

Abstract
Naturally occurring isothiocyanates (ITC) from cruciferous vegetables are widely studied for their cancer chemopreventive effects. Here we investigated the effects of ITC on Toll-like receptor (TLR) signaling, and found that two most promising ITCs, phenethyl isothiocyanate (PEITC) and D,L-sulforaphane (SFN), have differential effects on dsRNA mediated innate immune signaling through TLR3. PEITC preferentially inhibited TLR3 mediated IRF3 signaling and downstream gene expression in vivo and in vitro, whereas SFN caused inhibition of TLR3 mediated NF-κB signaling and downstream gene expression. Mechanistically, PEITC inhibited ligand (dsRNA) dependent dimerization of TLR3 resulting in inhibition of signaling through IRF3. On the other hand, SFN did not disrupt TLR3 dimerization indicating that it affects further downstream pathway resulting in NF-κB inhibition. In order to examine the biological significance of these findings in the context of anti-tumor activities of these compounds, we used two approaches: (a) first, we showed that dsRNA mediated apoptosis of tumor cells via TLR3 was inhibited in the presence of PEITC, whereas this response was augmented by SFN treatment; (b) second, in a separate assay measuring anchorage independent growth and colony formation by immortalized fibroblasts we made similar observations. Here again, PEITC antagonized dsRNA mediated inhibition of colony formation while SFN enhanced the inhibition. These results indicate biologically relevant functional differences between two structurally similar ITC and may provide important insights in therapeutic development of these compounds targeted to specific cancer.
INTRODUCTION
Numerous studies have shown that naturally occurring isothiocyanates (ITC) from cruciferous vegetables can suppress cancer initiation, promotion and progression (1, 2). Isothiocyanates are characterized by the sulfur containing functional group –N=C=S. They primarily exist as glucosinolates, which are hydrolyzed to ITC when the plant cells are disrupted releasing the enzyme myrosinases. ITC have been shown to affect many cellular functions related to tumorigenesis including cell cycle regulation, apoptosis, hormone signaling, angiogenesis, etc. (3, 4). Among the multitude of effects that these compounds exhibit on different tissues, the primary mechanistic focus has been on their role in activation of Nrf2, inhibition of NF-κB transcriptional activity and induction of apoptosis (1–3). However, except for a few studies focusing on the TLR4 response to SFN (5, 6), to the best of our knowledge effect of ITC on TLR signaling largely remains unknown.
The innate immune system recognizes conserved components of invading microbes through pattern recognition receptors (PRR) and helps to mount a response to protect the host (7). Activation of Toll-like Receptors (TLR) and other cytoplasmic receptors with their cognate ligands causes transcriptional induction of a set of genes, mainly via the NF-κB and/or interferon regulatory factor (IRF) family of transcription factors (8). Together they trigger the initial host-defense response and shape the later adaptive immune response. Toll-like receptor 3 (TLR3) is one of the primary sensors of double stranded (ds) RNA (9–11). TLR3 is activated either by natural dsRNA from virus infection, or synthetic dsRNA polyinosinic acid:polycytidylic acid (p(I):p(C)) applied externally. Upon dsRNA binding, TLR3 engages its unique adaptor TRIF (Toll-interleukin 1 receptor domain-containing adapter protein inducing interferon-beta) to propagate the signal leading to NF-κB or IRF3 mediated upregulation of a series of pro-inflammatory and cytokine genes.
We have recently shown natural variations of TLR3 mediated NF-κB activation between primary and metastatic head and neck cancer cells leading to their differential sensitivity to p(I):p(C) stimulated cell death (12). These findings have opened possibilities of differentially modulating TLR3 mediated IRF3 and NF-κB signaling for therapeutic purpose. Towards this we have previously identified and characterized small molecules that differentially modulate TLR3 mediated NF-κB and IRF3 activation (13). In the present study we have used naturally occurring ITC to study their effect on TLR3 signaling. We show that phenethyl isothiocyanate (PEITC, a constituent of watercress) potently inhibits IRF3 signaling, while a similar ITC, D,L-sulforaphane (SFN, a synthetic racemic analogue of broccoli constituent L-Sulforaphane) preferentially blocks NF-κB. Besides TLR3 mediated gene inductions, we also show the biological importance of these findings by demonstrating differential effects of these compounds on p(I):p(C) stimulated apoptosis and anchorage independent growth inhibition.
MATERIALS AND METHODS
Cell culture and reagents
HEK293 cells and HEK293 derived TLR3 expressing stable cells (Wt11), RL24 cells, HT1080 cells, RAW264.7 cells, SV40 T antigen expressing FVB MEF, and head and neck squemous cancer cells PCI15A were cultured in DMEM containing 10% FBS and penicillin/streptomycin. Prostate cancer LNCaP cells were cultured in RPMI 1640 supplemented with 10% fetal bovine serum (FBS), 2.4 mg/mL glucose, 1 mmol/L sodium pyruvate, and antibiotics. Synthetic dsRNA, polyinosinic:polycytidylic acid [poly(I):(C)] was obtained from GE Healthcare and dissolved in PBS. TLR3 signaling was induced by adding poly(I):(C) stock solution to the media as described before (13). Human IFNα was from PBL interferon source (Piscataway, NJ). Isothiocyanates (ITC) including phenethyl isothiocyanate (PEITC), erucin (ERU), erysolin (ERY), and sulforaphane (SFN) were purchased from LKT Laboratories (St. Paul, MN) and dissolved in DMSO. Anti-FLAG beads were from Sigma-Aldrich (St Louis, MO); anti-Cleaved-PARP was from Cell Signaling Technology Inc. (Beverly, MA); anti-HA, anti-α-tubulin, anti-β-actin were from Santa Cruz Biotechnology Inc. (Santa Cruz, CA); anti-TLR3 was from Imgenex (San Diego, CA). Anti-ISG54, ISG56, ISG60, anti-DRBP76, anti-IRF3 and anti-SV40 large T antigen antibodies have been described before (13, 14).
TLR3 cloning and stable cells for analysis of TLR3 oligomerization
TLR3 was PCR amplified from template plasmid pcDNA3.1 FLAG-human TLR3 using the primers: 5′-CACCATGTCTGCACTTCTGATCCTAGCTCTTGTTGGAGCTGCAGTTGCTTATCCGTATGATGTGCCGGATTATGCGCTTGGGATGCTGTGTGCATCC-3′ (with sequence of preprotrypin signal peptide and HA tag between ATG and TLR3 coding sequence) and 5′-TTACAGATCCTCTTCTGAGATGAGTTTTTGTTCATGTACAGAGTTTTTGGATCCAAGT GCTAC-3′ (with myc tag sequence between TLR3 coding sequence and the stop codon). The PCR product was cloned into pENTR/D-TOPO (Invitrogen) to obtain pENTR/HA-TLR3-myc. The expression vector pLenti CMV/HA-TLR3-myc was generated by LR recombination between pENTR/HA-TLR3-myc and pLenti CMV Puro DEST (Addgene) using Gateway LR Clonase II enzyme mix (Invitrogen) according to manufacturer’s guidelines. Wt11 cells which stably express FLAG-TLR3 were transfected with pLenti CMV/HA-TLR3-myc and selected with puromycin (1 μg/mL). The selected pool stable cells express FLAG tagged TLR3 as well as HA/myc-tagged TLR3.
Transfection and reporter assays
HEK293 cells were seeded into flat-bottomed white-wall 96-well cell culture plates at a density of 4 × 104 cells per well in 100 μL media. The cells were transfected the next day by Fugene 6 (Roche) with IFNβ-luciferase reporter (10 ng/well) and β-actin Renilla luciferase reporter (0.3 ng/well), together with IKKε, TBK1, IRF3-5D mutant, or vector control (20–40 ng/well). The total DNA per well was normalized to 50 ng by adding empty pcDNA3.1 vector. Wt11 cells were similarly transfected with NF-κB-luciferase reporter (10 ng/well) and β-actin Renilla luciferase reporter (0.3 ng/well). Twenty-four hours post-transfection, the cells were treated with PEITC and SFN respectively for 8 h or 1 h followed by 7 h, 50 μg/mL poly(I):(C) stimulation. Luciferase activities were measured using the Dual-Glo luciferase assay system (Promega, Madison, WI).
Nuclear fractionation and Co-immunoprecipitation and Western blotting
To isolate nuclear fractions, cells were suspended in the hypotonic buffer plus 0.1% Triton X-100, incubated on ice for 10 min, vortexed for 30 s, and centrifuged (12 000 g for 10 min at 4°C). The supernatants were used as cytoplasmic fractions. The nuclei pellet were thoroughly washed and lysed in lysis buffer (20 mM HEPES (pH 7.4), 1% Triton X-100, 150 mM NaCl, 1.5 mM MgCl2, 12.5 mM β-glycerophosphate, 2 mM EGTA, 10 mM NaF, 2 mM DTT, 1 mM Na3VO4, 1 mM PMSF and 1 × protease inhibitors). For immunoprecipitation, the treated cells were lysed in above lysis buffer, the cleared supernatants were incubated with anti-FLAG beads overnight, washed three times with lysis buffer, and boiled in 1 × SDS–PAGE loading buffer for elution. All the protein extracts were separated by SDS-PAGE and analyzed by immunoblotting as described.
Quantitative PCR analysis of gene expression
RNA was isolated from cells treated with ITC and/or stimuli by Trizol (Invitrogen, Carlsbad, CA), and cDNA was synthesized using iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA) and subjected to real-time PCR using a CFX96 Real Time System (Bio-Rad) according to manufacturer’s instructions. PCR amplification was normalized to RPL32. All qPCR primers information is available in our previous published paper (13, 15).
IL-8 ELISA
Cell supernatants were harvested and assayed for IL-8 protein amount by ELISA using the human IL-8 ELISA set reagent from BD Biosciences following manufacturer’s protocol as described before (13).
Immunohistochemistry (IHC) staining and quatitation
Immunohistochemistry was performed as previously described (Powolny et al., 2011). Briefly, prostate section were quenched with 3% hydrogen peroxide and blocked with normal serum. The sections were then probed with anti-ISG54 antibody, washed with Tris-buffered saline, and incubated with a HRP conjugated anti-rabbit secondary antibody. The characteristic brown color was developed by incubation with 3,3′-diaminobenzidine. The sections were counterstained with Meyers Hematoxylin (Sigma) and examined under a Leica microscope (Leica Microsystems, Bannockburn, IL). ISG54 expression was determined using Aperio Image Scope software which automatically counts blue-negative and brown-positive stained cells and categorizes them according to intensity (0, 1+, 2+ or 3+). At least ten non-overlapping images were captured from prostate sections of the mice fed control diet and PEITC-supplemented diet. Immunohistochemical images were analyzed using Aperio ImageScope software and the results are represented as H-score, a widely accepted method for semiquantitative analysis. H-score ranges from 0 to 300 based on intensity (0, 1+, 2+, and 3+) and percentage (0–100) of positively stained cells and is calculated using the following formula, H-score = (% of negative cells × 0) + (% 1+ cells × 1) + (% 2+ cells × 2) + (% 3+ cells × 3).
Soft agar assay
1% low-melting temperature agarose was melted in microwave oven and mixed with equal volume of 2 × MEM (Invitrogen) containing 20% FBS and 2 × antibiotics. The 0.5% agarose in 1 × MEM plus 10% FBS was added to 6-well plates (2 mL/well), and left at RT for more than 30 min to allow agarose to solidify as base agar. FVB MEF stably expressing SV40 large T and small T antigens in medium were mixed with above 0.5% agarose/MEM/FBS (volume ratio 2:3) to obtain 0.3% agarose and applied onto the top of 0.5% base agar in 6-well plates (1.6 × 104 cells of 2 mL agarose in each well). The plates were left at RT for more than 30 min to let the top agarose solidify before transferring into a cell culture incubator. Every two days, the p(I):p(C) (50 μg/mL) and/or PEITC (5 μM), SFN (5 μM) containing medium were added onto the surface of the top agar. After three weeks incubation in CO2 incubator, the cell colonies would be clearly visualized for assessment of anchorage independent cell growth.
RESULTS
Differential Effects of ITC on TLR3 signaling
Activation of TLR3 signaling directly induces a number of type I interferon (IFN) stimulated genes (ISG), such as ISG56 family genes, independent of IFN signaling (10). A number of cell lines of epithelial origin induces ISGs through TLR3 signaling pathway, when synthetic dsRNA analogue p(I):p(C) is added to the culture medium, as opposed to inducing RLR (RIG-I-like receptor) signaling (11, 16–18). Specifically, in HEK293 cells which do not express TLR3, this signaling is dependent on exogenous expression of TLR3 (19). To investigate the effects of ITC on dsRNA signaling initially we used a HEK293-cell based ISG56-promoter reporter assay (13). Using this system we tested effect of four naturally-occurring ITC: PEITC, Erucin (ERU), Erysolin (ERY) and SFN (Fig. 1A). PEITC is an aromatic ITC, whereas the remaining compounds belong to thioalkyl class and differ by oxidation state of the sulfur atom. We found that PEITC and ERU inhibited IRF3 mediated transcriptional activation of ISG56-promoter by p(I):p(C), while SFN and ERY did not show significant changes (Supplementary Fig. 1). In order to see the inhibition of endogenous gene induction we used quantitative RT-PCR (qRT-PCR) to measure mRNA induction for ISG56 and IFNB from HEK293 cells stably expressing TLR3 (HEK293-TLR3, also named as Wt11 cells). TLR3 mediated induction of both these genes, which are IRF3/IRF7 dependent, were inhibited by PEITC and ERU in a dose dependent manner (Fig. 1B and 1C). At the protein levels also we saw similar inhibition with PEITC and ERU for ISG56 and another ISG56 family protein ISG60 (Fig. 1D). On the other hand, SFN and ERY failed to cause inhibition of ISG56 or ISG60 induction (Fig 1D, lanes 7, 8, 9 and 10). In contrast, when we tested for the induction of genes that are induced by TLR3 signaling primarily via NF-κB signaling pathway, they were significantly more inhibited by ERY and SFN. As shown in Fig. 2A and 2B both IL-8 and TNFα mRNA induction were inhibited by ERY and SFN. However, PEITC and ERU did not inhibit IL-8 mRNA, and only partially inhibited TNFα mRNA, which may be due to the additional dependence of TNFα promoter on IRF3(20). Similar observations were made in experiments measuring IL-8 protein by ELISA (Fig. 2C), and NF-κB-reporter assay (Fig. 2D). Two important conclusions are discernible from these observations: (a) PEITC and SFN have differential response on TLR3 signaling, and (b) oxidation state of the sulfur in thioalkyl series strikingly changes its activity. Specifically, this study shows that sulfinyl analogue with SO moiety (SFN) and sulfonyl analogue with SO2 moiety (ERY) behave differently than the thio compound ERU. For example, TLR3-mediated induction of ISG56 is significantly inhibited in the presence of PEITC and ERU, but not SFN and ERY. We also found that the activity of ERU is similar to that exhibited by the aromatic ITC compound PEITC.
Fig. 1. ITC differentially affect TLR3 signaling and downstream gene expression.

(A) Chemical structures of various ITC used in this study. (B) and (C) HEK293-TLR3 cells (Wt11) were treated with ITC for 1h, then stimulated with 50 μg/mL p(I):p(C) for another 16 h. Cells were harvested for RNA extraction, and total RNA were analyzed by quantitative RT-PCR for ISG56 (B) and IFNβ (C). (D) HEK293-TLR3 cells were treated with the different ITC including PEITC, ERU, ERY and SFN at the indicated concentrations for 1h, then stimulated with 50μg/mL p(I):p(C) for another 6 h. Cell lysates were immunoblotted with anti-ISG56, ISG60, TLR3 and actin antibodies. NS is non-specific band. * p < 0.05 and ** p < 0.01, the ITC treated samples versus p(I):p(C) control sample in Panels B and C performed by one-way ANOVA analysis.
Fig. 2. ITC differentially affect TLR3 mediated NF-κB signaling and its downstream gene expression.

(A–B) Quantitative RT-PCR for NF-κB dependent gene IL-8 (A) and TNF-α (B) was performed as in Figure 1. (C) HEK293-TLR3 cells in 24-well plates were treated with PEITC or SFN for 1h, then stimulated with 50 μg/mL p(I):p(C) for another 24 h. Cell supernatants were harvested and IL-8 protein levels were detected by ELISA. (D) Wt11 cells were transfected with NF-κB-luciferase reporter and β-actin Renilla luciferase reporter. Twenty-four hours post-transfection, the cells were treated with PEITC or SFN for 1 h followed by another 7 h stimulation with 50 μg/mL poly(I):(C). The results were expressed as fold induction of NF-κB luciferase activity for poly(I):(C) stimulated cells relative to those of non-stimulated controls after normalization to Renilla luciferase. * p < 0.05 and ** p < 0.01, the ITC treated samples versus p(I):p(C) control sample performed by one-way ANOVA analysis.
Specific effects of PEITC on IRF3 signaling
In resting cells, IRF3 is predominantly present in the cytoplasm. Upon stimulation of various PRR signaling pathways IRF3 is phosphorylated by IKK family kinases TBK1 or IKKε leading to its nuclear translocation and transcriptional activation of IRF3 responsive genes (7). In order to investigate the mechanism of ITC mediated inhibition of IRF3 responsive gene induction we assayed IRF3 nuclear translocation (15). Nuclear fractions were prepared from HEK293-TLR3 cells treated with p(I):p(C) and various ITC, and immunoblotted with anti-IRF3 antibody. As shown in Fig. 3A, IRF3 was present in the cytoplasmic fraction (CP) (Fig. 3A, lane 11 from left), but was absent in the nuclear fraction prepared from untreated cells (Fig. 3A, lanes 1 from left), while its levels were significantly increased with p(I):p(C) treatment (Fig. 3A, lane 2 from left). However, pre-treatment of cells with PEITC or ERU at two different concentrations inhibited IRF3 nuclear translocation (Fig. 3A, lanes 3–6 from left), while the same were unchanged in ERY and SFN treated samples (Fig. 3A, lanes 7–10 from left). This indicated that PEITC affected TLR3-IRF3 signaling upstream of IRF3 activation. Further, to confirm the specificity of this effect we tested the effects of ITC on type I IFN mediated induction of ISG60. As discussed before, ISG56 family genes can be induced, independent of IRF3, by IFN signaling through JAK/STAT pathway via IRF9 (10). We took advantage of these two independent signaling pathway mediated induction of ISG60 and treated the HEK293-TLR3 cells with human IFNα. As shown in Fig. 3B, none of ITC tested showed any inhibition of IFN mediated ISG60 induction demonstrating the specificity of the ITC on TLR3-IRF3 pathway.
Fig. 3. PEITC inhibits TLR3 mediated IRF3 activation but not affects IFN signaling.
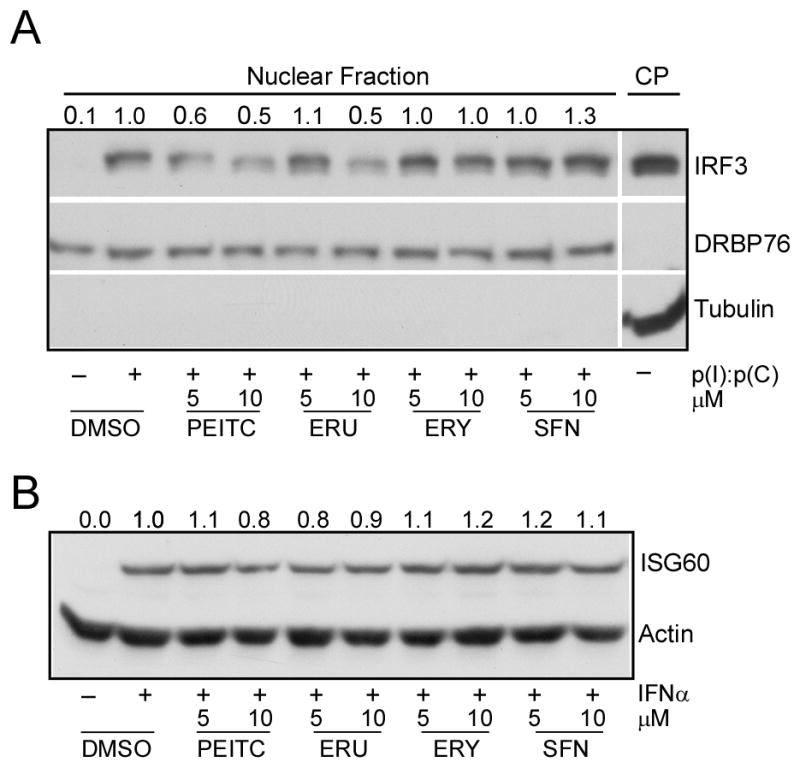
(A) HEK293-TLR3 cells were treated with PEITC or other ITC for 1h, then stimulated with 50 μg/mL p(I):p(C) for another 3 h. Cells were subjected to nuclear fractionation and the nuclear IRF3 detected by immunoblotting with the anti-IRF3 antibody. The effective nuclear and cytoplasmic (CP) fractionation was evidenced by specific detection of DRBP76 and tubulin respectively. The band intensities of nuclear IRF3 were normalized to DRBP76 band intensities and are shown on the top of each band. (B) HEK293-TLR3 cells were treated with PEITC or other ITC for 1 h, then stimulated with 500 U/mL human IFNα for 20 h. Cell lysates were detected by immunoblotting for ISG60 and actin. The band intensities of ISG60 after normalization to Actin were shown on the top of each band.
PEITC affects TLR3 oligomerization
To understand the mechanistic basis of this differential regulation, hereafter we focused on PEITC and SFN. In an attempt to identify the particular step in TLR3-IRF3 signaling pathway that was affected by PEITC, we used reporter assays. TLR3 signaling pathway can be activated without dsRNA or virus infection by artificially expressing either of several signaling components, such as IKKε (21), TBK1 (22), or constitutively active IRF3 (IRF3-5D mutant (23)) using transient transfection. As shown in Fig. 4, IFNβ-promoter driven luciferase activity was strongly induced by expression of IKKε (Fig. 4A), TBK1 (Fig. 4B) and IRF3-5D (Fig. 4C). However, both PEITC and SFN did not show any inhibition of this induction at two different concentrations. This was consistent with our previous findings that PEITC did not affect TLR3-IRF3 signaling downstream of IRF3 indicated by the inhibition of IRF3 nuclear translocation, rather it was further upstream.
Fig. 4. PEITC interferes with TLR3 oligomerization.

HEK293 cells in 96-well plates were cotransfected with IFNβ firefly luciferase and β-actin Renilla luciferase, together with 20 ng IKKε (A), 20 ng TBK1 (B) and 40 ng IRF3-5D mutant (C) for 24 h. The transfected cells were treated with PEITC and SFN at the indicated concentrations for an additional 8 h. The results were expressed as fold induction of IFNβ luciferase activity for IKKε, TBK1 or IRF3-5D transfected cells relative to those of vector transfected controls after normalization to Renilla luciferase. The results were representative of two similar experiments. No significant difference between PEITC or SFN treated samples and non-treated samples was found by one-way ANOVA analysis. (D) HEK293 cells stably expressing FLAG-TLR3 and HA-TLR3 were treated with PEITC and SFN at the indicated concentrations for 1h and then stimulated with 50 μg/mL p(I):p(C) for another 3 h. Cell lysates were immunoprecipitated with anti-FLAG and the immunoprecipitates together with cell lysate control detected by immunoblotting for HA-TLR3. (E) Band intensities of each lane from the top panel in (D) were quantified and normalized with corresponding those of the bottom panel (anti-HA bands in whole cell lysates) and plotted as relative intensities. The plotted graph represents the average band intensities plus standard errors from two similar experiments. * p < 0.05 and ** p < 0.01, the ITC treated samples versus p(I):p(C) control sample performed by one-way ANOVA analysis.
TLRs are known to oligomerize upon ligand binding (24), and due to the presence of the reactive –N=C=S group present in the ITC, it was suggested that SFN might affect TLR4 oligomerization (5). To test dsRNA mediated oligomerization of TLR3, we standardized an immunoprecipitation based assay. Two differentially tagged TLR3 expression constructs, one with FLAG, another with Hemagglutinin (HA) were both stably expressed in HEK293 cells. Immunoprecipitation of FLAG-tagged TLR3 from p(I):p(C) treated whole cell lysates showed a signal dependent enhancement in HA-tagged TLR3 co-precipitation (Fig. 4D, lanes 1, 2 from left). However, treatment of cells with PEITC inhibited HA-TLR3 co-precipitation, while SFN treatment did not strongly inhibit the oligomerization (Fig. 4D, lanes 3, 4, 5, 6 from left and Fig. 4E). These results indicated that unlike SFN, PEITC affected p(I):p(C) mediated oligomerization of TLR3, which might be a possible mechanism for its inhibition of TLR3-IRF3 signaling.
PEITC affects ISG expression in vivo
To investigate if the ITC mediated modulation of dsRNA signaling also occurs in cells with endogenous innate immune receptor expression we used mouse macrophage cell line RAW246.7 cells. As expected p(I):p(C) stimulated induction of ISG54 (a member of the mouse Isg56 family (25)) was inhibited by PEITC, while SFN did not show any inhibition (Fig. 5A). Previously, ISG54 was found to be induced in a number of tissues in mice by p(I):p(C) stimulation or Sendai virus infection (26). We chose to use mouse prostate tissue to investigate in vivo downregulation of ISG54 expression by PEITC. This was due to the demonstrated importance of TLR3 signaling in prostate cancer (27). Analysis of ISG54 protein level in the prostate of C57/BL6 mice fed a control diet or PEITC-supplemented diet (3 μmol PEITC/g diet) for 19 weeks (Powolny et al., 2011) showed significantly reduced ISG54 expression in both stromal cells and epithelial cells (Fig. 5B, 5C). These results indicated that PEITC was capable of inhibiting endogenous ISG54 expression in vivo.
Fig. 5. PEITC inhibits ISG 54 expression in vitro and in vivo.

(A) Mouse macrophage RAW264.7 cells were treated with PEITC and SFN at the indicated concentrations for 1h, then stimulated with 50 μg/mL p(I):p(C) for another 12 h. Cell lysates were detected by immunoblotting for ISG54 and actin. (B) Two representative immunohistochemical images depicting ISG54 expression in the prostate tissue sections from control and PEITC-treated normal mice (magnification, ×200). Scale bar = 100 μm. (C) Quantitative analysis of stromal and epithelial expression of ISG54 in the prostate tissue sections. At least ten randomly selected fields from each section were analyzed. Results shown are mean ± SD (n = 5 for both control and PEITC treatment groups). Statistical significance was determined by two-sided Student’s t test.
Effects of PEITC and SFN on dsRNA mediated cell death
Both PEITC and SFN are known to cause apoptosis by multiple mechanisms in cancer cells (1, 2). Among various mechanisms, involvement of ROS production, mitochondrial anti-apoptotic Bcl2 family proteins, NF-κB and Nrf2 pathway have been implicated to induce apoptosis in different cancer cells. However, the contribution of IRF signaling in ITC mediated apoptosis has not been described. We and others have shown that TLR3 activation by p(I):p(C) causes cell death through IRF3 pathway in a number of cancer cell lines (12, 28–30). As we observed inhibition of IRF3 signaling by PEITC, we hypothesized that PEITC might negatively impact p(I):p(C) induced apoptosis in cancer cells. To test this hypothesis, we treated two well characterized carcinoma derived cell lines: prostate cancer (PCa) cell line, LNCaP and head and neck squamous cell carcinoma (HNSCC) cell line, PCI15A either with p(I):p(C) alone or in combination with PEITC and SFN. As shown in Fig. 6A, treatment of LNCaP cells with p(I):p(C) induced ISG60 and mild apoptosis indicated by the appearance of cleaved PARP (Fig. 6A, lane 2), while treatment with PEITC alone resulted in strong induction of apoptosis (Fig. 6A, lane 3), as observed before (31). However, when these were combined, ISG60 induction was inhibited by PEITC, as well as levels of cleaved PARP was reduced (Fig. 6A, lane 4). Surprisingly, a higher dose of 10 μM PEITC induced substantial cytotoxicity in LNCaP cells indicated by the loss of β-actin. On the other hand treatment of LNCaP cells with SFN alone resulted in lower levels of cleaved PARP which was augmented by p(I):p(C) at low concentrations (Fig. 6A, lane 7, 8, 9, 10 from left and Fig. 6B). We saw a very similar pattern in PCI15A cells (Fig. 6C), where p(I):p(C) augmented SFN mediated apoptosis induction, but it inhibited PEITC mediated cell death, albeit weakly compared to LNCaP cells (Fig. 6D). Once again these results indicated another important distinction between PEITC and SFN in terms of their effects on p(I):p(C) induced apoptosis. SFN, which is known to inhibit NF-κB signaling (1), synergized with p(I):p(C) mediated apoptosis that is IRF3 dependent. On the other hand, PEITC affected p(I):p(C) mediated apoptosis in an opposite manner, due to its inhibitory effects on IRF3 signaling.
Fig. 6. PEITC inhibits TLR3 mediated ISG production as well as cell apoptosis in cancer cells.

(A) LNCaP and (C) PCI15A cells were treated with PEITC or SFN at the indicated concentrations for 1h and stimulated with 50 μg/mL p(I):p(C) for another 24 h. Cell lysates were analyzed by immunoblotting for the target proteins using anti- cleaved PARP (C-PARP), ISG56, ISG60 and β-actin antibodies. Apoptosis was quantified by measuring C-PARP band intensities for LNCaP (B) and PCI15A (D).
Modulation of dsRNA mediated inhibition of clonogenicity by ITC
Among various assays available to measure in vitro cellular transformation, anchorage independent growth of cells in soft agar is one of the hallmark characteristics of cellular transformation and uncontrolled cell growth. As the ITC are known for their cancer chemopreventive functions, we performed soft agar colony formation (clonogenicity) assay using SV40 T antigen transduced mouse embryonic fibroblasts (MEF) to assess how ITC in combination with p(I):p(C) modulated cellular transformation. T antigen transduced MEFs were responsive to p(I):p(C) by upregulating ISG54 expression (Fig. 7A). Culturing these cells in soft agar for 3 weeks generated large number of macroscopic colonies (Fig. 7B, panel 1). In presence of 5 μM PEITC or SFN, the numbers of colonies were reduced significantly (Fig. 7B, panel 2, 7C and 7D). In presence of 50 μg/mL of p(I):p(C), there was a substantial reduction in colony number, indicating anti-growth properties of TLR3 signaling (Fig. 7B, panel 3, 6C and 6D). However, when p(I):p(C) was combined with either PEITC or SFN, we saw a striking difference between these two ITC. While SFN synergized with p(I):p(C), PEITC exhibited antagonistic effects (Fig. 7B, panel 4, 7C and 7D). In presence of PEITC, number of colonies were increased compared to only p(I):p(C) treated samples. This result was consistent with our previous findings, and indicated that p(I):p(C) mediated inhibition of anchorage independent cell growth is at least partially mediated through IRF3 signaling, and the inhibition of IRF3 signaling by PEITC resulted in partial rescue of this inhibition. On the other hand, SFN probably promotes inhibition of NF-κB, which further enhances TLR3-IRF3 mediated inhibition of cell growth (Fig. 7E, and see later).
Fig. 7. PEITC and SFN differentially affect TLR3 induced inhibition of anchorage independent growth.

MEF derived from FVB mice transduced with SV40 T antigens were mixed with soft agar (1.6 × 104 cells in 0.3% agar) and applied onto the top of 0.5% base agar in 6-well plates. Every two days, medium containing p(I):p(C) (50 μM) and/or PEITC (5 μM), SFN (5μM) were applied onto the surface of the top agar. After three weeks incubation in CO2 incubator, the cell colonies were stained with crystal violet, visualized, and the cell colonies counted manually. (A) Induction of ISG54 and large T antigen in MEF stimulated with p(I):p(C) for 48 h and detected by immunoblotting. (B) Shown is the representative picture from one of the soft agar assay for PEITC and p(I):p(C) treated samples. (C) and (D) The cell colony numbers from four groups as indicated. The results were expressed as Mean ±SE. Statistical significance of the result was analyzed by two-tailed paired t test (* P < 0.05).
DISCUSSION
In this study we demonstrate differences in dsRNA signaling in presence of two structurally similar, naturally occurring ITC. Addition of poly(I):(C) to the medium engages TLR3 signaling in a large number of cells, resulting in activation of IRF3 and NF-κB. The other cytoplasmic dsRNA sensors RIG-I-like receptors, (RLR) are activated when the dsRNA ligand is delivered in the cytoplasm, experimentally by liposomal transfection of poly(I):(C), or in vitro transcribed dsRNA, or viral RNA (32). Although, the mechanism of poly(I):(C) uptake by different types of cells is not completely worked out, there are evidences that cell surface molecules such as CD11b (33), clathrin-mediated endocytosis (17) and lysosomal acidification (18) are uniquely important for this process. It is believed that poly(I):(C) taken up by these mechanisms, binds and signal through TLR3 from the endosome. Specifically, for HEK293 cells, which normally do not express TLR3, there are no IRF3 or NF-κB activation by poly(I):(C) added to the medium through RLR signaling. However, TLR3 expression enables them to signal with poly(I):(C) (19). As majority of our studies were carried out in cells of epithelial origin by adding poly(I):(C) in the medium, therefore we interpreted the results as ITC effects on TLR3 signaling. However, ITCs might have other effects on RLR signaling in a cell type specific manner, which we have not explored.
Among the two primary signaling pathways that are activated by dsRNA, the NF-κB is considered to be pro-inflammatory and growth promoting; whereas the IRFs are primarily involved in interferon (IFN) induction and enhance growth inhibitory and apoptotic pathway. With respect to specific cancer, the balance between these two pathways has been shown to be changed and determine the outcome (34). What remains to be understood is how to specifically modulate this balance, and use it to the therapeutic advantage for specific cancers. Results presented in this study provide us with new leads to develop such agents based on naturally occurring ITCs. We show that despite their structural similarities SFN and PEITC can have quite distinct effects on TLR3 signaling and cell death induction.
TLR3 signaling has been characterized in a number of model cancer cell lines (12, 35–41), as well as has been shown to have anti-cancer activity in vivo in prostate cancer model (27). The apoptotic activities of TLR3 ligand p(I):p(C) is dependent on IRF3 (25, 26, 42). On the other hand ITCs are also known to induce apoptosis in cancer cells by affecting multiple pathways. However, when in combination, ITCs differentially affected p(I):p(C) mediated apoptosis. While SFN augmented p(I):p(C) mediated apoptosis, PEITC showed opposite effect. Based on our results presented in Fig. 6 and supported by data in Fig. 7, we propose the following: IRF3 signaling positively contribute to dsRNA induced cell death and the inhibition of this pathway by PEITC leads to a reduction in net apoptosis by p(I):p(C). It has been shown that pro-apoptotic proteins Bax and Bak are involved in PEITC mediated apoptosis in prostate cancer cells (43). Our observation that in combination with p(I):p(C), PEITC mediated apoptosis is reduced may be due to the activation of pro-survival NF-κB pathway by TLR3. On the other hand, SFN enhances dsRNA mediated apoptosis by inhibiting NF-κB which primarily contributes to cellular survival. However, further experiments are needed to establish the biochemical details of PEITC on IRF3 signaling.
Evidence continues to accumulate to indicate that structural differences in ITC can profoundly affect their activity. For example, autophagy induction by SFN serves to protect against apoptosis (44). To the contrary, autophagy contributes to cell death induction by PEITC (31). The present study provides yet another example illustrating mechanistic differences in structurally-related ITC compounds (e.g., SFN and PEITC or the thioalkyl series). We show there are profound differences between SFN and PEITC effects on TLR3 signaling when used individually or in combination with TLR3 ligand. These observations underscore caution in extrapolation of mechanistic results between structurally-different ITC compounds.
The potential for using PEITC and SFN in tumor therapy has been demonstrated in a number of pre-clinical mouse models (45–51). On the other hand, synthetic dsRNA – analogues of TLR3 ligand are also being developed for various treatments including cancer (52). Our study provides a mechanistic basis for using them in combinatorial therapy. Accordingly, use of PEITC in combination with dsRNA may negatively affect their individual therapeutic potentials, while SFN and dsRNA combination may work synergistically to prevent tumor growth. In summary, our results show examples of mechanistic differences in the cellular response to two structurally similar ITC and may provide important insights in therapeutic development of these compounds targeted to specific cancer.
Supplementary Material
Acknowledgments
We thank Drs. Michael David and Dean Bacich for reagents and cell lines.
This work was supported in part by AI082673 from National Institute of Allergy and Infectious Diseases Grant (SNS), and CA101753 and CA115498 grants from the National Cancer Institute (SVS). This project used the UPCI core facilities and was supported in part by award P30CA047904.
ABBREVIATIONS
- p(I):p(C)
Polyinosinic acid:polycytidylic acid
- dsRNA
double stranded RNA
- ssRNA
single stranded RNA
- IFN
Interferon
- NF-κB
Nuclear Factor κB
- IRF
Interferon Regulatory Factor
- TLR
Toll-like Receptor
- ISG
Interferon Stimulated Gene
- TRIF
TIR-domain-containing adapter-inducing interferon-β
- RPL32
Ribosomal Protein L32
Footnotes
AUTHOR CONTRIBUTION
JZ designed and performed most of the experiments, AG designed and performed clonogenicity assays, EMC performed mouse cell experiments, ERH and JL performed immunohistochemistry, and SVS and SNS designed experiments and wrote the manuscript.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
References
- 1.Cheung KL, Kong AN. Molecular targets of dietary phenethyl isothiocyanate and sulforaphane for cancer chemoprevention. AAPS J. 2009;12:87–97. doi: 10.1208/s12248-009-9162-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stan SD, Kar S, Stoner GD, Singh SV. Bioactive food components and cancer risk reduction. J Cell Biochem. 2008;104:339–356. doi: 10.1002/jcb.21623. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Y. Allyl isothiocyanate as a cancer chemopreventive phytochemical. Mol Nutr Food Res. 2009;54:127–135. doi: 10.1002/mnfr.200900323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Higdon JV, Delage B, Williams DE, Dashwood RH. Cruciferous vegetables and human cancer risk: epidemiologic evidence and mechanistic basis. Pharmacol Res. 2007;55:224–236. doi: 10.1016/j.phrs.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Youn HS, Kim YS, Park ZY, Kim SY, Choi NY, Joung SM, Seo JA, Lim KM, Kwak MK, Hwang DH, Lee JY. Sulforaphane suppresses oligomerization of TLR4 in a thiol-dependent manner. J Immunol. 2010;184:411–419. doi: 10.4049/jimmunol.0803988. [DOI] [PubMed] [Google Scholar]
- 6.Lin W, Wu RT, Wu T, Khor TO, Wang H, Kong AN. Sulforaphane suppressed LPS-induced inflammation in mouse peritoneal macrophages through Nrf2 dependent pathway. Biochem Pharmacol. 2008;76:967–973. doi: 10.1016/j.bcp.2008.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 8.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 9.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 10.Sen GC, Sarkar SN. Transcriptional signaling by double-stranded RNA: role of TLR3. Cytokine Growth Factor Rev. 2005;16:1–14. doi: 10.1016/j.cytogfr.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 11.Vercammen E, Staal J, Beyaert R. Sensing of viral infection and activation of innate immunity by toll-like receptor 3. Clin Microbiol Rev. 2008;21:13–25. doi: 10.1128/CMR.00022-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Umemura N, Zhu J, Mburu YK, Forero A, Hsieh PN, Muthuswamy R, Kalinski P, Ferris RL, Sarkar SN. Defective NF-kappaB signaling in metastatic head and neck cancer cells leads to enhanced apoptosis by double-stranded RNA. Cancer Res. 2012;72:45–55. doi: 10.1158/0008-5472.CAN-11-1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu J, Smith K, Hsieh PN, Mburu YK, Chattopadhyay S, Sen GC, Sarkar SN. High-throughput screening for TLR3-IFN regulatory factor 3 signaling pathway modulators identifies several antipsychotic drugs as TLR inhibitors. J Immunol. 2010;184:5768–5776. doi: 10.4049/jimmunol.0903559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hein J, Boichuk S, Wu J, Cheng Y, Freire R, Jat PS, Roberts TM, Gjoerup OV. Simian virus 40 large T antigen disrupts genome integrity and activates a DNA damage response via Bub1 binding. J Virol. 2009;83:117–127. doi: 10.1128/JVI.01515-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu J, Coyne CB, Sarkar SN. PKC alpha regulates Sendai virus-mediated interferon induction through HDAC6 and beta-catenin. EMBO J. 2011;30:4838–4849. doi: 10.1038/emboj.2011.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garcia-Cattaneo A, Gobert FX, Muller M, Toscano F, Flores M, Lescure A, Del Nery E, Benaroch P. Cleavage of Toll-like receptor 3 by cathepsins B and H is essential for signaling. Proc Natl Acad Sci U S A. 2012;109:9053–9058. doi: 10.1073/pnas.1115091109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Itoh K, Watanabe A, Funami K, Seya T, Matsumoto M. The clathrin-mediated endocytic pathway participates in dsRNA-induced IFN-beta production. J Immunol. 2008;181:5522–5529. doi: 10.4049/jimmunol.181.8.5522. [DOI] [PubMed] [Google Scholar]
- 18.de Bouteiller O, Merck E, Hasan UA, Hubac S, Benguigui B, Trinchieri G, Bates EE, Caux C. Recognition of double-stranded RNA by human toll-like receptor 3 and downstream receptor signaling requires multimerization and an acidic pH. J Biol Chem. 2005;280:38133–38145. doi: 10.1074/jbc.M507163200. [DOI] [PubMed] [Google Scholar]
- 19.Sarkar SN, Smith HL, Rowe TM, Sen GC. Double-stranded RNA signaling by Toll-like receptor 3 requires specific tyrosine residues in its cytoplasmic domain. J Biol Chem. 2003;278:4393–4396. doi: 10.1074/jbc.C200655200. [DOI] [PubMed] [Google Scholar]
- 20.Werner SL, Barken D, Hoffmann A. Stimulus specificity of gene expression programs determined by temporal control of IKK activity. Science. 2005;309:1857–1861. doi: 10.1126/science.1113319. [DOI] [PubMed] [Google Scholar]
- 21.Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003;300:1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- 22.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 23.Grandvaux N, Servant MJ, tenOever B, Sen GC, Balachandran S, Barber GN, Lin R, Hiscott J. Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon-stimulated genes. J Virol. 2002;76:5532–5539. doi: 10.1128/JVI.76.11.5532-5539.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jin MS, Lee JO. Structures of the toll-like receptor family and its ligand complexes. Immunity. 2008;29:182–191. doi: 10.1016/j.immuni.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 25.Fensterl V, Sen GC. The ISG56/IFIT1 gene family. J Interferon Cytokine Res. 2011;31:71–78. doi: 10.1089/jir.2010.0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fensterl V, White CL, Yamashita M, Sen GC. Novel characteristics of the function and induction of murine p56 family proteins. J Virol. 2008;82:11045–11053. doi: 10.1128/JVI.01593-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chin AI, Miyahira AK, Covarrubias A, Teague J, Guo B, Dempsey PW, Cheng G. Toll-like receptor 3-mediated suppression of TRAMP prostate cancer shows the critical role of type I interferons in tumor immune surveillance. Cancer Res. 2010;70:2595–2603. doi: 10.1158/0008-5472.CAN-09-1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun R, Zhang Y, Lv Q, Liu B, Jin M, Zhang W, He Q, Deng M, Liu X, Li G, Li Y, Zhou G, Xie P, Xie X, Hu J, Duan Z. Toll-like receptor 3 (TLR3) induces apoptosis via death receptors and mitochondria by up-regulating the transactivating p63 isoform alpha (TAP63alpha) J Biol Chem. 2011;286:15918–15928. doi: 10.1074/jbc.M110.178798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Estornes Y, Toscano F, Virard F, Jacquemin G, Pierrot A, Vanbervliet B, Bonnin M, Lalaoui N, Mercier-Gouy P, Pacheco Y, Salaun B, Renno T, Micheau O, Lebecque S. dsRNA induces apoptosis through an atypical death complex associating TLR3 to caspase-8. Cell Death Differ. 2012 doi: 10.1038/cdd.2012.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salaun B, Coste I, Rissoan MC, Lebecque SJ, Renno T. TLR3 can directly trigger apoptosis in human cancer cells. J Immunol. 2006;176:4894–4901. doi: 10.4049/jimmunol.176.8.4894. [DOI] [PubMed] [Google Scholar]
- 31.Bommareddy A, Hahm ER, Xiao D, Powolny AA, Fisher AL, Jiang Y, Singh SV. Atg5 regulates phenethyl isothiocyanate-induced autophagic and apoptotic cell death in human prostate cancer cells. Cancer Res. 2009;69:3704–3712. doi: 10.1158/0008-5472.CAN-08-4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu M, Levine SJ. Toll-like receptor, RIG-I-like receptors and the NLRP3 inflammasome: key modulators of innate immune responses to double-stranded RNA viruses. Cytokine Growth Factor Rev. 2011;22:63–72. doi: 10.1016/j.cytogfr.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou H, Liao J, Aloor J, Nie H, Wilson BC, Fessler MB, Gao HM, Hong JS. CD11b/CD18 (Mac-1) Is a Novel Surface Receptor for Extracellular Double-Stranded RNA To Mediate Cellular Inflammatory Responses. J Immunol. 2013;190:115–125. doi: 10.4049/jimmunol.1202136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang B, Zhao J, Unkeless JC, Feng ZH, Xiong H. TLR signaling by tumor and immune cells: a double-edged sword. Oncogene. 2008;27:218–224. doi: 10.1038/sj.onc.1210904. [DOI] [PubMed] [Google Scholar]
- 35.Morikawa T, Sugiyama A, Kume H, Ota S, Kashima T, Tomita K, Kitamura T, Kodama T, Fukayama M, Aburatani H. Identification of Toll-like receptor 3 as a potential therapeutic target in clear cell renal cell carcinoma. Clin Cancer Res. 2007;13:5703–5709. doi: 10.1158/1078-0432.CCR-07-0603. [DOI] [PubMed] [Google Scholar]
- 36.Conforti R, Ma Y, Morel Y, Paturel C, Terme M, Viaud S, Ryffel B, Ferrantini M, Uppaluri R, Schreiber R, Combadiere C, Chaput N, Andre F, Kroemer G, Zitvogel L. Opposing effects of toll-like receptor (TLR3) signaling in tumors can be therapeutically uncoupled to optimize the anticancer efficacy of TLR3 ligands. Cancer Res. 2010;70:490–500. doi: 10.1158/0008-5472.CAN-09-1890. [DOI] [PubMed] [Google Scholar]
- 37.Weber A, Kirejczyk Z, Besch R, Potthoff S, Leverkus M, Hacker G. Proapoptotic signalling through Toll-like receptor-3 involves TRIF-dependent activation of caspase-8 and is under the control of inhibitor of apoptosis proteins in melanoma cells. Cell Death Differ. 2010;17:942–951. doi: 10.1038/cdd.2009.190. [DOI] [PubMed] [Google Scholar]
- 38.Matijevic T, Pavelic J. The dual role of TLR3 in metastatic cell line. Clin Exp Metastasis. 2011 doi: 10.1007/s10585-011-9402-z. [DOI] [PubMed] [Google Scholar]
- 39.Paone A, Starace D, Galli R, Padula F, De Cesaris P, Filippini A, Ziparo E, Riccioli A. Toll-like receptor 3 triggers apoptosis of human prostate cancer cells through a PKC-alpha-dependent mechanism. Carcinogenesis. 2008;29:1334–1342. doi: 10.1093/carcin/bgn149. [DOI] [PubMed] [Google Scholar]
- 40.Salaun B, Coste I, Rissoan M-C, Lebecque SJ, Renno T. TLR3 can directly trigger apoptosis in human cancer cells. Journal of immunology (Baltimore, Md : 1950) 2006;176:4894–4901. doi: 10.4049/jimmunol.176.8.4894. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Y, Sun R, Liu B, Deng M, Zhang W, Li Y, Zhou G, Xie P, Li G, Hu J. TLR3 activation inhibits nasopharyngeal carcinoma metastasis via downregulation of chemokine receptor CXCR4. Cancer biology & therapy. 2009:8. doi: 10.4161/cbt.8.19.9437. [DOI] [PubMed] [Google Scholar]
- 42.Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell. 2005;19:727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 43.Xiao D, Zeng Y, Choi S, Lew KL, Nelson JB, Singh SV. Caspase-dependent apoptosis induction by phenethyl isothiocyanate, a cruciferous vegetable-derived cancer chemopreventive agent, is mediated by Bak and Bax. Clin Cancer Res. 2005;11:2670–2679. doi: 10.1158/1078-0432.CCR-04-1545. [DOI] [PubMed] [Google Scholar]
- 44.Herman-Antosiewicz A, Johnson DE, Singh SV. Sulforaphane causes autophagy to inhibit release of cytochrome C and apoptosis in human prostate cancer cells. Cancer Res. 2006;66:5828–5835. doi: 10.1158/0008-5472.CAN-06-0139. [DOI] [PubMed] [Google Scholar]
- 45.Powolny AA, Bommareddy A, Hahm ER, Normolle DP, Beumer JH, Nelson JB, Singh SV. Chemopreventative potential of the cruciferous vegetable constituent phenethyl isothiocyanate in a mouse model of prostate cancer. J Natl Cancer Inst. 2011;103:571–584. doi: 10.1093/jnci/djr029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Singh SV, Powolny AA, Stan SD, Xiao D, Arlotti JA, Warin R, Hahm ER, Marynowski SW, Bommareddy A, Potter DM, Dhir R. Garlic constituent diallyl trisulfide prevents development of poorly differentiated prostate cancer and pulmonary metastasis multiplicity in TRAMP mice. Cancer Res. 2008;68:9503–9511. doi: 10.1158/0008-5472.CAN-08-1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Singh SV, Warin R, Xiao D, Powolny AA, Stan SD, Arlotti JA, Zeng Y, Hahm ER, Marynowski SW, Bommareddy A, Desai D, Amin S, Parise RA, Beumer JH, Chambers WH. Sulforaphane inhibits prostate carcinogenesis and pulmonary metastasis in TRAMP mice in association with increased cytotoxicity of natural killer cells. Cancer Res. 2009;69:2117–2125. doi: 10.1158/0008-5472.CAN-08-3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wattenberg LW. Inhibition of carcinogen-induced neoplasia by sodium cyanate, tert-butyl isocyanate, and benzyl isothiocyanate administered subsequent to carcinogen exposure. Cancer Res. 1981;41:2991–2994. [PubMed] [Google Scholar]
- 49.Stoner GD, Morrissey DT, Heur YH, Daniel EM, Galati AJ, Wagner SA. Inhibitory effects of phenethyl isothiocyanate on N-nitrosobenzylmethylamine carcinogenesis in the rat esophagus. Cancer Res. 1991;51:2063–2068. [PubMed] [Google Scholar]
- 50.Cheung KL, Khor TO, Huang MT, Kong AN. Differential in vivo mechanism of chemoprevention of tumor formation in azoxymethane/dextran sodium sulfate mice by PEITC and DBM. Carcinogenesis. 2010;31:880–885. doi: 10.1093/carcin/bgp285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hu R, Khor TO, Shen G, Jeong WS, Hebbar V, Chen C, Xu C, Reddy B, Chada K, Kong AN. Cancer chemoprevention of intestinal polyposis in ApcMin/+ mice by sulforaphane, a natural product derived from cruciferous vegetable. Carcinogenesis. 2006;27:2038–2046. doi: 10.1093/carcin/bgl049. [DOI] [PubMed] [Google Scholar]
- 52.Hennessy EJ, Parker AE, O’Neill LA. Targeting Toll-like receptors: emerging therapeutics? Nat Rev Drug Discov. 2010;9:293–307. doi: 10.1038/nrd3203. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.