approved protocol to investigate histological factors correlating with treatment success or failure. In a pilot effort, the slides of nine patients presenting with different stages of breast cancer, staged according to standard American Joint Committee on Cancer (AJCC) pathological criteria, were assessed: T1a (n = 3, more than 0.1 but less than 0.5 cm in greatest dimension), T1c (n = 3, more than 1.0 but less than 2.0 cm), and T2 (n = 3, more than 2.0 but less than 5.0 cm). It was not possible to stain for BrdU as proliferating cells must be exposed to the agent prior to harvesting and fixation, a precondition not available for clinical samples. We therefore stained the clinical samples for CENP-F, a kinetochore-binding protein that is maximally expressed during G2/M, and thus serves as an accurate marker of cells in G2/M.27,31 CENP-F is not expressed by quiescent G1/early S phase cells. The proportion of cells strongly expressing CENP-F was employed as an indicator of active cell cycling because this marker has been previously validated for cell cycle analysis.27
We began our studies by counting and calculating the average number of cells within at least five 1 mm2 areas of tumor tissue. It was readily apparent that the morphology and growth characteristics of tumors between patients could be distinguished (Fig. 4C). Stage T1a tumors showed an average of 105.3 ± 24 cells per 1 mm2, T1c tumors showed 443.3 ± 31 cells per 1 mm2, while the T2 tumors showed 760.7 ± 98 cells per 1 mm2. Therefore the number of cancer cells per given area—i.e., the cell density—increased proportionally with the increasing stage of disease. The average percentage of tumor cells of each mm2 area that strongly expressed CENP-F (i.e., were in G2/M) for each stage wereT1a: 22%, T1c: 8% and T2: 3%. Thus, the percentage of actively cycling cells relative to the entire tumor population was inversely proportional to advancing stage and cell density. It should be noted that because the more advanced stage tumors contained many more cancer cells, the overall number of cycling cells is higher as well. Nonetheless, these results supported a clinical correlation with our cell culture findings of decreased cell cycling in densely grown cancer cells.
Gene ontology suggests a critical role of cell density and cell-cell interactions in driving biological processes and cellular functions
Finally, we sought to determine the critical gene associations and the overriding ontology characteristics influenced by cell density conditions. To achieve this, we performed microarray analysis of cells grown at both low and high densities as in our previous experiments. Microarray data was processed with the GeneSifter (VizX Labs) microarray analysis system. We screened the data by eliminating genes that changed by less than 2-fold, and by only including genes that were flagged as “P” or “present” by the Affymetrix MAS5 algorithm. These filtering steps reduced the dataset to 5209 genes (Fig. 5). This subset was mined for biological information using the Z-score Report in GeneSifter to categorize genes according to their involvement in the following biological pathways: cellular component, biological process and molecular function. The Z-score associated with each pathway is an indication of the likelihood that association between the altered genes and that pathway occurred more or less frequently than expected. Large positive numbers (or negative) indicate that the pathway is significantly activated (or repressed). As shown in Table 1, (+) Z-scores indicating cellular components that were significantly activated in the cells grown at high density included those related to the regulation or structure of membrane and extracellular regions. Biological processes that were increased in cells grown at high density included those related to adhesion. Interestingly however, genes related to DNA damage repair and metabolism were repressed (Table 2). These results illustrated the dramatic influence of cell density on modulating gene expression profiles, with potential consequences for treatment response.
Figure 5.

Scatter plot of 38,500 genes derived from microarray analysis of cells grown at low (sparse) or high (Dense) density. Data was filtered from a raw data set of 38,500 genes. Five thousand, two hundred and nine differentially expressed genes were identified that met the filtering criteria. Genes that did not pass the filtering criteria are displayed in light gray. Genes that passed filtering criteria (5209 genes) are shown in black as upregulated (≥2-fold expression increase), or in dark gray as down-regulated (≥2-fold expression decrease).
Table 1.
Cellular component
| Rank | Z-score (+) | Z-score (−) | ||
|---|---|---|---|---|
| 1 | Integral to membrane | 11.27 | Intracellular | −9.1 |
| 2 | Intrinsic to membrane | 11.27 | Intracellular part | −8.66 |
| 3 | Membrane part | 10.84 | Nucleus | −7.04 |
| 4 | Membrane | 10.7 | Organelle | −6.95 |
| 5 | Plasma membrane part | 6.2 | Intracellular organelle | −6.93 |
| 6 | Plasma membrane | 6.14 | Membrane-bound organelle | −6.37 |
| 7 | Intrinsic to plasma membrane | 5.31 | Intracellular membrane-bound organelle | −6..34 |
| 8 | Integral to plasma membrane | 5.25 | Mitochondrion | −5.54 |
| 9 | Membrane fraction | 4.58 | Mitochondrial part | −4.75 |
| 10 | Extracellular region | 4.2 | Macromolecular | −4.74 |
Ontological categorization of the top ten Cellular Components up or downregulated in response to cell density as determined by Z-score.
Table 2.
Biological process
| Rank | Z-score (+) | Z-score (−) | ||
|---|---|---|---|---|
| 1 | Biological adhesion | 6.3 | Nucleic acid metabolic process | −6.16 |
| 2 | Cell adhesion | 6.3 | RNA metabolic process | −5.41 |
| 3 | Biomineral formation | 6.14 | Biopolymer metabolic process | −4.95 |
| 4 | Ossification | 6.14 | Cellular metabolic process | −4.71 |
| 5 | Multicellular organismal development | 5.68 | Macromolecular metabolic process | −4.55 |
| 6 | Tissue remodeling | 5.66 | Primary metabolic process | −4.26 |
| 7 | System development | 5.64 | Metabolic process | −4.24 |
| 8 | Bone remodeling | 5.59 | DNA metabolic process | −4.01 |
| 9 | Skeletal development | 5.53 | DNA repair | −3.87 |
| 10 | Developmental process | 5.0 | Translation | −3.82 |
Ontological categorization of the top ten Biological Processes up or downregulated in response to cell density as determined by Z-score.
Discussion
We report here that human cancer cells susceptible to rapid killing by microtubule-targeting drugs when grown sparsely became markedly more resistant to the same drugs when grown in higher density conditions. It is possible that the resistance associated with high cell growth density may in part be due to a lower fraction of cells actively undergoing cell-cycling, resulting in fewer cells undergoing mitotic catastrophe in the presence of these drugs. We may also speculate that the increased cell-cell interactions and cellular adhesion occurring under high density cellular growth conditions may alter gene expression patterns and which in turn alter the response to therapy. While the investigations described here were performed under normoxic conditions and neutral pH, these findings do not exclude the possibility that other physiological conditions may also confer drug resistance in clinical settings. Indeed, it seems also likely that any condition that limits cell cycling or that results in a portion of the tumor becoming temporarily quiescent would also decrease the susceptibility of cancer cells to rapidly killing by microtubule-targeting drugs. For example, high cell density has been found to attenuate the activation by DNA damage of p53,32 however, the SKBR3, PC3 and OVCAR-3 cells studied here are all functionally null for wildtype p53 activity, and p53 expression was not restored by treatment with either paclitaxel or vincristine (data not shown).
Paclitaxel has been associated with caspase-activation, which may contribute to cell death resulting from mitotic catastrophe.33–35 We confirmed that treatment with paclitaxel under conditions used in the studies described here leads to activation of caspase 3.36,37 Interestingly, we have found the activation of caspases is muted in cells growing at high density when treated with paclitaxel, compared to cells growing sparsely when treated (data not shown).
Our results merit confirmation in animal models of human cancers, and ultimately in human studies. Important questions that remain to be answered include whether there is a threshold tumor density or size beyond which the cancer cells within become drug resistant, and whether cellular growth density affects other aspects of the tumor microenvironment to mediate treatment resistance? Clinical experience suggests that while responses can still be obtained when solid malignancies have grown to larger sizes, cure with chemotherapy or radiation therapy alone is less likely. Technological advances that facilitate functional imaging of tumors such as FLT-PET38 or dynamic contrast MRI39 may give insight into intratumoral growth conditions such as cellular cycling or tumor physiology, and in turn allow the prediction of treatment response and outcome.
Materials and Methods
All cell lines cells were obtained from the American Type Culture Collection (ATCC) (Manassas, VA), and grown in DMEM media (Gibco/BRL) supplemented with 15% fetal bovine serum (FBS) at 37°C in 5% CO2. Nocodazole, paclitaxel, vincristine and cisplatin were obtained from Sigma (St. Louis, MO), and prepared as 1000X stock in DMSO. For experiments, these were diluted in media, and used at concentrations and conditions as previously described or noted in the figure legends.27,45 In all experiments, the media of all plates were changed to fresh prewarmed media simultaneously with the addition of drugs, to ensure that the results would not be affected by nutrient deprivation. Mock-treated controls were handled in an identical manner to experimental samples, with an identical amount of media added (without drug). Colony survival assays were performed by exposing cells to microtubule-targeting drugs for 6 h, after which the drugs were removed. The cells were thoroughly washed, and replated in fresh, drug-free plates in triplicate at serial dilutions and left to grow undisturbed for 10 d in the incubator. Colonies were subsequently fixed and stained with crystal violet in methanol, and scored as those containing 50 or more cells.
Immunofluorescence and fluorescence-activated cell sorting (FACS) analysis of DNA content was performed as previously described.27,45 During the FACS, no gating was performed on the propidium iodide (PI)-stained nuclei, in order that all cells were included in the analysis, including those with sub-G1 content representative of nuclear fragmentation. Prior to staining of the nuclei, cells with sub-G1 content were confirmed to be nonviable through Trypan blue and propidium iodide (PI) exclusion assays. For immunofluorescence, cells grown on coverslips were fixed in ice-cold acetone:methanol (50:50), washed in phosphate-buffered saline and KB buffer (50 mM TRIS-HCl pH 7.4, 150 mM NaCl, 0.1% BSA), prior to labeling with specific antibodies. The respective primary antibody that was then detected via the species-specific secondary antibody conjugated to Alexa Fluor 594 or 488 (Molecular Probes, Eugene, OR). DNA was stained with 0.1 mg/ml of 4′,6-diamidino-2-phenylindole (DAPI, Sigma). The coverslips were then mounted in 0.1% para-phenylenediamine in glycerol. Stained cells were examined with a x100 PlanNeofluor objective mounted on a Nikon TE-200 microscope equipped with epifluorescence optics. Images were captured with a Hammamatsu CCD camera that was controlled with IP LabSpectrum v2.0.1 (Scanalytics Inc.).
For immunohistochemistry, archived slides from an Institutional Review Board (IRB)-approved protocol were deparaffinized by immersion in 95°C 10 mM citric acid buffer (pH 6.0) for 15 m in a steamer, followed by treatment with hydrogen peroxide to block endogenous peroxides, and then blocked in 10% horse serum. The slides were then probed overnight with anti-CENP-F (Novus) at 1:1,000 in 4°C. A peroxidase labeled polymer (DakoCytomation Envision Plus Dual Link System Peroxidase, Carpinteria, CA) was applied at room temperature for 30 m. The slide was developed with Vector DAB Peroxidase Substrate Kit (SK-4100) and counterstained with hematoxylin. Calculations and statistical analysis for the colony survival assays, as well as the analyses based on immunofluorescence and immunohistochemistry, were performed with add-in statistical modules in Excel, in turn an integrated component of Microsoft Office Professional XP. Counts were taken from at least five separate microscopic fields, with cell counts rounded to the nearest whole number, and followed by standard error, e.g., 10.3 ± 24. Statistical differences were determined by Student’s t-test using the MedCalc (software version 10.4.8.0) statistical software package.
For microarray analysis, SKBR3 cells were harvested at low (10–30% confluency, approximately 10,000 cells per cm2) or high density (80–100% confluency, approximately 60,000 cells per cm2). Total RNA was isolated and purified from the cells using the RNeasy Mini Kit (QIAGEN, CA). RNA was amplified, labeled, and hybridized to the GeneChip Human Genome U133 Plus 2.0 Array (Affymetrix, Santa Clara, USA) per manufacturer’s instructions. The average intensity of all genes on the chip was adjusted to 150 to allow for comparison and analysis. Data was processed with the GeneSifter (VizX Labs) microarray analysis system. Data was normalized to the global median of gene expression. The biological pathway Z-score calculated by Genesifter was used for ontology rankings.
Conclusion
In conclusion, our findings may help provide a rationale for the selection and sequencing of drugs in anticancer treatment. For example, administering chemotherapy that kills cancer cells regardless of cell growth density may reduce cell number sufficiently to allow taxanes to be maximally effective in killing more cancer cells.40,41 Anticancer interventions whose efficacy might be independent of cell density include cisplatin, radiation therapy and, of course, surgery. A number of clinical studies have reported high response rates when cisplatin was combined with docetaxel.42–44 It remains to be determined whether efficacy can be further improved when the treatments are given sequentially versus concurrently. We hope the preliminary observations we report here might stimulate such studies.
Acknowledgments
We are grateful to present and past members of the Kao laboratory for indispensable and expert assistance, including Anil Magge, Shary Parker, Fang Liu, Arber Kodra, Elizabeth Gurney, Wes Baff, Katie Murphy and Jessica Liao. This work was supported by funds from the University of Pennsylvania Research Foundation and National Institutes of Health (CA107956-01) (to G.D.K.), Burroughs Wellcome Fund Career Award for Medical Scientists 1006792 (to J.F.D.). A portion of this research was supported by the Breast Cancer Research Foundation.
References
- 1.Schiff PB, Horwitz SB. Taxol stabilizes microtubules in mouse fibroblast cells. Proc Natl Acad Sci USA. 1980;77:1561–5. doi: 10.1073/pnas.77.3.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chabner BA, Horwitz SB, Clendennin NJ, Purvis JD. Vinca alkaloids. Cancer Chemother Biol Response Modif. 1991;12:67–73. [PubMed] [Google Scholar]
- 3.Hoebeke J, Van Nijen G, De Brabander M. Interaction of oncodazole (R 17934), a new antitumoral drug, with rat brain tubulin. Biochem Biophys Res Commun. 1976;69:319–24. doi: 10.1016/0006-291x(76)90524-6. [DOI] [PubMed] [Google Scholar]
- 4.Lee JC, Field DJ, Lee LL. Effects of nocodazole on structures of calf brain tubulin. Biochemistry. 1980;19:6209–15. doi: 10.1021/bi00567a041. [DOI] [PubMed] [Google Scholar]
- 5.Ngan VK, Bellman K, Hill BT, Wilson L, Jordan MA. Mechanism of mitotic block and inhibition of cell proliferation by the semisynthetic Vinca alkaloids vinorelbine and its newer derivative vinflunine. Mol Pharmacol. 2001;60:225–32. doi: 10.1124/mol.60.1.225. [DOI] [PubMed] [Google Scholar]
- 6.Chan GK, Yen TJ. The mitotic checkpoint: a signaling pathway that allows a single unattached kinetochore to inhibit mitotic exit. Prog Cell Cycle Res. 2003;5:431–9. [PubMed] [Google Scholar]
- 7.Jordan MA, Toso RJ, Thrower D, Wilson L. Mechanism of mitotic block and inhibition of cell proliferation by taxol at low concentrations. Proc Natl Acad Sci USA. 1993;90:9552–6. doi: 10.1073/pnas.90.20.9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kelling J, Sullivan K, Wilson L, Jordan MA. Suppression of centromere dynamics by Taxol in living osteosarcoma cells. Cancer Res. 2003;63:2794–801. [PubMed] [Google Scholar]
- 9.Musacchio A, Hardwick KG. The spindle checkpoint: structural insights into dynamic signalling. Nat Rev Mol Cell Biol. 2002;3:731–41. doi: 10.1038/nrm929. [DOI] [PubMed] [Google Scholar]
- 10.Yvon AM, Wadsworth P, Jordan MA. Taxol suppresses dynamics of individual microtubules in living human tumor cells. Mol Biol Cell. 1999;10:947–59. doi: 10.1091/mbc.10.4.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ravdin PM. Reflections on the development of resistance during therapy for advanced breast cancer. Implications of high levels of activity of docetaxel in anthracycline-resistant breast cancer patients. Eur J Cancer. 1997;33:7–10. doi: 10.1016/s0959-8049(97)90002-2. [DOI] [PubMed] [Google Scholar]
- 12.Ravdin PM, Burris HA, 3rd, Cook G, Eisenberg P, Kane M, Bierman WA, et al. Phase II trial of docetaxel in advanced anthracycline-resistant or anthracenedione-resistant breast cancer. J Clin Oncol. 1995;13:2879–85. doi: 10.1200/JCO.1995.13.12.2879. [DOI] [PubMed] [Google Scholar]
- 13.Valero V. Primary chemotherapy with docetaxel for the management of breast cancer. Oncology (Williston Park) 2002;16:35–43. [PubMed] [Google Scholar]
- 14.Petrylak DP, Tangen CM, Hussain MH, Lara PN, Jr, Jones JA, Taplin ME, et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med. 2004;351:1513–20. doi: 10.1056/NEJMoa041318. [DOI] [PubMed] [Google Scholar]
- 15.Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351:1502–12. doi: 10.1056/NEJMoa040720. [DOI] [PubMed] [Google Scholar]
- 16.Burkhart CA, Kavallaris M, Band Horwitz S. The role of beta-tubulin isotypes in resistance to antimitotic drugs. Biochim Biophys Acta. 2001;1471:1–9. doi: 10.1016/s0304-419x(00)00022-6. [DOI] [PubMed] [Google Scholar]
- 17.Cabral F, Abraham I, Gottesman MM. Isolation of a taxol-resistant Chinese hamster ovary cell mutant that has an alteration in alpha-tubulin. Proc Natl Acad Sci USA. 1981;78:4388–91. doi: 10.1073/pnas.78.7.4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giannakakou P, Gussio R, Nogales E, Downing KH, Zaharevitz D, Bollbuck B, et al. A common pharmacophore for epothilone and taxanes: molecular basis for drug resistance conferred by tubulin mutations in human cancer cells. Proc Natl Acad Sci USA. 2000;97:2904–9. doi: 10.1073/pnas.040546297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kavallaris M, Kuo DY, Burkhart CA, Regl DL, Norris MD, Haber M, et al. Taxol-resistant epithelial ovarian tumors are associated with altered expression of specific beta-tubulin isotypes. J Clin Invest. 1997;100:1282–93. doi: 10.1172/JCI119642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen GK, Duran GE, Mangili A, Beketic-Oreskovic L, Sikic BI. MDR 1 activation is the predominant resistance mechanism selected by vinblastine in MES-SA cells. Br J Cancer. 2000;83:892–8. doi: 10.1054/bjoc.2000.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Knutsen T, Mickley LA, Ried T, Green ED, du Manoir S, Schrock E, et al. Cytogenetic and molecular characterization of random chromosomal rearrangements activating the drug resistance gene, MDR1/P-glycoprotein, in drug-selected cell lines and patients with drug refractory ALL. Genes Chromosomes Cancer. 1998;23:44–54. doi: 10.1002/(sici)1098-2264(199809)23:1<44::aid-gcc7>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 22.Kuo DY, Mallick S, Shen HJ, DeVictoria C, Jones J, Fields AL, et al. Analysis of MDR1 expression in normal and malignant endometrium by reverse transcription-polymerase chain reaction and immunohistochemistry. Clin Cancer Res. 1996;2:1981–92. [PubMed] [Google Scholar]
- 23.Duan Z, Feller AJ, Penson RT, Chabner BA, Seiden MV. Discovery of differentially expressed genes associated with paclitaxel resistance using cDNA array technology: analysis of interleukin (IL) 6, IL-8, and monocyte chemotactic protein 1 in the paclitaxel-resistant phenotype. Clin Cancer Res. 1999;5:3445–53. [PubMed] [Google Scholar]
- 24.Tudor G, Aguilera A, Halverson DO, Laing ND, Sausville EA. Susceptibility to drug-induced apoptosis correlates with differential modulation of Bad, Bcl-2 and Bcl-xL protein levels. Cell Death Differ. 2000;7:574–86. doi: 10.1038/sj.cdd.4400688. [DOI] [PubMed] [Google Scholar]
- 25.Lee EA, Keutmann MK, Dowling ML, Harris E, Chan G, Kao GD. Inactivation of the mitotic checkpoint as a determinant of the efficacy of microtubule-targeted drugs in killing human cancer cells. Mol Cancer Ther. 2004;3:661–9. [PubMed] [Google Scholar]
- 26.Giovanella BC, Vardeman DM, Williams LJ, Taylor DJ, de Ipolyi PD, Greeff PJ, et al. Heterotransplantation of human breast carcinomas in nude mice. Correlation between successful heterotransplants, poor prognosis and amplification of the HER-2/neu oncogene. Int J Cancer. 1991;47:66–71. doi: 10.1002/ijc.2910470113. [DOI] [PubMed] [Google Scholar]
- 27.Kao GD, McKenna WG, Yen TJ. Detection of repair activity during the DNA damage-induced G2 delay in human cancer cells. Oncogene. 2001;20:3486–96. doi: 10.1038/sj.onc.1204445. [DOI] [PubMed] [Google Scholar]
- 28.Kartalou M, Essigmann JM. Recognition of cisplatin adducts by cellular proteins. Mutat Res. 2001;478:1–21. doi: 10.1016/s0027-5107(01)00142-7. [DOI] [PubMed] [Google Scholar]
- 29.Jensen R, Glazer PM. Cell-interdependent cisplatin killing by Ku/DNA-dependent protein kinase signaling transduced through gap junctions. Proc Natl Acad Sci USA. 2004;101:6134–9. doi: 10.1073/pnas.0400051101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blagosklonny MV, Fojo T. Molecular effects of paclitaxel: myths and reality (a critical review) Int J Cancer. 1999;83:151–6. doi: 10.1002/(sici)1097-0215(19991008)83:2<151::aid-ijc1>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 31.Liao H, Winkfein RJ, Mack G, Rattner JB, Yen TJ. CENP-F is a protein of the nuclear matrix that assembles onto kinetochores at late G2 and is rapidly degraded after mitosis. J Cell Biol. 1995;130:507–18. doi: 10.1083/jcb.130.3.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bar J, Cohen-Noyman E, Geiger B, Oren M. Attenuation of the p53 response to DNA damage by high cell density. Oncogene. 2004;23:2128–37. doi: 10.1038/sj.onc.1207325. [DOI] [PubMed] [Google Scholar]
- 33.Park SJ, Wu CH, Gordon JD, Zhong X, Emami A, Safa AR. Taxol induces caspase-10-dependent apoptosis. J Biol Chem. 2004;279:51057–67. doi: 10.1074/jbc.M406543200. [DOI] [PubMed] [Google Scholar]
- 34.Singh TR, Shankar S, Chen X, Asim M, Srivastava RK. Synergistic interactions of chemotherapeutic drugs and tumor necrosis factor-related apoptosis-inducing ligand/Apo-2 ligand on apoptosis and on regression of breast carcinoma in vivo. Cancer Res. 2003;63:5390–400. [PubMed] [Google Scholar]
- 35.von Haefen C, Wieder T, Essmann F, Schulze-Osthoff K, Dorken B, Daniel PT. Paclitaxel-induced apoptosis in BJAB cells proceeds via a death receptor-independent, caspases-3/-8-driven mitochondrial amplification loop. Oncogene. 2003;22:2236–47. doi: 10.1038/sj.onc.1206280. [DOI] [PubMed] [Google Scholar]
- 36.Kim M, Murphy K, Liu F, Parker SE, Dowling ML, Baff W, et al. Caspase-mediated specific cleavage of BubR1 is a determinant of mitotic progression. Mol Cell Biol. 2005;25:9232–48. doi: 10.1128/MCB.25.21.9232-9248.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim M, Liao J, Dowling ML, Voong KR, Parker SE, Wang S, et al. TRAIL inactivates the mitotic checkpoint and potentiates death induced by microtubule-targeting agents in human cancer cells. Cancer Res. 2008;68:3440–9. doi: 10.1158/0008-5472.CAN-08-0014. [DOI] [PubMed] [Google Scholar]
- 38.Everitt S, Hicks RJ, Ball D, Kron T, Schneider-Kolsky M, Walter T, et al. Imaging cellular proliferation during chemo-radiotherapy: a pilot study of serial 18F-FLT positron emission tomography/computed tomography imaging for non-small-cell lung cancer. Int J Radiat Oncol Biol Phys. 2009;75:1098–104. doi: 10.1016/j.ijrobp.2008.12.039. [DOI] [PubMed] [Google Scholar]
- 39.Zahra MA, Hollingsworth KG, Sala E, Lomas DJ, Tan LT. Dynamic contrast-enhanced MRI as a predictor of tumour response to radiotherapy. Lancet Oncol. 2007;8:63–74. doi: 10.1016/S1470-2045(06)71012-9. [DOI] [PubMed] [Google Scholar]
- 40.Hutcheon AW, Heys SD, Sarkar TK. Neoadjuvant docetaxel in locally advanced breast cancer. Breast Cancer Res Treat. 2003;79:19–24. doi: 10.1023/a:1024333725148. [DOI] [PubMed] [Google Scholar]
- 41.Valero V, Holmes FA, Walters RS, Theriault RL, Esparza L, Fraschini G, et al. Phase II trial of docetaxel: a new, highly effective antineoplastic agent in the management of patients with anthracycline-resistant metastatic breast cancer. J Clin Oncol. 1995;13:2886–94. doi: 10.1200/JCO.1995.13.12.2886. [DOI] [PubMed] [Google Scholar]
- 42.Bamias A, Deliveliotis C, Karayiannis A, Varkarakis I, Zervas I, Pantazopoulos D, et al. Neoadjuvant chemotherapy with docetaxel and cisplatin in patients with high-risk resectable bladder carcinoma: long term results. Eur Urol. 2004;46:344–50. doi: 10.1016/j.eururo.2004.04.024. [DOI] [PubMed] [Google Scholar]
- 43.Decatris MP, Sundar S, O’Byrne KJ. Platinum-based chemotherapy in metastatic breast cancer: current status. Cancer Treat Rev. 2004;30:53–81. doi: 10.1016/S0305-7372(03)00139-7. [DOI] [PubMed] [Google Scholar]
- 44.Dieras V, Guastalla JP, Ferrero JM, Cure H, Weber B, Winckel P, et al. A multicenter phase II study of cisplatin and docetaxel (Taxotere) in the first-line treatment of advanced ovarian cancer: a GINECO study. Cancer Chemother Pharmacol. 2004;53:489–95. doi: 10.1007/s00280-004-0762-9. [DOI] [PubMed] [Google Scholar]
- 45.Kao GD, McKenna WG, Guenther MG, Muschel RJ, Lazar MA, Yen TJ. Histone deacetylase 4 interacts with 53BP1 to mediate the DNA damage response. J Cell Biol. 2003;160:1017–27. doi: 10.1083/jcb.200209065. [DOI] [PMC free article] [PubMed] [Google Scholar]