

Abstract
The functional plasticity of CD8+ T cells in an atopic environment, encompassing a spectrum from IFN-γ- to IL-13-producing cells, is pivotal in the development of allergic airway hyperresponsiveness (AHR) and inflammation and yet remains mechanistically undefined. We demonstrate that CD8+ T cell IL-13 induction proceeded through a series of distinct IL-4/GATA3-regulated stages characterized by gene expression and epigenetic changes. In vivo, CD8+ T cells exposed to an environment rich in IL-4 displayed epigenetic changes at the GATA3 and IL-13 promoter indicative of transcriptional activation and IL-13 production. In vitro, IL-4 triggered the step-wise molecular conversion of CD8+ T cells from IFN-γ to IL-13 production. During the initial stage, IL-4 suppressed T-bet and induced GATA3 expression, characterized by enhanced activating histone modifications and RNA Pol II recruitment to the GATA3 locus. Notably, recruitment of GATA3 and RNA Pol II to the IL-13 promoter was also detected at this initial stage. However, enhanced IL-13 transcription only occurred at a later stage following TCR stimulation, indicating that IL-4 induced GATA3 recruitment poises the IL-13 locus for TCR-mediated transcription. Thus, both in vivo and in vitro an atopic (IL-4) environment poises CD8+ T cells via step-wise epigenetic and phenotypic mechanisms for pathogenic conversion to IL-13 production, which is ultimately triggered via an allergen-mediated TCR stimulus.
Keywords: CD8 T cells, IL-4, plasticity, IL-13, asthma
Introduction
The role of CD4+ T cells and the production of Th2 related cytokines in asthma have been substantiated in many studies and different species (1–4). In contrast, the role of CD8+ T cells is less clear with conflicting results showing a protective (5, 6), enhancing (7–9), or dual role (10–12) depending on environmental conditions. In asthmatics, CD8+ T cells have been correlated with reduced lung function (7). In mice, CD8+ T cell depletion resulted in reduced airway responses (13). A unique subset of CD8+ T cells, CD8 effector memory T cells (TEM), played a critical role in experimental asthma (14–16). Unlike central memory CD8+ T cells, they expressed a high affinity receptor for leukotriene B4 (LTB4), BLT1, which was essential for their recruitment and accumulation in the lung (17). The central role of CD8+BLT1+ T cells in the lung was attributed to their capacity for IL-13 production (9). Effector CD8+ T cells are typically defined by their ability to secrete IFN-γ and to produce molecules involved in cytolysis such as perforin and granzyme B (18, 19). The mechanism(s) by which effector CD8+ T cells can be converted into pathogenic IL-13-producing cells in the allergic lung has not been defined.
T helper cell lineage commitment and differentiation have been viewed largely in a unidirectional manner with non-reversible terminal endpoints having distinct effector functions. Recognition of T cell subsets beyond T helper 1 (Th1) and Th2 has prompted a re-examination of this concept. It appears that subsets are more “plastic,” with instability of lineage-specific transcription factor expression providing flexibility in differentiation options (20). Most studies have focused on the plasticity of CD4+ T cells, demonstrating a capacity for redirecting their functional programs. These differentiation decisions are dictated primarily by cytokines in the microenvironment; for example, reciprocal involvement of IL-4 and IL-12/IFN-γ in CD4+ T cell polarization has been well characterized (21, 22). In contrast, little has been reported on the functional plasticity of CD8+ T cells (23, 24).
Epigenetic mechanisms regulate CD4+ T cell lineage differentiation from Th0 to Th1, Th2, Treg and Th17 subsets (25–27). Th2 polarization is regulated by association of the permissive histone modification tri-methylation of lysine 4 of histone 3 (H3K4me3) with the IL-4 promoter and repressive tri-methylation of lysine 27 of histone 3 (H3K27me3) with the IFN-γ promoter (26). Alterations in H3K4me3, H3K27me3 and histone acetylation regulate CD8+ T cell memory development (27–29) and permissive histone modifications poise cytokine gene promoters for rapid transcription following stimulation. Little is known about the interrelationships of histone regulation, asthma pathophysiology, and CD8+ T cell plasticity in Tc1 and Tc2 phenotypes.
Here, we examined the mechanisms controlling IL-4-mediated CD8+ T cell conversion to an asthma-associated phenotype. Adoptive transfer of IFN-γ-producing CD8+ T cells into an atopic environment demonstrated their plasticity via an acquired ability in vivo to produce IL-13 and to induce AHR and inflammation in sensitized and challenged recipients. Histone modifications and changes in recruitment of Pol II at lineage specific effector cytokine and transcription factor loci characterized both the in vitro and in vivo lineage conversion of CD8+ T cells. These studies begin to define the mechanisms through which CD8+ T cells contribute to AHR and inflammation and suggest novel ways to interfere with these processes in airway allergic diseases.
Materials and Methods
Animals
OT-1 TCR transgenic (OT-1) mice and homozygous CD8-deficient mice were bred in the animal facility at National Jewish Health (Denver, CO). OT-1 mice (C57BL/6 strain) express a transgenic TCR specific for SIINFEKL peptide (OVA257–264) as analyzed by FACS staining of peripheral blood cells with antibodies against Vα2 and Vβ5 subunits. CD8-deficient mice were generated by targeting the CD8α-chain gene in C57BL/6 mice (14). Animal experiments in this study were conducted under the protocol approved by the Institutional Animal Care and Use Committee of National Jewish Health.
CD8+ T cell culture
CD8+ effector memory T cells were generated in vitro, as previously described (14). Spleens were obtained from OT-1 mice and processed into mononuclear cells (MNCs) using histopaque (Sigma, Saint Louis, MO). 1 µg/ml SIINFEKL peptide (OVA257–264) was used to stimulate cells for 1.5 hrs. Two days later, living cells were re-isolated using histopaque and cultured in complete RPMI 1640 medium that contained recombinant mouse IL-2 (20 ng/ml) (R&D, Minneapolis, MN) or IL-2+IL-4 (20 ng/ml) (Peprotech, Rocky Hill, NJ) or IL-2+IL-5 (20 ng/ml) (Peprotech, Rocky Hill, NJ). Medium with cytokines was changed every day for a further four days. The cells were then re-stimulated with 1 µg/ml SIINFEKL in medium containing IL-2 (20 ng/ml) and 2 µM monensin (Calbiochem, La Jolla, CA) for 4 hrs (Supplemental Fig. 1A).
Sensitization and airway challenge
CD8-deficient mice were sensitized with 20 µg of OVA (Calbiochem, La Jolla, CA) emulsified in 2.25 mg of alum (AlumImuject; Pierce, Rockford, IL) on day 1 and day 14 by intraperitoneal injection (14). Mice were challenged with 1% OVA for 20 min on days 28, 29 and 30 using an ultrasonic nebulizer (model NE-U07; Omron Healthcare, Kyoto, JP). Airway function was measured and samples were collected on day 32.
Adoptive transfer
CD8+ T cells (1×106) generated in medium containing IL-2 or IL-2+IL-4 were injected into OVA-sensitized CD8-deficient mice intravenously on day 28, followed by three challenges on days 28, 29 and 30. CD8+ T cells (5×106) generated in medium containing IL-2 were injected as a positive control (Supplemental Fig. 1B).
Anti-mouse IL-4 antibody administration
Anti-mouse IL-4 (200 µg) was injected intravenously into sensitized CD8-deficient mice on days 26–30. Control, rat IgG antibody was administered in the same manner. Sensitized mice received 5×106 CD8+ T cells on day 28 intravenously followed by three challenges on days 28, 29 and 30. Airway function was measured and samples were collected on day 32 (Supplemental Fig. 1C).
Assessment of airway function
Airway function was assessed as described previously (14, 15) by measuring changes in airway resistance (RL) in response to increasing doses of inhaled methacholine (MCh) (Sigma, Saint Louis, MO). Data were presented as percentage change from the baseline RL values after saline inhalation.
Bronchoalveolar lavage (BAL) analysis
After measurement of AHR, lungs were lavaged via the tracheal tube with 1 ml of HBSS. The supernatants were collected and IFN-γ, IL-4, IL-5 and IL-13 (eBiosicence, San Diego, CA) levels were measured by ELISA (11, 14). Total leukocyte numbers were counted and differentiated (11, 14).
Lung histology
Lungs were isolated and fixed in 10% formalin, then embedded in paraffin and cut into 5 µm thick tissue sections. Sections were stained with periodic acid-Schiff (PAS). Mucus-containing cells were quantitated as previously described (30).
Recovery of and in vitro stimulation of adoptively transferred CD8+ T cells
Lung cells from sensitized and challenged CD8-deficient mice which received CD8+ T cells were isolated by collagenase (GIBCO, Carlsbad, CA) digestion and enriched using nylon wool columns (14). CD8+ T cells were further purified using MACS beads (Miltenyi Biotec, Auburn, CA). Isolated CD8+ T cells (1×106/ml) were stimulated with 50 ng/ml PMA (Calbiochem, La Jolla, CA) and 1 µM ionomycin (Calbiochem, La Jolla, CA) in the presence of 2 µM monensin for 4 hrs. Cells were collected and washed twice with PBS containing 1% BSA for IFN-γ and IL-13 intracellular staining as described below.
Flow cytometric analysis
For surface staining, cells were washed with PBS containing 1% BSA twice, then incubated with anti-mouse CD16/CD32 (2.4G2) (BD Bioscience, San Jose, CA) at 4°C for 5 min and stained with PE labeled anti-mouse IL-4Rα (mIL4R-M1) (BD Bioscience) or PE labeled anti-mouse IL-5Rα (T21) (BD Bioscience). Isotype-matched antibodies were used as controls. For intracellular staining, 1×106/ml cells were washed with PBS containing 1% BSA twice, then stimulated with 1 µg/ml SIINFEKL in the presence of 2 µM monensin at 37°C for 4 hrs. After fixation with 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) and permeabilization with 0.1% saponin (Sigma, Saint Louis, MO), cells were stained with FITC-labeled anti-mouse IFN-γ (XMG 1.2) (eBioscience), PE labeled anti-mouse IL-13 (eBio13A) (eBioscience) or Alexa Flour 647-labeled anti-mouse Eomes (Dan11mag) (eBioscience). Cell staining was monitored on a FACSCalibur (BD Bioscience) and analyzed using Flowjo software (Tree Star, Inc, Ashland, OR).
RNA preparation and analyses
Total RNA was extracted from 5×106 differentiated CD8+ T cells using the RNeasy Mini kit (Qiagen, Valencia, CA). For Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR), 1 µg of total RNA was converted into cDNA using iScript cDNA Synthesis kit (Bio-Rad, Hercules, CA). Quantitative real-time PCR was performed using the ABI 7700 sequence detection system (Applied Biosystems, Foster City, CA). Fold-changes were determined using the 2 −ΔΔCt method, with normalization to expression of mouse GAPDH.
Chromatin immunoprecipitation (ChIP)
ChIP assays were performed according to manufacturer protocols (31) with ChIP assay kit reagents (Active Motif, Carlsbad, CA). Briefly, chromatin was crosslinked by the addition of methanol-free formaldehyde to target cells at a final concentration of 1% at room temperature (RT). Crosslinking was stopped by the addition of 0.125 M glycine at RT. Following cell lysis, the cross-linked chromatin was sheared using a Covaris S2 focused energy isothermal sonicator to an average size of 300–500 base pairs, with highest density at 500 bp. Chromatin immunoprecipitation was performed according to manufacturer's protocols with magnetic beads and the following antibodies: anti-H3K4me3, anti-H3K27me3, anti-RNA Pol II (Abcam, Cambridge, MA), anti-RNA Pol II pS5CTD (Active Motif, Carlsbad, CA), anti-GATA3 (Santa Cruz Biotech, Santa Cruz, CA). The immunoprecipitated genomic DNA was analyzed via SYBR green quantitative PCR (qPCR). A matched isotype antibody was used as a negative control. The following promoter specific primers were used: GATA3, (forward) 5’-ggttgcagtttccttgtgct-3’ (reverse) 5’-cgacgcaacttaaggaggtt-3’; T-bet, (forward) 5’-aacttcctgggggagagaaa-3’ (reverse) 5’-gaattcgcttttggtgagga-3’; IL-13, (forward) 5’-ccaccgtggaaataaaccac-3’ (reverse) 5’-tctctgctttgttgggcatt-3’; IFN-γ, (forward) 5’-agagcccaaggagtcgaaag-3’ (reverse) 5’-tacctgatcgaaggctcctc-3’. Quantitative PCR data were analyzed via the %input methodology: (2^ (CT of total input – CT of specific IP)) ×100. ChIP genomic DNA from anti-GATA3 IP was also examined using IL-13 promoter specific primers via end-point PCR followed by agarose gel electrophoresis. The ChIP-PCR data was analyzed via Image J densitometry.
Statistical analysis
All data were representative of at least three independent experiments with 4 mice/group. Results were expressed as the mean±SEM. Student’s two-tailed t test was used to determine the level of difference between two groups. ANOVA was used to determine the levels of difference among more than three groups. The Mann-Whitney and T-test statistical tests were performed for analysis of the ChIP data taking into account variability in the assays across multiple biological repeats.
Results
IL-4 promotes CD8+ T cell-mediated AHR and inflammation in vivo
CD8-deficient mice develop lower AHR and eosinophilia than WT mice following sensitization and challenge in asthma models (14) and adoptive transfer of 5×106 CD8+ T cells restores the full extent of AHR, eosinophilia and goblet cell metaplasia (14). Thus, in response to asthma-related stimuli, skewed CD8+ T cells contribute to AHR and inflammation in vivo. To define the role of IL-4, a prototypical asthma-associated cytokine, in CD8+ T cell-mediated regulation of AHR and inflammation, 1×106 CD8+ T cells differentiated in IL-2 or IL-2+IL-4 in vitro (Supplemental Fig. 1A) were transferred into sensitized CD8-deficient mice prior to airway challenge (Supplemental Fig. 1B). The results demonstrated that 1×106 CD8+ T cells differentiated in IL-2+IL-4 fully restored AHR and airway inflammation (Figs. 1A and B). In contrast, 1×106 CD8+ T cells differentiated in IL-2 alone could not restore these responses, although a higher dose of 5×106 cells was sufficient. IFN-γ levels were significantly lower and IL-4, IL-5 and IL-13 levels were significantly higher in the BAL fluid of mice which received CD8+ T cells differentiated in IL-2+IL-4 compared to those which received CD8+ T cells differentiated in IL-2 alone (Figs. 1C-F). Lung sections were analyzed by PAS staining and showed that the recipients of CD8+ T cells differentiated in IL-2+IL-4, but not IL-2 alone, had significantly increased numbers of PAS+ mucus-containing goblet cells and an increased accumulation of inflammatory cells (Fig. 1G). IL-4+IL-2, but not IL-2 alone, was sufficient to convert CD8+ T cells to a pro-asthmatic phenotype.
Figure 1. Limiting numbers of CD8+ T cells differentiated in IL-2+IL-4 restore AHR and inflammation in CD8-deficient recipients.
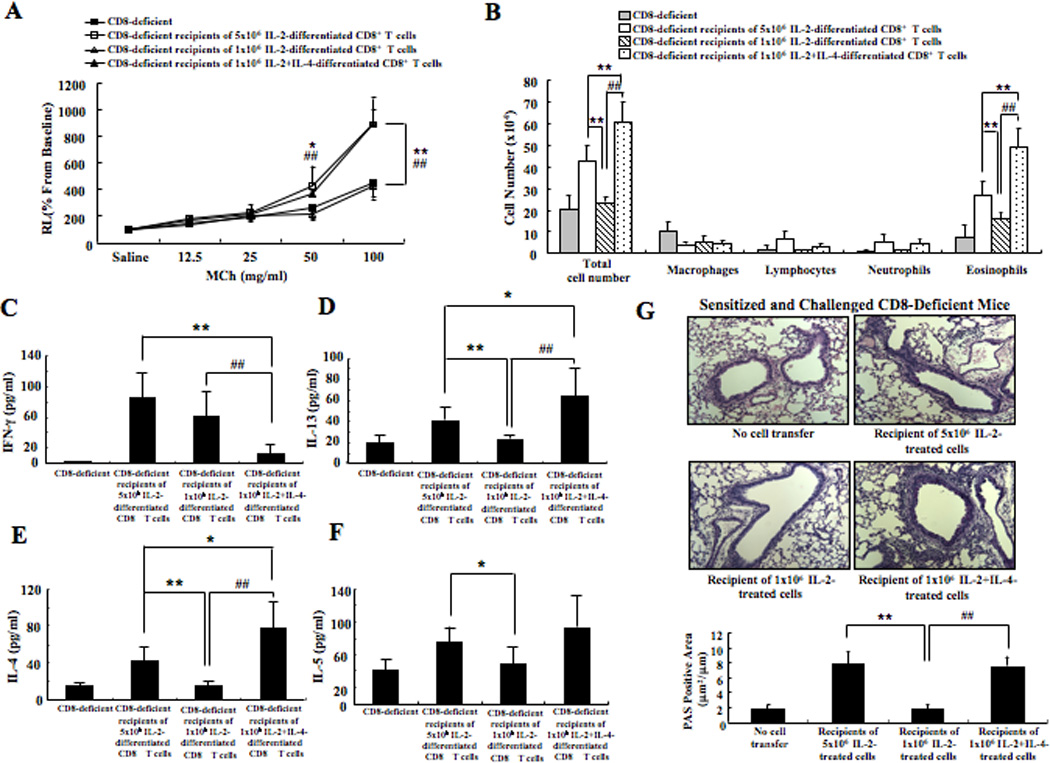
CD8-deficient mice were sensitized and challenged and received no cells, 1×106, or 5×106 CD8+ T cells differentiated in IL-2 alone or IL-2+IL-4. (A) Changes in airway resistance (RL) were measured in response to increasing concentrations of methacholine. (B) Cell composition in BAL fluid. Cytokine levels in BAL fluid. The top 2 graphs show IFN-γ (C) and IL-13 (D) levels and the bottom 2 graphs show IL-4 (E) and IL-5 (F) levels. (G) Representative photomicrographs of lung histology (×200). Quantitative analysis of goblet cells was as described in Materials and Methods. Data (mean±SEM) are from 2–3 experiments with 3–4 mice/experiment. **p<0.01 and *p<0.05 compared to sensitized and challenged CD8-deficient recipients of 5×106 IL-2-differentiated cells. ##p<0.01 compared to sensitized and challenged CD8-deficient recipients of 1×106 IL-2-differentiated cells.
IL-4 is essential for the functional conversion of transferred CD8+ T cells in sensitized and challenged recipients
Given the ability of IL-4 to skew CD8+ T cells in vitro, we examined the role of IL-4 in mediating in vivo CD8+ T cell conversion. Anti-mouse IL-4 or isotype control was administered to sensitized CD8-deficient mice prior to transfer of 5×106 CD8+ T cells (differentiated in IL-2 alone) as outlined in Supplemental Figure 1C. Mice treated with anti-IL-4 had lower AHR and less eosinophilia in BAL fluid, similar to levels observed with sensitized and challenged CD8-deficient mice that did not receive any cells (Figs. 2A and B). Levels of IL-4, IL-5, and IL-13 in BAL fluid were lower and IFN-γ was significantly higher in recipient mice treated with anti-IL-4 (Figs. 2C-F). Histological examination showed that numbers of PAS+ mucus-containing goblet cells were significantly lower in the recipients that received anti-IL-4 (Fig. 2G).
Figure 2. Anti-IL-4 treatment of CD8-deficient recipients of CD8+ T cells prevents restoration of AHR and inflammation.
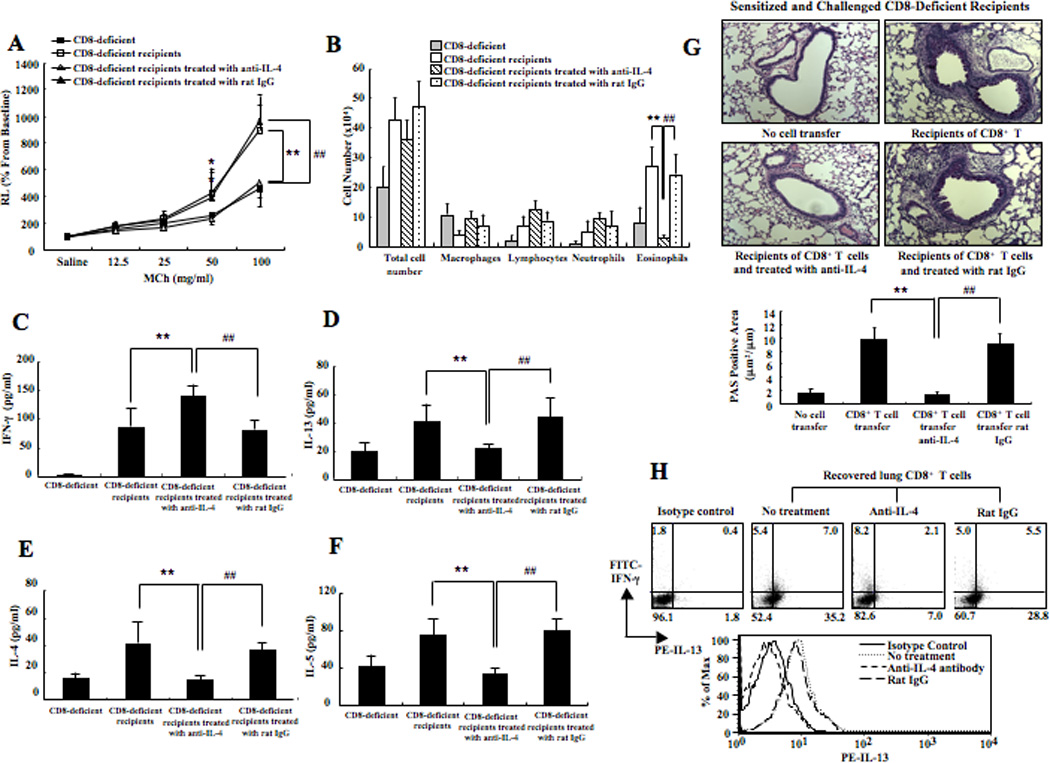
(A) Changes in airway resistance (RL) were measured in response to increasing concentrations of methacholine. (B) Cell composition in BAL fluid. Cytokine levels in BAL fluid. The top 2 graphs show IFN-γ (C) and IL-13 (D) levels and the bottom 2 graphs show IL-4 (E) and IL-5 (F) levels. (G) Representative photomicrographs of lung histology (×200). Quantitative analysis of goblet cells was as described in Materials and Methods. (H) IFN-γ and IL-13 expression in recovered lung CD8+ T cells. Data (mean±SEM) were from at least 6–10 mice. **p<0.01 and *p<0.05 compared to sensitized and challenged CD8-deficient recipients of 5×106 IL-2-differentiated CD8+ T cells group. ##p<0.01 compared to sensitized and challenged CD8-deficient recipients of 5×106 IL-2-differentiated CD8+ T cells treated with an isotype control.
Lung CD8+ T cells were recovered after challenge of CD8-deficient mice that had received CD8+ T cells and treated with anti-IL-4. The cells were stimulated with PMA and ionomycin in the presence of monensin for 4 hrs. The number of IL-13 single positive cells was dramatically lower and IFN-γ single positive cells were higher in the CD8+ T cells recovered from the lungs of mice treated with anti-IL-4 compared to those treated with isotype control antibody or mice not treated with antibody (Fig. 2H). Thus, IL-4 was required for the in vivo conversion of CD8+ T cells to an IL-13-secreting, Tc2 phenotype.
Transfer of Tc1 skewed CD8+ T cells to an allergen-sensitized host engenders their Tc2 epigenetic conversion
IL-13 expression is regulated in part by the lineage-specific transcription factor GATA3 (32). However, the role of IL-4 in regulating GATA3-mediated IL-13 expression by asthma-promoting CD8+ T cells remains unexplored. Utilizing ChIP-qPCR, histone modifications were compared at the promoter regions of IL-13, IFN-γ, GATA3, and T-bet in CD8+ T cells differentiated in vitro with IL-2 or recovered from the lungs following adoptive transfer (of 5×106 IL-2-treated cells) to allergen-sensitized and challenged recipients, as illustrated in Figures 3A-D. Association of the permissive H3K4me3 histone modification at the IL-13 promoter increased 2-fold in recovered lung CD8+ T cells while the reciprocal repressive H3K27me3 histone modification decreased 11-fold compared to pre-transfer cells (Fig. 3A). Association of the permissive H3K4me3 histone modification at the GATA3 promoter also increased 3-fold in recovered lung CD8+ T cells compared to pre-transfer CD8+ T cells, while reciprocal repressive H3K27me3 modification decreased 9-fold (Fig. 3C). In contrast, reciprocal changes in the permissive and repressive histone modifications were not observed at the IFN-γ and T-bet promoters. While H3K27me3 decreased at the IFN-γ and T-bet promoters in recovered lung cells vs. pre-transfer, no change in H3K4me3 was observed (Figs. 3B and D, respectively). These data indicate that in vivo Tc2 plasticity of CD8+ T cells was associated with epigenetic regulation of the key induced, lineage-specific, promoter loci.
Figure 3. ChIP analysis of histone modifications in pre-transfer IL-2-differentiated CD8+ T cells compared to recovered lung CD8+ T cells.
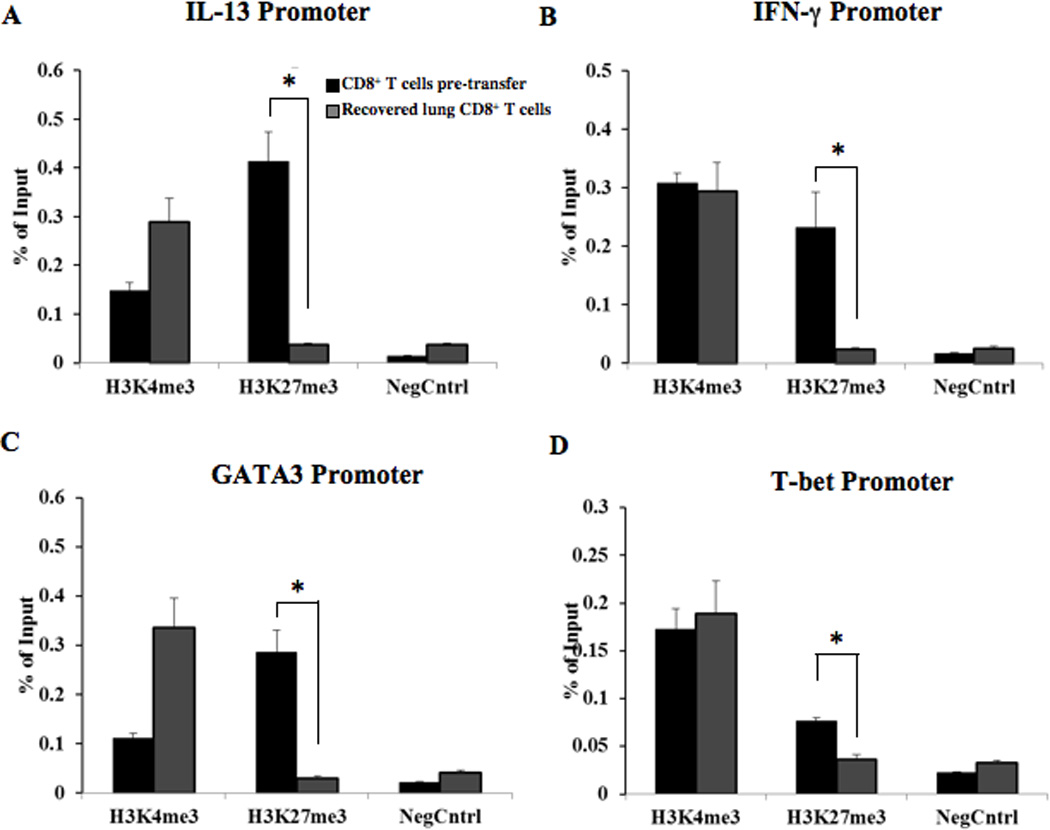
ChIP analysis of CD8+ T cells to determine changes in permissive (H3K4me3) and repressive (H3K27me3) histone modifications from transferred CD8 T cells to recovered lung CD8+ cells post-adoptive transfer. ChIP was coupled with qPCR using primers specific for the core promoter regions (−500 relative to the transcription start site) of IL-13 (A), IFN-γ (B), GATA3 (C), and T-bet (D). Two sets of stimulated CD8+ T cells were analyzed for each IP and promoter locus: pre-adoptive transfer, in vitro IL-2-differentiated CD8+ T cells and post-adoptive transfer CD8+ T cells recovered from the lungs of sensitized and challenged CD8-deficient recipients. The ChIP isolated genomic DNA was used as the template for qPCR analysis with the promoter locus specific primers. An isotype-matched antibody was used as a negative control for each sample and primer set. The data were calculated using the % total genomic input method. The data are from cells pooled from the lungs of 10 mice. Statistical significance was calculated using the Mann-Whitney test and T-test, *p<0.05.
IL-4 alters the phenotype of CD8+ T cells for IL-13 production in vitro
We used a staged in vitro differentiation system to more closely define the mechanisms through which IL-4 regulates CD8+ T cell conversion. The protocol for differentiation of CD8+ T cells in vitro is illustrated in Supplemental Figure 1A. Approximately 25% of the cells were positive for IL-4 receptor on day 0 and increased on days 2 and 4 in the IL-2-treated group (Table 1 and Supplemental Fig. 2A). In the presence of IL-2+IL-4, the percentage of IL-4 receptor-positive cells was further increased. As a control, there was little change in the number of IL-5 receptor-positive cells in any of the groups (Supplemental Fig. 2B).
Table 1.
% Positive Cells
| Day 0 | Day 2 | Day 4 | |||
|---|---|---|---|---|---|
| IL-2 | IL-2+IL-4 | IL-2 | IL-2+IL-4 | ||
| %IL-4Rα | 24.5±1.6 | 38.2±3.8 | 50.6±5.5** | 75.2±2.9 | 90±5.6## |
| %IL-5Rα | 11.4±1.7 | 15.3±3.3 | 15.8±2.5 | 12.8±3.8 | 15.7±5.2 |
Kinetics of IL-4 and IL-5 receptor expression in CD8+ T cells differentiated in IL-2 or IL-2+IL-4 in vitro. The data shown are representative of at least 9 independent experiments.
p<0.01 compared to the IL-2 group on day 2,
p<0.01 compared to the IL-2 group on day 4.
CD8+ T cells were differentiated in the presence of IL-2, with or without IL-4 or IL-5 as a control. After 6 days in culture, cells were re-stimulated with SIINFEKL in the presence of monensin for 4 hrs. As shown in Figure 4A and Table 2, virtually all of the cells were negative for both IFN-γ and IL-13 in the IL-2-treated group without antigen re-stimulation; about 90% of the cells were identified as IFN-γ-producing after SIINFEKL re-stimulation. Few cells were IL-13-producing after SIINFEKL re-stimulation. Among cells cultured in medium containing IL-2+IL-4, few cells (~1%) were IFN-γ-producing and small numbers (~6%) produced IL-13. Following SIINFEKL re-stimulation, there was a significant decrease in the numbers of IFN-γ-producing cells compared to that seen in IL-2-treated cultures and ~40% of the cells were now IL-13-producing. A substantial fraction of cells were identified as double negatives in the IL-2+IL-4 treated group even after re-stimulation with SIINFEKL. CD8+ T cells which were differentiated in medium containing IL-2+IL-5 showed a similar phenotype as IL-2 treated cells (data not shown).
Figure 4. IFN-γ and IL-13 expression in CD8+ T cells differentiated in IL-2 or IL-2+IL-4 in vitro.
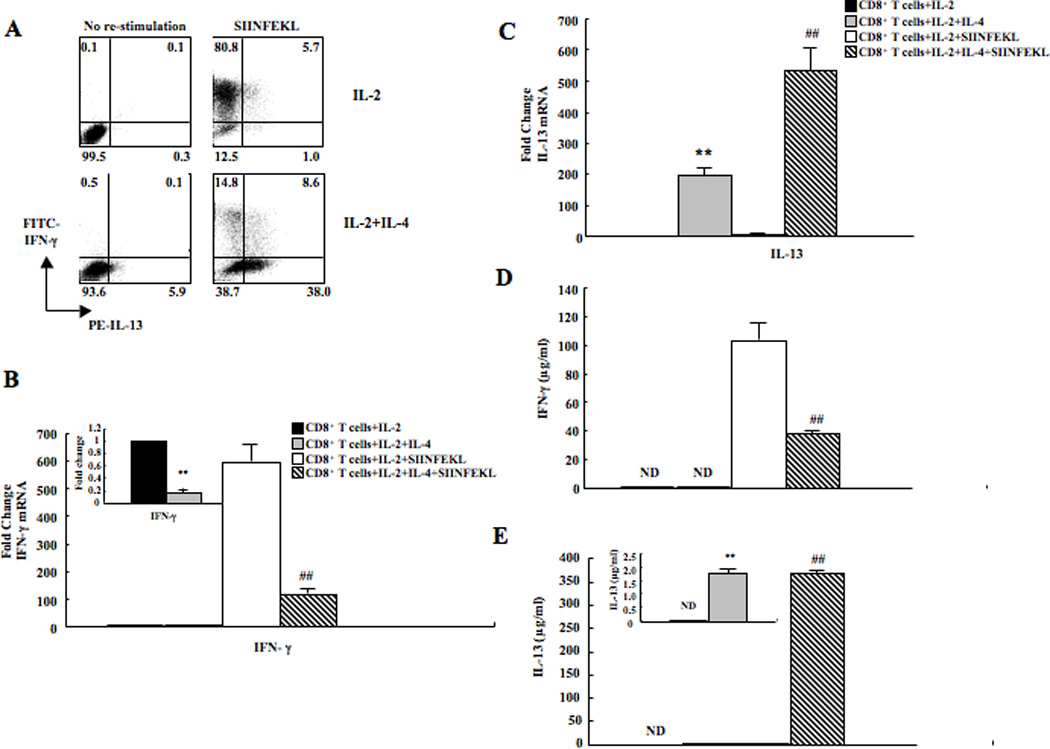
(A) Representative results of intracellular staining of IFN-γ and IL-13 expression in CD8+ T cells with or without SIINFEKL re-stimulation. (B) IFN-γ mRNA levels in CD8+ T cells. (C) IL-13 mRNA levels. (D) IFN-γ protein levels. (E) IL-13 protein levels. Data (mean±SEM) are from 6–10 independent experiments. **p<0.01, *p<0.05 compared to the IL-2 group, ##p<0.01 compared to the IL-2+SIINFEKL group.
Table 2.
% Positive Cells
| IL-2 | IL-2 +IL-4 |
IL-2+ SIINFEKL |
IL-2+IL-4 +SIINFEKL |
|
|---|---|---|---|---|
| IFN-γ single positive cells |
0.2±0.1 | 0.8±0.3** | 85.4±4.8 | 16.7±9.0## |
| IL-13 single positive cells |
0.2±0.1 | 5.6±1.4** | 0.5±0.4 | 33.3±8.7## |
| IFN-γ+ IL-13+ | 0 | 0.1 | 3.2±2.4 | 7.5±2.5## |
| IFN-γ− IL-13− | 99.6±0.2 | 94±2.4** | 10.9±4.6 | 42.4±8.1## |
IFN-γ and IL-13 expression in CD8+ T cells differentiated in IL-2 or IL-2+IL-4. Intracellular staining of IFN-γ and IL-13 in CD8+ T cells with or without SIINFEKL re-stimulation. Data (mean±SEM) showing % positive cells were from at least 9 independent experiments.
p<0.01 compared to the IL-2 group,
p<0.01 compared to the IL-2+SIINFEKL group.
Cells from each group were assessed by quantitative RT-PCR. IFN-γ mRNA levels were significantly lower in cells treated with IL-2+IL-4 compared to cells treated with IL-2 alone. After re-stimulation with SIINFEKL for 4 hrs, IFN-γ mRNA levels remained lower in IL-2+IL-4-treated cells (Fig. 4B). In contrast, IL-13 mRNA levels were significantly higher in cells treated with IL-2+IL-4 compared to cells treated with IL-2 alone and the cells showed the same pattern after re-stimulation with SIINFEKL for 4 hrs (Fig. 4C). IFN-γ and IL-13 protein levels were determined by ELISA. IFN-γ levels were below limits of detection in cells treated either with IL-2 or IL-2+IL-4. After re-stimulation with SIIFNFEKL, IFN-γ levels produced by IL-2-treated cells were significantly increased. IFN-γ production was significantly lower in the cells differentiated in the presence of IL-2+IL-4 (Fig. 4D). In contrast, IL-13 levels were significantly higher in cultures of cells grown with IL-2+IL-4 compared with IL-2 alone. The same pattern of increased IL-13 production was seen after re-stimulation with SIINFEKL for 4 hrs except that the levels produced by cells grown in IL-2+IL-4 were much higher after antigen re-stimulation (Fig. 4E). IL-4 and IL-5 protein levels were also evaluated and the data showed that both were increased in cultures treated with IL-2+IL-4 compared to IL-2 alone (data not shown). Overall, CD8+ T cells shifted their cytokine profile from exclusively IFN-γ-producing to predominantly Th2 cytokine-producing in the presence of IL-4.
IL-4 alters the levels of lineage specific transcription factor expression in CD8+ T cells in vitro
Expression of IFN-γ and IL-13 in T cells is regulated by the transcription factors T-bet and GATA3, respectively (33, 34). Repressor of GATA (ROG) is a transcription factor that controls the expression of GATA3 (35, 36). CD8+ T cells cultured with IL-2+IL-4 expressed significantly lower levels of T-bet and ROG and significantly higher levels of GATA3 compared to cells cultured with IL-2 alone both before and after re-stimulation with SIINFEKL (Fig. 5A).
Figure 5. Transcription factor expressions in CD8+ T cells.
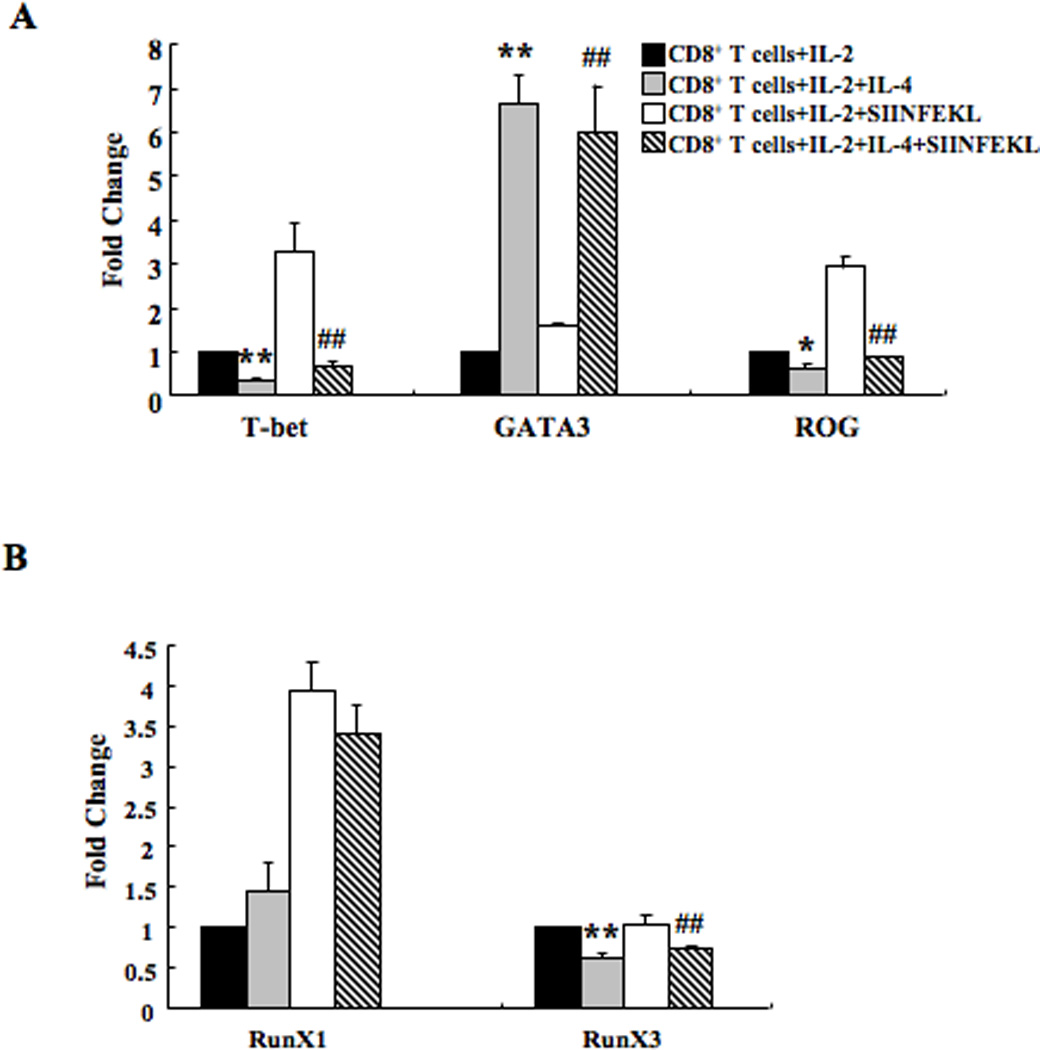
(A) T-bet, GATA3, and ROG expression in CD8+ T cells differentiated in IL-2 or IL-2+IL-4 with or without SIINFEKL re-stimulation. (B) Runx1 and Runx3 expression levels. Data (mean±SEM) are from 3–5 independent experiments and expressed as the mean±SEM. **p<0.01, *p<0.05 compared to the IL-2 group, ##p<0.01 compared to the IL-2+SIINFEKL group.
Runx3 augments Th1 and down-modulates the Th2 phenotype by attenuating GATA3 expression (37). Runx1 inhibits the differentiation of naive CD4+ T cells into the Th2 lineage by repressing GATA3 expression (38). Runx3 but not Runx1 expression levels were significantly lower in cells treated with IL-2+IL-4 compared to cells treated with IL-2 alone, with or without re-stimulation by SIINFEKL (Fig. 5B).
Eomesodermin (Eomes) is a member of the same subfamily of T-box factors as T-bet and regulates IFN-γ expression in CD8+ T cells (39, 40). The addition of IL-4 to the IL-2-containing medium resulted in a lower percentage of cells stained positive for Eomes, both before and after re-stimulation with either SIINFEKL (Supplemental Fig. 3). Thus, the changes induced in the CD8+ T cell cytokine profile by IL-4 were accompanied by changes in the levels of transcription factors known to be “lineage-specific” differentiation factors.
IL-4 regulates GATA3 recruitment, histone modifications, and RNA Pol II recruitment in CD8+ T cells
As described above (Fig. 3), T cell lineage conversion is associated with post-translational modifications to histone proteins at the IL-13, IFN-γ, GATA3, and T-bet promoters during in vivo Tc2 conversion (25, 26, 41). ChIP-qPCR assays were performed using the step-wise differentiation model of CD8+ Tc2 conversion, which enabled a multi-stage examination of IL-4-regulated re-programming of CD8+ T cells for IL-13 production (Supplemental Fig. 1A). The analysis revealed a 30-fold increase in GATA3 recruitment to the IL-13 promoter in IL-2+IL-4 treated CD8+ T cells vs. IL-2 alone (Fig. 6A). Moreover, a significant decrease in IL-13 promoter-associated GATA3 recruitment was observed in IL-2+IL-4 cells following treatment with SIINFEKL, suggesting temporal, spatial and/or TCR-mediated regulation of the promoter region. In contrast, no significant recruitment of GATA3 to the IL-13 promoter was observed in cells that had been differentiated in IL-2 alone with or without SIINFEKL. This dynamic GATA3 recruitment was confirmed by ChIP-PCR followed by gel electrophoresis (Supplemental Fig. 4).
Figure 6. ChIP analysis of GATA3 recruitment, histone modifications, and Pol II recruitment in differentiated CD8+ T cells.
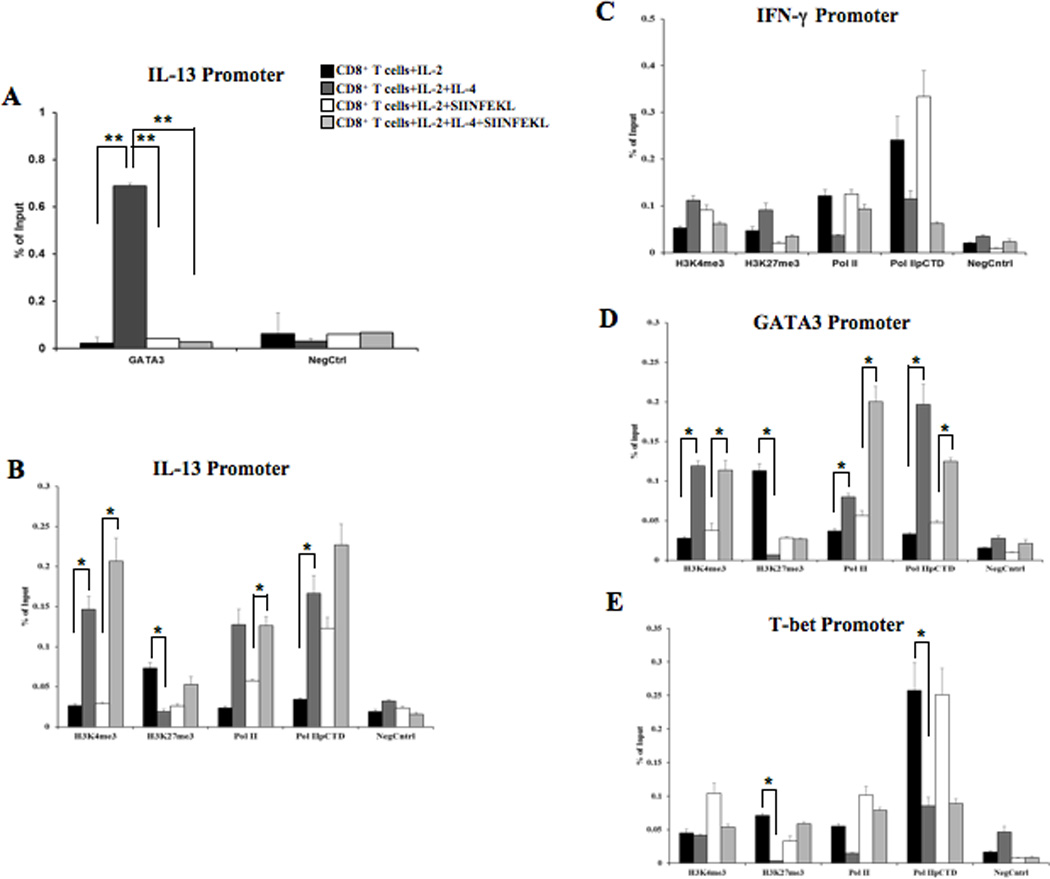
Four groups of stimulated CD8+ T cells were analyzed for each IP and promoter locus: IL-2 alone, IL-2+IL-4, IL-2+SIINFEKL (TCR stimulus), IL-2+IL-4+SIINFEKL. The ChIP isolated genomic DNA was used as the template for qPCR analysis with the promoter locus specific primers. An isotype-matched antibody was used as a negative control for each sample and primer set. Recruitment of GATA3 to the IL-13 promoter was examined via ChIP (A). Histone modifications (permissive H3K4me3 and repressive H3K27me3), recruitment of the RNA Pol II complex (Pol II), and recruitment of the Pol II initiation complex (phosphorylated serine 5 in the C-terminal domain, Pol IIpCTD) were also examined (B-E). ChIP was coupled with qPCR using primers specific for the core promoter regions (−500 relative to the transcription start site) of IL-13 (A-B), IFN-γ (C), GATA3 (D), and T-bet (E). The data were calculated using the % total genomic input method. The data represent the averages and SEM across three independent biological experiments. Statistical significance was calculated using the Mann-Whitney and/or T-test, **p<0.01, *p<0.05.
We next examined epigenetic regulation of the promoter regions of IL-13 and IFN-γ at various stages of in vitro CD8+ T cell differentiation. Association of the permissive histone modification H3K4me3 at the IL-13 promoter increased 5-fold in IL-2+IL-4 vs. IL-2 treated CD8+ T cells, while H3K27me3 decreased 4-fold (Fig. 6B), corresponding to the period of GATA3 promoter-recruitment and association (Fig. 6A). ChIP was also used to analyze recruitment of the RNA polymerase II complex (Pol II) given the evidence for Pol II-histone methylation interaction and regulation (42, 43). IL-2+IL-4 treatment increased recruitment of Pol II and Pol II with a phosphorylated serine 5 of the C-terminal domain (Pol IIpCTD; the initiation complex) to the IL-13 promoter approximately 5-fold vs. IL-2 alone (Fig. 6B), despite low levels of IL-13 transcription and very low protein production at this stage (Fig. 4), suggesting that IL-2+IL-4 treatment poised the IL-13 locus for active transcription, which then occurred after T-cell receptor stimulation. No significant differences in histone modifications or Pol II recruitment were observed at the IFN-γ promoter in IL-2+IL-4 treated CD8+ T cells vs. IL-2 alone (Fig. 6C).
We also examined the GATA3 and T-bet promoter regions via ChIP. IL-2+IL-4 treatment increased association of permissive H3K4me3 at GATA3 by more than 4-fold compared to IL-2 alone (Fig. 6D). A similar pattern was observed following SIINFEKL stimulation. IL-2+IL-4 also induced a 16-fold decreased association of repressive H3K27me3 at GATA3 compared with IL-2 alone (Fig. 6D). There were no significant differences detected in H3K27me3 between the treatment groups following SIINFEKL stimulation. IL-2+IL-4 treatment increased Pol II recruitment more than 2-fold and increased recruitment of Pol IIpCTD more than 6-fold compared with IL-2 treatment alone (Fig. 6D). A similar pattern was observed following SIINFEKL stimulation. While the recruitment of Pol II and Pol IIpCTD at the T-bet promoter was greater in IL-2 vs. IL-2+IL-4 treated cells, the differences in histone modifications between the treatment groups were not significant (Fig. 6E). Thus, histone modifications were co-regulated with Pol II recruitment at the IL-13 and GATA3 promoters but not the IFN-γ or T-bet promoters. We observed that IL-4-regulated GATA3 recruitment coincided with histone modifications at IL-13 promoter, although significant IL-13 protein production required SIINFEKL stimulation.
Discussion
Substantial evidence supports the notion that CD8+ T cells, in concert with CD4+ T cells, play important roles in allergic airway disease, through production of IL-13 and other inflammatory cytokines (9, 13, 14). IL-4-induced skewing from conventional IFN-γ-producing effector cells to pathogenic IL-13-producing cells was shown to proceed in a step-wise manner characterized initially by changes in expression of the lineage specific transcription factors GATA3 (increased) and T-bet (decreased) and then of IL-13 and IFN-γ. These changes were accompanied by histone modifications and RNA Pol II recruitment at the proximal promoters, especially for those regulating GATA3 and IL-13. Following TCR re-stimulation, increased expression of IL-13 RNA and protein levels occurred. Functionally, limited numbers of in vitro-converted CD8+ T cells fully restored allergen-induced AHR and inflammation following adoptive transfer into CD8-deficient mice. When non-converted CD8+ T cells (i.e., those grown in IL-2 alone) were transferred and then recovered from sensitized and challenged CD8-deficient mice, they recapitulated and confirmed the molecular changes seen in vitro, with conversion to IL-13 production in an IL-4 dependent manner.
IL-4 stimulates a spectrum of intracellular signaling cascades in CD8+ T cells (44). IL-4 and IFN-γ reciprocally regulate type 2 polarization of CD8+ T cells. IL-4 was shown to induce effector CD8+ T cells that express type 2 as well as type 1 cytokines. Finkelman et al. demonstrated that endogenously produced IL-4 was a direct, non-redundant stimulator of CD8+ T cell proliferation in antigen-induced CD8+ T cell responses and these stimulatory effects were enhanced in a model of airway disease (24). Th1 effector memory cells were able to turn off IFN-γ expression in vivo and “re-differentiate” under the right conditions (45). In a tumor model, tumor-derived IL-4 induced the expression of type 2 cytokines and GATA3 by responding CD8+ T cells, reducing anti-tumor efficacy (46).
In the present study, we have proposed that CD8 T cells committed to IFN-γ production, when exposed to IL-4 in vitro or an atopic environment in vivo, transit distinct stages characterized by changes in transcription factor expression, histone modifications, and ultimately, following antigen stimulation, transcription, and translation of IL-13. In a different system, Hayashi et al. (47) similarly showed that Th1 cells, committed to IFN-γ production, can also make Th2 cytokines such as IL-13 under specific conditions. Our data are compatible with the notion that IFN-γ-positive cells can acquire the capacity to produce IL-13 when expanded in vitro with IL-4 or exposed to IL-4 in vivo. What remains unclear is whether these cells have lost the ability to produce IFN-γ and thus, are clearly "converted" to IL-13 production or rather emerge from CD8+ IFN-γ-negative cells. As reviewed by O'Shea and Paul (22), it is still unclear what is meant by the terms lineage stability and plasticity/conversion and how they can be distinguished. Moreover, such findings do support arguments for flexibility in cytokine production and less likely a strict and fixed lineage commitment model in CD8 T cell differentiation. At the present time due to technological issues, it remains difficult to ascertain the frequency and origin of converted cells.
Based on our earlier findings of the role of CD4+IL-4+ T cells in the initial activation of CD8+ T cells (14), we examined the role of IL-4 during the functional differentiation of CD8+ T cells in a carefully regulated and staged in vitro system and identified mechanisms controlling the plasticity of CD8+ T cells, focusing initially on the functional development of CD8+ T cells into IFN-γ or IL-13 producers in vitro. To examine the mechanisms involved in this “step-wise” conversion, expression of the lineage-specific master regulatory transcription factors which control T cell differentiation were monitored. Prior to TCR re-stimulation but following IL-4 treatment, GATA3 expression levels significantly increased in T cells, while T-bet and repressor of GATA3 (ROG) levels decreased. Differences in Runx3 and Eomes expression were also observed. Significantly, changes in lineage-specific transcription factors required only IL-4 and were not dependent on TCR re-stimulation. In contrast, IL-4 mediated induction of IL-13 RNA and protein expression required TCR re-stimulation. Thus, IL-4 appeared to play different roles at the stages of CD8+ T cell differentiation and activation. IL-4 alone was sufficient to alter lineage-specific transcription factor levels during CD8+ T cell differentiation, poising these cells to produce cytokines following TCR engagement.
We observed IL-4 induced epigenetic programming of CD8+ T cells which poised the IL-13 locus for transcription following TCR re-stimulation in vitro or following adoptive transfer and allergen challenge in vivo (28, 29, 47). H3K4me3 at the GATA3 and IL-13 promoters was dependent on IL-4 treatment and occurred in the absence of TCR re-stimulation, while H3K4me3 histone modifications at the T-bet and IFN-γ promoters were not regulated by IL-4. Interestingly, loss of the repressive histone mark, H3K27me3, was observed at the GATA3, IL-13, T-bet, and IFN-γ promoters in CD8+ T cells recovered following adoptive transfer in vivo. However, only the GATA3 promoter exhibited a significant loss of H3K27me3 in the parallel in vitro comparisons, IL-2 vs. IL-2+IL-4 and TCR re-stimulation. Addition of IL-4 in the absence of TCR re-stimulation did result in significant loss of the repressive H3K27me3 mark at the GATA3, IL-13, and T-bet promoters but not at the IFN-γ promoter. The differences observed between the in vivo and in vitro histone modification patterns likely reflect the inherent complexities of in vivo inflammation, which is characterized by multiple cytokine pathways and other microenvironment signals. It is interesting to note that although the IFN-γ promoter remained intransigent to the effects of IL-4 in vitro, maintenance of permissive and loss of repressive histone marks in vivo suggested plasticity of CD8+ T cell effector function in the in vivo setting. Nevertheless, the in vivo and in vitro data demonstrated that IL-4 alone, in the absence of TCR re-stimulation, resulted in epigenetic poising of the IL-13 locus through gain of permissive and loss of repressive histone modifications, which was co-regulated with recruitment of Pol II (48).
IL-4 was also required for GATA3 expression in CD8+ T cells and we observed an IL-4 dependent recruitment of GATA3 protein to the IL-13 promoter. In a previous study of in vitro polarized CD4+ T cells, GATA3 binding throughout the genome was dependent on lineage development, was associated with histone modifications, and was not always associated with transcriptional changes (32). Monitoring the step-wise CD8+ T cell skewing reported here, we observed that IL-4 mediated GATA3 promoter recruitment coincided with histone modifications and Pol II recruitment but was temporally separated from the enhancement of IL-13 transcription and protein production. A SIINFEKL TCR stimulus was required for IL-13 protein production, but interestingly, GATA3 was not associated with the IL-13 promoter during the TCR re-stimulation stage of the in vitro system. This observation required application of the staged in vitro system and could not have been made with end-point in vitro polarized cells or in vivo recovered, differentiated cells, given the complexity of the interactions. Wei et al. demonstrated that the activity of GATA3 at a gene-specific level was dependent on the lineage-identity of CD4+ T cells (32). Their study revealed that the specific GATA3 genomic binding loci differed greatly between Th1, Th2, Th17, and iTreg cells and that the binding of GATA3 to a specific gene locus was not always associated with a change in transcription (32). As expected, deletion of GATA3 did affect gene expression in their study, but none of the genes regulated by GATA3 were shared among any of the CD4+ T cell lineages (32). On the other hand, GATA3 did regulate methylation of the histone lysines H3K4 and H3K27 at many of its gene targets, including IL-13, T-bet, and IFN-γ in CD4+ T cells (32). Evaluation of similar events using the staged in vitro system enabled the demonstration that GATA3 activity is also dependent on the temporal activation-phase of T cells, albeit with the recognition that Tc1 and Tc2 CD8+ T cell lineages were examined. Similar to CD4+ T cells, the binding of GATA3 at the IL-13 locus in CD8+ T cells did correspond with changes in histone methylation. Binding of GATA3 was associated with significantly enhanced permissive H3K4me3 and significantly decreased repressive H3K27me3 histone marks at the IL-13 locus in CD8+ T cells treated with IL-4. However, during the TCR re-stimulation stage, when GATA3 was no longer associated with the IL-13 promoter, only the enhanced H3K4me3 permissive mark remained significant. This may imply that in CD8+ T cells, the presence of GATA3 is required for the inhibition of repressive H3K27me3 but is not required for sustaining the permissive H3K4me3 mark. Thus, enhanced IL-13 transcription and protein production in CD8+ T cells were temporally associated with sustained permissive H3K4me3 but not necessarily sustained loss of repressive H3K27me3. We propose a model in which IL-4 mediated GATA3 recruitment regulates histone modifications at the promoter, which then poise the IL-13 locus for transcription following a TCR stimulus. In the absence of IL-4, GATA3 was not recruited to the IL-13 promoter and subsequently, TCR re-stimulation did not result in cytokine production. As a result, an allergic inflammatory lung microenvironment containing IL-4 supports asthma pathogenesis through epigenetically poising CD8+ T cells for Tc2 conversion via differential histone modifications at key promoters. Our observations indicate that the Tc2 pathway requires an extrinsic stimulus, IL-4, while the Tc1 pathway may represent a default pathway, since addition of IL-4 did not greatly affect the histone modifications at T-bet or IFN-γ. This represents an obvious difference from the Th2 lineage pathway characterized by GATA3-mediated epigenetic repression of T-bet and IFN-γ in CD4+ T cells (32), emphasizing the cell-type intrinsic activities of GATA3 and histone modifications. This may also suggest that Tc2 conversion as observed here, reflects functional plasticity in the gain of an epigenetically permissive IL-13 locus rather than a permanent repression of the IFN-γ locus. Further understanding of the molecular mechanisms by which IL-4 and GATA3 regulate the plasticity and/or stable conversion of CD8+ T cells should suggest novel therapeutic strategies and new targets for the treatment of asthma.
Supplementary Material
Acknowledgments
We thank Diana Nabighian for her assistance in preparation of the manuscript.
Grant Support: This work was supported by NIH grants AI-77609, HL-36577, and HL-61005 and by the Walter Scott Foundation. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NHLBI or the NIH.
References
- 1.Busse WW, Lemanske RF., Jr Asthma. New Engl. J. Med. 2001;344:350–362. doi: 10.1056/NEJM200102013440507. [DOI] [PubMed] [Google Scholar]
- 2.Grünig G, Warnock M, Wakil AE, Venkayya R, Brombacher F, Rennick DM, Sheppard D, Mohrs M, Donaldson DD, Locksley RM, Corry DB. Requirement for IL-13 independently of IL-4 in experimental asthma. Science. 1998;18:2261–2263. doi: 10.1126/science.282.5397.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lloyd CM, Hessel EM. Functions of T cells in asthma: more than just T(H)2 cells. Nat. Rev. Immunol. 2010;10:838–848. doi: 10.1038/nri2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim HY, DeKruyff RH, Umetsu DT. The many paths to asthma: phenotype shaped by innate and adaptive immunity. Nat. Immunol. 2010;11:577–584. doi: 10.1038/ni.1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomas MJ, Noble A, Sawicka E, Askenase PW, Kemeny DM. CD8 T cells inhibit IgE via dendritic cell IL-12 induction that promotes Th1 T cell counter-regulation. J. Immunol. 2008;168:216–223. doi: 10.4049/jimmunol.168.1.216. [DOI] [PubMed] [Google Scholar]
- 6.Allakhverdi Z, Lamkhioued B, Olivenstein R, Hamid Q, Renzi PM. CD8 depletion-induced late airway response is characterized by eosinophilia, increased eotaxin, and decreased IFN-gamma expression in rats. Am. J. Respir. Crit. Care Med. 2000;162:1123–1131. doi: 10.1164/ajrccm.162.3.9910001. [DOI] [PubMed] [Google Scholar]
- 7.van Rensen EL, Sont JK, Evertse CE, Willems LN, Mauad T, Hiemstra PS, Sterk PJ the AMPUL Study Group. Bronchial CD8 cell infiltrate and lung function decline in asthma. Am. J. Respir. Crit. Care Med. 2005;172:837–841. doi: 10.1164/rccm.200504-619OC. [DOI] [PubMed] [Google Scholar]
- 8.Miyahara N, Takeda K, Miyahara S, Taube C, Joetham A, Koya T, Matsubara S, Dakhama A, Tager AM, Luster AD, Gelfand EW. Leukotriene B4 (BLT1) is essential for allergen-mediated recruitment of CD8+ T cells and airway hyperresponsiveness. J. Immunol. 2005;174:4979–4984. doi: 10.4049/jimmunol.174.8.4979. [DOI] [PubMed] [Google Scholar]
- 9.Miyahara N, Takeda K, Kodama T, Joetham A, Taube C, Park JW, Miyahara S, Balhorn A, Dakhama A, Gelfand EW. Contribution of antigen-primed CD8+T cells to the development of airway hyperresponsiveness and inflammation is associated with IL-13. J. Immunol. 2004;172:2549–2558. doi: 10.4049/jimmunol.172.4.2549. [DOI] [PubMed] [Google Scholar]
- 10.Takeda K, Dow SW, Miyahara N, Kodama T, Koya T, Taube C, Joetham A, Park JW, Dakhama A, Kedl RM, Gelfand EW. Vaccine-induced CD8+ T cell-dependent suppression of airway hyperresponsiveness and inflammation. J. Immunol. 2009;183:181–190. doi: 10.4049/jimmunol.0803967. [DOI] [PubMed] [Google Scholar]
- 11.Koya T, Miyahara N, Takeda K, Matsubara S, Matsuda H, Swasey C, Balhorn A, Dakhama A, Gelfand EW. CD8+ T cell-mediated airway hyperresponsiveness and inflammation is dependent on CD4+IL-4+ T cells. J. Immunol. 2007;179:2787–2796. doi: 10.4049/jimmunol.179.5.2787. [DOI] [PubMed] [Google Scholar]
- 12.Wells JW, Cowled CJ, Giorgini A, Kemeny DM, Noble A. Regulation of allergic airway inflammation by class I-restricted allergen presentation and CD8 T-cell infiltration. J. Allergy Clin. Immunol. 2007;119:226–234. doi: 10.1016/j.jaci.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 13.Hamelmann E, Oshiba A, Paluh J, Bradley K, Loader J, Potter TA, Larsen GL, Gelfand EW. Requirement for CD8+T cells in the development of airway hyperresponsiveness in a murine model of airway sensitization. J. Exp. Med. 1996;183:1719–1729. doi: 10.1084/jem.183.4.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miyahara N, Swanson BJ, Takeda K, Taube C, Miyahara S, Kodama T, Dakhama A, Ott V, Gelfand EW. Effector memory CD8+ T cells mediate inflammation and airway hyper-responsiveness. Nat. Med. 2004;10:865–869. doi: 10.1038/nm1081. [DOI] [PubMed] [Google Scholar]
- 15.Miyahara N, Takeda K, Miyahara S, Matsubara S, Koya T, Joetham A, Krishnan E, Dakhama A, Haribabu B, Gelfand EW. Requirement for the leukotriene B4 receptor-1 in allergen-induced airway hyperresponsiveness. Am. J. Respir. Crit. Care Med. 2005;172:161–167. doi: 10.1164/rccm.200502-205OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Isogai S, Taha R, Tamaoka M, Yoshizawa Y, Hamid Q, Martin JG. CD8 (+) alpha beta T cells can mediate late airway responses and airway eosinophilia in rats. J. Allergy Clin. Immunol. 2004;114:1345–1352. doi: 10.1016/j.jaci.2004.09.021. [DOI] [PubMed] [Google Scholar]
- 17.Miyahara N, Ohnishi H, Miyahara S, Takeda K, Matsubara S, Matsuda H, Okamoto M, Loader JE, Joetham A, Tanimoto M, Dakaham A, Gelfand EW. Leukotriene B4 release from mast cells in IgE-mediated airway hyperresponsiveness and inflammation. Am. J. Respir. Cell Mol. Biol. 2009;40:672–682. doi: 10.1165/rcmb.2008-0095OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Belz GT, Kallies A. Effector and memory CD8+T cell differentiation: toward a molecular understanding of fate determination. Curr. Opin. Immunol. 2010;222:279–285. doi: 10.1016/j.coi.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 19.Badovinac VP, Twinnereiem AR, Harty JT. Regulation of antigen-specific CD8+T cell homeostasis by perforin and interferon-γ. Science. 2000;290:1354–1358. doi: 10.1126/science.290.5495.1354. [DOI] [PubMed] [Google Scholar]
- 20.Wan YY, Flavell RA. How diverse – CD4 effector T cells and their functions. J. Mol. Cell. Biol. 2009;1:20–36. doi: 10.1093/jmcb/mjp001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou L, Chong M, Littman D. Plasticity of CD4+ T cell lineage differentiation. Immunity. 2009;30:646–655. doi: 10.1016/j.immuni.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 22.O’Shea J, Paul W. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cell. Science. 2010;327:1098–1102. doi: 10.1126/science.1178334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Erard F, Wild MT, Garcia-Sanz JA, Le Gros G. Switch of CD8 T cells to noncytolytic CD8-CD4- cells that make Th2 cytokines and help B cells. Science. 1993;260:1802–1805. doi: 10.1126/science.8511588. [DOI] [PubMed] [Google Scholar]
- 24.Morris SC, Heidorn SM, Herbert DR, Perkins C, Hildeman DA, Khodoun MV, Finkelman FD. Endogenously produced IL-4 nonredundantly stimulates CD8+ T cells proliferation. J. Immunol. 2009;182:1429–1438. doi: 10.4049/jimmunol.182.3.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roh TY, Cuddapah S, Cui K, Zhao K. The genomic landscape of histone modifications in human T cells. Proc. Natl. Acad. Sci. USA. 2006;103:15782–15827. doi: 10.1073/pnas.0607617103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wei G, Wei L, Zhu J, Zang C, Hu LJ, Yao Z, Cui K, Kanno Y, Roh TY, Watford WT, Schones DE, Peng W, Sun HW, Paul WE, O'Shea JJ, Zhao K. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity. 2009;30:155–167. doi: 10.1016/j.immuni.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Araki Y, Wang Z, Zang C, 3rd, Wood H, Schones D, Cui K, Roh TY, Lhotsky B, Wersto RP, Peng W, Becker KG, Zhao K, Weng NP. Genome-wide analysis of histone methylation reveals chromatin state-based regulation of gene transcription and function of memory CD8+ T cells. Immunity. 2009;30:912–925. doi: 10.1016/j.immuni.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Araki Y, Fann M, Wersto R, Weng NP. Histone acetylation facilitates rapid and robust memory CD8 T cell response through differential expression of effector molecules (eomesodermin and its targets: perforin and granzyme B) J. Immunol. 2008;180:8102–8108. doi: 10.4049/jimmunol.180.12.8102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Northrop JK, Wells AD, Shen H. Cutting edge: chromatin remodeling as a molecular basis for the enhanced functionality of memory CD8 T cells. J. Immunol. 2008;181:865–868. doi: 10.4049/jimmunol.181.2.865. [DOI] [PubMed] [Google Scholar]
- 30.Tomkinson A, Cieslewicz G, Duez C, Larson KA, Lee JJ, Gelfand EW. Temporal association between airway hyperresponsiveness and airway eosinophilia in ovalbumin-sensitized mice. Am. J. Respir. Crit. Care Med. 2001;163:721–730. doi: 10.1164/ajrccm.163.3.2005010. [DOI] [PubMed] [Google Scholar]
- 31.Greer SF, Zika E, Conti B, Zhu XS, Ting JP. Enhancement of CIITA transcriptional function by ubiquitin. Nat. Immunol. 2003;2003(4):1074–1082. doi: 10.1038/ni985. [DOI] [PubMed] [Google Scholar]
- 32.Wei G, Abraham BJ, Yagi R, Jothi R, Cui K, Sharma S, Narlikar L, Northrup DL, Tang Q, Paul WE, Zhu J, Zhao K. Genome-wide analyses of transcription factor GATA3-mediated gene regulation in distinct T cell types. Immunity. 2011;35:299–311. doi: 10.1016/j.immuni.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sundrud MS, Nolan MA. Synergistic and combinatorial control of T cell activation and differentiation by transcription factors. Curr. Opin. Immunol. 2010;22:286–292. doi: 10.1016/j.coi.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 34.Amsen D, Spilianakis CG, Flavell RA. How are T(H)1 and T(H)2 effector cells made? Curr. Opin. Immunol. 2009;21:153–160. doi: 10.1016/j.coi.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miaw SC, Choi A, Yu E, Kishikawa H, Ho IC. ROG, repressor of GATA, regulates the expression of cytokine genes. Immunity. 2000;12:323–333. doi: 10.1016/s1074-7613(00)80185-5. [DOI] [PubMed] [Google Scholar]
- 36.Hirahara K, Yamashita M, Iwamura C, Shinoda K, Hasegawa A, Yoshizawa H, Koseki H, Gejyo F, Nakayama T. Repressor of GATA regulates Th2-driven allergic airway inflammation and airway hyperresponsiveness. J. Allergy Clin. Immunol. 2008;122:512–520. doi: 10.1016/j.jaci.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 37.Kohu K, Ohmori H, Wong WF, Onda D, Wakoh T, Kon S, Yamashita M, Nakayama T, Kubo M, Satake M. The Runx3 transcription factor augments Th1 and down-modulates Th2 phenotypes by interacting with and attenuating GATA3. J. Immunol. 2009;183:7817–7824. doi: 10.4049/jimmunol.0802527. [DOI] [PubMed] [Google Scholar]
- 38.Komine O, Hayashi K, Natsume W, Watanabe T, Seki Y, Seki N, Yagi R, Sukzuki W, Tamauchi H, Hozumi K, Habu S, Kubo M, Satake M. The Runx1 transcription factor inhibits the differentiation of naive CD4+ T cells into the Th2 lineage by repressing GATA3 expression. J. Exp. Med. 2003;198:51–61. doi: 10.1084/jem.20021200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, Mullen AC, Gasink CR, Kaech SM, Miller JD, Gapin L, Ryan K, Russ AP, Lindsten T, Orange JS, Goldrath AW, Ahmed R, Reiner SL. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat. Immunol. 2005;6:1236–1244. doi: 10.1038/ni1268. [DOI] [PubMed] [Google Scholar]
- 40.Pearce EL, Mullen AC, Martins GA, Krawczyk CM, Hutchins AS, Zediak VP, Banica M, DiCioccio CB, Gross DA, Mao CA, Shen H, Cereb N, Yang SY, Lindsten T, Rossant J, Hunter CA, Reiner SL. Control of effector CD8+ T cell function by the transcripton factor Eomesodermin. Science. 2003;302:1041–1043. doi: 10.1126/science.1090148. [DOI] [PubMed] [Google Scholar]
- 41.Lavenu-Bombled C, Trainor CD, Makeh I, Romeo PH, Max-Audit I. Interleukin-13 gene expression is regulated by GATA-3 in T cells: role of a critical association of a GATA and two GATG motifs. J. Biol. Chem. 2002;277:18313–18321. doi: 10.1074/jbc.M110013200. [DOI] [PubMed] [Google Scholar]
- 42.Laribee RN, Krogan NJ, Xiao T, Shibata Y, Hughes TR, Greenblatt JF, Strahl BD. BUR kinase selectively regulates H3 K4 trimethylation and H2B ubiquitylation through recruitment of the PAF elongation complex. Curr. Biol. 2005;15:1487–1493. doi: 10.1016/j.cub.2005.07.028. [DOI] [PubMed] [Google Scholar]
- 43.Ng HH, Robert F, Young RA, Struhl K. Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol. Cell. 2003;11:709–719. doi: 10.1016/s1097-2765(03)00092-3. [DOI] [PubMed] [Google Scholar]
- 44.Acacia de Sa Pinheiro A, Morrot A, Chakravarty S, Overstreet M, Bream JH, Irusta PM, Zavala F. IL-4 induces a wide-spectrum intracellular signaling cascade in CD8+ T cells. J. Leukoc. Biol. 2007;81:1102–1110. doi: 10.1189/jlb.0906583. [DOI] [PubMed] [Google Scholar]
- 45.Harrington LE, Janowski KM, Oliver JR, Zajac AJ, Weaver CT. Memory CD4 T cells emerge from effector T-cell progenitors. Nature. 2008;2008(452):356–360. doi: 10.1038/nature06672. [DOI] [PubMed] [Google Scholar]
- 46.Apte SH, Groves P, Olver S, Baz A, Doolan DL, Kelso A, Kienzle N. IFN-gamma inhibits IL-4-induced type 2 cytokine expression by CD8 T cells in vivo and modulates the anti-tumor response. J. Immunol. 2010;185:998–1004. doi: 10.4049/jimmunol.0903372. [DOI] [PubMed] [Google Scholar]
- 47.Hayashi N, Yoshimoto T, Izuhara K, Matsui K, Tanaka T, Nakanishi K. T helper 1 cells stimulated with ovalbumin and IL-18 induce airway hyperresponsiveness and lung fibrosis by IFN-gamma and IL-13 production. Proc. Natl. Acad. Sci. USA. 2007;104:14765–14770. doi: 10.1073/pnas.0706378104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.DiSpirito JR, Shen H. Quick to remember, slow to forget: rapid recall responses of memory CD8+ T cells. Cell Res. 2010;20:13–23. doi: 10.1038/cr.2009.140. [DOI] [PubMed] [Google Scholar]
- 49.Denton AE, Russ BE, Doherty PC, Rao S, Turner SJ. Differentiation-dependent functional and epigenetic landscapes for cytokine genes in virus-specific CD8+ T cells. Proc. Natl. Acad. Sci. USA. 2011;108:15306–15311. doi: 10.1073/pnas.1112520108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.