

Abstract
Nucleolar stress results when ribosome biogenesis is disrupted. An excellent example is the human Treacher Collins syndrome in which the loss of the nucleolar chaperone, Treacle, leads to p53-dependent apoptosis in embryonic neural crest cells and ultimately to craniofacial birth defects. Here, we show that depletion of the related nucleolar phosphoprotein, Nopp140, in Drosophila melanogaster led to nucleolar stress and eventual lethality when multiple tissues were depleted of Nopp140. We used TEM, immuno-blot analysis and metabolic protein labeling to show the loss of ribosomes. Targeted loss of Nopp140 in larval wing discs caused Caspase-dependent apoptosis which eventually led to defects in the adult wings. These defects were not rescued by a p53 gene deletion, as the craniofacial defects were in the murine model of TCS, thus suggesting that apoptosis caused by nucleolar stress in Drosophila is induced by a p53-independent mechanism. Loss of Nopp140 in larval polyploid midgut cells induced premature autophagy as marked by the accumulation of mCherry-ATG8a into autophagic vesicles. We also found elevated phenoloxidase A3 levels in whole larval lysates and within the hemolymph of Nopp140-depleted larvae vs. hemolymph from parental genotype larvae. Phenoloxidase A3 enrichment was coincident with the appearance of melanotic tumors in the Nopp140-depleted larvae. The occurrence of apoptosis, autophagy and phenoloxidase A3 release to the hemolymph upon nucleolar stress correlated well with the demonstrated activation of Jun N-terminal kinase (JNK) in Nopp140-depleted larvae. We propose that JNK is a central stress response effector that is activated by nucleolar stress in Drosophila larvae.
Keywords: Nopp140, nucleolus, apoptosis, autophagy, Drosophila
Introduction
The nucleolar phosphoprotein Nopp1401,2 likely functions as a molecular chaperone for small nucleolar ribonucleoprotein particles (snoRNPs), either in their assembly, transport to the nucleolus from Cajal bodies or in their association with pre-rRNA as these snoRNPs mediate site-specific 2′-O-methylation (box C/D snoRNPs) or pseudouridylation (box H/ACA snoRNPs)3 (reviewed in ref. 4). Drosophila melanogaster expresses two isoforms of Nopp140 by alternative slicing.5 The isoforms are identical up to amino acid residue 584, but then differ in their carboxy termini; one (Nopp140-True) is the canonical ortholog of mammalian Nopp140, while the other (Nopp140-RGG) has a Gly and Arg rich domain common to many RNA-associated proteins. Previously, we used RNAi to target the common 5′ end of both splice variant transcripts. Nopp140 mRNA levels depleted by ≥ 50% caused larval and pupal lethality, while depletions by only ~30% produced viable adults, but with defective legs, wings, thoracic bristles and abdominal cuticles.6 These adult structures normally derive from larval imaginal discs and histoblasts which presumably maintain high demands for ribosome biogenesis and thus protein synthesis.
Related to Nopp140 in structure and function is Treacle, a nucleolar protein found thus far in vertebrates along with Nopp140. TCOF1 is the gene on human chromosome 5 that encodes Treacle. Haplo-insufficiencies in TCOF1 are closely associated with the Treacher Collins-Franceschetti syndrome (TCS), which is marked by craniofacial malformations during fetus development.7,8 TCS arises from the loss of neural crest cells that normally migrate to populate branchial arches I and II on about day 24 of human embryogenesis.9 Loss of Treacle in these particular neural crest cells results in abnormal ribosome biogenesis (referred to as nucleolar stress) and thus a loss in protein synthesis leading ultimately to cellular stress and p53-dependent apoptosis.10 Using the murine system, Jones et al.10 showed that depleting p53 or blocking p53 function prevented apoptosis in these neural crest cells; they were thus able to rescue the craniofacial abnormalities normally associated with the TCS.
Malformations in adult flies resulting from the partial loss of Nopp140 in Drosophila larval progenitor tissues were reminiscent of the human TCS. The first goal of this study was to determine what cell stress responses resulted from the depletion of Nopp140 by RNAi expression in Drosophila larval cells leading either to lethality or to malformations in adult structures. Apoptosis in larval imaginal disc cells and histoblasts could well explain the morphological defects observed in adult structures. The second goal was to determine if p53 is required for the Drosophila nucleolar stress response as it is in the mammalian system. The third goal was to identify principal stress effectors that respond to nucleolar stress responses in Drosophila.
Results
We previously used the GAL4 system in Drosophila to express RNAi that targeted mRNAs encoding both Nopp140 isoforms.6 We showed that larvae depleted of Nopp140 mRNAs to ~50% of wild type levels died in the late third instar larval and pupal stages. Conversely, when reduced Nopp140 transcripts were depleted by only ~30%, the larvae survived to adults, but their wings, legs, thoracic bristles or abdominal cuticles were malformed.6 Essentially two RNAi-expressing transgenes, UAS-C3 and UAS-C4.2, were used in the following studies. Both effectively reduce Nopp140 levels when induced by GAL4. UAS-C4.2 maps on the second chromosome and UAS-C3 maps on the third chromosome; both transgenes reside in the original UAS-C4 line.6 For the sake of simplicity, we refer to UAS-C4.2, UAS-C3 or the original UAS-C4 as Nopp140-RNAi in the text, but figure legends differentiate which transgene was actually used in the respective experiments.
Nucleolar stress caused by the loss of Nopp140
Heterozygous Act5C > Nopp140-RNAi lacked Nopp140 within most, but not all of their imaginal diploid cells (Fig. 1A and B) and polyploid cells (Fig. 1C and D). Polyploid cells lacking Nopp140 condense their chromatin (Fig. 1C).6 These larvae died in the late larval and early pupal stages when reared at 27−28°C. While we used w1118 as our wild type control in Figure 1E−H, homozygous parental larvae develop normally (they are viable and fertile) with no detectable loss of Nopp140 in their tissues.

Figure 1. Selective loss of Nopp140 in Act5C > UAS-C4.2 larvae. Expression of RNAi targeting Nopp140 transcripts caused depletion of Nopp140 in most tissues as shown by DAPI staining and immuno-fluorescence microscopy using an antibody directed against a unique sequence within the carboxy terminal domain of Nopp140-RGG.6(A and B) Nucleoli in most small diploid imaginal disc cells from Act5C > UAS-C4.2 larvae lacked Nopp140-RGG. A few cells retained detectable Nopp140-RGG in their nucleoli (B), due presumably to insufficient RNAi expression in these particular cells. (C and D) Nucleoli in giant polyploid cells from Act5C > UAS-C4.2 larvae also lacked Nopp140-RGG. Polyploid cells lacking Nopp140 contained condensed chromatin.6(E and F) Nucleoli in most imaginal disc cells from wild type larvae stained with anti-Nopp140-RGG. (G and H) Nucleoli in polyploid midgut cells and the smaller imaginal island cells from wild type larvae stained well with the anti-Nopp140-RGG antibody. Scale bar: 25 µm for all images.
We compared tissues from Act5C > Nopp140-RNAi larvae to the same tissues from wild type (w1118) or parental type larvae by transmission electron microscopy, immuno-blot analysis and metabolic protein labeling to confirm that ribosome synthesis was disrupted due to Nopp140 depletion (i.e., nucleolar stress). A diploid cell from an Act5C > Nopp140-RNAi larval wing disc (Fig. 2A) contained some rough endoplasmic reticulum, but the number of free ribosomes in the cytosol was greatly reduced compared with that in the wild type disc cell (Fig. 2B). In addition, the nuclei (Nu) in the Act5C > Nopp140-RNAi diploid cells contained numerous virus-like particles (arrow in Fig. 2A). These particles likely arise from copia retrotransposons11 that can be induced by environmental stress,12 aging13 or perturbations in poly(ADP-ribose) polymerase.14 In preparing tissues for TEM analysis, we noted the small size of the imaginal discs isolated from Act5C > Nopp140-RNAi larvae. We have yet to determine if reduced disc size is due to smaller cells or failure to produce sufficient cell numbers. A polyploid midgut cell from an Act5C > Nopp140-RNAi larva (Fig. 2C) contained relatively very few ribosomes as compared with the same cell type from a wild type larva (Fig. 2D). Again, the presence of virus-like bodies the nucleus (arrow in Fig. 2C) indicated these Nopp140-depleted polyploid cells were under stress. While Act5C > Nopp140-RNAi imaginal discs failed to grow, brains from these larvae appeared normal in size; the brain cells showed normal levels of cytoplasmic ribosomes, and their nuclei were clear of virus-like bodies (image not shown). This was surprising since the endogenous Act5C gene is normally expressed at high levels in the larval central nervous system, and we expected the Act5C-GAL4 transgene would be similarly expressed. One possibility is that Nopp140 expression levels may be sufficiently high in the larval central nervous system48 (see http://flybase.org/reports/FBgn0037137.html) such that Nopp140-RNAi expression cannot effectively deplete enough Nopp140 to disrupt ribosome synthesis.

Figure 2. TEM, immuno-blot analysis and metabolic protein labeling demonstrate loss of ribosomes and protein synthesis. (A−D) Separate tissues were isolated by hand and prepared for TEM analysis. Imaginal wing disc cells from Act5C > UAS-C4.2 larvae (A) contained fewer ribosomes as compared with similar cells from wild type (w1118) larvae (B). Polyploid midgut cells from Act5C > UAS-C4.2 larvae (C) contained far fewer ribosomes than did similar, identical cells from wild type (w1118) larvae (D). Nuclei (Nu) in Act5C > UAS-C4.2 larvae (panels A and C) contained numerous copia virus-like particles indicating stress. Scale bar: 1 µm for all TEM images. (E) Specific antibodies showed a loss of RpL23a and RpL34 in lysates from Act5C > UAS-C4.2 larvae compared with parental type larvae. Lane 1: homozygous UAS-C4.2 parental lysate; Lane 2: heterozygous Act5C-GAL4/CyO-GFP parental lysate; Lane 3: RNAi-expressing UAS-C4.2/Act5C-GAL4 lysate; Lane 4: a separate RNAi-expressing UAS-C4.2/Act5C-GAL4 lysate. (F) Metabolic protein labeling was significantly reduced in Nopp140-RNAi-expressing UAS-C4.2/Act5C-GAL4 larvae compared with larval parental controls. The entire experiment was repeated three times. Absorbance values were determined in triplicate. Graphs were assembled using Microsoft Excel; two-tailed P values were equated using t-Test analysis (two sample assuming unequal variances).
By immuno-blot analysis (Fig. 2E), we saw a loss of ribosomal proteins L23A and L34 in third instar Act5C > Nopp140-RNAi larvae (lanes 3 and 4) compared with the two respective parental larval types (lanes 1 and 2). We note, however, that younger Act5C > Nopp140-RNAi larvae that had yet to show obvious defects such as melanotic tumors (see below) contained near normal levels of ribosomal proteins (lane not shown). These younger larvae likely maintained sufficient quantities of maternal Nopp140 and ribosomes that dwindle as these larvae advance further into the third instar stage.
To verify the effects of ribosome loss in third instar larvae, we compared metabolic protein labeling in parental control larvae vs. Act5C > Nopp140-RNAi larvae depleted for Nopp140. The loss of Nopp140 leading to the loss of ribosomes, caused a reduction in protein synthesis to ≤ 40% (Fig. 2F). In this assay, we analyzed two separate samples of Act5C > Nopp140-RNAi. One sample (*) was older with a greater number of melanotic masses compared with the other, yet we saw similar protein labeling profiles in the two samples. We conclude from the various assays in Figure 2 that third instar Act5C > Nopp140-RNAi larvae had a pronounced loss of ribosomes and thus protein synthesis, and this resulted in lethality by the late third larval instar-early pupal stages.
Selective depletion of Nopp140 in larval wing discs
To assess the loss of Nopp140 without inducing lethality, we used the larval wing disc driver, A9-GAL4, on the X chromosome to induce the Nopp140-RNAi gene (UAS-C4.2 on the second chromosome) (A9 > UAS-C4.2). We set up the cross such that all male progeny displayed normal wings. Conversely, female progeny (A9 > Nopp140-RNAi) expressed RNAi in their larval wing discs, which eventually led to malformed adult wings. The wing phenotype in these females varied from a relatively mild upward curl along the anterior and posterior edges of the wing to severely shriveled (vestigial-like) wings (see Table 1). Large fluid-filled blisters were common in the wings of newly eclosed females. Their wings were left badly malformed as the blisters receded over time. Table 1 compares the frequency of the severe wing phenotype to that of the less severe curled wing phenotype. While all the male progeny had normal wings, 47% of the A9 > Nopp140-RNAi females showed wings with the mild defect, and the remaining 53% showed the severe defect. None of the females had normal wings. We conclude that A9 > Nopp140-RNAi provides a non-lethal phenotype that we can score to access nucleolar stress in diploid imaginal (progenitor) wing disc cells.
Table 1.X and/or Y chromosome, second chromosome and third chromosome designations are separated by semi-colons. Male larvae (w1118/Y) lacked the A9-GAL4 driver and therefore did not express RNAi from UAS-C4.2 on their second chromosome. Female larvae (A9/w1118) expressed RNAi to deplete Nopp140 in their wing discs. Two crosses were performed, with (+) and without (∆) the p53 gene on the third chromosome. Abnormal wing phenotypes appeared in only the females as either a mild wing edge curl or a more severe vestigial-like (shriveled) wing. Phenotype frequencies were comparable with or without the p53 gene.
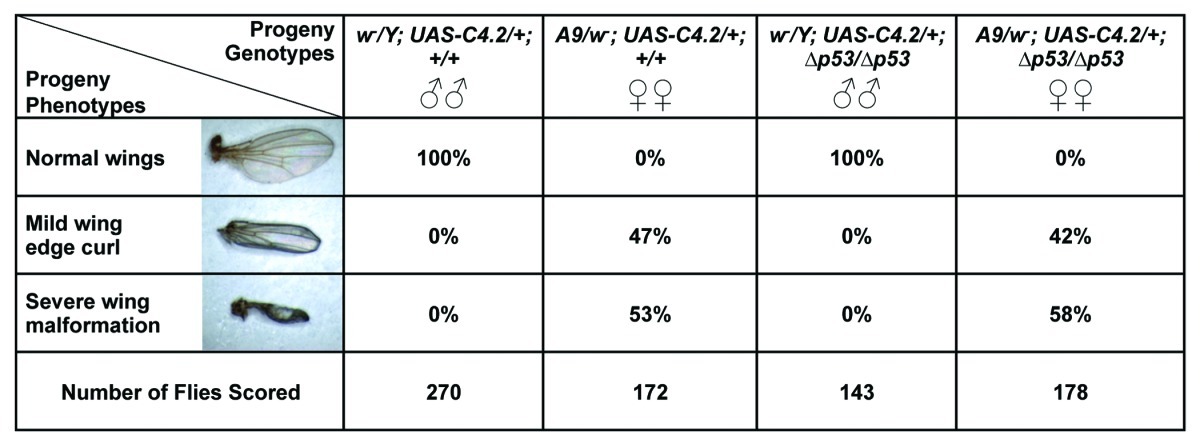
TCS arises in mammals when specialized embryonic neural crest cells undergo apoptosis due to the loss of Treacle.10 To determine if nucleolar stress caused by the loss of Nopp140 in Drosophila wing discs also induces apoptosis, we reversed the original cross for Table 1 such that homozygous A9-GAL4 driver females were crossed to males homozygous for Nopp140-RNAi. All larval progeny from this cross expressed Nopp140-RNAi in their wing discs. We probed these wing discs with anti-cleaved Caspase 3 (Asp175) from Cell Signaling Technology. Although the precise antigen remains unknown, this particular antibody provides a good marker for Caspase-9-like DRONC activity in apoptotic Drosophila cells.15 A wing disc from an A9 > Nopp140-RNAi larva was heavily labeled by anti-Caspase (Fig. 3A−C). Higher magnification showed that the apoptotic cells contained condensed chromatin within their nuclei (Fig. 3D) and that anti-Caspase 3 labeled the cytoplasm (Fig. 3E) as expected. Wing discs from wild type larvae (w1118) showed minimal anti-Caspase labeling (Fig. 3F-H). Therefore, as with the loss of Treacle in mammalian neural crest cells, selective loss of Nopp140 in Drosophila imaginal wing disc cells induced apoptosis leading to malformed wings in adult flies.

Figure 3. Loss of Nopp140 in imaginal wing discs of A9 > UAS-C4.2 larvae resulted in apoptosis. Phase contrast (A) and DAPI staining (B) of a wing disc isolated from an A9 > UAS-C4.2 larva. (C) The wing disc stained heavily with anti-Caspase 3 indicating apoptosis. (D and E) Higher magnification of panels B and C showed anti-Caspase 3 staining in the cytoplasm as expected. Cells with copious anti-Caspase 3 labeling showed condensed chromatin by DAPI staining (arrows). (F−H) Wing discs from control (w1118) larvae were probed with anti-Caspase 3 antibody. They showed minimal background labeling.
Apoptosis induced by nucleolar stress in Drosophila is p53-independent
Jones et al.10 showed that apoptosis induced by nucleolar stress in mouse embryonic neural crest cells was p53-dependent. The deletion of the mouse p53 gene or chemical inactivation of p53 suppressed this apoptosis, reducing the severity of phenotypes typically associated with TCS. To test if apoptosis in Drosophila imaginal wing discs resulting from depletion of Nopp140 is p53-dependent, we repeated the original cross described in Table 1 such that only female progeny would display wing defects, but this time the two parental types (the A9-GAL4 driver and the Nopp140-RNAi-expressing lines) were homozygous for the p53 gene deletion (p53[5A-1–4]) on the third chromosome (referred to simply as p53∆). This deletion was originally described by Rong et al.16; homozygous deletion flies are viable and fertile, but they display reduced apoptosis in response to DNA damage caused by ionizing radiation.17-19
Genomic PCRs verified that the modified parental lines were in fact deleted for p53 (Fig. 4A). The wild type p53 gene in w1118 flies produced the expected 7.3 kbp PCR product (lane 1), while the modified A9-GAL4 driver (lane 2), the original p53∆/p53∆ stock from the Bloomington Stock Center (lane 3) and the Nopp140-RNAi line (lane 4) produced the expected 4.0 kbp product indicating homozygous deletion of the p53 gene (see Flybase, http://flybase.org/reports/FBrf0151688.html for descriptions of the expected PCR products). Thus, all A9 > Nopp140-RNAi progeny from this second cross should be homozygous deficient for the p53 gene. While the male progeny from this cross showed normal wings as expected (Fig. 4B, left wing), all female progeny again displayed malformed wings (Fig. 4B, right wing). Table 1 again summarizes the results: of the female progeny obtained, 42% displayed the less severe curled wing edge phenotype, while 58% showed the more severe shriveled wing phenotype (Table 1). This compares well to 47% and 53%, respectively, for those female progeny that were homozygous for the wild type p53 gene. We note that variability in wing phenotype in both crosses (with and without p53) may be caused to slight temperature fluctuations affecting GAL4-mediated induction of UAS-transgenes. To minimize this effect, all crosses were performed at 27–28°C.

Figure 4. Verifying the p53 gene deletion in parental stocks and establishing p53-independent apoptosis in Nopp140-depleted wing discs. (A) The p53 gene deletion was verified using genomic PCR. The 7.3 kb band denotes presence of the wild type p53 gene while the 4.0 kb band denotes its deletion. Tested genotypes were: w1118 stock (wild type) (lane 1), A9-GAL4/A9-GAL4; +/+; p53∆/p53∆ (the GAL4 driver) (lane 2), the original p53∆/p53∆ stock (Bloomington 6815) (lane 3), w1118/w1118; UAS-C4.2/UAS-C4.2; p53∆/p53∆ (RNAi) (lane 4). (B) The cross reported in Table 1 was repeated using the A9-GAL4 and UAS-C4.2 lines that were homozygous for p53∆. Adult male progeny (w1118/Y; UAS-C4.2/+; p53∆/p53∆) again produced normal wings (left wing), while female progeny (A9-GAL4/w1118; UAS-C4.2/+; p53∆/p53∆) expressed RNAi to deplete Nopp140 and again displayed malformed wings (right wing). (C and D) Apoptosis was evident in the larval wing discs of A9 > UAS-C4.2; p53∆/p53∆ larvae as shown by anti-Caspase 3 labeling within the cytoplasm. White arrows point to corresponding nuclei containing condensed chromatin.
We again reversed the cross such that all progeny were A9 > Nopp140-RNAi; p53∆/p53∆. These larvae also showed apoptosis in wing discs by anti-Caspase 3 labeling (Fig. 4C and D). We performed these crosses several times to be certain that the presence or absence of p53 had no effect on wing malformations due to Nopp140 depletion. The combined data in Table 1 indicates that there were no appreciable differences in the abnormal wing phenotypes due to Nopp140 depletion when p53 was present or deleted. We conclude, therefore, that larvae selectively depleted for Nopp140 in their wing discs induce apoptosis in these discs, but in a p53-independent manner.
Autophagy in larval polyploidy midgut cells depleted of Nopp140
In earlier studies to examine the loss of Nucleostemin I in Drosophila larvae, we observed by TEM premature autophagy in larval polyploid midgut cells.20 Here, we ectopically co-expressed mCherry-ATG8a21 to determine if excessive autophagy occurred in midgut cells of Nopp140-depleted (da > Nopp140-RNAi; UAS-mCherry-ATG8a) third instar larvae. We first expressed mCherry-ATG8a in the midgut of a second instar larva using the da-GAL4 driver, but in an otherwise wild type background; mCherry-ATG8a distributed diffusely throughout these midgut cells (Fig. 5A). As a positive control for autophagy, we starved similar second instar larvae to induce autophagy.22 As expected, mCherry-ATG8 aggregated into vesicles within the cytoplasm of these midgut polyploid cells (Fig. 5B). We interpret the appearance of these vesicles as autophagosomes and lysosomes.23 Similar vesicles with mCherry-ATG8 appeared prematurely and in great abundance in the midgut of Nopp140-depleted larvae (da-GAL4 > Nopp140-RNAi; UAS-mCherry-ATG8a) (Fig. 5C). Thus, the loss of Nopp140 and nucleolar function induces premature autophagy in the polyploid midgut cells.

Figure 5. Autophagy was prominent in larval polyploid cells depleted for Nopp140. (A and D)da > mCherry-ATG8 in a wild type background. Labeling remained diffuse throughout the cell. (B and E) As a positive control for autophagy, similar da > mCherry-ATG8a larvae were starved of amino acids. The accumulation of mCherry-ATG8a into autophagosomes and lysosomes (red speckling in the cytoplasm) indicated autophagy as expected due to starvation. (C and F) Well fed da > UAS-C4.2; mCherry-ATG8a larvae displayed similar accumulations of mCherry-ATG8a into cytoplasmic vesicles within their midgut cells indicating premature autophagy in response to the loss of Nopp140.
Accumulation of phenoloxidase A3 in response to Nopp140 depletion
Coomassie-stained SDS-gels showed an abundant 70 kDa protein in whole lysates of Act5C > Nopp140-RNAi larvae vs. parental control larvae (Fig. 6A). At first, we thought the accumulated protein was a classic heat shock protein (Hsp70) or perhaps a heat shock cognate protein (Hsc70) that was induced upon nucleolar stress. However, antibodies against Hsp70 and Hsc70 failed to show significant accumulations of either protein in the Nopp140-depleted larvae. LC-MS/MS finally identified the protein as phenoloxidase A3 encoded by PO45 (conceptual gene CG8193) in Drosophila melanogaster.

Figure 6. Phenoloxidase A3 accumulated in Nopp140-depleted larvae. (A) Whole larval lysates from parental control larvae (Act5C-GAL4/CyO, lane 1; homozygous UAS-C4.2, lane 2) were compared with those from Nopp140-depleted larvae (UAS-C4.2/Act5C-GAL4, lane 3; UAS-C4/Act5C-GAL4, lane 4). UAS-C4 is the original stock containing UAS-C4.2 on the 2nd chromosome and UAS-C3 on the 3rd chromosome. The prominent 70 kDa protein in the lysates of Nopp140-depleted larvae (arrow) was identified by LC-MS/MS as phenoloxidase A3. (B) Hemolymph proteins were resolved on a native polyacrylamide gel and stained with Coomassie Blue. Parental control larvae were homozygous UAS-C4.2 (lane 1) and Act5C/CyO-GAL4 (lane 2). Nopp140-RNAi-expressing UAS-C4.2/Act5C-GAL4 larvae (lane 3) with only a few melanotic masses were compared with similar UAS-C4.2/Act5C-GAL4* larvae (lane 4) that had excess melanotic masses. (C) A native gel similar to that shown in panel B but stained for phenoloxidase activity using tyrosine as substrate. Arrows in B and C mark the position of the most prominent phenoloxidase activity. (D) Melanotic masses were found mostly in the midgut of Act5C > UAS-C4.2 larvae. (E) Melanotic masses also occurred at other sites within Act5C > UAS-C4.2 larvae. This large mass accumulated at the tips of the dorsal arms of the pharyngeal sclerite.
Phenoloxidases in Drosophila are released into the hemolymph from circulating crystal cells, usually as part of an innate immune response to parasitic infection. Upon infection, the phenoloxidases convert phenols to quinones that polymerize to form melanin aggregates that then encapsulate the parasite.24 To test if phenoloxidases were released to the hemolymph of Nopp140-depleted larvae, we isolated larval serum proteins,25 resolved them on native polyacrylamide gels and then either stained the gels with Coomassie Blue (Fig. 6B) or stained the gels for phenoloxidase activity using tyrosine as a substrate26 (Fig. 6C). Compared with parental types, Act5C > Nopp140-RNAi larvae showed greater amounts of phenoloxidase in their hemolymph, indicating another phenotype associated with the loss of nucleolar function. In collecting the Act5C > Nopp140-RNAi larvae, we purposely pooled larvae with numerous melanotic masses. These larvae are designated with an asterisk in Figure 6, and they showed more phenoloxidase in their hemolymph than did Act5C > Nopp140-RNAi larvae with only a few melanotic masses.
We noted previously the accumulation of melanotic masses (tumors) in Nopp140-depleted larvae and pupae.6 These masses seem to arise preferentially in the midgut (Fig. 6D), but we also found melanotic masses scattered throughout the larva; for example, Figure 6E shows melanotic masses at the ends of the dorsal arms of the pharyngeal sclerite. The observed rise in phenoloxidase abundance in whole lysates and its accumulation in the hemolymph correlate well with the appearance of melanotic masses in Nopp140-depleted larvae.
JNK and pro-apoptotic Hid are activated in response to nucleolar failure
Thus far we have documented three stress response phenotypes that arise due to the loss of nucleolar function: apoptosis in imaginal wing discs, autophagy in polyploid midgut cells and the formation of melanotic masses due to the release of phenoloxidase from crystal cells. JNK (also known as the stress-activated protein kinase, SAPK) is a principal cell stress response effector27,28 that could link all three phenotypes. Activated Drosophila JNK (also referred to as basket) is known to induce apoptosis by activating Hid29 and autophagy by inducing dFoxo.30,31 Activated JNK also induces the rupture of crystal cells to release phenoloxidases.32 Therefore, we tested if Drosophila JNK is activated in Nopp140-depleted larvae (Fig. 7B). Activation of human JNK is marked by the phosphorylation of residues Thr183 and Tyr185. These residues are conserved in the one isoform of JNK expressed in D. melanogaster. The rabbit monoclonal antibody 81E11 from Cell Signaling cross reacted well with activated Drosophila JNK on immuno-blots (Fig. 7B). Whole lysates from Act5C > Nopp140-RNAi larvae showed an elevated level of activated JNK vs. lysates from parental controls. We conclude that activated JNK accumulates in Nopp140-depleted larvae.
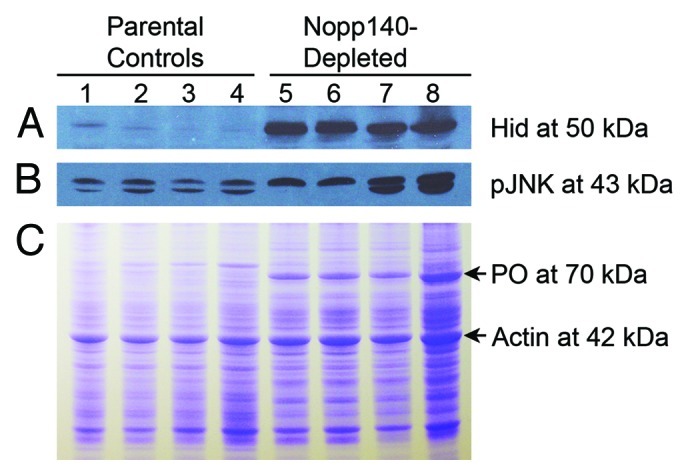
Figure 7. Pro-apoptotic Hid and activated JNK were both upregulated in Nopp140-depleted larvae. (A) An immuno-blot containing lysates from parental control larvae (lanes 1–4) and Act5C > Nopp140-RNAi larvae (lanes 5–8) was probed with rabbit anti-Hid.34 Hid at ~50 kDa was substantially increased in the Nopp140-depleted larvae as compared with parental larvae. Lane 1: homozygous UAS-C4.2; Lane 2: Act5C-GAL4/CyO; Lane 3: Act5C-GAL4/+; Lane 4: CyO-GFP/+, Lanes 5 and 7: UAS-C4.2/Act5C-GAL4; Lanes 6 and 8: UAS-C4/Act5C-GAL4. UAS-C4 is the original transgenic stock that contains UAS-C4.2 on the 2nd chromosome and UAS-C3 on the 3rd chromosome. (B) A companion blot with the same lysates was probed with rabbit monoclonal antibody 81E11 (Cell Signaling #4668) directed against phospho-JNK at 43 kDa. (C) Equal amounts of the same lysates as used in panels A and B were resolved on a Coomassie-stained SDS-gel. Phenoloxidase A3 (PO) is at 70 kDa.
The anti-pJNK immuno-blot (Fig. 7B) showed two bands in some lysate preparations from both parental types and from the Nopp140-depleted larvae, while we saw only the higher molecular weight band in other separately prepared lysates. At this time we have no clear explanation for the appearance of the bottom band other than partial proteolysis.
In their model of p53-independent apoptosis in Drosophila, McNamee and Brodsky33 proposed that JNK induces Hid protein expression that then induces apoptosis. To test if Hid was induced, we probed the same lysates used in Figure 7B with a rabbit polyclonal antibody directed against Drosophila Hid.34 We found Hid levels notably increased in Actin5C > Nopp140-RNAi larvae vs. parental controls (Fig. 7A).
Discussion
The human TCS results from mutations in the TCOF1 gene which encodes the nucleolar phosphoprotein, Treacle. Subsets of embryonic neural crest cells are particularly sensitive to the loss of Treacle, a nucleolar chaperone for ribosome biosynthesis. These cells normally migrate to populate embryonic branchial arches I and II that then give rise to adult craniofacial structures. Without Treacle, these neural crest cells fail to meet a relatively high threshold requirement for functional ribosomes. As a result these cells are stressed and undergo p53-dependent apoptosis. Their loss ultimately leads to the craniofacial malformations associated with the TCS. Jones et al.10 could alleviate the syndrome by blocking p53 function either by a p53 gene deletion or by a specific p53 protein inhibitor.
Vertebrate Nopp140 and Treacle are related in structure and function.4 Both proteins are over 100 kDa in size, both contain LisH dimerization motifs in their N-terminal region, and both proteins contain a large central domain consisting of alternating acidic and basic motifs with similar amino acid compositions; the acidic motifs are rich in Glu, Asp and phospho-Ser, while the basic motifs are rich in Lys and Pro. Both proteins are believed to function as chaperones for snoRNP complexes as these complexes direct site-specific methylation and pseudouridylation of pre-rRNA within the dense fibrillar regions of eukaryotic nucleoli. While most snoRNAs appear to be non-essential when individually deleted, failure to collectively modify pre-rRNA by the loss of a snoRNP chaperone would likely produce defective or partially functional ribosomes in all cells. Embryonic progenitor cells with high demands for protein synthesis would be most susceptible to the loss of functional ribosomes. Loss of these cells would lead to lethality or developmental defects. Thus Treacle and Nopp140 are required for normal development. Despite their multiple similarities, Nopp140 and Treacle differ in their carboxy termini, suggesting that each protein maintains its own unique interactions and functions. Interestingly, while Drosophila expresses two isoforms of Nopp140 by alternative splicing,5 Drosophila lacks a Treacle ortholog.
We showed that larval cells depleted for Nopp140 undergo a disruption in ribosome biosynthesis (nucleolar stress). Larval tissues expressing Nopp140-RNAi, both diploid and polyploid, have fewer ribosomes than wild type (Fig. 2A−D). These larvae also have reduced amounts of ribosomal proteins L23 and L34 (Fig. 2E). Metabolic labeling verified these findings, revealing that Nopp140-depleted larvae synthesize significantly reduced amounts of protein (≤ 40%) compared with parental controls (Fig. 2F). As larval tissues develop, maternally supplied ribosomes and Nopp140 diminish, and cells expressing Nopp140-RNAi are unable to supply either functional ribosomes and/or sufficient protein synthesis. This nucleolar stress thus leads to distinctive cell death pathways.
Here we showed that the loss of Drosophila Nopp140 specifically in imaginal wing disc cells lead to apoptosis and impaired wing development, analogous to the loss of Treacle in embryonic neural crest cells. However, while Jones et al.10 rescued the TCS in mice by employing a p53 gene deletion, the similar rescue failed in Drosophila as apoptosis occurred in the absence of the p53 gene (Fig. 3). Induction of apoptosis in the absence of p53 has been well documented in Drosophila.18,33
Besides apoptosis, we documented a strong and premature induction of autophagy in larval midgut polyploid cells that were depleted for Nopp140 (Fig. 5C). As with apoptosis, JNK activation is reportedly linked to autophagy induction22 by way of the downstream activation of dFoxo.30 We note, however, that this stress-induced autophagy is in contrast to a JNK-independent induction of autophagy that normally occurs during development or in response to starvation resulting in the inhibition of Tor signaling.22,23
We also showed that formation of excess melanotic masses in Nopp140-depleted larvae was coincident with the accumulation of phenoloxidase A3 both in whole larval lysates (Fig. 6A) and in the larval hemolymph (Fig. 6B and C). While phenoloxidases are normally synthesized and stored in crystal cells, they are not secreted in a typical secretory pathway; rather, crystal cells rupture in a JNK-dependent manner releasing the phenoloxidases to the hemolymph.24,32 How Nopp140-depleted larvae synthesize excess amounts of phenoloxidase when nucleolar function (ribosome biosynthesis) is impaired remains a perplexing question. Since the Nopp140-depleted larvae are developmentally delayed, we speculate that phenoloxidase A3 may simply amass in abundance over time despite a slow rate of protein synthesis as compared with wild type larvae. This could explain the excess accumulations of phenoloxidase in whole larval lysates as shown by Coomassie-stained gels (Fig. 6A). The activation of JNK in the Nopp140-depeleted larvae may then release the phenoloxidase to the hemolymph as shown in the hemolymph enrichments (Fig. 6B and C).
Many of the melanotic masses we saw seemed to reside within non-hematopoietic tissues such as the midgut, trachea and salivary glands.35 One possibility suggested by Bidla et al.32 is that apoptosis followed by secondary necrosis triggers formation of melanotic masses. If so, we would expect to see the formation of multiple melanotic masses at non-hematopoietic sites undergoing heavy apoptosis and secondary necrosis.
Nucleolar stress in Drosophila vs. mammals
Our working hypothesis is that depletion of Nopp140 in Drosophila cells disrupts normal ribosome biogenesis, a term now referred to as “nucleolar stress.”36,37 Studies on nucleolar stress in mammalian cells have focused primarily on the nucleolar protein ARF (p19ARF in mice, p14ARF in humans), its interactions with MDM2 which is the E3 ubiquitin ligase for p53 and the role that nucleolar protein B23 plays in sequestering ARF to the nucleolus.37 Upon stress in mammalian cells, ARF indirectly activates p53 by interacting with MDM2 to suppress its ubiquitylation of p53. Under these conditions, p53 accumulates and activates genes necessary for cell cycle arrest, senescence or apoptosis. Interestingly, Drosophila lacks identifiable ARF, MDM2 and B23. Thus nucleolar stress in Drosophila likely plays out by an alternative mechanism.
From our work, JNK appears to be a common link between Hid (apoptosis), dFoxo (autophagy)22,30 and the release of phenoloxidases from crystal cells as these cells rupture in JNK-dependent manner.32 We showed by immuno-blot analysis that Hid rises (Fig. 7A) coincidently with activated JNK (Fig. 7B) in Nopp140-depleted larvae. While JNK activation has been reported to be delayed in the p53-independent mechanism,18,38 once activated, JNK is thought to induce Hid expression and thus apoptosis.33 While we have yet to assess the abundance and activities of dFoxo and dFos in the Nopp140-depleted larvae, others have shown that Hid induction results from JNK activation via these two transcription factors.29,39 From the combined evidence, we propose that JNK is a central effector activated in response to perturbations in Drosophila ribosome biogenesis.
JNK is a member of the MAP kinase superfamily, and while mammals express multiple forms of JNK, Drosophila melanogaster expresses only one version of JNK (basket). Precisely how Drosophila JNK is activated in response to nucleolar failure remains unknown. As a clue, Moreno et al.40 showed that JNK signaling is required for the apoptotic elimination of slowly proliferating Minute-/+ cells in larval wing discs where Minute- is a dominant, haplo-insufficient mutation in a gene encoding a ribosomal protein.41 McNamee and Brodsky33 proposed that gene haplo-insufficiencies in these same ribosomal protein genes could induce expression of brinker (brk) which encodes a transcription factor in larval wing discs, and that brk is sufficient to activate JNK, thus leading to apoptosis. Also upstream of Drosophila JNK are at least two JNKKs (Hemipterous and MKK4) and several JNKKKs. Puckered is the Drosophila protein phosphatase that is also induced by JNK to deactivate JNK in a negative feedback loop. How Hemipterous and Puckered are regulated upon nucleolar stress in Drosophila remains unknown. Our future efforts in linking nucleolar stress to JNK activation will examine the expression levels of brk, hemipterous and puckered.
Finally, JNK signaling has been implicated in direct nucleolar function. For example, mammalian JNK2 phosphorylates the RNA polymerase I transcription factor, TIF-IA, to downregulate rRNA synthesis.42 JNK activity was also required for the release of B23 and p19ARF from mammalian nucleoli upon UV radiation (DNA-damage).43 Conversely, Mialon et al.44 showed that upon genetic or chemical inhibition of JNK signaling, the mammalian nucleolar RNA helicase, DDX21, is partially redistributed to the nucleoplasm and that rRNA processing is inhibited. Their observation suggests that besides stress-induced JNK-activation, JNK signaling maintains homeostasis for the nucleolus in non-stressed cells. Thus the effects of activated JNK on the localization of Drosophila nucleolar components during normal cell growth and upon stress remain largely unknown, but subject for future investigation.
Materials and Methods
Fly lines
Fly lines expressing short hairpin RNA specific for the common 5′ end of mRNAs that encode the two Nopp140 isoforms were P{w[+mC] = UAS-Nopp140.dsRNA}C3 and P{w[+mC] = UAS-Nopp140.dsRNA}C4.2 as described previously.6 Here we refer to them simply as UAS-C3 and UAS-C4.2. UAS-C3 resides on the third chromosome. The original UAS-C4 line contained two RNAi-expressing transgenes: one on the second chromosome and the other on the third chromosome. The transgene on the third chromosome is UAS-C3. We isolated the second chromosome from UAS-C4 and refer to the resulting line as UAS-C4.2. Both UAS-C3 and UAS-C4.2 lines are homozygous viable and fertile. The P{UAS-mCherry-Atg8a} line, D169 (third chromosome), was a kind gift from Thomas Neufeld.21 Four additional lines came from the Bloomington Drosophila Stock Center; stock 6815 is a deletion for the p53 gene (p53[5A-1–4]).16 We refer to this deletion simply as p53∆ throughout this report. GAL4 driver lines included P{w[+mC] = Actin5C-GAL4}25FO1/CyO (stock 4414, referred to simply as Act5C-GAL4), the homozygous daughterless-GAL4 line on the third chromosome (stock 8641, referred to as da-GAL4) and the wing disc-specific P{w[+m*] = GAL4}A9 on the X chromosome (stock 8761, referred to here as A9-GAL4). A9-GAL4 expresses GAL4 strongly in larval wing and haltere discs.45 We previously used the w1118 line (Bloomington stock 3605) to construct our RNAi-expressing transgenic lines,6 and we use it here as our “wild type” control. All stocks were reared at 22–24°C on standard Drosophila medium. Genetic crosses using GAL4 drivers were kept at 27–28°C.
Genomic PCR to verify p53∆
Genomic DNA from adult flies was prepared according to E. Jay Rehm of the Berkeley Drosophila Genome Project (http://www.fruitfly.org/about/methods/inverse.pcr.html). Primers for verifying the p53 gene deletion (p53[5A-1–4]) were described by K. Golic in a reference report to FlyBase (http://flybase.org/reports/FBrf0151688.html). The forward primer is P53-C1: 5′-AGCTAATGTGACTTCGCATTGAACAAA-3′, and the reverse primer is P53-C2: 5′-TCGATAAACATTGGCTACGGCGATTGT. These primers were prepared by Integrated DNA Technologies. The deletion should produce a 4.0 kbp product, while the wild type allele should produce a 7.3 kbp product. We used i-MAXTM II from iNtRON Biotechnology for the long distance genomic PCRs.
Immuno-fluorescence and transmission electron microscopy
For immuno-fluorescence microscopy, larval tissues were fixed in a phosphate buffered solution containing freshly prepared paraformaldehyde (2% final concentration), washed, blocked and incubated with antibodies as described.46 We used a previously characterized isoform-specific guinea pig antiserum6 at 1/100 and an Alexa Fluor® 546-conjugated goat anti-guinea pig antibody (A11074, Molecular Probes) at 1/250 to demonstrate the loss of Nopp140-RGG in RNAi-expressing larvae. To demonstrate apoptosis in imaginal wing discs, we used rabbit anti-cleaved Caspase-3 (Asp175) (#9661, Cell Signaling Technology) at 1/200. This particular antibody has been well described as a marker for Caspase-9-like DRONC activity in apoptotic Drosophila cells.15 The secondary antibody was the Alexa Fluor® 488-conjugated goat anti-rabbit from Molecular Probes (A11034) at 1/250. Tissues were stained with DAPI at 1 µg/mL in the final wash buffer and viewed with a Zeiss Axioskop equipped with a SPOT RT/SE CCD camera (Diagnostic Instruments).
Transmission electron microscopy of wild type and Nopp140-RNAi-expressing larval tissues was performed as described.20 Diploid tissues (imaginal discs and brains) and polyploid tissues (mid-gut and Malpighian tubules) were hand isolated, fixed and flat-embedded separately so we could section and analyze one known tissue at a time. From Nopp140-RNAi-expressing larvae, we captured 16 images of polyploid cells and 20 images of diploid cells. About half as many images were captured from the same wild type tissues. We used a JEOL 100CX operating at 80 KV and a magnification of 16,000×. Negatives (8 × 10 cm) were scanned at 1200 dpi. Resulting positive images were prepared using Photoshop.
Immuno-blots
Third instar larval lysates were prepared by using small plastic pestles designed for Eppendorf tubes. Larvae were homogenized in 267 µL of Laemmli sample buffer to which we added 3 µL of 0.1 M PMSF dissolved in isopropanol, 15 µL of a protease inhibitor cocktail (P-8340, Sigma-Aldrich Corp.) and 15 µL of 2-mercaptoethanol. The homogenate was then sonicated by 5 bursts using a Branson Digital Sonifier; each burst was for 5 sec at 40% maximum power. The resulting lysates were centrifuged for 30 min. The supernatant was boiled for 5 min. and re-centrifuged for an additional 30 min. Lysate proteins were resolved by standard SDS-PAGE and blotted to nitrocellulose using a Bio-Rad Trans-Blot semi-dry transfer cell. Blots were blocked with 5% non-fat dry milk in TTBS. We used rabbit antibodies against ribosomal proteins RpL34 (center) and RpL23A (C-terminus) (Abgent; catalog numbers AP13207c-ev20 and AP1939b, respectively) at 1/2000 in 5% non-fat dry milk. We used the 81E11 rabbit monoclonal antibody directed against phospho-JNK (Thr183/Tyr185; Cell Signaling Technologies, #4668) and a rabbit anti-Hid antibody that was kindly provided by Hyung Don Ryoo34 at New York University Medical School diluted to 1/1000 in TTBS.
Mass spectroscopy
Proteins resolved on SDS-polyacrylamide gels were identified by mass spectroscopy at Louisiana State University’s Pennington Biomedical Research Center. Briefly, 2 mm gel plugs were automatically excised using a ProteomeWorks spot cutter (Bio-Rad) and deposited in a 96-well plate. In-gel tryptic digestion was performed using an automated robotic workstation (MassPrep, Waters Corp.). Gel plugs were washed with NH4HCO3 and CH3CN followed by reduction and alkylation using 10 mM DTT and 55 mM iodoacetamide, respectively. Proteins were cleaved by sequencing grade trypsin (150 ng/gel plug) at 37°C overnight. The peptide fragments were extracted with 2% CH3CN, 1% HCOOH in water and directly analyzed using liquid chromatography (LC) – tandem mass spectrometry (MS/MS). The peptides from each digested band were separated and analyzed using a capillary LC system coupled online with a nanospray quadrupole time-of-flight (Q-TOF) MS (Waters Corp.) as described.47 Tandem mass spectra were analyzed using the ProteinLynx Global Server 2.5 software (Waters Corp.) against a Swiss Prot database. The database search settings included one missed tryptic cleavage and fixed carbamidomethylation of cysteine residues.
Hemolymph phenoloxidase assays
Relative hemolymph phenoloxidase levels were determined using native gels and staining techniques with tyrosine as substrate as described previously.26
Metabolic labeling
Twenty third-instar larvae, either parental controls or progeny expressing RNAi to deplete Nopp140, were added to ~300 µL of Robb’s A + B solution49 in disposable plastic depression slides and gently torn open to expose internal tissues. Excess fluid was removed leaving approximately 50 µL to which we added 10 μL of EXPRE35S35S Protein Labeling Mix, [35S]-EasyTag™ (Perkin-Elmer, NEG772002M, 10.0 mCi/ml). The depression slides were placed in a moist chamber, and the tissues were incubated for 30 min at room temperature. Tissues were then washed once with Robb’s A + B solution. Lysates were prepared by adding 25 μL of a protease inhibitor cocktail (Sigma-Aldrich, P-8340), 445 μL of Laemmli sample buffer, 25 μL 2-mercaptoethanol and 5 μL 0.1 M PMSF dissolved in isopropanol. Tissues were homogenized in Eppendorf tubes with disposal plastic pestles, boiled for five minutes and centrifuged for one hour. Protein concentrations were determined in triplicate using a modified Lowry assay.50 Absorbance values were determined using a Perkin-Elmer Lambda 3B spectrophotometer. To determine incorporated CPM, 200 μL of larval lysate was combined with 100 μL of ddH2O, 100 μL BSA (2 mg/ml) and 80 μL 100% TCA (16.7% final concentration). Samples were mixed and incubated on ice for 30 min. Each sample was passed through a vacuum filter assembly (Sigma-Aldrich, Z290475) containing a Whatman 25 mm GFC glass microfiber filter which was then rinsed with cold 10% TCA and 100% EtOH. Filters were air-dried and placed separately into 10 mL of Scintiverse (TM) BD Cocktail (Fisher Scientific). CPM was determined using a Beckman LS 6000IC scintillation counter. Ratios of CPM per microgram of protein were determined for each sample. The entire experiment was repeated three times. We also verified the relative protein concentrations and CPM values for each sample by resolving lysate proteins on SDS-10% polyacrylamide gels; Coomassie blue staining verified the relative protein concentrations. The gels were dried and exposed to Kodak X-OMAT LS film; resulting autoradiograms verified the relative scintillation counts.
Acknowledgments
We thank Dr Thomas Neufeld (University of Minnesota) for the mCherry-ATG8a transgenic line and Dr Hyung Don Ryoo (New York University Medical School) for the rabbit anti-Hid antibody. We thank Dr Indu Kheterpal and her staff at LSU’s Pennington Biomedical Research Center for performing the mass spectroscopy. We thank Dr Ying Xiao for embedding and thin sectioning tissue samples for TEM analysis. We also thank Louisiana State University’s S-STEM Scholars Program for supporting Renford Cindass and the Louisiana Biomedical Research Network (LBRN) for the summer fellowship awarded to Dana Mayer. This work was supported by the National Science Foundation, award MCB0919709.
Glossary
Abbreviations:
- Nopp140
nucleolar phosphoprotein of 140 kDa
- TCS
Treacher Collins-Franceschetti syndrome
- RNAi
RNA interference
- TEM
transmission electron microscopy
- snoRNPs
small nucleolar ribonucleoprotein particles
- UAS
Upstream Activation Sequence
- JNK
Jun-amino terminal kinase
- LC-MS/MS
liquid chromatography-tandem mass spectroscopy
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/nucleus/article/23944
References
- 1.Meier UT. Comparison of the rat nucleolar protein Nopp140 with its yeast homologue SRP40. Differential phosphorylation in vertebrates and yeast. J Biol Chem. 1996;271:19376–84. [PubMed] [Google Scholar]
- 2.Isaac C, Yang Y, Meier UT. Nopp140 functions as a molecular link between the nucleolus and the coiled bodies. J Cell Biol. 1998;142:319–29. doi: 10.1083/jcb.142.2.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang Y, Isaac C, Wang C, Dragon F, Pogačić V, Meier UT. Conserved composition of mammalian box H/ACA and box C/D small nucleolar ribonucleoprotein particles and their interaction with the common factor Nopp140. Mol Biol Cell. 2000;11:567–77. doi: 10.1091/mbc.11.2.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.He F, DiMario P. Structure and function of Nopp140 and Treacle. In: Olson MOJ, ed. The Nucleolus, Protein Reviews. New York, NY: Springer, 2011:253-278. [Google Scholar]
- 5.Waggener JM, DiMario PJ. Two splice variants of Nopp140 in Drosophila melanogaster. Mol Biol Cell. 2002;13:362–81. doi: 10.1091/mbc.01-04-0162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cui Z, DiMario PJ. RNAi knockdown of Nopp140 induces Minute-like phenotypes in Drosophila. Mol Biol Cell. 2007;18:2179–91. doi: 10.1091/mbc.E07-01-0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dixon MJ. Treacher Collins syndrome. Hum Mol Genet. 1996;5(Spec No):1391–6. doi: 10.1093/hmg/5.supplement_1.1391. [DOI] [PubMed] [Google Scholar]
- 8.Trainor PA, Dixon J, Dixon MJ. Treacher Collins syndrome: etiology, pathogenesis and prevention. Eur J Hum Genet. 2009;17:275–83. doi: 10.1038/ejhg.2008.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dixon J, Jones NC, Sandell LL, Jayasinghe SM, Crane J, Rey J-P, et al. Tcof1/Treacle is required for neural crest cell formation and proliferation deficiencies that cause craniofacial abnormalities. Proc Natl Acad Sci U S A. 2006;103:13403–8. doi: 10.1073/pnas.0603730103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones NC, Lynn ML, Gaudenz K, Sakai D, Aoto K, Rey J-P, et al. Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nat Med. 2008;14:125–33. doi: 10.1038/nm1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shiba T, Saigo K. Retrovirus-like particles containing RNA homologous to the transposable element copia in Drosophila melanogaster. Nature. 1983;302:119–24. doi: 10.1038/302119a0. [DOI] [PubMed] [Google Scholar]
- 12.Strand DJ, McDonald JF. Copia is transcriptionally responsive to environmental stress. Nucleic Acids Res. 1985;13:4401–10. doi: 10.1093/nar/13.12.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gartner LP, Gartner RC. Nuclear inclusions: a study of aging in Drosophila. J Gerontol. 1976;31:396–404. doi: 10.1093/geronj/31.4.396. [DOI] [PubMed] [Google Scholar]
- 14.Tulin A, Stewart D, Spradling AC. The Drosophila heterochromatic gene encoding poly(ADP-ribose) polymerase (PARP) is required to modulate chromatin structure during development. Genes Dev. 2002;16:2108–19. doi: 10.1101/gad.1003902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fan Y, Bergmann A. The cleaved-Caspase-3 antibody is a marker of Caspase-9-like DRONC activity in Drosophila. Cell Death Differ. 2010;17:534–9. doi: 10.1038/cdd.2009.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rong YS, Titen SW, Xie HB, Golic MM, Bastiani M, Bandyopadhyay P, et al. Targeted mutagenesis by homologous recombination in D. melanogaster. Genes Dev. 2002;16:1568–81. doi: 10.1101/gad.986602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jaklevic BR, Su TT. Relative contribution of DNA repair, cell cycle checkpoints, and cell death to survival after DNA damage in Drosophila larvae. Curr Biol. 2004;14:23–32. doi: 10.1016/j.cub.2003.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wichmann A, Jaklevic B, Su TT. Ionizing radiation induces caspase-dependent but Chk2- and p53-independent cell death in Drosophila melanogaster. Proc Natl Acad Sci U S A. 2006;103:9952–7. doi: 10.1073/pnas.0510528103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wichmann A, Uyetake L, Su TT. E2F1 and E2F2 have opposite effects on radiation-induced p53-independent apoptosis in Drosophila. Dev Biol. 2010;346:80–9. doi: 10.1016/j.ydbio.2010.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosby R, Cui Z, Rogers E, deLivron MA, Robinson VL, DiMario PJ. Knockdown of the Drosophila GTPase nucleostemin 1 impairs large ribosomal subunit biogenesis, cell growth, and midgut precursor cell maintenance. Mol Biol Cell. 2009;20:4424–34. doi: 10.1091/mbc.E08-06-0592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang Y-Y, Neufeld TP. An Atg1/Atg13 complex with multiple roles in TOR-mediated autophagy regulation. Mol Biol Cell. 2009;20:2004–14. doi: 10.1091/mbc.E08-12-1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu H, Wang MC, Bohmann D. JNK protects Drosophila from oxidative stress by trancriptionally activating autophagy. Mech Dev. 2009;126:624–37. doi: 10.1016/j.mod.2009.06.1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neufeld TP. Autophagy and cell growth--the yin and yang of nutrient responses. J Cell Sci. 2012;125:2359–68. doi: 10.1242/jcs.103333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang H. Regulation and function of the melanization reaction in Drosophila. Fly (Austin) 2009;3:105–11. doi: 10.4161/fly.3.1.7747. [DOI] [PubMed] [Google Scholar]
- 25.Roberts DB, Wolfe J, Akam ME. The developmental profiles of two major haemolymph proteins from Drosophila melanogaster. J Insect Physiol. 1977;23:871–8. doi: 10.1016/0022-1910(77)90013-0. [DOI] [PubMed] [Google Scholar]
- 26.Asano T, Takebuchi K. Identification of the gene encoding pro-phenoloxidase A(3) in the fruitfly, Drosophila melanogaster. Insect Mol Biol. 2009;18:223–32. doi: 10.1111/j.1365-2583.2008.00858.x. [DOI] [PubMed] [Google Scholar]
- 27.Lin A. Activation of the JNK signaling pathway: breaking the brake on apoptosis. Bioessays. 2003;25:17–24. doi: 10.1002/bies.10204. [DOI] [PubMed] [Google Scholar]
- 28.Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Cell Biol. 2007;19:142–9. doi: 10.1016/j.ceb.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 29.Bilak A, Su TT. Regulation of Drosophila melanogaster pro-apoptotic gene hid. Apoptosis. 2009;14:943–9. doi: 10.1007/s10495-009-0374-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Juhász G, Puskás LG, Komonyi O, Érdi B, Maróy P, Neufeld TP, et al. Gene expression profiling identifies FKBP39 as an inhibitor of autophagy in larval Drosophila fat body. Cell Death Differ. 2007;14:1181–90. doi: 10.1038/sj.cdd.4402123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang Y-Y, Neufeld TP. Autophagy takes flight in Drosophila. FEBS Lett. 2010;584:1342–9. doi: 10.1016/j.febslet.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bidla G, Dushay MS, Theopold U. Crystal cell rupture after injury in Drosophila requires the JNK pathway, small GTPases and the TNF homolog Eiger. J Cell Sci. 2007;120:1209–15. doi: 10.1242/jcs.03420. [DOI] [PubMed] [Google Scholar]
- 33.McNamee LM, Brodsky MH. p53-independent apoptosis limits DNA damage-induced aneuploidy. Genetics. 2009;182:423–35. doi: 10.1534/genetics.109.102327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ryoo HD, Gorenc T, Steller H. Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Dev Cell. 2004;7:491–501. doi: 10.1016/j.devcel.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 35.Minakhina S, Steward R. Melanotic mutants in Drosophila: pathways and phenotypes. Genetics. 2006;174:253–63. doi: 10.1534/genetics.106.061978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mayer C, Grummt I. Cellular stress and nucleolar function. Cell Cycle. 2005;4:1036–8. doi: 10.4161/cc.4.8.1925. [DOI] [PubMed] [Google Scholar]
- 37.Tollini LA, Frum RA, Zhang Y. The role of the nucleolus in the stress response. In: Olson MOJ, ed. The Nucleolus, Protein Reviews. New York, NY: Springer, 2011:281-299. [Google Scholar]
- 38.Gowda PS, Zhou F, Chadwell LV, McEwen DG. p53 binding prevents phosphatase-mediated inactivation of diphosphorylated c-Jun N-terminal kinase. J Biol Chem. 2012;287:17554–67. doi: 10.1074/jbc.M111.319277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luo X, Puig O, Hyun J, Bohmann D, Jasper H. Foxo and Fos regulate the decision between cell death and survival in response to UV irradiation. EMBO J. 2007;26:380–90. doi: 10.1038/sj.emboj.7601484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moreno E, Basler K, Morata G. Cells compete for decapentaplegic survival factor to prevent apoptosis in Drosophila wing development. Nature. 2002;416:755–9. doi: 10.1038/416755a. [DOI] [PubMed] [Google Scholar]
- 41.Marygold SJ, Roote J, Reuter G, Lambertsson A, Ashburner M, Millburn GH, et al. The ribosomal protein genes and Minute loci of Drosophila melanogaster. Genome Biol. 2007;8:R216. doi: 10.1186/gb-2007-8-10-r216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mayer C, Bierhoff H, Grummt I. The nucleolus as a stress sensor: JNK2 inactivates the transcription factor TIF-IA and down-regulates rRNA synthesis. Genes Dev. 2005;19:933–41. doi: 10.1101/gad.333205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yogev O, Saadon K, Anzi S, Inoue K, Shaulian E. DNA damage-dependent translocation of B23 and p19 ARF is regulated by the Jun N-terminal kinase pathway. Cancer Res. 2008;68:1398–406. doi: 10.1158/0008-5472.CAN-07-2865. [DOI] [PubMed] [Google Scholar]
- 44.Mialon A, Thastrup J, Kallunki T, Mannermaa L, Westermarck J, Holmström TH. Identification of nucleolar effects in JNK-deficient cells. FEBS Lett. 2008;582:3145–51. doi: 10.1016/j.febslet.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 45.Haerry TE, Khalsa O, O’Connor MB, Wharton KA. Synergistic signaling by two BMP ligands through the SAX and TKV receptors controls wing growth and patterning in Drosophila. Development. 1998;125:3977–87. doi: 10.1242/dev.125.20.3977. [DOI] [PubMed] [Google Scholar]
- 46.de Cuevas M, Lee JK, Spradling AC. alpha-spectrin is required for germline cell division and differentiation in the Drosophila ovary. Development. 1996;122:3959–68. doi: 10.1242/dev.122.12.3959. [DOI] [PubMed] [Google Scholar]
- 47.Zvonic S, Lefevre M, Kilroy G, Floyd ZE, DeLany JP, Kheterpal I, et al. Secretome of primary cultures of human adipose-derived stem cells: modulation of serpins by adipogenesis. Mol Cell Proteomics. 2007;6:18–28. doi: 10.1074/mcp.M600217-MCP200. [DOI] [PubMed] [Google Scholar]
- 48.Chintapalli VR, Wang J, Dow JAT. Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat Genet. 2007;39:715–20. doi: 10.1038/ng2049. [DOI] [PubMed] [Google Scholar]
- 49.Robb JA. Maintenance of imaginal discs of Drosophila melanogaster in chemically defined media. J Cell Biol. 1969;41:876–85. doi: 10.1083/jcb.41.3.876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peterson GL. Determination of total protein. Methods Enzymol. 1983;91:95–119. doi: 10.1016/S0076-6879(83)91014-5. [DOI] [PubMed] [Google Scholar]