

Abstract
Cancer is arguably the ultimate complex biological system. Solid tumors are micro-structured soft matter that evolves as a consequence of spatio-temporal events at the intracellular (e.g., signaling pathways, macromolecular trafficking), intercellular (e.g., cell-cell adhesion/communication), and tissue (e.g., cell-extracellular matrix interactions, mechanical forces) scales. To gain insight, tumor and developmental biologists have gathered a wealth of molecular, cellular and genetic data, including immunohistochemical measurements of cell type-specific division and death rates, lineage tracing, and gain-of-function/loss-of-function mutational analyses. These data are empirically extrapolated to a diagnosis/prognosis of tissue-scale behavior, e.g., for clinical decision. Integrative Physical Oncology (IPO) is the science that develops physically consistent mathematical approaches to address the significant challenge of bridging the nano (nm)-micro (μm) to macro (mm, cm) scales with respect to tumor development and progression. In the current literature, such approaches are referred to as multiscale modeling. In the present review, we attempt to assess recent modeling approaches on each separate scale and critically evaluate the current “hybrid-multiscale” models used to investigate tumor growth in the context of brain and breast cancers. Finally, we provide our perspective on the further development and the impact of Integrative Physical Oncology.
INTRODUCTION
A wealth of qualitative evidence links disease progression with tumor morphology, invasion and metastasis. Brain tumors are the 10th most common tumor in adults and the 7th leading cause of death in developed countries. Glioblastoma is the most deadly, with life expectancy of 15-18 months after diagnosis. Brain tumors are graded, not staged. The WHO classification system, despite being almost uniformly accepted, is an imperfect grading system, since tumors within the WHO Grade IV classification have drastically different prognosis, from a high 5-year survival for medulloblastoma to short-term mortality for glioblastoma (1). The new WHO approach incorporates and interrelates morphology, with a few cytogenetic, molecular genetic, and immunologic markers, in an attempt to construct a cellular classification (2). For instance, diagnostic morphology for Grade IV includes cellular atypia and nuclear pleomorphism, necrosis, vascular or endothelial proliferation, and pseudo-palisading. Diffuse infiltration of stroma is always present, with tumors cells as far as several centimeters away from the radiologically identified lesion. Similarly in breast cancer, the 2nd most prevalent cancer among women in the USA, pathologic criteria are broadly defined and widely varying response to therapy and outcomes for tumors with the same diagnosis are common (3). For example, ~80% of breast cancers are diagnosed as “no special type” and are lumped together as “infiltrating ductal carcinoma”. Yet these tumors are associated with differing morphological features, gene expression profiles (4, 5), responses to therapy, and patient survival.
In the past ten years, a large amount of new molecular data has emerged from genome-wide association studies of glioblastoma (6, 7), and breast cancer (8, 9). From a clinical point of view, broad histopathologic criteria are used to diagnose these tumors, made on fixed specimens, and prognose actual tumor behavior in the living patient that evolves over time. However, the variability of tumor progression and response to therapy demonstrate that more detail about individual tumors is critically needed. For instance, the abundance of microscale phenotype data (cell architecture, mitotic rates, etc) has not been integrated into a comprehensive picture of individual tumor behavior. The gap between the microscopic underlying processes of cancer cell behavior and the emerging macroscopic tumor growth and progression must be urgently addressed. This includes the need for a better understanding of the interplay between a tumor and its micro-/macroenvironment, which influences growth and treatment response and remains poorly understood (10, 11).
A main objective of Integrative Physical Oncology (IPO) is to employ mathematical modeling to develop biophysically sound mechanistic links among the multi-modal, multi-dimensional and multi-scalar phenomena involved in tumor progression. Mathematical modeling provides rigorous tools to link and quantify the multi-factorial connections between variables governing growth, prognosis and treatment. The resulting unified model of tumor behavior can provide a deeper fine-grained diagnosis, thus leading to more accurate and definitive predictions of treatment response and survival. To date, models have been developed at each of the relevant scales, and were partially successful in answering specific questions on tumor development. In the following, we briefly review some of these recent efforts at the sub-cellular, cellular and tissue scales applied to breast and brain cancer, and also recent attempts at hybrid-multiscale modeling. A critical analysis reveals crucial issues and outlines future directions and applications of IPO, including the novel approach of “mathematical pathology”.
SUB-CELLULAR SCALE
Tumors arise initially from a single cell. A normal cell (a.k.a. cell-of-origin) transforms step-by-step into a tumor cell due to various genetic and epigenetic changes (12-15). The ways in which this happens are manifold, as are the biological components and signaling pathways involved (12). Among the best studied key molecules/pathways directly or indirectly associated with cancer are Ras/ERK, PI3K/Akt/mTOR, VEGF, Rb, p53, and Wnt, each of which has been intensively targeted by drug development efforts.
Depending in particular on the cell-of-origin, its potency, the number and kinds of carcinogenic mutations, tumors can develop largely varying characteristics with respect to their cellular morphology, proliferative activity, and therapeutic response (15). Moreover, populations of cells within a single tumor are often heterogeneous, suggesting distinct dynamics at the single cell level (15). Their concerted action, together with influences from the microenvironment, gives rise to a specific tumor phenotype. Hence, identification and understanding of the tumor-specific biochemical mechanisms at the sub-cellular scale can greatly aid researchers in the development of tailored therapeutic strategies.
Key Issues and Modeling Efforts
Accurate modeling and simulation of single cell dynamics is a challenging task, due to the vast number of biochemical species involved, the often heterogeneous distribution of molecules inside the cell, and the discrete and stochastic nature of biochemical reactions. Moreover, the intracellular environment is geometrically complex and involves a plethora of correlated spatiotemporal processes that are, per se, multi-scaled. Typical time scales of interest range between microseconds (molecular diffusion) to weeks/years (cell lifespan), while spatial scales range from angstroms or nanometers (molecules) to tens of micrometers (cell size).
When choosing an appropriate modeling description, one has to decide (for each scale) if a given sub-cellular system can be best described as discrete or continuous, spatially homogeneous or heterogeneous, deterministic or stochastic, and how hybrid or multiscale modeling approaches can be constructed starting from the single cell level (16). Aside from equation-based models (ordinary, stochastic, partial and delay differential equations), accelerated simulation algorithms based on master equations, such as delay (17, 18) or spatial Monte-Carlo (19), or even highly resolved particle-tracking methods (20) have become popular for spatiotemporal modeling of intracellular processes. Recently, agent-based modeling and simulation methods (ABMs) have also shown great promise for understanding phenomena in biology and medicine (21).
Alternative approaches that have been used in the context of cancer modeling are rule-based models and Boolean networks. Rule-based modeling involves the representation of molecules as structured objects and their interactions as rules for transforming the attributes of these objects (22). Boolean network models have been suggested for problems where no quantitative information on reaction rates and initial conditions is available. Among other applications, Boolean networks have been used for modeling receptor crosstalk in endothelial cells, mapping environmental cues to cells (23).
During the last decade, many modeling efforts have addressed specific signaling pathways, including the aforementioned carcinogenic and related types (24-27). However, many models in the literature assume a closed system, often devoid of the crosstalk between pathways. Moreover, most models follow a mechanistic, continuous deterministic approach and assume spatial homogeneity in the distribution of participating molecules. Inevitably, this ignores any potential discrete or stochastic effects, while there has been increasing evidence that spatial heterogeneities significantly affect the dynamics (28, 29).
There are several issues that, to the best of our knowledge, have not yet been considered. For instance, a single mutated allele (as opposed to mutation of both alleles) in tumor-supressor genes may be sufficient for cancer progression (30). Logically, the mix of mutated and non-mutated copies of a gene might introduce additional complexity, as each allele would express different products. Also, there are many known epigenetic effects related to various types of cancer (31-34). For instance, low level DNA methylation in tumor cells (as compared to DNA methylation levels in normal cells) was one of the first epigenetic abnormalities observed in human cancer cells (14). Changes in the epigenome are also linked to a higher metastatic potential in many tumor types (14).
From Intracellular to Tissue Level
Tumor cells, like normal cells, communicate with their local environment. Each cell receives a multitude of signals from its surroundings, processes these signals with a complex network of highly intertwined pathways, and in turn signals to other cells. The latter usually happens via direct contact or over short distances by secreting signaling molecules. In tumor cells, these signals can affect cell-cell and cell-ECM (extracellular matrix) interactions, eventually causing loss of cellular adhesion, induction of angiogenesis, cell migration, tissue invasion, and metastasis. At this stage, the initially local single-cell defect has developed into a multi-cellular process at the tissue, organ, or even organism scale.
CELLULAR SCALE
We illustrate key issues and progress in cell-scale cancer modeling with a specific discussion of ductal carcinoma in situ (DCIS), a significant precursor to invasive breast carcinoma whose growth is confined to the duct lumen. DCIS is commonly detected as a subtle pattern of calcifications in mammograms; mammograms are also used to plan surgical resection (lumpectomy) of the tumor, but multiple surgeries are often required to fully eliminate DCIS. This highlights deficiencies in current surgical planning and, more generally, an insufficient understanding of the biophysical underpinnings of DCIS. See (35, 36) and the references therein. Please note that as cell-scale modeling is a broad topic, we cannot possibly cover all major modeling groups; see other recent reviews of interest (37-39).
Key Issues and Modeling Efforts
Stem/progenitor cell hierarchies
In DCIS, proliferation, apoptosis, and stem/progenitor cell differentiation cell dynamics are dysregulated, leading to cell overproliferation (40). In (41) a cellular automaton (CA) model—where the cells are uniformly-sized and restricted to a Cartesian mesh— was developed to study the impact of stem cell-progenitor cell hierarchies on DCIS development, finding that these hierarchies increase genetic heterogeneity and accelerate DCIS evolution.
Cell polarization, complex microarchitectures
Non-cancerous breast duct epithelial cells are polarized: integrins on the cells’ basal surfaces adhere to ligands on the basement membrane (BM), E-cadherins on the cells’ lateral sides adhere to E-cadherin on neighboring cells, and the cells’ apical sides are typically devoid of adhesion receptors. When cells lose adhesion to the BM, they commit anoikis, a specialized type of apoptosis that is triggered by loss of integrin signaling ((36) and references therein). In DCIS, cell polarization and anoikis are dysregulated, and complex microstructures (e.g., micropapillary and cribriform DCIS) can form in the viable rim. In (42-45), an immersed boundary model of DCIS was developed, where each cell’s morphology is evolved under the balance of adhesive and fluid mechanical forces. The authors explicitly modeled the cells’ polarized adhesion, with functional ties to proliferation and apoptosis via simplified signaling models. The model produced complex micropapillary-like structures (Fig. 1). The work in (46) developed a lattice-free, agent-based model of DCIS, where each cell is a sphere that moves under the balance of adhesive and repulsive forces. Cells were assumed to adhere to at most two neighbors (as a simplified phenomenological model of polarization). The model reproduced micropapillary-like and cribriform-like microstructures (Fig. 1). The authors hypothesized that the cribriform subtype arises from micropapillary DCIS when overproliferation causes micropapillae to connect around “microlumens.” In these modeling efforts, mechanistic cell-scale models that recapitulate complex known tumor microstructures have given key insight on their biomechanical underpinnings.
Fig. 1. Mathematical modeling of complex DCIS microstructures.
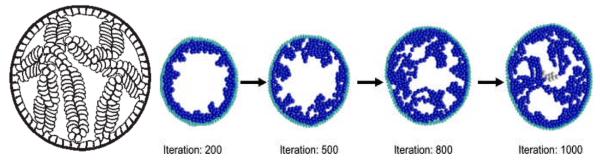
(Left) An immersed boundary model produced micropapillary-like DCIS structures when cell polarization was assumed. Reproduced by permission from (45). (Right) An agent-based model predicted that polarized DCIS cells form micropapillary structures (iterations 200 and 500) that merge into cribriform -like structures (iterations 800 and onward). Reproduced by permission from (46).
These results could be improved by more accurately modeling the cells’ individual morphologies, which is possible using a cellular Potts model (CPM). In a CPM, each cell occupies a finite, simply connected set of grid points in a Cartesian mesh, which enables the model to capture and characterize dynamic changes in a cell’s size, shape, and location. This multicellular system is updated by a Monte Carlo technique to reduce the total system energy, which includes separate terms to model motility, growth, and cell-cell and cell-BM adhesion. Key signaling events (e.g., anoikis) can readily be implemented in each cell. See (47-49) and the references therein, and the Hybrid-Multiscale models subsection below. The cellular Potts approach has been used to model the 3-D avascular tumor growth response to microenvironmental survival signals (47), and also to elucidate the impact of cell-ECM interactions on glioma invasion in non-uniform ECM structures (48).
Hypoxia and Invasion
Growth substrates (particularly oxygen and glucose) can only reach a (necessarily avascular) DCIS tumor by diffusion; as the tumor grows, oxygen gradients form, leading to hypoxia and eventually (comedo) necrosis. Over long time scales, microcalcifications can form in the necrotic debris ((36) and references therein). In (50-52), a cellular automaton model was employed to investigate the impact of hypoxia, glycolysis (a form of anaerobic metabolism), and acidosis (a buildup of acidic glycolysis byproducts) on DCIS invasion. The authors found that sustained hypoxia can select for aggressive DCIS sub-clones that colonize the viable rim and eventually invade the stroma, thus providing new hypotheses on DCIS invasion.
Necrotic core/calcification biomechanics
The authors in (35, 39, 36, 53) recently developed an agent-based model of DCIS, which included detailed necrotic cell volume changes and the first model of calcification. After a careful calibration to the biological literature and patient data, the model predicted that the mechanical separation between the viable rim and the necrotic core arises due to the fast time scales of necrotic cell swelling and lysis. The authors determined that necrotic cell lysis acts as a major biomechanical stress relief; as a result, much of the proliferative cell flux is directed towards the duct center, rather than along the duct. An additional consequence is the formation of an “age structured” necrotic core, with the oldest (often calcified) material in center, and the newest material on the perinecrotic boundary. These results are consistent with patient histopathology (Fig. 2). The model also predicted that DCIS tumors grow linearly at 7-10 mm/year, and that the tumor’s mammographic (calcification) size linearly correlates with the tumor’s pathologic size. Both these results are in excellent quantitative agreement with the clinical literature. These results show that rigorously calibrated mechanistic cell-scale models can explain macro-scale observations in DCIS histopathology and radiology, and may eventually assist surgical planning by augmenting mammography with model-predicted surgical margins.
Fig. 2. Patient-calibrated DCIS simulation.

(Top): After calibration to patient immunohistochemistry and morphometric measurements, an agent model correctly reproduced the solid-type DCIS microstructure: an 80 μm viable rim with most frequent proliferation (green cells) on the outermost edge and apoptosis (red cells) throughout, a mechanical separation at the perinecrotic boundary, and an “age-structured” necrotic core with increasing pyknosis (nuclear degradation) and calcification (progression indicated by the shade of red) towards the duct center. The bright red central region is a radiologically detectable casting-type microcalcification. (Bottom): These features are seen in patient hematoxylin and eosin stained histopathology, including the mechanical gap (black arrows; increased by tissue dehydration), increasing pyknosis towards the duct center (red arrows show more intact nuclei; green arrows show largely degraded nuclei), and central calcium phosphate microcalcifications (white arrows). Adapted with permission from (36).
Integration with Multiscale Modeling Frameworks
As the intermediate scale between the molecular (intracellular) and tissue scales, cell-scale models will play an essential role in emerging multiscale modeling frameworks (37, 38). The intracellular scale can be directly incorporated into cellular-scale models by including a molecular-scale model in each cell (37). Cell-scale effects are currently incorporated into tissue-scale models through hybrid techniques (see below) (37, 38, 35). Equally importantly, cell-scale models can be directly compared with clinical measurements, making them ideal for calibrating multiscale frameworks (39, 36). Using the rigorous agent model calibration developed in (36) along with a novel upscaling argument, the authors of (35) calibrated a steady-state continuum model of DCIS volume to immunohistochemical and morphometric data from several patients. The model accurately predicted the overall tumor volume in 14 of the 17 cases, thus demonstrating the potential for cell-scale models to rigorously calibrate multiscale frameworks to molecular and cellular data, by dynamic upscaling procedures.
TISSUE SCALE
To a large extent, the tissue scale is the clinically relevant scale of the disease. It is the scale at which first symptoms are noticed, e.g., by finding an abnormal mass during physical examination (organ level) or by discovering elevated amount of chemicals such as tumor markers or abnormal immune system proteins in blood test (systemic level). It is also the scale at which secondary imaging investigations are performed, such as MRI or mammography for breast tumor. MRI is particularly helpful in determining macroscopic characteristics of tumors such as the approximate size, and morphology, which are necessary information for tumor grade classification and more accurate treatment planning. The tissue scale is the relevant one for surgical and chemo-/radio-therapy planning.
Key Issues and Modeling Efforts
At the tissue scale, a tumor is not only the macroscopic manifestation of the underlying processes at smaller scales, but also the result of its interactions with the surrounding healthy tissue. While the molecular and cellular mechanisms result in carcinogenesis, tumor growth also depends in a complex manner on biochemical and biophysical transport where continuum modeling (e.g., mechanics) is required. On an individual patient basis, cancer modeling at the tissue scale may translate into an improved understanding of the time scale of tumor growth, and consequently, provide answers to practical clinical questions such as: where and what to resect; where to target and how to optimize radiotherapy; how drugs, antibodies or small molecules are transported through tissues into neoplastic cells and how this transport affects therapeutic response.
Modeling and computational approaches for macroscopic tumor growth require the description of the dynamics of billions of tumor cells. Such a description is possible only by averaging single cell behaviors into macroscopic quantities that characterize observables of tissue growth, such as tumor size and growth rate, cell density, mechanical pressure and stress. At this level of description, approaches can be split into cell population dynamics and continuum models. Both approaches are based on phenomenological functional relationships that describe particular macroscopic features in terms of model parameters, most of which are not direct measurable quantities at the cellular scale but account for average cell behavior at the tissue scale. Typical time scales range from days (e.g., for in vivo growth of multi-cellular spheroids) to months (mean life expectancy is 12-15 months for glioma) and years (breast cancer is often a 20-year long disease process), while spatial scales are of the order of mm and cm or more when considering the whole human body.
Cell population dynamics models are used when no clear spatial structure emerges or it is not taken into account. This may be the case when the system of interest looks very well mixed with respect to its various components (e.g., different cell populations) or in the absence of pertinent morphological and structural tissue data. Such systems are considered spatially homogeneous and are modeled by selecting the most appropriate description among the large variety of differential equations, i.e. ordinary, delay, stochastic, age-structured. Examples of work are the modeling of carcinogenesis (54) and the interactions between a malignant tumor and the immune system (55). The assumption of spatial homogeneity can also be used to investigate the fate of anticancer drug delivered within the whole organism, when the description of the complex geometry would be too challenging and would not increase the understanding of drug fate, e.g., for pharmacological control based on pharmacokinetics and pharmacodynamics (56).
Continuum models are based on conservation laws of physics and use deterministic partial differential equations as a spatiotemporal modeling framework to account for spatial heterogeneities in both tumors and their microenvironment. One particular interest of these models is their ability to include mechanical effects on tumor growth, as for breast (57) and brain (58) tumors described as an elastic soft tissue. Tumors can also be represented by more complex material with dissipative regimes generated by cell reorganization (59). All these models have in common the need for biophysical constitutive laws to describe tumor mechanical properties (60).
A simplified approach consists in describing tumor growth by a mass balance equation where mass changes occur in time and space due to tumor cell proliferation and migration. This simplified description by partial differential reaction-diffusion equations (RDE) has been used for gliomas, in combination with pretreatment MRIs to quantify patient-specific cell proliferation and invasion rates that are prognostically significant (61), and to simulate surgical resection, radiotherapy and chemotherapy (62). More sophisticated models including the effect of vascularization and angiogenesis have also been developed by using RDEs to investigate therapeutic strategies (63) or multiphase-mixture modeling to predict drug response in breast cancer (64). In the multiphase-mixture representation (65), tumor and host regions are described as a mixture of multiple solid phases (tumor and stroma cells, ECM, substrates, etc.) and the interstitial fluid. This approach is flexible enough to account for interaction forces generated by the extracellular matrix (66) and computationally efficient to predict growth and morphological changes of tumor spheroids that result from heterogeneities of the microenvironment (67). See Fig. 3.
Fig. 3. 3D computer model predicts gross morphologic features of a growing glioblastoma.

Viable (light gray) and necrotic (dark gray) tissue regions and vasculature (mature blood-conducting vessels in red; new non-conducting vessels in blue) are shown. The simulations reveal that the morphology is affected by neovascularization, vasculature maturation, and vessel cooption. Adapted with permission from (67).
Integration with Multiscale Modeling Frameworks
One major criticism to the tissue level models is that direct calibration of parameters at this scale is not possible in general and relies on fitting, which makes the models mostly unreliable outside the range of parameters over which they have been calibrated. Other important issues to consider are as follows: Since individual cell activity can ultimately be responsible for generation of a macroscopic structure, such as a mammary ductal tree or a breast tumor (68), how can a tissue model be informed from single cell activity? Vice-versa, since mechanical and other phenomena in a tissue may lead, at the individual cell level, to phenotypic adaptation generated by physical forces such as hydrostatic pressure and shear stress (69), how can we account for tissue scale information in a cell-scale model? These issues require the development of multiscale mathematical tools capable of bridging the gap between scales and thus arguably the current gap in oncology.
HYBRID-MULTISCALE MODELS
Due to the intrinsic multiscale nature of cancer, a deeper understanding requires the development of models that integrate and combine the phenomena spanning the multiple scales involved. Hybrid continuum-discrete implementations, which typically seek to combine the best of the tissue (continuum) and cellular (discrete) scale approaches while minimizing their limitations, are a very promising modeling approach. Many published methods claim to be multiscale because they are based on a hybrid description of the tumor components, typically by using a discrete representation of the various cell populations and continuum fields to describe cell substrates (e.g., nutrients, oxygen and diffusible factors). These models incorporate processes at multiple scales but can hardly capture the non-trivial interactions among scales that are responsible for the growth of malignant tissue. In a truly multiscale model, one employs: 1) different representations to describe the same quantity of interest at different scales, e.g., using cell density at the tissue scale and a number of discrete cell agents at the cellular scale; 2) direct calibration/validation of the cell-scale parameters and equations from individual measurements; and finally 3) rigorous upscaling techniques to “close” the continuum equations at the tissue scale, thus providing an accurate description of the processes thereby. Only a few recent studies provide an implementation of discrete and continuum descriptions to simultaneously account for single cell and tissue behaviors, and the interactions between the two scales. This defines the class of hybrid-multiscale models.
In a recent study (70) the authors treat tumor’s necrotic and quiescent areas as a continuous viscoelastic medium coupled to an ABM of tumor cells located at the tumor periphery where proliferative activity is artificially constrained to occur. The ABM allows for the detailed description of single cells by including their intercellular dynamics, and is coupled to the continuum description via a balance of forces between single cells and the tumor (quiescent) tissue. Another group developed a hybrid continuum-discrete implementation that combines an ABM for invasive tumor cells, which is directly coupled to the continuum model used for the tumor bulk by balancing transfers of mass and momentum between the two representations (71-73). There is also indirect coupling between the continuum and discrete tumor representations through the (reaction-diffusion) equations for cell substrates (e.g., oxygen, glucose) and the ECM since both types of cell representation take up nutrients and growth factors, and remodel the extracellular matrix. See Fig. 4. Rules are posed to describe the conditions for switching between discrete and continuum representations without artificially imposing where such transitions occur. Briefly, discrete cells are released in hypoxic regions, to model the epithelial-to-mesenchymal transition, and discrete cells are converted back to the continuum description when their local population exceeds a threshold.
Fig. 4. Individual (discrete) palisading glial cells invasion into vascularized tissue.
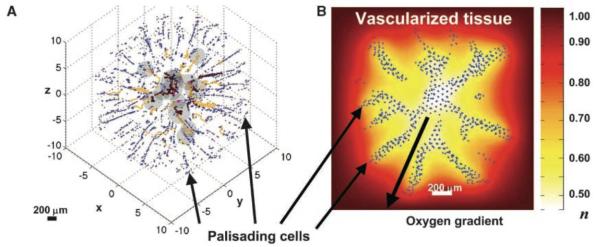
(A) Computer simulation from a hybrid-multiscale model showing palisading cells escaping from the perinecrotic region (dark gray) by up-regulating motility and down-regulating adhesion and proliferation. This phenotypic change is driven by hypoxia as the selective evolutionary force (see discussion section). Cell migration occurs via chemotaxis and haptotaxis in response to gradients of oxygen and ECM concentration, respectively. Brown: conducting vessels; yellow: non-conducting. (B) Background shows distribution of oxygen concentration (n=1 in vascularized tissue and n<1 in the tumor white/yellow perinecrotic region). Adapted with permission from (71).
Although both examples above utilize co-existing continuum and discrete tumor cell representations, the overall approach is still phenomenological since the functional relationships between the parameters and variables used at the continuum level involve quantities that are not directly obtained from the cell scale. Further, the rules for determining whether to convert discrete cells to the continuum representation and vice-versa are empiric.
The multiple-scale processes involved in tumor angiogenesis also present mathematical and computational challenges, even though vasculature formation and remodelling involves a smaller ensemble of (endothelial) cells compared to a whole tri-dimensional tumor. As an example of angiogenesis modeling at the interface between molecular and multi-cellular phenomena, a novel approach was introduced in (74, 75) to reduces the number of phenomenological rules needed. At the cellular scale, a cellular Potts model was implemented to describe cellular growth, proliferation, migration, apoptosis, and restructuring of the extracellular matrix. This cellular model was integrated with a partial differential equation model describing the spatio-temporal evolution of VEGF and with a Boolean network model of intracellular signaling that considered VEGF-specific receptors, integrin receptors and signaling initiated by cell-cell contact.
DISCUSSION: INTEGRATIVE PHYSICAL ONCOLOGY
From our arguments, the reader may realize that the development of predictive tumor growth models involves major mathematical challenges. While theoreticians and experimentalists have developed numerous models to investigate tumor development and growth and the underlying processes at various scales, mathematical multiscale models have only recently attempted to bridge the gap between the various spatiotemporal scales in the quest for predictive models of cancer. The heart of the matter lies in the difficulty of deriving a mathematical framework that provides the tools for a biophysically sound approach that is also physically consistent across the scales, a requirement of utmost importance for Integrative Physical Oncology. Indeed, such a framework would allow, through upscaling techniques, the use of directly measurable quantities at the cellular scale to inform the model parameters at the tissue scale, in principle making multiscale models predictive because the data used for calibration exist at a different scale (and are of different nature) from those used for validation.
The field of Integrative Physical Oncology must address complex mathematical issues to appropriately model tumor development and better understand the emergence of macroscopic tumor characteristics as a result of molecular and cellular phenomena. However, IPO may have a broader impact by also producing novel biological hypotheses on previously overlooked phenomena. Biologists tend to consider the single cell as the Fundamental Tissue Unit (cFTU, where “c” stands for cell). This reductionistic approach has proven very helpful in improving the current understanding of sub-cellular and cellular processes that lead to normal and pathologic tissue development. However, based on the principles of evolution, it is well accepted that progression in cancer is not only a consequence of intrinsic instability of the cell genome but also the result of extrinsic influences acting as selective forces upon tumor cells. Therefore, we argue that there must exist another fundamental tissue unit, herein named mFTU (where “m” stands for microenvironmental). The mFTU spans a length scale across which physical transport of substrates (e.g., oxygen diffusion) occurs and generates selective stress that ultimately leads to the prevalence of one phenotype over another. We believe that a typical order of magnitude of the mFTU should be the length scale at which transport phenomena lead to the establishment of tangible concentration gradients of substrates (oxygen) and we suggest considering at least 10 cell-lengths as a characteristic mFTU length scale (i.e., an associated volume of approximately 103 cells or 1003 um3). This length scale will of course change depending on the problem.
The need for an extended FTU to understand the role of the environment on normal and malignant tissue development is already recognized by the biological community, e.g., (76, 77). However, the complexity of coupled multi-factorial processes is prohibitive for a systematic experimental exploration of the phenomena at the mFTU scale, a statement exacerbated when one considers in vivo investigations, for which costs, in terms of time and financial support, as well as technical challenges, are strongly limiting factors. Via multiscale mathematical methods, IPO offers a framework that allows for a systematic approach to investigate the mFTU that can also be used to sharpen further experimental focus. Indeed, IPO proposes a mathematical representation of the system of interest (i.e., a very small tissue sample) in which through parameter-sensitivity perturbation studies: 1) the influence of each component can be tested independently and experimentally validated; and 2) the behavior of the entire system can be theoretically predicted to provide specific directions for future experimental investigations. Then, predictions at the tissue level would be the result of the coupled non-linear dynamics of a mosaic of interacting mFTUs, the behavior of each of them being understood as a single piece of the puzzle and assembled via biophysical laws and boundary conditions.
One example of the necessity of the mFTU as the minimal scale of importance is cancer invasion, e.g., tumor fingering into surrounding host tissue, which occurs due to the interplay of sub-cellular processes leading, among other malfunctions, to dysregulated cell proliferation and adhesion. As shown in (67, 78), these cell-scale (cFTU) effects are mediated and driven by physical transport phenomena at the mFTU scale, e.g., diffusion gradients of cell substrates pointing away from the tumor bulk, and modulate the features of the invasion process; but cFTU effects alone would not lead to organized and effective (clinically relevant) invasion of the surrounding stroma, but rather to trivial random walks of cells in the neighborhood of the tumor/stroma interface with no average direction (67). A second example is tumor resistance to chemotherapy. A traditional approach that focuses only on drug treatment failure at the molecular and cellular (cFTU) levels typically and significantly underestimates resistance in vivo because it cannot account for processes at larger scales such as the phenomenon of “diffusion barriers” associated with limited drug and cell substrate penetration into the bulk of a tumor (79). By extending the scale of investigation to the mFTU and considering drug (and oxygen) transport by diffusion through the tissue, it has been shown that chemo-resistance is significantly driven by the environment and often to an extent larger than the intrinsic resistance of single cells (64), thus leading to in-vivo/in-vitro IC50 ratios significantly larger than one (80). Our final example is the investigation of stem cell niches and their role in breast cancer development. From a purely biological point of view, the functional definition of a niche is a set of microenvironmental components (e.g., ECM, signaling molecules, blood vessels) and biological processes (e.g., juxtacrine and paracrine cell signaling) that regulate stem cell behavior. From a physical point of view, we suggest reconsidering the definition of niche within the context of mFTU, so that regulatory effects of the physical transport processes of oxygen and other species on stem cell behavior may be properly accounted for.
A clinical implication of the mFTU concept in the context of IPO lies in the definition and use of histopathologic criteria to diagnose and classify (brain and breast) tumors. These criteria are based on the analysis of small parts of tumor specimens (biopsies) and aim to help pathologists formulate prognosis of tumor progression, in other words, to extrapolate the spatiotemporal behavior of the whole tumor from a small tissue sample. From a physical point of view, as we emphasize above, the macroscopic tumor features result from the complex dynamics at the mFTU level, which suggests a minimal size necessary for biopsies to capture the physical transport phenomena. Multiscale modeling would allow for the construction of a “functional mapping” fro m a range of histopathological data of phenotypic and stromal properties into predicted macroscopic tumor features of translational relevance such as growth rate, fingering growth rate, drug response, etc., thus producing a new set of mathematical pathologic criteria. A simplified example of such an approach is the recent work by (35), in which the underlying physics of transport across the mFTU accurately connects immunohistopathology measurements from biopsies to patient-specific translational quantities such as the surgical volume of breast tumors (Fig. 5). In doing so, the authors demonstrated that the size of breast tumors does not correlate with grade, but rather with a specific mathematical functional of both mitotic and apoptotic indices in the breast ducts. Hence, we claim that IPO may have a major impact via the identification of robust predictors of tumor progression based on molecular-scale data (71), which might not require multiple time-point measurements from patients, thus helping bridge the gap discussed at the beginning of this paper through feasible incorporation within the current clinical practice. This is the foundation of mathematical pathology.
Figure 5. Validation of model predictions against pathology-determined DCIS tumor sizes.

Surgical tumor size vs. parameter A that is related to the ratio of tumor apoptotic and mitotic indices in the breast ducts. The dotted curve represents the theoretical predictions by a continuum model. Symbols are DCIS tumor size measurements from individual patient histopathology and are sub-classified by their grade. The shaded region indicates the standard deviation in the measurement of A in individual duct. Reproduced from (35) with permission.
Acknowledgement
We acknowledge the NIH for support through the Physical Sciences in Oncology Center grants 1U54CA143837 and 1U54CA143907 (VC), the Integrative Cancer Biology Program grant 1U54CA149196 for the Center for Systematic Modeling of Cancer Development (VC, ML, AC), and finally NINDS NS062184, NS046810, NIGMS GM47368 and P50GM085273 (ELB). We also acknowledge the National Science Foundation (NSF) for support under grant DMS-0818104 (VC).
Footnotes
STATEMENT OF INDIVIDUAL CONTRIBUTIONS: All authors contributed revisions on the entire paper.
HH and AC contributed to the overall design, formulation and critical discussion of the concepts in the manuscript and wrote the manuscript except for specific sections as described below.
AB wrote the paragraphs on Boolean network and rule-based models, cellular Potts models and angiogenesis.
TML and AL wrote the section ‘Sub-cellular scale’.
ML and ELB contributed to the critical discussion and the writing of the biological and translational concepts in Introduction and Discussion.
PM wrote the section ‘Cellular Scale’.
VC originated the overall design and contributed formulation and critical discussion of the concepts in the manuscript.
Contributor Information
Haralampos Hatzikirou, Dept. of Pathology, University of New Mexico, Albuquerque, USA.
Arnaud Chauviere, Dept. of Pathology, University of New Mexico, Albuquerque, USA achauviere@salud.unm.edu.
Amy L. Bauer, Los Alamos National Laboratory, Los Alamos, USA
André Leier, STMC, University of New Mexico, Albuquerque, USA.
Michael T. Lewis, Lester and Sue Smith Breast Center, Depts. of Molecular and Cellular Biology and Radiology, Baylor College of Medicine, Houston, USA
Paul Macklin, Division of Mathematics, University of Dundee, United Kingdom.
Tatiana T. Marquez-Lago, STMC, University of New Mexico, Albuquerque, USA
Elaine L. Bearer, Dept. of Pathology, University of New Mexico, Albuquerque, USA
Vittorio Cristini, Depts. of Pathology and Chemical Engineering, University of New Mexico, Albuquerque, USA vcristini@salud.unm.edu.
References
- 1.Kleihues P, Burger PC, Scheithauer BW. The new WHO classification of brain tumours. Brain Pathol. 1993;3(3):255–68. doi: 10.1111/j.1750-3639.1993.tb00752.x. [DOI] [PubMed] [Google Scholar]
- 2.Cunliffe CH, Fischer I, Parag Y, Fowkes ME. State-of-the-art pathology: new WHO classification, implications, and new developments. Neuroimaging Clin N Am. 2010;20(3):259–71. doi: 10.1016/j.nic.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 3.Diab SG, Clark GM, Osborne CK, Libby A, Allred DC, Elledge RM. Tumor Characteristics and Clinical Outcome of Tubular and Mucinous Breast Carcinomas. Journal of Clinical Oncology. 1999;17(5):1442–8. doi: 10.1200/JCO.1999.17.5.1442. [DOI] [PubMed] [Google Scholar]
- 4.O’Brien KM, Cole SR, Tse C-K, Perou CM, Carey LA, Foulkes WD, et al. Intrinsic breast tumor subtypes, race, and long-term survival in the Carolina breast cancer study. Clinical Cancer Research. 2010;16(24):6100–10. doi: 10.1158/1078-0432.CCR-10-1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prat A, Parker J, Karginova O, Fan C, Livasy C, Herschkowitz J, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Research. 2010;12(5):R68. doi: 10.1186/bcr2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rickman DS, Bobek MP, Misek DE, Kuick R, Blaivas M, Kurnit DM, et al. Distinctive molecular profiles of high-grade and low-grade gliomas based on oligonucleotide microarray analysis. Cancer Res. 2001;61(18):6885–91. [PubMed] [Google Scholar]
- 7.Vitucci M, Hayes DN, Miller CR. Gene expression profiling of gliomas: merging genomic and histopathological classification for personalised therapy. Br J Cancer. 2010;104(4):545–53. doi: 10.1038/sj.bjc.6606031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang HY, Nuyten DS, Sneddon JB, Hastie T, Tibshirani R, Sorlie T, et al. Robustness, scalability, and integration of a wound-response gene expression signature in predicting breast cancer survival. Proc Natl Acad Sci U S A. 2005;102(10):3738–43. doi: 10.1073/pnas.0409462102. PMCID: 548329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hedenfalk I, Duggan D, Chen Y, Radmacher M, Bittner M, Simon R, et al. Gene-expression profiles in hereditary breast cancer. N Engl J Med. 2001;344(8):539–48. doi: 10.1056/NEJM200102223440801. [DOI] [PubMed] [Google Scholar]
- 10.Jordan BF, Runquist M, Raghunand N, Baker A, Williams R, Kirkpatrick L, et al. Dynamic contrast-enhanced and diffusion MRI show rapid and dramatic changes in tumor microenvironment in response to inhibition of HIF-1alpha using PX-478. Neoplasia. 2005;7(5):475–85. doi: 10.1593/neo.04628. PMCID: 1501160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liotta LA, Kohn EC. The microenvironment of the tumour-host interface. Nature. 2001;411:375–9. doi: 10.1038/35077241. [DOI] [PubMed] [Google Scholar]
- 12.Bronchud MH, Foote M, Giaccone G, Olopade O, Workman P, editors. Principles of molecular oncology. 3rd ed Humana Press Inc.; 2008. [Google Scholar]
- 13.Croce CM. Oncogenes and Cancer. New England Journal of Medicine. 2008;358(5):502–11. doi: 10.1056/NEJMra072367. [DOI] [PubMed] [Google Scholar]
- 14.Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005;6(8):597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- 15.Visvader JE. Cells of origin in cancer. Nature. 2011;469(7330):314–22. doi: 10.1038/nature09781. [DOI] [PubMed] [Google Scholar]
- 16.Burrage K, Burrage P, Leier A, Marquez-Lago TT, Nicolau DV. Stochastic simulation for spatial modelling of dynamic processes in a living cell. In: Koeppl H, Setti G, di Bernardo M, Densmore D, editors. Design and Analysis of Bio-molecular Circuits. Springer; 2011. [Google Scholar]
- 17.Leier A, Marquez-Lago TT, Burrage K. Generalized binomial tau-leap method for biochemical kinetics incorporating both delay and intrinsic noise. J Chem Phys. 2008;128(20):205107. doi: 10.1063/1.2919124. [DOI] [PubMed] [Google Scholar]
- 18.Marquez-Lago TT, Leier A, Burrage K. Probability distributed time delays: integrating spatial effects into temporal models. BMC Syst Biol. 2010;4:19. doi: 10.1186/1752-0509-4-19. PMCID: 2847994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marquez-Lago TT, Burrage K. Binomial tau-leap spatial stochastic simulation algorithm for applications in chemical kinetics. J Chem Phys. 2007;127(10):104101. doi: 10.1063/1.2771548. [DOI] [PubMed] [Google Scholar]
- 20.Andrews SS, Addy NJ, Brent R, Arkin AP. Detailed simulations of cell biology with Smoldyn 2.1. PLoS Comput Biol. 2010;6(3):e1000705. doi: 10.1371/journal.pcbi.1000705. PMCID: 2837389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pogson M, Smallwood R, Qwarnstrom E, Holcombe M. Formal agent-based modelling of intracellular chemical interactions. Biosystems. 2006;85(1):37–45. doi: 10.1016/j.biosystems.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 22.Blinov ML, Faeder JR, Goldstein B, Hlavacek WS. BioNetGen: software for rule-based modeling of signal transduction based on the interactions of molecular domains. Bioinformatics. 2004;20(17):3289–91. doi: 10.1093/bioinformatics/bth378. [DOI] [PubMed] [Google Scholar]
- 23.Bauer AL, Jackson TL, Jiang Y, Rohlf T. Receptor cross-talk in angiogenesis: Mapping environmental cues to cell phenotype using a stochastic, Boolean signaling network model. Journal of Theoretical Biology. 2010;264(3):838–46. doi: 10.1016/j.jtbi.2010.03.025. [DOI] [PubMed] [Google Scholar]
- 24.Birtwistle MR, Hatakeyama M, Yumoto N, Ogunnaike BA, Hoek JB, Kholodenko BN. Ligand-dependent responses of the ErbB signaling network: experimental and modeling analyses. Mol Syst Biol. 2007;3:144. doi: 10.1038/msb4100188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Borisov N, Aksamitiene E, Kiyatkin A, Legewie S, Berkhout J, Maiwald T, et al. Systems-level interactions between insulin-EGF networks amplify mitogenic signaling. Mol Syst Biol. 2009;5:256. doi: 10.1038/msb.2009.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hsieh M-Y, Yang S, Raymond-Stinz MA, Steinberg S, Vlachos DG, Shu W, et al. Stochastic simulations of ErbB homo and heterodimerisation: potential impacts of receptor conformational state and spatial segregation. IET Syst Biol. 2008;2(5):256–72. doi: 10.1049/iet-syb:20070073. [DOI] [PubMed] [Google Scholar]
- 27.Walker DC, Georgopoulos NT, Southgate J. From pathway to population-a multiscale model of juxtacrine EGFR-MAPK signalling. BMC Syst Biol. 2008;2:102. doi: 10.1186/1752-0509-2-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kholodenko BN, Kolch W. Giving space to cell signaling. Cell. 2008;133(4):566–7. doi: 10.1016/j.cell.2008.04.033. [DOI] [PubMed] [Google Scholar]
- 29.Tian T, Harding A, Inder K, Plowman S, Parton RG, Hancock JF. Plasma membrane nanoswitches generate high-fidelity Ras signal transduction. Nat Cell Biol. 2007;9(8):905–14. doi: 10.1038/ncb1615. [DOI] [PubMed] [Google Scholar]
- 30.Morgan DO. The cell cycle: principles of control. New Science Press, Ltd.; 2007. [Google Scholar]
- 31.Ballestar E, Esteller M. Epigenetic gene regulation in cancer. Adv Genet. 2008;61:247–67. doi: 10.1016/S0065-2660(07)00009-0. [DOI] [PubMed] [Google Scholar]
- 32.Bell DW. Our changing view of the genomic landscape of cancer. J Pathol. 2010;220(2):231–43. doi: 10.1002/path.2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berdasco M, Esteller M. Aberrant epigenetic landscape in cancer: how cellular identity goes awry. Dev Cell. 2010;19(5):698–711. doi: 10.1016/j.devcel.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 34.Kristensen LS, Nielsen HM, Hansen LL. Epigenetics and cancer treatment. Eur J Pharmacol. 2009;625(1-3):131–42. doi: 10.1016/j.ejphar.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 35.Macklin P, Edgerton ME, Cristini V, Cristini V, Lowengrub JS. Multiscale Modeling of Cancer. Cambridge University Press; 2010. Agent-based cell modeling: application to breast cancer; pp. 206–34. [Google Scholar]
- 36.Macklin P, Edgerton ME, Thompson AM, Cristini V. Patient-calibrated agent-based modelling of ductal carcinoma in situ (DCIS): From microscopic measurements to macroscopic predictions of clinical progression. J Theor Biol. 2011 doi: 10.1016/j.jtbi.2012.02.002. (accepted pending revision) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deisboeck TS, Wang Z, Macklin P, Cristini V. Multiscale Cancer Modeling. Annu Rev Biomed Eng. 2011;13 doi: 10.1146/annurev-bioeng-071910-124729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lowengrub J, Frieboes HB, Jin F, Chuang Y-L, Li X, Macklin P, et al. Nonlinear modeling of cancer: Bridging the gap between cells and tumors. Nonlinearity. 2010;23(1):R1–R91. doi: 10.1088/0951-7715/23/1/r01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Macklin P, Edgerton ME, Lowengrub JS, Cristini V. Discrete Cell Modeling. In: Cristini V, Lowengrub JS, editors. Multiscale Modeling of Cancer: An Integrated Experimental and Mathematical Modeling Approach. Cambridge University Press; Cambridge, UK: 2010. pp. 88–122. [Google Scholar]
- 40.Allred DC, Wu Y, Mao S, Nagtegaal ID, Lee S, Perou CM, et al. Ductal Carcinoma In situ and the Emergence of Diversity during Breast Cancer Evolution. Clinical Cancer Research. 2008;14(2):370–8. doi: 10.1158/1078-0432.CCR-07-1127. [DOI] [PubMed] [Google Scholar]
- 41.Bankhead A, III, Magnuson NS, Heckendorn RB. Cellular automaton simulation examining progenitor hierarchy structure effects on mammary ductal carcinoma in situ. Journal of Theoretical Biology. 2007;246(3):491–8. doi: 10.1016/j.jtbi.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 42.Rejniak KA. An immersed boundary framework for modelling the growth of individual cells: An application to the early tumour development. Journal of Theoretical Biology. 2007;247(1):186–204. doi: 10.1016/j.jtbi.2007.02.019. [DOI] [PubMed] [Google Scholar]
- 43.Rejniak KA, Anderson ARA. A computational study of the development of epithelial acini: I. Sufficient conditions for the formation of a hollow structure. Bull Math Biol. 2008;70(3):677–712. doi: 10.1007/s11538-007-9274-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rejniak KA, Anderson ARA. A computational study of the development of epithelial acini: II. Necessary conditions for structure and lumen stability. Bull Math Biol. 2008;70(5):1450–79. doi: 10.1007/s11538-008-9308-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rejniak KA, Dillon RH. A Single Cell-Based Model of the Ductal Tumour Microarchitecture. Comput Math Meth Med. 2007;8(1):51–69. [Google Scholar]
- 46.Norton KA, Wininger M, Bhanot G, Ganesan S, Barnard N, Shinbrot T. A 2D mechanistic model of breast ductal carcinoma in situ (DCIS) morphology and progression. J Theor Biol. 2010;263(4):393–406. doi: 10.1016/j.jtbi.2009.11.024. PMCID: 2839055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiang Y, Pjesivac-Grbovic J, Cantrell C, Freyer JP. A Multiscale Model for Avascular Tumor Growth. Biophysical Journal. 2005;89(6):3884–94. doi: 10.1529/biophysj.105.060640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rubenstein BM, Kaufman LJ. The Role of Extracellular Matrix in Glioma Invasion: A Cellular Potts Model Approach. Biophysical Journal. 2008;95(12):5661–80. doi: 10.1529/biophysj.108.140624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shirinifard A, Gens JS, Zaitlen BL, Popławski NJ, Swat M, Glazier JA. 3D Multi-Cell Simulation of Tumor Growth and Angiogenesis. PLoS One. 2009;4(10):e7190. doi: 10.1371/journal.pone.0007190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gatenby RA, Smallbone K, Maini PK, Rose F, Averill J, Nagle RB, et al. Cellular adaptations to hypoxia and acidosis during somatic evolution of breast cancer. Br J Cancer. 2007;97(5):646–53. doi: 10.1038/sj.bjc.6603922. PMCID: 2360372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Silva AS, Gatenby RA, Gillies RJ, Yunes JA. A quantitative theoretical model for the development of malignancy in ductal carcinoma in situ. J Theor Biol. 2010;262(4):601–13. doi: 10.1016/j.jtbi.2009.10.031. [DOI] [PubMed] [Google Scholar]
- 52.Smallbone K, Gatenby RA, Gillies RJ, Maini PK, Gavaghan DJ. Metabolic changes during carcinogenesis: potential impact on invasiveness. J Theor Biol. 2007;244(4):703–13. doi: 10.1016/j.jtbi.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 53.Macklin P, Kim J, Tomaiuolo G, Edgerton ME, Cristini V. Agent-Based Modeling of Ductal Carcinoma in Situ: Application to Patient-Specific Breast Cancer Modeling. In: Pham T, editor. Computational Biology: Issues and Applications in Oncology. Springer; New York, NY: 2009. pp. 77–112. [Google Scholar]
- 54.Gatenby RA, Gillies RJ. A microenvironmental model of carcinogenesis. Nature Reviews Cancer. 2007;8(1):56–61. doi: 10.1038/nrc2255. [DOI] [PubMed] [Google Scholar]
- 55.Eftimie R, Bramson JL, Earn DJD. Interactions between the immune system and cancer: A brief review of non-spatial mathematical models. Bulletin of Mathematical Biology. 2011;73(1):2–32. doi: 10.1007/s11538-010-9526-3. [DOI] [PubMed] [Google Scholar]
- 56.Clairambault J. Modelling Physiological and Pharmacological Control on Cell Proliferation to Optimise Cancer Treatments. Mathematical Modelling of Natural Phenomena. 2009;4(3):12–67. [Google Scholar]
- 57.Pathmanathan P, Gavaghan DJ, Whiteley JP, Chapman SJ, Brady JM. Predicting tumor location by modeling the deformation of the breast. IEEE transactions on bio-medical engineering. 2008;55(10):2471–80. doi: 10.1109/TBME.2008.925714. [DOI] [PubMed] [Google Scholar]
- 58.Hogea C, Biros G, Abraham F, Davatzikos C. A robust framework for soft tissue simulations with application to modeling brain tumor mass effect in 3D MR images. Physics in medicine and biology. 2007;52(23):6893–908. doi: 10.1088/0031-9155/52/23/008. [DOI] [PubMed] [Google Scholar]
- 59.Ambrosi D, Preziosi L. Cell adhesion mechanisms and stress relaxation in the mechanics of tumours. Biomechanics and modeling in mechanobiology. 2009;8(5):397–413. doi: 10.1007/s10237-008-0145-y. [DOI] [PubMed] [Google Scholar]
- 60.Unnikrishnan GU, Unnikrishnan VU, Reddy JN, Lim CT. Review on the Constitutive Models of Tumor Tissue for Computational Analysis. Applied Mechanics Reviews. 2010;63(4):040801. [Google Scholar]
- 61.Wang C, Rockhill J, Mrugala M, Peacock D, Lai A, Jusenius K, et al. Prognostic significance of growth kinetics in newly diagnosed glioblastomas revealed by combining serial imaging with a novel biomathematical model. Cancer Research. 2009;69:9133. doi: 10.1158/0008-5472.CAN-08-3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Eikenberry SE, Sankar T, Preul MC, Kostelich EJ, Thalhauser CJ, Kuang Y. Virtual glioblastoma: growth, migration and treatment in a three-dimensional mathematical model. Cell Proliferation. 2009;42(4):511–28. doi: 10.1111/j.1365-2184.2009.00613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McDougall SR, Anderson ARA, Chaplain MAJ. Mathematical modelling of dynamic adaptive tumour-induced angiogenesis: clinical implications and therapeutic targeting strategies. Journal of Theoretical Biology. 2006;241(3):564–89. doi: 10.1016/j.jtbi.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 64.Frieboes HB, Edgerton ME, Fruehauf JP, Rose FR, Worrall LK, Gatenby RA, et al. Prediction of drug response in breast cancer using integrative experimental/computational modeling. Cancer Research. 2009;69(10):4484–92. doi: 10.1158/0008-5472.CAN-08-3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Byrne H, Preziosi L. Modelling solid tumour growth using the theory of mixtures. Mathematical medicine and biology : a journal of the IMA. 2003;20(4):341–66. doi: 10.1093/imammb/20.4.341. [DOI] [PubMed] [Google Scholar]
- 66.Preziosi L, Tosin A. Multiphase modelling of tumour growth and extracellular matrix interaction: mathematical tools and applications. Journal of Mathematical Biology. 2009;58(4-5):625–56. doi: 10.1007/s00285-008-0218-7. [DOI] [PubMed] [Google Scholar]
- 67.Frieboes HB, Lowengrub JS, Wise S, Zheng X, Macklin P, Elaine LBD, et al. Computer simulation of glioma growth and morphology. Neuroimage. 2007;37:S59–S70. doi: 10.1016/j.neuroimage.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kordon EC, Smith GH. An entire functional mammary gland may comprise the progeny from a single cell. Development. 1998;125(10):1921–30. doi: 10.1242/dev.125.10.1921. [DOI] [PubMed] [Google Scholar]
- 69.Butcher D, Alliston T, Weaver V. A tense situation: forcing tumour progression. Nature Reviews Cancer. 2009;9(2):108–22. doi: 10.1038/nrc2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stolarska M, Kim Y, Othmer H. Multi-scale models of cell and tissue dynamics. Philosophical Transactions of the Royal Society A: Mathematical, Physical and Engineering Sciences. 2009;367(1902):3525–53. doi: 10.1098/rsta.2009.0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bearer EL, Lowengrub JS, Frieboes HB, Chuang YL, Jin F, Wise SM, et al. Multiparameter Computational Modeling of Tumor Invasion. Cancer Research. 2009;69(10):4493–501. doi: 10.1158/0008-5472.CAN-08-3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Frieboes HB, Jin F, Chuang YL, Wise SM, Lowengrub JS, Cristini V. Three-dimensional multispecies nonlinear tumor growth-II: Tumor invasion and angiogenesis. Journal of Theoretical Biology. 2010;264(4):1254–78. doi: 10.1016/j.jtbi.2010.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jin F, Chuang Y-L. Hybrid continuum-discrete tumor models. Multiscale Modeling of Cancer: An Integrated Experimental and Mathematical Modeling Approach. 2010:127–58. [Google Scholar]
- 74.Bauer AL, Jackson TL, Jiang Y. A Cell-Based Model Exhibiting Branching and Anastomosis during Tumor-Induced Angiogenesis. Biophysical Journal. 2007;92(9):3105–21. doi: 10.1529/biophysj.106.101501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bauer AL, Jackson TL, Jiang Y. Topography of Extracellular Matrix Mediates Vascular Morphogenesis and Migration Speeds in Angiogenesis. PLoS Comput Biol. 2009;5(7):e1000445. doi: 10.1371/journal.pcbi.1000445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bissell M, Aggeler J. Dynamic reciprocity: how do extracellular matrix and hormones direct gene expression? Prog Clin Biol Res. 1987;249:251–62. [PubMed] [Google Scholar]
- 77.Tanos T, Brisken C. What signals operate in the mammary niche? Breast Disease. 2008;29(1):69–82. doi: 10.3233/bd-2008-29108. [DOI] [PubMed] [Google Scholar]
- 78.Hatzikirou H, Basanta D, Simon M, Schaller K, Deutsch A. “Go or Grow”: the key to the emergence of invasion in tumour progression? Mathematical Medicine and Biology. 2010 doi: 10.1093/imammb/dqq011. [DOI] [PubMed] [Google Scholar]
- 79.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog Signaling Enhances Delivery of Chemotherapy in a Mouse Model of Pancreatic Cancer. Science. 2009;324(5933):1457–61. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sinek JP, Sanga S, Zheng XM, Frieboes HB, Ferrari M, Cristini V. Predicting drug pharmacokinetics and effect in vascularized tumors using computer simulation. Journal of Mathematical Biology. 2009;58(4-5):485–510. doi: 10.1007/s00285-008-0214-y. [DOI] [PMC free article] [PubMed] [Google Scholar]