

Abstract
In this Letter we report on the advances in our NPBWR1 antagonist program aimed at optimizing the 5-chloro-2-(3,5-dimethylphenyl)-4-(4-methoxyphenoxy)pyridazin-3(2H)-one lead molecule previously obtained from a high-throughput screening (HTS)-derived hit. Synthesis and structure–activity relationships (SAR) studies around the 3,5-dimethylphenyl and 4-methoxyphenyl regions resulted in the identification of a novel series of non-peptidic submicromolar NPBWR1 antagonists based on a 5-chloro-4-(4-alkoxyphenoxy)-2-(benzyl)pyridazin-3(2H)-one chemotype. Amongst them, 5-chloro-2-(9H-fluoren-9-yl)-4-(4-methoxyphenoxy)pyridazin-3(2H)-one 9h (CYM50769) inhibited NPW activation of NPBWR1 with a submicromolar IC50, and displayed high selectivity against a broad array of off-targets with pharmaceutical relevance. Our medicinal chemistry study provides innovative non-peptidic selective NPBWR1 antagonists that may enable to clarify the biological role and therapeutic utility of the target receptor in the regulation of feeding behavior, pain, stress, and neuroendocrine function.
Keywords: NPBWR1 (GPR7) antagonists, Anorexia, Analgesia, Stress
Neuropeptide W (NPW) and neuropeptide B (NPB) bind and activate two G-protein coupled receptors (GPCRs), namely NPBWR1 (GPR7) and NPBWR2 (GPR8).1 NPB mRNA is widely distributed throughout the mouse brain and is present in the hippocampus, hypothalamin nucleus, Edinger–Westphal nucleus (EW), locus coerulius, inferior olive and lateral parabrancheal nucleus.1,2 The expression of NPW is more confined to EW, ventral tegmental area (VTA), periaqueductal gray and dorsal raphe nucleus.3
Human NPBWR1 was mapped to chromosome 10q11.2-121.1 and NPBWR2 to chromosome 20q13.3. NPBWR1 and NPBWR2 share 64% sequence homology and decrease intracellular cAMP by coupling to the Gi-class of GPCRs.1,4
In situ hybridization and tissue binding studies showed that NPBWR1 mRNA is localized in the extended amygdala (central nucleus of the amygdala, bed nucleus of the stria terminalis) and the hypothalamus (suprachiasmatic nucleus, VTA).1,5
This neuropeptide system has been hypothesized to play an important role in modulating feeding behavior.6,1 Intracerebroventricular (i.c.v.) administration of NPW to fasting rats or free-feeding rats before dark phase suppressed food intake and increased both heat production and body temperature.7 Furthermore, NPBWR1 knockout mice developed adult-onset obesity.8
Intrathecal (i.t.) administration of NPW produced analgesic effects in inflammatory pain in the mouse formalin test, but not mechanical or thermal pain.9 Interestingly, samples taken from patients with inflammatory/immunomediated neuropathies showed a higher expression of NPBWR1 restricted to myelin-forming Schwan cells. Similarly, animal models of acute inflammatory and trauma induced neuropathic pain exhibited an increased myelin-associated expression of NPBWR1, suggesting a central role of this receptor in analgesia.10
Remarkably, i.c.v. administration of NPW in the brain reduced seizure activity in mice, suggesting that NPWR1 may be a suitable target for epilepsy.11
I.c.v. injection of NPB in male rats elevated prolactin and corticosterone, and decreased growth hormone levels in circulation, suggesting that this peptide may play a physiological role in the neuroendocrine response to stress via the activation of the hypothalamus-pituitary-adrenal axis.12 NPBWR1 knockout mice showed abnormality in the contextual fear condition test in addition to an increased autonomic and neuroendocrine response to physical stress, suggesting that the impairment of NPBWR1 leads to stress vulnerability.13 A recent study provided evidence that human genetic differences in NPBWR1 modulate emotional responses to facial expressions, particularly towards angry facial expression; it is speculated that this behavior may be linked to the role of NPBWR1 in modulating fear.14
Recently, we reported the identification, design and structure–activity relationships (SAR) of a novel non-peptidic NPBWR1 antagonist chemotype obtained from a high-throughput screening (HTS) of the Molecular Libraries-Small Molecule Repository (MLSMR) library. The 5-chloro-2-(3,5-dimethylphenyl)-4-(4-methoxyphenoxy)pyridazin-3(2H)-one 1 (CYM50557) was identified as a lead molecule in our NPBWR1 antagonist program (Fig. 1).15 Its sub-micromolar potency and excellent selectivity against a panel of pharmacologically relevant off-target proteins prompted us to further optimize 1 in an effort to develop an in vivo pharmacological tool to better elucidate the biological role of NPBWR1. In this letter we report on the expansion of the SAR studies around the coil region a and the pendant 4-methoxyphenyl moiety b (Fig. 1).
Figure 1.
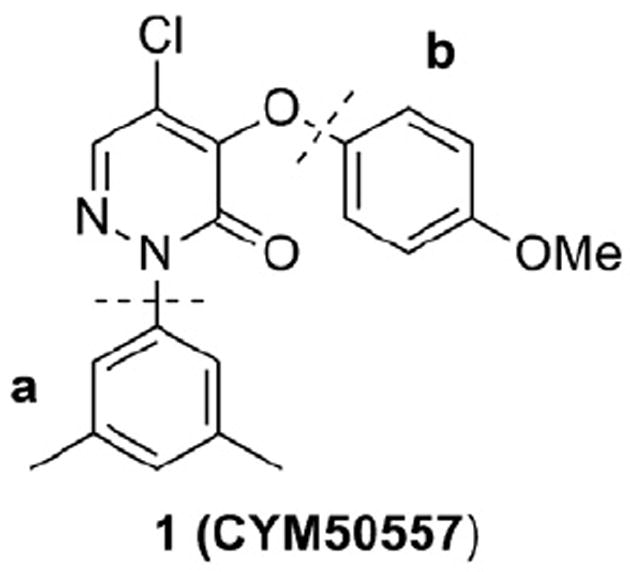
NPBWR1 lead molecule.
NPBWR1, IC50 (μM) = 0.27
Our SAR studies on 1 started by exploring the region a. The synthesis of 7a–7al is depicted in Schemes 1 and 2 and their biological results are listed in Table 1.16 MOM-protection of N-2 followed by substitution on position 4 of the pyridazinone using the 4-metoxyphenoxide furnished 4. MOM-deprotection of 4 using boron tribromide (BBr3) yielded the key intermediate 5. Alkylation of 5 with a series of electronically and structurally diverse alkyl halides 6a-6ag and 6aj–6al furnished the final compounds 7a–7ag and 7aj–7al (Scheme 1). Compound 7ag was reduced with lithium borohydride (LiBH4) to give 7ah which was transformed to 7ai using acetyl chloride (Scheme 2).
Scheme 1.
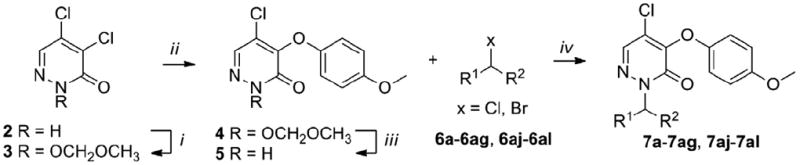
Synthesis of 7a–7ag, 7aj–7al.
Reagents and conditions: (i) 2 (1 equiv.), MOMCl (1.2 equiv.), DMAP (0.1 equiv.), DIPEA (1.4 equiv.) CH2Cl2, 0°C to rt, overnight, 85%; (ii) 3 (1 equiv.), 4-methoxyphenol (1.1 equiv.), NaH (1.1 equiv.), 1,4-dioxane, 15°C to rt, overnight, 65%; (iii) 4 (1 equiv.), BBr3 (1.1 equiv.), CH2Cl2, -78°C to rt, 1h, 93%; (iv) 5 (1 equiv.), 6a-6ag, 6aj-6al (1.2 equiv.), K2CO3 (1.5 equiv.), DMF, rt, overnigt, 40-95%.
Scheme 2.
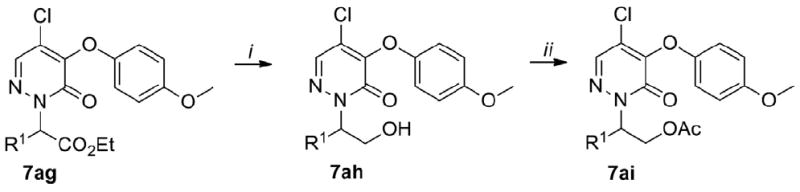
Synthesis of 7ah–7ai.
Reagents and conditions: (i) 7ag (1 equiv.), LiBH4 (1 equiv.), THF, reflux, 2h, 28%; (ii) 7ah (1 equiv.), AcCl (1 equiv.), CH2Cl2, rt, 2h, 95%.
Table 1.
NPBWR1 antagonist activity of compounds 7a–7al (IC50 μM)
| Compound | R1 | R2 | IC50 |
|---|---|---|---|
| 7a | Cyclopentyl | H | 3.70 |
| 7b | Phenyl | H | 0.78 |
| 7c | 2-Methylphenyl | H | 0.37 |
| 7d | 3-Methylphenyl | H | 1.40 |
| 7e | 4-Methylphenyl | H | 1.70 |
| 7f | 2-Trifluoromethylphenyl | H | 0.37 |
| 7g | 3-Trifluoromethylphenyl | H | 1.80 |
| 7h | 4-Trifluoromethylphenyl | H | 1.00 |
| 7i | 2,6-Dimethylphenyl | H | 0.24 |
| 7j | 3,5-Dimethylphenyl | H | 0.41 |
| 7k | 3,4-Dimethylphenyl | H | 0.61 |
| 7l | 4-Methoxyphenyl | H | 3.80 |
| 7m | 4-Phenylphenyl | H | 9.40 |
| 7n | 4-Isopropylphenyl | H | 10.0 |
| 7o | 4-Chlorophenyl | H | 0.59 |
| 7p | 2-Ethylphenyl | H | 0.26 |
| 7q | 2-Isopropylphenyl | H | 0.64 |
| 7r | 2-Methoxyphenyl | H | 0.67 |
| 7s | 2-(Methoxycarbonyl)phenyl | H | 1.20 |
| 7t | 2-Cyanophenyl | H | 1.40 |
| 7u | (1,1′-Biphenyl)-2-yl | H | 0.54 |
| 7v | 2-(Pyrazol-1-yl)phenyl | H | 0.22 |
| 7w | Anthracen-9-yl | H | 0.32 |
| 7x | Naphtalen-2-yl | H | 1.50 |
| 7y | Naphtalen-1-yl | H | 0.14 |
| 7z | Naphthalen-1-ylmethyl | H | 4.20 |
| 7aa | 4-Fluoronaphtalen-1-yl | H | 0.36 |
| 7ab | 2-Methylnaphtalen-1-yl | H | 0.22 |
| 7ac | 2-Methoxynaphtalen-1-yl | H | 0.35 |
| 7ad | 4-Bromonaphtalen-1-yl | H | 2.00 |
| 7ae | 4-Methylnaphtalen-1-yl | H | 0.37 |
| 7af | Quinolin-5-yl | H | 3.40 |
| 7ag | Naphtalen-1-yl | CO2Et | 0.45 |
| 7ah | Naphtalen-1-yl | CH2OH | 2.50 |
| 7ai | Naphtalen-1-yl | CH2OAc | 11.60 |
| 7aj | Naphtalen-1-yl | Me | 1.90 |
| 7ak | Naphtalen-1-yl | Phenyl | 18.00 |
| 7al | Phenyl | COPh | 0.58 |
Data are reported as mean of n = 3 determinations.
The cyclopentyl analog 7a was ~14-fold less potent than 1. Interestingly, the benzyl derivative 7b was only ~3-fold less potent than 1 indicating that the insertion of a methylene between the aromatic nucleus and the pyridazinone is tolerated in region a of the chemotype. The attachment of a methyl group in the ortho position of the phenyl ring (7c) led to twofold increase in potency compared to the unsubstituted 7b, whereas a ~2-fold decrease of potency was observed for the meta- and para-methyl substituted analogs (7d, 7e). Similarly, the 2-trifluoromethyl analog 7f was twofold more potent than 7b, whereas a mild decrease of potency was observed for the 3-trifluoromethyl 7g and 4-trifluoromethyl 7h analogs. Interestingly, an increase of potency versus the mono-substituted counterparts was observed when a second methyl group was added in the ortho, meta or para position of 7c, 7d, 7e. Indeed, the 2,6-dimethyl 7i and 3,5-dimethyl 7j analogs were ~1.5- and ~3.4-fold more potent than 7c and 7d, respectively, while the 3,4-dimethyl analog 7k was ~2- and ~3-fold more potent than 7d and 7e, respectively. Furthermore, the 4-methoxy 7l, 4-phenyl 7m and 4-isopropyl 7n analogs were less potent than 7b by ~5-, ~12- and ~13-fold, respectively, thus showing that installing electron-donating, aromatic or alkylic groups in para position is detrimental for the potency. The potency was impacted to a lesser extent when a chlorine was inserted in para position, with 7o being slightly more potent than 7b. Further exploring the ortho position showed that increasing the size from a methyl (7c) to an ethyl group (7p) led to a small increase in potency, while the isopropyl derivative 7q was slightly less potent than 7p. The 2-methoxy analog 7r was nearly equipotent to the isopropyl analog 7q. Interestingly, the methyl ester 7s and cyano 7t derivatives were 3- to 4-fold less potent than 7c. Notably, the biphenyl derivative 7u was only 1.5-fold less potent than 7c, while the pyrazole 7v was slightly more potent than 7c and 7p. Taken together this data suggests that both steric and electronic factors in the ortho position modulate the potency. Next, we explored the installation of bicyclic and tricyclic aromatic systems in the region a. The anthracene 7w was ~2-fold more potent than 7b and slightly less potent than the pyrazole 7v. Remarkably, the naphtalen-2-yl analog 7x was ~4-fold less potent than 7c, while the naphtalen-1-yl 7y (CYM50719) was ~3-fold more potent than 7c. Based on these results we explored the SAR around the naphthalen-1-yl moiety. Introducing an additional methylene spacer between the pyridazinone and the naphthalenyl ring (7z) led to 30-fold loss of potency. Installing a methyl in position 2 (7ab) led to a small decrease in potency compared to 7y, and a 2- to 3-fold loss of potency was observed for the 2-methoxy analog 7ac. The installation of a fluorine (7aa), methyl (7ae) and bromine (7ad) at position 4 led to ~3-, ~3- and ~14-fold loss in potency, respectively, confirming that substitutions in this position are not tolerated. Interestingly, the quinoline 7af was ~24-fold less potent than the naphthalene 7y indicating that the basic atom in this position is detrimental for the potency. Next, a series of analogs with disubstituted benzylic position was explored keeping, first, the naphtalen-1-yl as the constant moiety. Interestingly, the ethyl ester 7ag was ~3-fold less potent than the non-substituted 7y. Surprisingly, the acetate 7ai and the primary alcohol 7ah were ~83- and ~18-fold less potent than 7y, respectively. Interestingly, the methyl 7aj and phenyl 7ak substituted analogs were ~13- and ~128-fold less potent than 7y. Additionally, the phenyl ketone 7al was slightly more potent than the non-substituted 7b. This data showed that steric interactions in this portion of the molecule are important for the potency, and indicated only a minor decrease of the potency when the second substituent contains a carbonyl group immediately attached to the benzylic carbon (7ag, 7al). We speculated that the partial ketoenol tautomerization could positively impact the potency by forcing the benzylic substituents into a quasi-planar conformation. Based on this working hypothesis, we synthesized planar or planar-like tricyclic structures (9a–9h). The synthesis of these derivatives is depicted in Schemes 3 and 4. Furthermore, the biphenyl system was opened and a carbonyl group was inserted to obtain the quasi-planar ketone 9j and the amide 9k. Additionally, the bicyclic amide 9i was studied. The synthesis of 9i–9k is depicted in Scheme 5. Coupling of pyridazinone 5 with a series of tricyclic systems 8a–8e using Ullman conditions led to the products 9a–9e (Scheme 3). Alkylation of intermediate 5 with benzylchlorides 10a–10c using sodium hydride as the base led to the formation of 9f–9h. Alkylation of 5 with the α-halo carbonyl 11, 12a and 12b using potassium carbonate as the base furnished 9i–9k. The biological data of 9a–9k is reported in Table 2.16
Scheme 3.
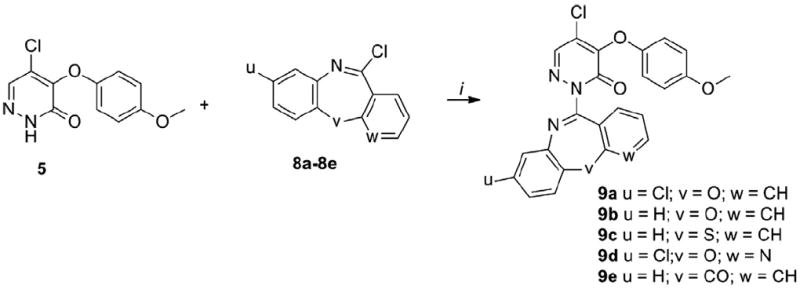
Synthesis of 9a–9e.
Reagents and conditions: (i) 5 (1 equiv.), 8a-8e (1.3 equiv.), Cul (0.1 equiv.), K2CO3 (1.2 equiv.), DMF, 110°C, 8h, 30-65%.
Scheme 4.
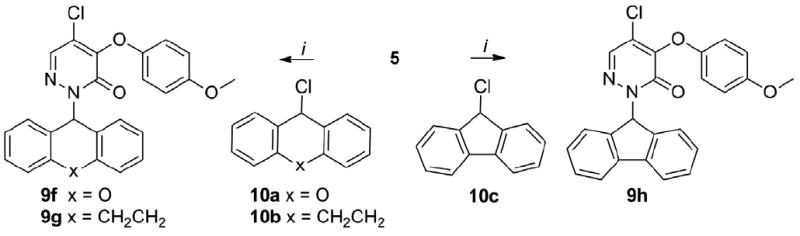
Synthesis of 9f–9h.
Reagents and conditions: (i) 5 (1 equiv.), 10a, 10b, 10c (1.4 equiv.), NaH (1.1 equiv.), DMF, 0°C to rt, 30-50%.
Scheme 5.
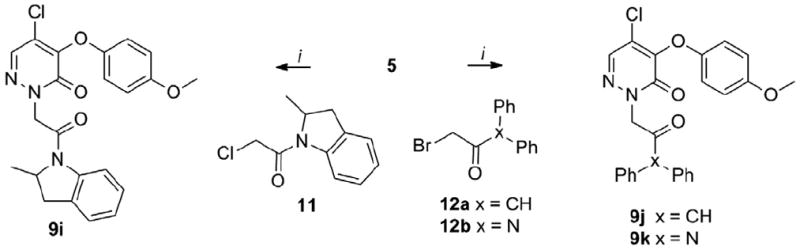
Synthesis of 9i–9k.
Reagents and conditions: (i) 5 (1 equiv.), 11, 12a, 12b (1.2 equiv.), K2CO3 (1.5 equiv.), DMF, rt, overnight, 40-95%.
Table 2.
NPBWR1 antagonist activity of compounds 9a–9k (IC50 μM)
| Compound | IC50 (μM)a |
|---|---|
| 9a | 0.37 |
| 9b | 0.49 |
| 9c | >25 |
| 9d | 0.53 |
| 9e | 2.20 |
| 9f | 3.10 |
| 9g | 18.00 |
| 9h | 0.12 |
| 9i | 0.78 |
| 9j | 0.43 |
| 9k | 0.28 |
Data are reported as mean of n = 3 determinations.
Remarkably, the dibenzoxazepines 9a and 9b were slightly more potent than the ketone 7at. Surprisingly, the dibenzothiazepine 9c was >50-fold less potent than 9b. Interestingly the benzopyridoxazepine 9d was less than twofold less potent than the carba-analog 9a, showing that the insertion of a basic center in this series was tolerated. When the oxygen from 9b was exchanged for a carbonyl group (9e) the potency decreased by ~4-fold. Removing the imine from the tricyclic system led to the tricyclic compounds 9f–9h. Remarkably, the fluorene 9h (CYM50769) was slightly more potent than the naphtalen-1-yl derivative 7y. Increasing the middle-ring size from 5 to 7 members (9g) led to 150-fold loss in potency. The xanthene 9f was ~26-fold less potent than 9h. Interestingly, the elongated bicyclic analog 9i was only ~6-fold less potent than the fluorene 9h indicating that is possible to increase the distance between the pyridazinone and the aromatic rings without major decrement in the potency. Furthermore, the biphenyl ketone 9j was nearly equipotent to the tricyclic system 9b and slightly more potent than 7at. Interestingly, the biphenylamide 9k was slightly more potent than the carba-analog 9j.
Next, we explored the region b while selecting three moieties from the region a. The synthesis of 15a–15w is depicted in Scheme 6. The results are listed in Table 3. 16 Alkylation of pyridazinone 2 with chlorides 6c, 6ac and 10c using potassium carbonate yielded the intermediates 13a–13c. Substitution with a series of phenols in position 4 of pyridazinones 13a–13c using sodium hydride as the base yielded the final products 15c–15w and the intermediates 16a, 16b which were deprotected to the primary alcohols 15a, 15b using tetrabutyl ammonium fluoride (TBAF).
Scheme 6.
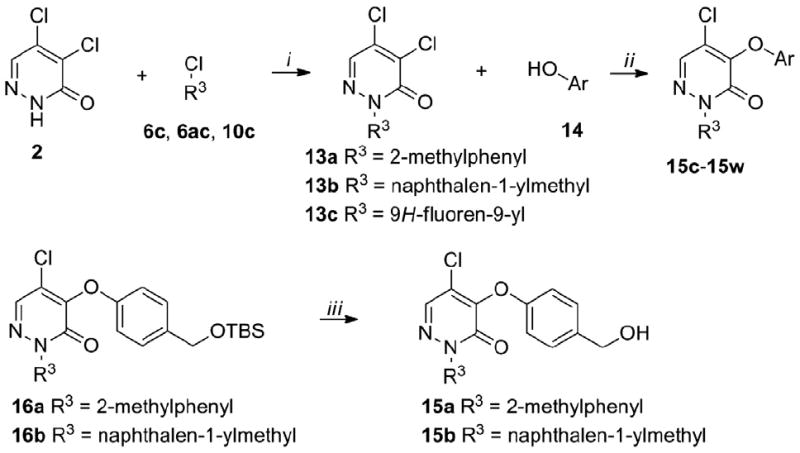
Synthesis of 15a–15w.
Reagents and conditions: (i) 2 (1 equiv.), 6c, 6ac, 10c (1.2 equiv.), K2CO3 (1.5 equiv.), DMF, rt, overnight, 40-95%; (ii) 13a-c (1 equiv.), 14 (1.1 equiv.), NaH (1.1 equiv.), 1,4-dioxane, 15°C to rt, overnight, 30-70%; (iii) 16a,b (1 equiv.), TBAF (2 equiv.), THF, 0°C, 1h, 60-90%.
Table 3.
NPBWR1 antagonist activity of compounds 15a–15w
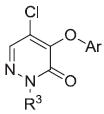
| |||
|---|---|---|---|
| Compound | R3 | Ar | IC50a (μM) |
| 15a | 2-Methylbenzyl | 4-(Hydroxymethyl)phenyl | 0.73 |
| 15b | Naphthalen-1-ylmethyl | 4-(Hydroxymethyl)phenyl | 4.40 |
| 15c | Naphthalen-1-ylmethyl | 2-Flouro-4-methoxyphenyl | 0.64 |
| 15d | Naphthalen-1-ylmethyl | 3-Fluoro-4-methoxyphenyl | 0.36 |
| 15e | Naphthalen-1-ylmethyl | 4-Ethoxyphenyl | 0.10 |
| 15f | Naphthalen-1-ylmethyl | 4-Propoxyphenyl | 1.10 |
| 15g | Naphthalen-1-ylmethyl | 4-Ethylphenyl | 0.25 |
| 15h | Naphthalen-1-ylmethyl | 4-Trifluoromethoxyphenyl | 5.50 |
| 15i | Naphthalen-1-ylmethyl | 4-Isopropoxyphenyl | 0.53 |
| 15j | Naphthalen-1-ylmethyl | 4-(Methoxycarbonyl)phenyl | >20.00 |
| 15k | Naphthalen-1-ylmethyl | 4-(Methylthio)phenyl | 0.43 |
| 15l | Naphthalen-1-ylmethyl | 3-Methoxyphenyl | 0.61 |
| 15m | Naphthalen-1-ylmethyl | 3-Ethoxyphenyl | 0.98 |
| 15n | Naphthalen-1-ylmethyl | 3-Ethylphenyl | 8.50 |
| 15o | Naphthalen-1-ylmethyl | 3,4-Dimethoxyphenyl | 0.29 |
| 15p | Naphthalen-1-ylmethyl | 3,4,5-Trimethoxyphenyl | 1.80 |
| 15q | Naphthalen-1-ylmethyl | Benzofuran-5-yl | 0.82 |
| 15r | Naphthalen-1-ylmethyl | 2,3-Dihydrobenzofuran-5-yl | 0.43 |
| 15s | Naphthalen-1-ylmethyl | 2,3-Dihydro-1H-inden-5-yl | 0.83 |
| 15t | 9H-Fluoren-9-yl | 4-Ethoxyphenyl | 0.25 |
| 15u | 9H-Fluoren-9-yl | 4-Ethylphenyl | 0.24 |
| 15v | 9H-Fluoren-9-yl | 4-Trifluoromethoxyphenyl | >20.00 |
| 15w | 9H-Fluoren-9-yl | 4-Propoxyphenyl | >20.00 |
Data are reported as mean of n = 3 determinations.
First, we attached solubility-enhancing groups in this portion of the molecule. Interestingly the 2-methylbenzyl primary alcohol 15a was only ~2-fold less potent than the 4-methoxy counterpart 7c, while the naphthalenyl alcohol 15b was ~31-fold less potent than 7y indicating that the 2-methylbenzyl and naphthalenyl series differ in their SAR. We further explored the region b while keeping the naphthalene in the region a. Adding a fluorine in position 2 (15c) or 3 (15d) of the phenyl ring, decreased the potency by ~4- and ~2-fold, respectively compared to 7y, indicating that changing the dipolar moment in the phenyl ring influences negatively the antagonist activity. Next, the SAR at position 4 was explored. The ethoxy analog 15e (CYM50775) was slightly more potent than 7y. Conversely, the n-propoxy 15f and isopropoxy 15i were ~8- and ~4-fold less potent than 7y, respectively, suggesting that steric factors modulate the potency. Remarkably, the ethyl 15g was less than twofold less potent than 7y indicating that the hydrogen bond acceptor capability in this position is not fundamental for the functional activity. Furthermore, the trifluoromethoxy 15h was ~39-fold less potent than 7y, and a dramatic loss of potency was observed for the methyl ester 15j, while changing the oxygen of 7y for sulfur (15k) decreased the potency only by threefold. Next, we explored the position 3 of the phenyl ring. The methoxy 15l, ethoxy 15m and ethyl 15n analogs were ~4-, 10- and 34-fold less potent than 7y, 15e and 15g respectively. Remarkably, the 3,4-dimethoxy 15o and 3,4,5-trimethoxy 15p were 2- and ~13-fold less potent than 7y suggesting that the negative effect of polisubtitution on the aromatic ring is additive. Since the methoxy on position 3 of 15o was tolerated, we speculated that bicyclic systems may improve the potency. However, the benzofuran 15q was ~3-fold less potent than 15o, while the dihydrobenzofurane 15r was less than ~2-fold less potent than 15o and ~2-fold more potent than the indene 15s. Different SAR was observed within the fluoren-9-yl series. The ethoxy 15t and ethyl 15u analogs were equipotent and twofold less potent than 9h, while a dramatic decrease in potency was observed for the 4-trifluoromethoxy 15v and 4-propoxy 15w.
Amongst the synthesized compounds, 9h (CYM50769) was selected for further characterization.17 The solubility of 9h in a phosphate buffered saline (PBS) at pH 7.4 is 0.17 μM. The compound is non-cytotoxic to U2OS cells at 20 μM and chemically stable in PBS at pH 7.4 with half-life higher than 48 h. The selectivity profile was investigated against the Ricerca panel of off-target proteins including GPCRs, enzymes, transporters and ion channels at a concentration of 30 μM. Remarkably, out of 35 tested targets only CYP450 1A2, 5-HT2B and CYP450 2C19 showed 67%, 63% and 51% inhibition, respectively.
In summary, we have reported the synthesis and SAR studies around the coil and 4-methoxyphenyl regions (a, b) of novel non-peptidic NPBWR1 antagonists based on a 5-chloro-4-(4-alkoxyphenoxy)-2-(benzyl)pyridazin-3(2H)-one chemotype. Small changes in region b had a negative impact on the potency, while the region a was found to interact with a liphophilic pocket that can accommodate a great variety of bulky quasi-planar substituents. Our studies resulted in the identification of a novel series of submicromolar NPBWR1 antagonists including 7y (CYM50719), 9h (CYM50769) and 15e (CYM50775) endowed with greater potency than our previously reported lead 1. Amongst them, 9h was further profiled and found to be highly selective against a broad array of off-targets with pharmaceutical relevance, making this compound suitable for further development. Our medicinal chemistry advances around this chemotype will be communicated in due course.
Acknowledgments
This work was supported by the National Institute of Health Molecular Library Probe Production Center Grant U54 MH084512 (Peter Hodder, Hugh Rosen).
References and notes
- 1.Tanaka H, Yoshida T, Miyamoto N, Motoike T, Kurosu H, Shibata K, Yamanaka A, Williams SC, Richardson JA, Tsujino N, Garry MG, Lerner MR, King DS, O’Dowd BF, Sakurai T, Yanagisawa M. Proc Natl Acad Sci U S A. 2003;100:6251. doi: 10.1073/pnas.0837789100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jackson VR, Lin SH, Wang Z, Nothacker HP, Civelli O. J Comp Neurol. 2006;497:367. doi: 10.1002/cne.20989. [DOI] [PubMed] [Google Scholar]
- 3.Kitamura Y, Tanaka H, Motoike T, Ishii M, Williams SC, Yanagisawa M, Sakurai T. Brain Res. 2006;1093:123. doi: 10.1016/j.brainres.2006.03.041. [DOI] [PubMed] [Google Scholar]
- 4.O’Dowd BF, Scheideler MA, Nguyen T, Cheng R, Rasmussen JS, Marchese A, Zastawny R, Heng HH, Tsui L, Shi X, Asa S, Puy L, George SR. Genomics. 1995;28:84. doi: 10.1006/geno.1995.1109. [DOI] [PubMed] [Google Scholar]
- 5.Lee DN, Nguyen T, Porter CA, Cheng R, George SR, O’Dowd BF. Brain Res Mol Brain Res. 1999;71:96. doi: 10.1016/s0169-328x(99)00171-0. [DOI] [PubMed] [Google Scholar]
- 6.Shimomura Y, Harada M, Goto M, Sugo T, Matsumoto Y, Abe M, Watanabe T, Asami T, Kitada C, Mori M, Onda H, Fujino M. J Biol Chem. 2002;277:35826. doi: 10.1074/jbc.M205337200. [DOI] [PubMed] [Google Scholar]
- 7.Mondal MS, Yamaguchi H, Date Y, Shimbara T, Toshinai K, Shimomura Y, Mori M, Nakazato M. Endocrinology. 2003;144:4729. doi: 10.1210/en.2003-0536. [DOI] [PubMed] [Google Scholar]
- 8.Ishii M, Fei H, Friedman JM. Proc Natl Acad Sci U S A. 2003;100:10540. doi: 10.1073/pnas.1334189100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamamoto T, Saito O, Shono K, Tanabe S. Brain Res. 2005;1045:97. doi: 10.1016/j.brainres.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 10.Zaratin PF, Quattrini A, Previtali SC, Comi G, Hervieu G, Scheideler MA. Mol Cell Neurosci. 2005;28:55. doi: 10.1016/j.mcn.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 11.Green BR, Smith M, White KL, White HS, Bulaj G. ACS Chem Neurosci. 2011;2:51. doi: 10.1021/cn1000974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Samson WK, Baker JR, Samson CK, Samson HW, Taylor MM. J Neuroendocrinol. 2004;16:842. doi: 10.1111/j.1365-2826.2004.01239.x. [DOI] [PubMed] [Google Scholar]
- 13.Nagata-Kuroiwa R, Furutani N, Hara J, Hondo M, Ishii M, Abe T, Mieda M, Tsujino N, Motoike T, Yanagawa Y, Kuwaki T, Yamamoto M, Yanagisawa M, Sakurai T. PLoS ONE. 2011;6:e16972. doi: 10.1371/journal.pone.0016972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Watanabe N, Wada M, Irukayama-Tomobe Y, Ogata Y, Tsujino N, Suzuki M, Furutani N, Sakurai T, Yamamoto M. PLoS ONE. 2012;7:e35390. doi: 10.1371/journal.pone.0035390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Urbano M, Guerrero M, Zhao J, Velaparthi S, Saldanha SA, Chase P, Civelli O, Hodder P, Schaeffer M, Brown S, Rosen H, Roberts E. Bioorg Med Chem Lett. 2000 doi: 10.1016/j.bmcl.2012.09.074. http://dx.doi.org/10.1016/j.bmcl.2012.09.074. [DOI] [PMC free article] [PubMed]
- 16.The biological inhibition assay employed a chimeric cell line that forces the receptor to use Gqi3; therefore the assay readout was calcium release. HEK cells stably co-transfected with the human NPBWR1 and Gqi3 (hGPR7 HEK293T/Gqi3 cell line) were used for this study. Cells were plated at 10,000 cells/well of a 384 well plate in 25 μL media and incubated overnight. Next, 25 μL of Fluo8 NW (ABD Bioquest) was added to all wells and the assay plate incubated for 50 min at 37 °C, 5% CO2 and 95% relative humidity. Test compounds were added and the assay plate was incubated for 15 min at room temperature. The assay was started by performing a basal read of fluorescence (495 nm excitation and 515 nm emission) for 15 s on the FLIPR Flexstation II 384 (Molecular Devices). Next, 5.5 μl of GPR7 agonist (20 nM final concentration = EC80) in FLIPR Buffer (HBSS/20 mM Hepes/0.1% BSA) or FLIPR Buffer alone were dispensed to the appropriate wells. Then a real time fluorescence measurement was immediately performed for the remaining 45 s of the assay. Tested compounds were assayed in triplicate in an 8-point 1:3 dilution series starting at a nominal concentration of 20 μM. For each test compound, percent inhibition was plotted against the log of the compound concentration. A three parameter equation describing a sigmoidal dose–response curve was then fitted using GraphPad Prism (GraphPad Software Inc) normalized from 0 to 100 for each assay. In cases where the highest concentration tested (i.e. 20 μM) did not result in greater than 50% activation, the IC50 was determined manually as greater than 20 μM.
- 17.http://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=1880