

Abstract
Angiotensin II (Ang II) is a major contributor to the progression of renal fibrosis. Wang and colleagues provide evidence that signaling through the prolyl-4-hydroxylase domain (PHD)–hypoxia-inducible factor-1 (HIF-1) pathway mediates profibrotic effects of Ang II in rat renal medullary interstitial cells under normoxic conditions, thus placing the HIF oxygen-sensing pathway into the center of an Ang II-induced profibrotic signaling cascade.
Aside from its potent hemodynamic effects, angiotensin II (Ang II) stimulates growth of different renal cell types and increases the expression and synthesis of extracellular matrix proteins and various cytokines and chemokines, thus promoting inflammation and fibrosis.1 In the context of renal injury, it is a major contributor to the progression of chronic kidney disease. Ang II signals through two specific G protein-coupled receptors, the Ang II receptor type 1 (AT1) and the Ang II receptor type 2 (AT2). Signaling through the AT1 receptor mediates vasoconstriction and aldosterone release, stimulates tubular sodium transport, and promotes growth, inflammation, and fibrosis, whereas AT2 signaling has been proposed to antagonize these AT1-mediated effects and is believed to protect from progression of chronic kidney disease. Another major effect of AT1-receptor activation is the generation of reactive oxygen species (ROS). This occurs predominantly through activation of membrane-bound NADPH oxidase, which converts NADPH and molecular oxygen to NADP+ and superoxide anions (reviewed by Sachse and Wolf 2).
Now, Wang and colleagues3 (this issue) demonstrate that Ang II-induced ROS generation leads to non-hypoxic stabilization of the oxygen-sensitive α-subunit of hypoxia-inducible factor (HIF)-1 in rat renal medullary interstitial cells, which in turn is required for Ang II-stimulated collagen I/III and tissue inhibitor of metal-loproteinases (TIMP)-1 synthesis, thereby placing HIF-1 into the center of an Ang II-induced profibrotic signaling cascade (Figure 1). HIF-1α stabilization was, furthermore, necessary for Ang II-induced growth and vimentin expression. While treatment with Ang II also led to increased HIF-2α expression, inactivation of HIF-2α, in contrast to HIF-1α knockdown, did not significantly affect Ang II-induced TIMP-1 or collagen I /III levels.
Figure 1. Angiotensin II stabilizes hypoxia-inducible factor-1α in renal medullary interstitial cells.
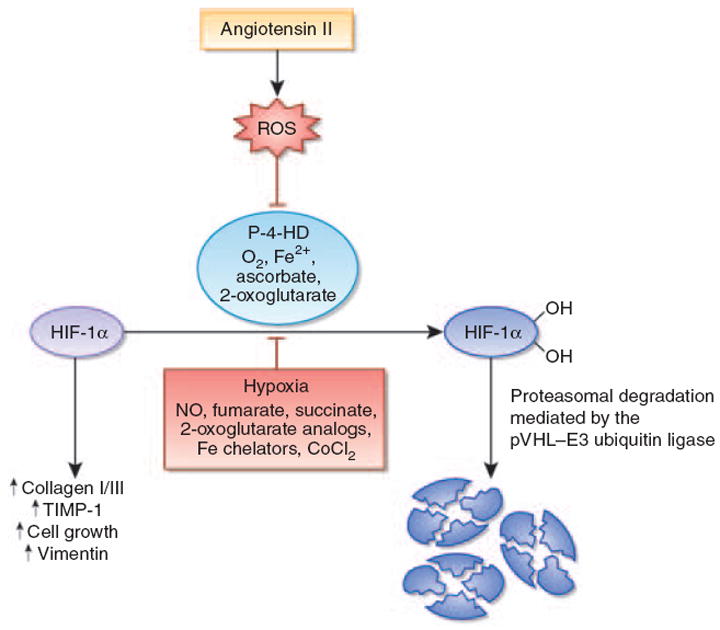
Angiotensin II (Ang II) stimulates the production of reactive oxygen species (ROS). This leads to the inhibition of the hypoxia-inducible factor (HIF)-hydroxylation reaction and subsequent stabilization of HIF-1α. HIF-1α is required for Ang II-induced profibrotic gene expression and proliferation of renal medullary interstitial cells. Under normoxia, HIF-1α is normally hydroxylated by prolyl-4-hydroxylases and targeted for proteasomal degradation by the von Hippel-Lindau (pVHL)–E3 ubiquitin ligase complex. When prolyl-4-hydroxylation is inhibited, HIF-1α is stabilized and translocates to the nucleus, where it heterodimerizes with the aryl hydrocarbon receptor nuclear translocator (ARNT). HIF-1α–ARNT heterodimers bind to the HIF consensus-binding site RCGTG, followed by transactivation of target genes. In addition to ROS, nitric oxide, the Krebs cycle metabolites succinate and fumarate, cobalt chloride, and iron chelators such as desferrioxamine inhibit HIF prolyl-4-hydroxylases in the presence of oxygen. CoCl2, cobalt chloride; Fe2+, ferrous iron; NO, nitric oxide; P-4-HD, prolyl-4-hydroxylases; TIMP-1, tissue inhibitor of metalloproteinases-1.
HIF-1 and HIF-2 are basic helix-loop-helix transcription factors that consist of an oxygen-sensitive α-subunit and a constitutively expressed β-subunit, also known as the aryl hydrocarbon receptor nuclear translocator (ARNT). Although HIF-α is continuously synthesized, it is rapidly degraded under normoxia. This requires hydroxylation of specific proline residues within the oxygen-dependent degradation domain of HIF-α, enabling interaction with the von Hippel–Lindau–E3 ubiquitin ligase complex, which then targets HIF for rapid proteasomal degradation (reviewed by Kaelin and Ratcliffe4) (Figure 1). The hydroxylation reaction is dependent on molecular oxygen, ferrous iron, and ascorbate and is carried out by 2-oxoglutarate-dependent dioxygenases (prolyl-4-hydroxylase domain (PHD) proteins). Three major HIF-hydroxylating enzymes have been identified, PHD1, PHD2, and PHD3, of which PHD2 is the main dioxygenase for HIF degradation under normoxia. The three PHD proteins differ with regard to their cellular expression levels, intracellular localization, hypoxia-inducibility, and biochemical behavior (reviewed by Kaelin and Ratcliffe4). In the kidney, all three enzymes are expressed in a cell type-dependent manner.5
The role of HIF-1 in renal injury is highly context- and cell type-dependent. Whereas HIF-1 confers cytoprotection in the acute setting, there is now growing evidence that HIF-1 activation under certain chronic disease conditions can promote fibrosis in the kidney and in other organs.6 This was recently demonstrated in transgenic and conditional knockout mice7,8 and may occur through increased expression of extracellular matrix-modifying genes, such as lysyl-oxidases and plasminogen activator inhibitor-1; functional cooperation with transforming growth factor-β1; promotion of epithelial-to-mesenchymal transition; and the modulation of renal inflammation. Increased HIF expression has been found in animal models of chronic kidney disease and in renal biopsy material from patients with diabetic nephropathy and other forms of renal disease, where it correlates with disease severity.7,9,10 Stabilization of HIF-α in this context is thought to be due to hypoxia, which results from a combination of structural and functional changes that are commonly found in fibrotic kidneys. These include decreased peritubular blood flow associated with glomerular injury, capillary rarefaction, vasoconstriction, luminal narrowing of atherosclerotic vessels, increased oxygen demand from hyperfiltration and tubular hypertrophy, limited oxygen diffusion as a consequence of extracellular matrix expansion, and renal anemia.9
It has been known for some time that Ang II can act as a hypoxia mimetic, stabilizing HIF-1α in vascular smooth muscle cells under normoxic conditions.11 This results primarily from HIF prolyl-4-hydroxylase inhibition, which is due to increased ROS generation and diminished intracellular ascorbate levels, leading to the oxidation of a critical iron atom that is positioned in the center of the PHD catalytic domain.12 Although Ang II-generated ROS appear to be largely derived from the activation of NADPH oxidase,2 more recent studies suggest that Ang II-induced HIF-1α stabilization may be mediated by mitochondrial ROS.13 Although initially heavily debated, the importance of mitochondrial ROS in the modulation of molecular oxygen sensing and regulation of HIF activity has been recently demonstrated by elegant genetic, pharmacological, and RNA interference-based studies. 14-16 Wang and colleagues3 (this issue) take the Ang II–HIF connection one step further and investigate whether the PHD–HIF pathway is part of an Ang II-induced profibrotic signaling cascade in the kidney. Although their studies are mainly in vitro-based, the authors provide supporting in vivo evidence and show that Ang II infusion over a period of 2 weeks resulted in HIF-1α stabilization in renal medullary cells, which was associated with an increase in α-smooth muscle actin expression. However, additional studies are warranted that assess the relative contribution of Ang II-induced hemodynamic alterations to HIF-1α stabilization in the kidney.17 Once activated, HIF-1α either acts as a transcription factor that binds to a specific DNA sequence, which is contained within a hypoxia-response element (HRE), or it interacts with other signaling molecules outside the nucleus. 6 Although Wang and colleagues3 demonstrate that HIF-1α is required for Ang II-induced profibrotic effects, it remains to be determined to what degree this involves HRE-mediated transcription; for example, TIMP-1 contains a HIF-binding site in its regulatory region.18 Furthermore, it would be important to define which other HIF-controlled biological responses are activated by Ang II alone as compared to hypoxic stimulation. In this context it is worth pointing out that hypoxia and Ang II do not appear to act synergistically in the study by Wang and colleagues. 3
In a second set of experiments, Wang and colleagues 3 demonstrate that Ang II reduces HIF-1α hydroxylation and investigate whether manipulations of PHD2 expression levels modulate Ang II-induced collagen I/III and TIMP-1 synthesis. PHD2 is critically important for HIF degradation under normoxia, as genetic inactivation of PHD2 results in embryonic lethality.4 As predicted, an increase in PHD2 levels reduced collagen I/III and TIMP-1 expression in the presence of Ang II, whereas PHD2 knockdown had the opposite effect. Whether pharmacological inhibition of PHD enzymes, which is currently being evaluated in clinical trials as a new modality for the treatment of renal anemia, has the same effect on the renal medullary interstitial cells in vivo is unclear and warrants further investigation. Interestingly, immunohistochemical analysis of renal PHD expression did not visualize PHD2 protein in non-endothelial interstitial cells, whereas both PHD1 and PHD3 enzymes were easily detectable.5 While it is unclear whether there are regional differences in interstitial PHD2 expression, it is plausible that, with regard to HIF stabilization, cells that express little or reduced amounts of PHD2 become more susceptible to changes in intracellular ascorbate and/or ROS concentrations. To what degree PHD1 and PHD3 contribute to Ang II-stimulated collagen I/III and TIMP-1 synthesis remains to be investigated. This is particularly important with regard to PHD3, which is hypoxia-inducible and HIF-regulated and is part of a negative-feedback loop. PHD3 protein levels are predicted to increase when HIF-1 is activated, which could lead to a reduction in collagen I/III and TIMP-1 expression over time. A time course study should be able to clarify this issue. Differences in PHD levels between various renal cell types may also explain why, in contrast to hypoxia and iron chelation, treatment of human renal cortical fibroblasts with H2O2 did not produce an increase in TIMP-1 or collagen I expression. 18
In summary, Wang and colleagues3 link profibrotic effects of Ang II in the kidney to the PHD–HIF molecular oxygen-sensing pathway. Their findings not only provide novel insights into angiotensin biology but also lend further support to the concept that activation of HIF signaling in the kidney can occur in the absence of significant hypoxia. The paper by Wang and colleagues3 provokes future studies that investigate the Ang II–PHD–HIF axis in other renal cell types and in the context of acute and chronic kidney injury.
Acknowledgments
VHH is supported by the Krick-Brooks Chair in Nephrology and by grants from the National Institutes of Health.
Footnotes
DISCLOSURE
The author declared no conflict of interest.
References
- 1.Mezzano SA, Ruiz-Ortega M, Egido J. Angiotensin II and renal fibrosis. Hypertension. 2001;38:635–638. doi: 10.1161/hy09t1.094234. [DOI] [PubMed] [Google Scholar]
- 2.Sachse A, Wolf G. Angiotensin II-induced reactive oxygen species and the kidney. J Am Soc Nephrol. 2007;18:2439–2446. doi: 10.1681/ASN.2007020149. [DOI] [PubMed] [Google Scholar]
- 3.Wang Z, Tang L, Zhu Q, et al. Hypoxia-inducible factor-1α contributes to the profibrotic action of angiotensin II in renal medullary interstitial cells. Kidney Int. 2011;79:300–310. doi: 10.1038/ki.2010.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 5.Schodel J, Klanke B, Weidemann A, et al. HIF-prolyl hydroxylases in the rat kidney: physiologic expression patterns and regulation in acute kidney injury. Am J Pathol. 2009;174:1663–1674. doi: 10.2353/ajpath.2009.080687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haase VH. Hypoxia-inducible factors in the kidney. Am J Physiol Renal Physiol. 2006;291:F271–F281. doi: 10.1152/ajprenal.00071.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Higgins DF, Kimura K, Bernhardt WM, et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest. 2007;117:3810–3820. doi: 10.1172/JCI30487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kimura K, Iwano M, Higgins DF, et al. Stable expression of HIF-1alpha in tubular epithelial cells promotes interstitial fibrosis. Am J Physiol Renal Physiol. 2008;295:F1023–F1029. doi: 10.1152/ajprenal.90209.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fine LG, Norman JT. Chronic hypoxia as a mechanism of progression of chronic kidney diseases: from hypothesis to novel therapeutics. Kidney Int. 2008;74:867–872. doi: 10.1038/ki.2008.350. [DOI] [PubMed] [Google Scholar]
- 10.Neusser MA, Lindenmeyer MT, Moll AG, et al. Human nephrosclerosis triggers a hypoxia-related glomerulopathy. Am J Pathol. 2010;176:594–607. doi: 10.2353/ajpath.2010.090268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richard DE, Berra E, Pouyssegur J. Nonhypoxic pathway mediates the induction of hypoxia-inducible factor 1alpha in vascular smooth muscle cells. J Biol Chem. 2000;275:26765–26771. doi: 10.1074/jbc.M003325200. [DOI] [PubMed] [Google Scholar]
- 12.Page EL, Chan DA, Giaccia AJ, et al. Hypoxia-inducible factor-1alpha stabilization in nonhypoxic conditions: role of oxidation and intracellular ascorbate depletion. Mol Biol Cell. 2008;19:86–94. doi: 10.1091/mbc.E07-06-0612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patten DA, Lafleur VN, Robitaille GA, et al. Hypoxia-inducible factor-1 activation in nonhypoxic conditions: the essential role of mitochondrial-derived reactive oxygen species. Mol Biol Cell. 2010;21:3247–3257. doi: 10.1091/mbc.E10-01-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guzy RD, Hoyos B, Robin E, et al. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1:401–408. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 15.Mansfield KD, Guzy RD, Pan Y, et al. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab. 2005;1:393–399. doi: 10.1016/j.cmet.2005.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brunelle JK, Bell EL, Quesada NM, et al. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005;1:409–414. doi: 10.1016/j.cmet.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 17.Norman JT, Stidwill R, Singer M, Fine LG. Angiotensin II blockade augments renal cortical microvascular pO2 indicating a novel, potentially renoprotective action. Nephron Physiol. 2003;94:39–46. doi: 10.1159/000071289. [DOI] [PubMed] [Google Scholar]
- 18.Norman JT, Clark IM, Garcia PL. Hypoxia promotes fibrogenesis in human renal fibroblasts. Kidney Int. 2000;58:2351–2366. doi: 10.1046/j.1523-1755.2000.00419.x. [DOI] [PubMed] [Google Scholar]