

Abstract
Hantaviruses use αvβ3 integrins on the surface of human host cells as a gateway to invasion, hence compounds that target this receptor may be used as antiviral agents. To accomplish this aim, new peptidomimetic compounds were selected based on similarity to a cyclic peptide known to bind the αvβ3 receptor. This first round of biological screening identified peptidomimetic molecules which were effective hantavirus inhibitors in the low micromolar range, two thousand times more potent than the original cyclic peptide. Pharmacophore models were built to broaden the structural diversity of the second set of compounds screened. Structure-activity relationships (SAR) were drawn from the entire dataset. Further characterization by dose-response studies revealed that three compounds had potency in the nanomolar range. Selectivity assays with a panel of hantaviruses supported the mechanism of inhibition by targeting the αvβ3 receptor, through the β3 integrin.
Keywords: anti-hantavirus, Sin Nombre Virus (SNV), peptidomimetics, Structure-Activity Relationships, entry inhibitor
Hantavirus cardiopulmonary syndrome (HCPS) and hantavirus induced hemorrhagic fever with renal syndrome (HFRS) are diseases of global concern. Hantaviruses are from the family Bunyaviridae, and are classified according to their geographic distribution and disease presentation as either New World or Old World hantaviruses. New World hantaviruses, such as Sin Nombre virus (SNV) found in North America and Andes virus (ANDV) of South America, are causative agents of HCPS. Old World hantaviruses such as Hantaan (HTNV) and Seoul viruses (SEOV) cause HFRS and can be found in Eurasia.1 SNV, which is endemic to the southwestern United States, causes HCPS with a mortality rate approaching 40 %.
These and other pathogenic hantaviruses are carried by wild rodents of the family Muridae. Hantaviruses are enveloped, negative-sense RNA viruses. The hantavirus genome consists of three segments designated L (large; ~6,500 nucleotides (nt)), M (middle; 3,600 to 3,700 nt) and S (small; 1,700 to 2,100 nt) which encode, respectively, an RNA-dependent RNA polymerase (RdRp), a glycoprotein precursor (GPC) that is processed into Gn and Gc transmembrane glycoproteins, and a nucleocapsid protein (N).
There are currently no therapeutic agents with proven efficacy to specifically treat SNV infection. One effective way to block viral infection may stem from the inhibition of viral recognition of the host cell surface entry receptor. Along these lines, a successful anti-HIV drug, Maraviroc, was designed based on a key early step in the viral entry process.2 A similar approach can be used to develop anti-hantavirus compounds since αvβ3 integrin is known to be a host cell surface receptor for entry of pathogenic hantavirus.3,4 We previously reported specific disulfide cyclized nonapeptides, identified by use of a combinatorial peptide phage display library, which bind αvβ3 and inhibit SNV entry into αvβ3-expressing cells.5 From this previous work, one of the most potent peptides, cyclo-[CPFVKTQLC], was analyzed by a combination of alanine-scanning and residue deletions to identify the residues critical to activity. We used the 3D (three-dimensional) structure obtained by NMR studies in 6 of the resulting peptide, cyclo-[CPFVC] as a template to search for peptidomimetic molecules to block SNV infection, in a ligand based virtual screening (LBVS) approach. Dose-response results showed that these molecules are concentration-dependent inhibitors and a panel of hantaviruses screened further support the mechanism of these molecules through the β3 integrin receptor.
Round one of shape and electrostatics similarity studies and biological results
In order to begin identifying potential peptidomimetic compounds which could functionally mimic cyclo-[CPFVC], similarity studies using 3D shape and electrostatics of the entire ChemDiv library were performed using Rapid Overlay of Chemical Structures (ROCS v. 2.3.1)7 based on the three dimensional structure of this disulfide cyclised pentapeptide obtained by NMR.6 The overall best hits scored around 1.2 using the Combo index, which corresponds to 60 % of combined shape and electrostatics similarities. As seen in Figure 1, it is apparent that the low similarity score came from the lack of superposition of the small molecules to parts of the cyclic peptide, specifically the disulfide bond used for cyclization. Small molecules with drug-like properties were devised and, in this respect, the aim was not to identify molecules with a high degree of overall similarity to the peptide, but instead to identify molecules that could work as surrogates of parts of its structure with a potency increment.
Figure 1.
Superposition of selected compounds to the cyclic peptide CPFVC. ChemDiv compounds identified by ROCS as being similar in shape and electrostatics to cyclo-[CPFVC] were superimposed onto the pentamer. Stick representations of the molecules are shown in magenta (CPFVC), green (C481-1998), blue (C481-0157) and orange (E945-0123).
Molecular similarity with ROCS was pursued once some studies showed that this ligand-based approach had a hit rate (or enrichment factor) at least as good as docking, if not better, perhaps due to poor performance of the docking scoring function.8 Additional comparative work showed ROCS to be the best approach among LBVS methods, providing further support for its use.9 Additionally, ROCS could easily be applied to the entire ChemDiv library. The top-ranked compounds identified by ROCS were further evaluated by using a set of drug-like filters.10 We considered using docking as a follow-up step to further narrow down the list of candidate compounds for experiments, but several limitations made this less practical. In particular, the binding site is at the surface, thus the compounds do not bind in a deep pocket, but rather in a shallow crevice. This is probably the worst case scenario to expect highly accurate compound selection results out of a docking method.11
In the first round of screening,12 49 compounds were selected for biological testing, mainly representing 3 distinct structural families (Table 1 and supporting information). The ability of the compounds to inhibit SNV infection of Vero E6 cells compared to untreated controls was determined by immunofluorescent assay (IFA),13 as previously described.3 The following were used as controls: ReoPro (80 µg/ml), a commercially available Fab fragment of a humanized monoclonal antibody that binds to the αvβ3 receptor and partially blocks the hantavirus entry in vitro, a random cyclic nonapeptide, the original cyclic nonapeptide CPFVKTQLC5 and the cyclic pentapeptide CPFVC.6 Peptides were used at a concentration of 2 mM, while small molecules were tested at 1 µM. For reasons that are unclear, neither ReoPro nor other antibodies targeting integrin β3 completely block viral infection.5 As the maximum effectiveness of ReoPro in inhibiting hantavirus entry in vitro approaches 80 %, we set this as a threshold for maximal expected efficacy for normalization rather than overinflating the data by normalization to 100%. This allowed for consistent comparison of peptide and small molecule activity.
Compounds, sorted by structural family, are shown in Table 1 along with percent inhibition of SNV infection compared to untreated controls. Of the 49 tested compounds, one, G319-0078, inhibited SNV infection by greater than 40 %, three compounds by 30–40 % (E945-0123, C481-0157 and C481-1998), one compound by 20-30 % (G319-0007) and seven compounds by 10–20 %. The remaining thirty-seven compounds inhibited cell infection by 10 % or less at the same concentration. These preliminary tests provided striking results, especially considering that the peptidomimetics were tested in micromolar (1 µM) concentrations, which is two thousand times less compared to the reference peptides CPFVC and CPFVKTQLC (2 mM). Moreover, visual inspection of the host cells (Vero E6) revealed that the new compounds tested were not cytotoxic at this concentration.
Table 1.
Initial IFA screen of ChemDiv peptidomimetics for inhibition of SNV (round 1).a
 | |||||
|---|---|---|---|---|---|
| ChemDiv compound |
%Inhib. (±SD)b |
Substituents | ChemDiv compound |
%Inhib. (±SD)b |
Substituents |
| Family 1A | |||||
| C481-0157 | 35 (±14) | C481-1998 | 34 (±11) | ||
| Family 1B | |||||
| G319-0078 | 46.5 (±6.8) | G319-0007 | 22.5 (±4.5) | ||
| Family 1C | Family 2 | ||||
| K938-0016 | 14 (±11) |  |
8006-8225 | 8 (±11) |  |
| Family 3 | Singleton | ||||
| E945-0123 | 39 (±18) | 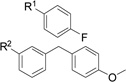 |
8007-7686 | 16.1 (±9.1) | 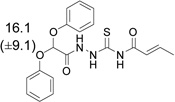 |
| Controls | |||||
| ReoPro | 80.2 (±4.5) | Random | 5 (±11) | ||
| CPFVC | 53 (±11) | DMSO | 1.6 (±6.8) | ||
| CPFVKTQLC | 54.6 (±4.5) | Media | 0 (±14) | ||
See the supporting information to retrieve the whole dataset.
% Inhib.: Percentage of inhibition. SD: Standard Deviation for at least 4 replicates. Peptides were used at a concentration of 2 mM, while small molecules were tested at 1 µM.
In family 1A, substituents with aromatic or aliphatic amines were more potent than the corresponding analogues (C481-0157, C481-1998, C481-0192, C481-1669, C481-0290, C481-0444, C481-0304, C481-1718, Table 1). Despite bearing the amine, compound C481-1893 inhibited by less than 10 %, however its longer substituent at the ortho position may account for its reduced potency. Likewise, C481-2222 showed poor inhibition which may be explained by its bearing an alkyl-aryl amine which is not protonated at the physiological pH due to the conjugation of the electron pair to the benzene ring.
Family 1B is a series closely related to family 1A, but here molecules bearing aromatic rings with aryl-alkyl ether were the most potent of the series. One important exception is compound C737-1938, which has a cyclohexane. However, the 1B profile is by no means similar to the results of family 1A. The most striking difference between Families 1A and 1B comes from the comparison of C481-0633 and G319-0078 that have the same benzodioxolane moiety. While G319-0078 is one of the most potent compounds, with 47 % inhibition, C481-0633 is inactive. This suggests that molecules from these two families may not bind in the same way.
Compounds from family 1C do not have the fused rings attached to the sulfonamide as seen in families 1A and 1B. This may account for its weak potency in relation to 1A and 1B, especially when considering compounds pairs that have the same branches: K938-1906 and C481-0157, as well as K938-1842 and G319-0078.
Family 2 members showed little to no inhibition and these scaffolds were not pursued. In contrast, family 3 contained a single molecule, E945-0123, in this first set which showed promising inhibition (38.5 %), leading us to select other members of this class to assay in the second round of screening. Finally, no singleton was considered sufficiently to elicit further consideration.
Pharmacophore identification, round two selection and biological screening
Guided by these initial compelling results, a second set of molecules was devised to further improve the chemical diversity. The core structures from the first round were used as the basis for the selection of additional compounds using the Unity software.14 From the pool of family 1 compounds exhibiting cell infection inhibition greater than 20 %, ROCS alignment was used to guide the selection of a four-point pharmacophore model with a partial match directive to points 3 and 4. Key components of the pharmacophores included; five and six membered rings (lactams, maleimides and conjugated hydrazides) at positions 1, 2 and 3, with a positive nitrogen atom located in position 4 (Figure 2). The positive charge at point 4 came from the aliphatic amines seen in the most potent members of family 1A in Table 1. As shown in Figure 1, the five and six membered rings of the pharmacophore match the proline and phenylalanine residues of the original search molecule, cyclo-[CPFVC], which previously shown to be important amino acids to the binding of cyclopeptides.6
Figure 2.
Pharmacophore model from round one screening. A four-point pharmacophore was identified by comparison of cyclo-[CPFVC] and the most potent ChemDiv compounds identified by ROCS in Table 1. The key elements of the pharmacophores model are: points 1, 2 and 3: 5 and 6 member rings, including lactams, maleimides and conjugated hydrazides. Point 4: positive nitrogen atom.
The second round of screening resulted in 68 molecules that were selected and tested to gain additional information about the families tested in the first round of screening, and to seek different scaffolds that may have with similar inhibitory properties. Results of inhibition assays from this second round of screening are shown in Table 2 and the supporting information.
The second batch of family 1A compounds provided some bioactive molecules with lone pairs (oxygen or sulfur atoms in or in the vicinity of aromatic rings) like C481-1375, C481-1844, C481-1690 and C481-1256. It is interesting to note that similar molecules, such as C481-1375 and C481-0465; C481-0906 and C481-1690, had such disparate potencies. However, the SAR for this second set is not straightforward because of the considerable standard deviation values. This is likely a function of the solubility of the compounds, which will be further addressed below. Finally, a cyclic aliphatic amine proved to be a poor substituent since C481-2082 was inactive, while C481-1998 was one of the most potent compounds of this family.
The majority of the family 1B compounds from round 2 were not more potent than the family members assayed in round 1. The sole exception being round 2 compound K906-2276 which, with the addition of a chlorine atom in the ortho position, inhibited SNV infection by 21 % compared to round 1 compound C737-2034 which inhibited by only 11 %.
New molecules from Family 1C did not provide any high potency hits (above 20 % inhibition), further confirming that the fused rings from families 1A and 1B are important to the bioactivity. Dimethoxybenzyl in the context of compounds K938-0195, 4191-0187 and K938-0016 showed similar potency (14–15 %), but were still less potent than G319-007 (22 %) or its cyclic analogue G319-0078 (47 %) from Family 1B. Likewise, K938-1941 showed the same trend in relation to K938-1906 and C481-0157.
The best comparative results from family 1C came from 4191-0187 and K938-0195, which showed similar potency to other methoxyphenyl and methoxybenzyl compounds from family 1B, specifically C737-2042 & G319-0139 (Table 1). These results support the hypothesis that the fused rings are critical to the potency of families 1A and 1B, and likely serve as mimics of the proline and phenylalanine residues of the pentamer CPFVC in interacting with the hantavirus entry receptor, αvβ3 integrin receptor. Further experimental structural work will be required to definitively address this hypothesis.
Family 3 compounds were expanded from a single compound from round 1 (E945-0123, 39 % inhibition). Two of the eleven family 3 compounds tested in round 2 gave inhibition greater than 20 % (E945-0166 at 24 % and E945-0147 at 28 %). By repositioning the chlorine from para (E945-0166) to meta (E945-0156), potency dropped from 24 % to 6 %. Other analogues with a methyl group replacing the fluorine at the heteroaromatic fused ring were not active, including one with a basic amine group (E990-0470). The methyl group may be linked to steric hindrance, as demonstrated by the inactivity of the methyl analogue E990-0606 compared to its fluorinated derivative, E945-0147.
New families selected based on the pharmacophore model developed from round 1 were also tested in this second round of screening. Each of the additional families (family 4 through family 7), were composed of two to five structures. Of the singletons tested in this second round of screening, four compounds showed promising results. These include compounds 5408-0644 and 8012-0652 (both with 29 % inhibition), 4133-0409 (33 % inhibition) and 0091–0142 (44 % inhibition). The diversity of these chemotypes from families 1–7 and the singletons complicates any SAR analyses. These compounds may be used in future work to drive the discovery of novel derivatives of the original peptide inhibitor.
It is important to note that some inter-experimental variation seen with the peptidomimetics in Tables 1 and 2 is likely due to the relative poor solubility of these compounds in aqueous solution. The computational procedures provided means of defining them as highly DMSO soluble, but the solubility in water, defined as low to medium, was worse than expected. To address this issue, we have begun testing a number of delivery vehicles (BASF, Chicago, IL) to identify means of improving solubility, and possibly, efficacy. In preliminary experiments, we have identified one delivery vehicle which improves inhibition by compounds 8012-0652 and G319-0078 by 2.5- and 1.7-fold, respectively (data not shown). These preliminary results show promise that the potency and solubility of the hantavirus entry inhibitors identified here may be improved by optimization of the screening method or vehicle used for delivery. The next step towards the solubility increment of these leads can be accomplished by altering the chemical structures through a specific optimization step.
Table 2.
Expanded dataset of peptidomimetic compounds (round 2).a
 | |||||
|---|---|---|---|---|---|
| ChemDiv compound |
%Inhib. (±SD)b |
Substituents | ChemDiv compound |
%Inhib. (±SD)b |
Substituents |
| Family 1A | Family 1B | ||||
| C481-1256 | 29 (±22) | K906-2276 | 21.4 (±9.0) | ||
| Family 3 | Family 4A | ||||
| E945-0147 | 28 (±16) | 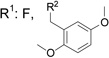 |
C260-1054 | 15 (±15) | 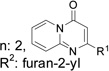 |
| Family 4B | Family 5 | ||||
| C241-1447 | 12.2 (±7.2) |  |
E786-2220 | 2.3 (±6.3) | R1 N, R2 2-fluorophenyl |
| Family 6 | Family 7 | ||||
| K292-0689 | 41 (±11) |  |
K784-7791 | 26 (±21) | R1 methyl, R2 furan-2-yl |
| Singleton | |||||
| 0091-0142 | 44.2 (±9.0) |  |
|||
See the supporting information to retrieve the whole dataset.
% Inhib.: Percentage of inhibition. SD: Standard Deviation for at least 4 replicates. Molecules were tested at 1 µM.
Dose-response and selectivity of viral inhibition
We selected the following compounds for further characterization: C481-1844 (family 1A, round 1), G319-0078 (family 1B, round 1), C418-1256 (family 1A, round 2), K292-0689 (family 6, round 2), 8012-0652 (singleton, round 2) and 5408-0644 (singleton, round 2). These compounds were further tested for the ability to inhibit SNV entry in a dose-dependent manner. Three of the six molecules showed dose-dependent inhibition of SNV infection in Vero E6 cells as determined by IFA (Figure 3). These molecules, 8012-0652, C481-1256 and G319-0078, could partially inhibit the SNV infection with EC50 of 1.3(±0.6), 2.5(±0.8) and 1.6(±0.6) nM, respectively. Among other compounds, G319-0078 and C481-1256 are racemates, and further work will be done to identify the most potent enantiomer.
Figure 3.

Dose-response of peptidomimetics in inhibiting SNV infection in vitro. IFA was used to determine the dose-dependent inhibition of SNV infection in VeroE6 cells by ChemDiv compounds 8012-0652, C481-1256 and G319-0078. Controls included cyclo-CPFVC and a random cyclic peptide at 2 mM. Data points represent the mean and standard error of N = 4.
In order to determine the selectivity of molecules 8012-0652, C481-1256 and G319-0078 towards SNV, further tests were conducted to assess their ability to block infection of other pathogenic hantaviruses which enter cells via integrin β3, Andes (ANDV) and Hantaan viruses (HTNV), compared to the non-pathogenic hantavirus, Prospect Hill (PHV), which is known to enter by integrin β1.3,15 As shown in Figure 4, both 8012-0652 and G319-0078 showed comparable inhibition to cyclo-CPFVC, but at 100-fold lower concentrations (10 µM compared to 1 mM), with C481-1256 giving slighter lower inhibition. As expected, neither cyclo-CPFVC nor the peptidomimetics were effective in blocking entry by the integrin β1-dependent PHV. As was the case with the pentapeptide CPFVC,6 these results speak to the selectivity of these small molecules in blocking cellular entry of pathogenic hantaviruses via host integrin β3. Hence these molecules can be considered protein-protein inhibitors.16 This cell-based assay provides support for our hypothesis that these molecules act through the β3 subunit, like ReoPro. Further studies are underway to better address the binding mode and mechanism of action.
Figure 4.

Selectivity of peptidomimetics for inhibiting viral entry via integrin β3. Focus reduction assay demonstrating the selectivity of cyclo-[CPFVC] and ChemDiv peptidomimetics in inhibiting hantavirus entry via integrin β3 (SNV, ANDV and HTNV) and not by hantavirus that utilize integrin β1 (PHV).
Comparative docking
Structural similarities between cyclo-CPFVC and the peptidomimetics are made clear when the molecules are superimposed (see Fig. 1). The structure of one of the best peptidomimetics identified, G319-0078, superimposed on the NMR structure of cyclo-CPFVC is shown in Figure 5a. In addition, comparative docking (Autodock)6 of cyclo-CPFVC and G319-0078 onto the ReoPro binding site of the three dimensional crystal structure of integrin β3 (PDB ID 1U8C)17 shows substantial superposition of the small molecule to the cyclopeptide (Fig. 5b). However, docking and ROCS results showed some disagreement. According to the ROCS superposition, the cyclic peptide rings with proline and phenylalanine amino acid residues should align with the G319-0078 rings. As shown in Figure 5, there is a slight mismatch of the docking poses which may be due to incorrect posing of cyclo-CPFVC. As previously mentioned, proline and phenylalanine are known to be enriched at protein interaction interfaces,18 as such these side chains should interact with the amino acids of the receptor. In addition, these residues were previously shown by alanine scanning to be crucial for the inhibitory activity of CPFVC,6 being very important for peptide recognition of the receptor. Therefore, it is counterintuitive that the docking pose placed these amino acids residues outside the binding side, facing the solvent. This may have occurred because the docking tool did not take into account the solvent; hence the penalty for solvent exposure of hydrophobic rings was likely underestimated. This is not the case for the small molecule, where the rings of the peptidomimetics are facing the surface of β3 (Fig. 5). Additional work using biophysical methods such as X-ray crystallography or molecular dynamics/Monte Carlo with explicit solvent and different starting geometries, would be required to better address this issue.19
Figure 5.

Comparative docking of cyclo-[CPFVC] and ChemDiv compound G319-0078. ROCS (a) and docking (b) results of G319-0078 (pink) and its comparison with cyclic peptide CPFVC (yellow). Docking was performed with Autodock 3.0 using the three-dimensional crystal structure of β3 integrin (PDB ID 1U8C) as the receptor. The green surface in (b) represents the boundaries of the binding site.
SAR is known for a myriad of αvβ3 inhibitors that target the RGD (Arginine-Glycine-Aspartate) binding site.20 However, this structural information may not be useful for the optimization of the new chemicals reported here, as they were designed to bind at a different site where ReoPro interacts. While the RGD binding pocket is located at the interface of the αvβ3 heterodimer,20 the ReoPro binding pocket is located on the β3 subunit.21 The selectivity assays in Figure 4 support the binding at the β3 subunit, but further studies are required to determine the mechanism.
New entry inhibitors of hantaviruses that selectively target αvβ3 integrin were designed using LBVS and cell-based assays. These bioactive compounds were able to bind the receptor at host cell plasma membrane, thus preventing viral recognition at the cell surface. The approach successfully achieved the goal to identify these protein-protein interaction inhibitors, despite the fact that they probably bind to a shallow crevice at the cell surface receptor composed by a β3 integrin subunit. The most potent representatives were at least 2000 times better than the original cyclo-peptide (2 mM). Furthermore, an in vitro selectivity assay with a panel of hantaviruses corroborated the mechanism proposed by demonstrating that the peptidomimetics are selective towards integrin β3 versus integrin β1. Compelling results were achieved with a unique set of chemical scaffolds, leading to novel inhibitors of interest for the next phases of the drug discovery process. The therapeutic potential of these compounds needs to be further refined by the optimization of solubility along with the pharmacokinetics, as well as in vivo studies using animal models.
Supplementary Material
Acknowledgements
This work was supported by NCMR grant “Integrated Network of Ligand-based Autonomous Bioagent Detectors” and by Public Health Service grants U01 AI 56618 (BH), U01 AI054779 (BH), R56 AI034448 (RSL) and 1 CO6 RR012511. PH was supported by a National Institute of Allergy and Infectious Disease grant T32 AI07538-06 (BH) and by an NIH Ruth L. Kirschstein National Research Service Awards grant F32 AI074246-01A1. PH is currently funded by a Veterans Affairs Career Development Award 2 (NMVAHCS).
References and Notes
- 1.Schmaljohn CS, Nichol ST. In: Fields Virology. Knipe DM, Howley PM, Griffin MD, Lamb RA, Martin MA, Roizman B, Strauss SE, editors. Philadelphia: Lippincott Williams & Wilkins; 2007. pp. 1741–1789. [Google Scholar]
- 2.Kuritzkes D, Kar S, Kirkpatrick P. Nat. Rev. Drug Discov. 2008:7 15. [Google Scholar]
- 3.Gavrilovskaya IN, Shepley M, Shaw R, Ginsberg MH, Mackow ER. Proc. Natl. Acad. Sci. USA. 1998;95:7074. doi: 10.1073/pnas.95.12.7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raymond T, Gorbunova E, Gavrilovskaya IN, Mackow ER. Proc. Natl. Acad. Sci. USA. 2005;102:1163. doi: 10.1073/pnas.0406743102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Larson RS, Brown DC, Ye C, Hjelle BJ. Virol. 2005;79:7319. doi: 10.1128/JVI.79.12.7319-7326.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Hall PR, Malone L, Sillerud LO, Ye C, Hjelle BL, Larson RS. Chem. Biol. Drug Des. 2007;69:180. doi: 10.1111/j.1747-0285.2007.00489.x. [DOI] [PubMed] [Google Scholar]; (b) Bharadwaj M, Lyons CR, Wortman IA, Hjelle B. Vaccine. 1999;17:2836. doi: 10.1016/s0264-410x(99)00096-1. [DOI] [PubMed] [Google Scholar]; (c) Botten J, Mirowsky K, Kusewitt D, Bharadwaj M, Yee J, Ricci R, Feddersen RM, Hjelle B. Proc. Natl. Acad. Sci. USA. 2000;97:10578. doi: 10.1073/pnas.180197197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.More than six hundred thousand compounds from the ChemDiv library were subjected to the virtual screening analyzes in a stepwise procedure. First, Omega software v. 2.2.1 (OpenEye Scientific Software, Inc., Santa Fe, NM, USA) was used to generate up to 400 conformations of each compound in the database with the default parameters. Then, Rapid Overlay of Chemical Structures (ROCS v. 2.3.1) software was chosen to superimpose the ChemDiv compounds onto the cyclic pentapeptide CPFVC based on a combined score compounded by structural (shape) and electrostatics (colors) properties. ImplicitMillsDean and optchem flags were enabled to define the type of color force field and optimization based on chemistry. The Tanimoto coefficient was used to select the best matches among the ChemDiv database in relation to the cyclopeptide. The three-dimensional structure of the cyclic pentapeptide CPFVC used on this step was obtained from NMR measurements and deposited in the RCSB PDB with the accession code 2P7R. The selected compounds were grouped into families according to their scaffolds. VIDA (v. 3.0.0 OpenEye), Sybyl (v. 8.0) and PyMOL (v. 0.99) were used to visualize the results.
- 8.Hawkins PC, Skillman AG, Nicholls A. J. Med. Chem. 2007;50:74. doi: 10.1021/jm0603365. [DOI] [PubMed] [Google Scholar]
- 9.Tresadern G, Bemporad D, Howe T. J. Mol. Graph. Model. 2009;27:860. doi: 10.1016/j.jmgm.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 10.The putative solubility and pharmacokinetic properties of all molecules were estimated by ClogP from Biobyte Corporation and built-in VolSurf (Molecular Discovery Ltd.) models of solubility and permeability
- 11.Leach AR, Shoichet BK, Peishoff CE. J. Med. Chem. 2006;49:5851. doi: 10.1021/jm060999m. [DOI] [PubMed] [Google Scholar]
- 12.Virus strains used in the inhibition assays included SNV strain SN77734, previously described by this laboratory, Hantaan virus strain HTN 76/118, provided by Ho Wang Lee, ANDV strain CHI-7913, provided by Hector Galeno (Instituto Salud Publica, Santiago, Chile) and Prospect Hill virus (PHV-1), provided by Ric Yanagihara (University of Hawaii). Cysteine-constrained peptides were synthesized by Biopeptide (San Diego, CA), solubilised in 10 mM phosphate-buffered saline (PBS), pH 7.4, and stored at −80 °C until use. ReoPro, a Fab, was purchased from Eli Lilly and Company (Indianapolis, IN) and suspended in 0.01 M sodium phosphate, 0.15 M sodium chloride, and 0.001 % polysorbate 80, pH 7.2, and used at a final working concentration of 80 µg/ml. Fluorescein isothiocyanate (FITC)-conjugated anti-rabbit immunoglobulin G (IgG) was purchased from Boehringer Mannheim (Indianapolis, IN). Vero E6 cells (ATCC CRL 1586) were purchased from the American Type Culture Collection (Manassas, VA). All small molecules were purchased from ChemDiv Inc. (San Diego, CA) and were at least 90 % pure according to NMR analysis.
- 13.Procedures requiring live virus were conducted in a biosafety level 3 facility, Centers for Disease Control registration number C20041018-0267. All compounds were tested by immunofluorescence assay (IFA) as previously described for the ability to prevent SNV infection of Vero E6 cells. Briefly, duplicate wells of 104 Vero E6 cells/well were plated onto Lab-Tek 16-well chamber slides (Fisher Scientific, Pittsburgh, PA) using minimal essential medium (MEM) (GIBCO, Grand Island, NY) with 2.5 % fetal bovine serum (FBS), and incubated at 37 °C for 24 h. Confluent VeroE6 cells were washed with PBS, then treated by adding media, peptide, ReoPro or small molecules in media in a volume of 50 µl. Following 1 h incubation at 37 °C, virus (500–1,000 focus-forming units/ml) was added in media in a volume of 50 µl and allowed to incubate an additional hour. We then removed the virus/treatment solution by aspiration, and after two washes in PBS, fresh media was added. After 24 to 36 h, we fixed the cells in 300 µl/well ice-cold methanol/acetone (50:50) and stored the slides at 4 °C until staining. The cells were washed twice with PBS before staining and then overlaid with 100 µl of a 1:10,000 dilution of polyclonal rabbit anti-SNV recombinant N antibody in PBS in a humidified chamber for 1 h at 37 °C. 50 µl of 1:500 FITC-conjugated anti-rabbit IgG in PBS was added, and then the cells were washed three times in PBS. Finally, the cells were counterstained with 200 µl of 1:105 Evans Blue (wt/vol) for 3 to 5 min. then excess dye was removed by rinsing in water. After mounting the slides, foci were counted by fluorescent microscopic examination. The percent inhibition was calculated as the number of infected cells compared to untreated controls, and normalized to inhibition by ReoPro (80 µg/ml). As the maximum effectiveness of ReoPro in inhibiting hantavirus entry in vitro approaches 80 %, we set this as a threshold for maximal expected efficacy for normalization rather than overinflating the data by normalization to 100%. In general, ReoPro normalized data was mathematical corrected by the appropriate multiplication factor to place ReoPro activity at 80%. We did not use ribavirin as a control since it does not block infection by preventing viral entry via the host receptor and, therefore, would not offer further insight with respect to identification of novel viral inhibitors of the αvβ3 surface receptor for hantavirus infections.
- 14.We used Unity software (Sybyl v. 8.0) to filter the second set of molecules through a pharmacophore model based on aligned compounds from the first set that inhibited infection by at least 20 %. Tight spatial constraints (0.3 Å) were used to avoid obtaining an excessive number of molecules out of the original ChemDiv database, while at the same time optimizing for chemical diversity. A 4-point derived model had a partial match directive to centers 3 & 4. It means that the new compounds selected could bear both pharmacophore points or only one of them.
- 15.Gavrilovskaya IN, Brown EJ, Ginsberg MH, Mackow ER. J. Virol. 1999;73:3951. doi: 10.1128/jvi.73.5.3951-3959.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Betzi S, Guerlesquin F, Morelli X. Comb. Chem. High Throughput Screening. 2009;12:968. doi: 10.2174/138620709789824736. [DOI] [PubMed] [Google Scholar]
- 17.Xiong JP, Stehle T, Goodman SL, Arnaout MA. J. Biol. Chem. 2004;279:40252. doi: 10.1074/jbc.C400362200. [DOI] [PubMed] [Google Scholar]
- 18.Sillerud LO, Larson RS. Curr. Protein Pept. Sci. 2005;6:151. doi: 10.2174/1389203053545462. [DOI] [PubMed] [Google Scholar]
- 19.Moitessier N, Henry C, Maigret B, Chapleur Y. J. Med. Chem. 2004;47:4178. doi: 10.1021/jm0311386. [DOI] [PubMed] [Google Scholar]
- 20.(a) Letourneau JJ, Liu J, Ohlmeyer MHJ, Riviello C, Rong Y, Li H, Appell KC, Bansal S, Jacob B, Wong A, Webb ML. Bioorg. Med. Chem. Lett. 2009;19:352. doi: 10.1016/j.bmcl.2008.11.074. [DOI] [PubMed] [Google Scholar]; (b) Elliot D, Henshaw E, MacFaul PA, Morley AD, Newham P, Oldham K, Page K, Rankine N, Sharpe P, Ting A, Wood CM. Bioorg. Med. Chem. Lett. 2009;19:4832. doi: 10.1016/j.bmcl.2009.06.041. [DOI] [PubMed] [Google Scholar]; (c) Henry C, Moitessier N, Chapleur Y. Mini Rev. Med. Chem. 2002;2:531. doi: 10.2174/1389557023405602. [DOI] [PubMed] [Google Scholar]; (d) Coleman PJ, Duong LT. Expert Opin. Ther. Patents. 2002;12:1009. [Google Scholar]; (e) Binder M, Trepel M. Expert Opin. Drug Discov. 2009;4:229. doi: 10.1517/17460440902751581. [DOI] [PubMed] [Google Scholar]
- 21.Artoni A, Li J, Mitchell B, Ruan J, Takagi J, Springer TA, French DL, Coller BS. Proc. Natl. Acad. Sci. USA. 2004;101:13114. doi: 10.1073/pnas.0404201101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.