

Abstract
In vivo control of Mycobacterium tuberculosis (Mtb) reflects the balance between host-immunity and bacterial evasion strategies. Effector TH1 cells that mediate protective immunity by depriving the bacterium of its intracellular niche are regulated to prevent over exuberant inflammation. One key immunoregulatory molecule is Tim3. Although Tim3 is generally recognized to down regulate TH1 responses, we recently described that its interaction with Galectin-9 expressed by Mtb infected macrophages stimulates IL-1β secretion, which is essential for survival in the mouse model. Why IL-1β is required for host resistance to Mtb infection is unknown. Here we show that IL-1β directly kills Mtb in murine and human macrophages and does so through the recruitment of other antimicrobial effector molecules. IL-1β directly augments TNF signaling in macrophages through the upregulation of TNF secretion and TNFR1 cell surface expression, and results in activation of caspase-3. Thus, IL-1β and downstream TNF production leads to caspase-dependent restriction of intracellular Mtb growth.
INTRODUCTION
Host resistance to Mycobacterium tuberculosis (Mtb) relies on the cooperation between innate and adaptive immunity. The factors that drive this cooperation involve cytokines secreted by TH1 cells through cell-contact dependent signals and myeloid cells that are activated by TH1 cells to produce antimicrobial effector molecules. Of particular note, interferon-γ (IFNγ) and tumor necrosis factor (TNF) are produced by Mtb-specific TH1 cells and activate infected macrophages (Mϕ) to induce intracellular mediators such as nitric oxide (NO) and promote changes in intracellular physiology including phagolysosomal fusion (1, 2). Both IFNγ−/− and NOS2−/− mice are extremely susceptible to Mtb, which indicates the crucial role of IFNγ and NO in immunity against tuberculosis (3–5). TNF plays a key role in granuloma formation thereby molding the extracellular milieu in which Mtb infected Mϕ interact with Mtb-specific T cells. TNF blockade in Mtb infected wild type (WT) mice or latently infected humans exacerbate disease (6, 7). Together IFNγ and TNF play an important role in shaping the unique microenvironment in lung granulomas and differentially modulate effector T cell immune reactivity.
Following resolution and clearance of infection, effector T cells are deleted, which prevents excessive tissue inflammation and development of immunopathology. The expression of cell surface inhibitory receptors, such as Tim3, negatively regulates effector TH1 cells (8). In addition to its role in T cell exhaustion, we previously described that Tim3 expressed by T cells interacts with Gal9 expressed by infected Mϕ to promote a program of cellular activation indicated by cytokine secretion and increased anti-mycobacterial activity (9). Cytokine secretion induced by Tim3/Gal9 interaction was reliant on the caspase-1 dependent on IL-1β secretion, suggesting that autocrine feedback by IL-1β further promotes Mϕ activation and antimicrobial activity (9). Interestingly, both IFNγ and interleukin-1β (IL-1β) induce Galectin-9 (Gal9), the ligand for Tim3 that upon binding to Tim3 transduces a signal to the T cells that triggers apoptosis resulting in clonal contraction and/or deletion of effector TH1 cells (10–13). Thus, Tim3 and Gal9 define a bidirectional regulatory pathway that results in two distinct cellular outcomes – activation of Mϕ and deactivation of T cells. While such a mechanism may be appropriate for acute infection, it appears to be detrimental in the case of persistent pathogens such as HIV, HCV, and tuberculosis.
As the antibacterial activity induced by Tim3 is mediated by IL-1β, we became interested in how IL-1β promotes intracellular control of Mtb replication. IL-1αβ−/− and IL-1R−/− mice are extremely susceptible to low dose aerosol Mtb infection and die nearly as rapidly as IFNγ, IFNγR, and TNF knockout mice, despite elevated levels of IFNγ and TNF in their lungs (14–18). These compelling data highlight the important contribution of IL-1β in defense against tuberculosis. The biological activity of IL-1β is tightly regulated (19). Regulation occurs at the level of (a) gene expression, (b) post-transcriptional activation of an inactive proform by proteolytic cleavage, and (c) competition with decoy receptors and soluble IL-1R antagonists (19). Although production of IL-1β by Mϕ in vitro generally requires both TLR signaling and inflammasome/caspase-1, IL-1β production during the early host response to Mtb infection appears to be independent of these two factors (16, 19, 20).
Despite the abundance of data on the importance of IL-1β in defense against tuberculosis, the molecular mechanism by which IL-1β enhances host resistance is unknown. In our low MOI model, fewer than 10% of Mϕ are infected and IL-1β secretion is not detected (9). We evaluated how IL-1β restricts Mtb replication under conditions that induce IL-1β (e.g., Tim3) or by directly treating infected macrophages with recombinant IL-1β. We report that IL-1β activates Mtb infected macrophages to restrict the intracellular bacterial replication. The molecular basis for the IL-1β mediated Mtb control requires the recruitment of the TNF, up regulation of cell-surface TNFR expression and caspase-3 activation. These data support a model by which IL-1β promotes Mϕ apoptosis, which inhibits the growth of intracellular Mtb.
MATERIALS AND METHODS
Ethics Statement
All animal work has been conducted according to relevant U.S. guidelines. The animals were housed in Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)-approved animal facilities and meets National Institute of Health standards as set forth in the Guide for the Care and Use of Laboratory Animals (Revised, 2010). The Institutional Animal Care and Use Committee of the Dana Farber Cancer Research Institute (Animal Welfare Assurance A3431-01) approved all experimental procedures. The institution also accepts as mandatory the PHS Policy on Humane Care and Use of Laboratory Animals by Awardee Institutions and NIH Principles for the Utilization and Care of Vertebrate Animals Testing, Research, and Training. Human blood collected from healthy donors was purchased from Research Blood Components (Boston, MA) and its use was approved by the Institutional Review Board of Brigham and Women’s Hospital (Human Subjects Assurance FWA00000484). Written informed consent was obtained from healthy volunteers recruited at National Institute of Respiratory Diseases (INER), Mexico City.
Materials
Reagents were as follows: anti-mouse CD120a (TNFR1/p55; Clone# 55R-286; Biolegend), anti-mouse CD120b (TNFR2/p75; TR75-89; Biolegend), anti-mouse CD11b (M1/70; Biolegend), anti-mouse F4/80 (BM8; Biolegend), rat anti-mouse CD16/CD32 (Fc-Block; Biolegend), CD11b microbeads (Miltenyi Biotec), human IgG1κ (I5154; Sigma Aldrich), Human serum (Gemini Bioproducts), Caspase-3 Inhibitor II (Z-DEVD-Fmk; 264155; Calbiochem), Caspase-8 Inhibitor II (Z-IETD-Fmk; 218759; Calbiochem), Caspase-9 Inhibitor I (Z-LEHD-Fmk; 218761; Calbiochem), Caspase inhibitor negative control (Z-FA-Fmk; 342000; Calbiochem), Green FLICA Caspases 3 & 7 Assay Kit (Immunochemistry Technologies LLC), recombinant mouse interleukin-1α (rIL-1α; R&D Systems), recombinant mouse interleukin-1β (rIL-1β; R&D Systems), recombinant mouse Tumor Necrosis Factor alpha (rTNFα; R&D Systems), recombinant human interleukin-1β (rIL-1β; R&D Systems), recombinant human Tumor Necrosis Factor alpha (rTNF; R&D Systems), anti-mouse IL-1β antibody (B122; eBiosciences), anti- human IL-1β antibody (H1b-27; Biolegend), anti-mouse TNFα antibody (MP6-XT22; Biolegend), anti-human TNFα antibody (MAb1; Biolegend), Armenian hamster IgG (eBio299Arm; eBiosciences), Rat IgG1 (RTK2071, Biolegend), cytotoxicity detection kitplus (lactate dehydrogenase, LDH; 04744926001, Roche), Full length and soluble Tim3-immunoglobulin fusion protein (Tim3-Ig; V. Kuchroo), anti-Tim4 antibody (anti-phosphatidyl serine receptor, clone# 4G3; V. Kuchroo), Mouse TNF-α ELISA MAX Standard (430901; Biolegend), Ficoll-Paque PLUS (GE Healthcare Life Sciences).
Mice
Six- to ten-week old C57BL/6J, IL-1R−/−, TNF−/−, TNFR1−/−, TNFR2−/− mice were obtained from Jackson laboratories; MyD88−/− mice were obtained from K. Kobayashi, Harvard Medical School (21).
Human peripheral blood
Human blood collected from healthy donors were bought from Research Blood Components, Boston, MA. Research Blood Components tests each donor’s blood for standard blood-borne pathogens prior to the initial donation, and at the time of each subsequent donation. Only blood samples negative for HIV, HTLV, HCV and HBV were used. Healthy donors were also recruited at National Institute of Respiratory Diseases (INER), Mexico City. All healthy donors from Mexico City had received bacillus Calmette-Guérin (BCG) vaccination at birth. Written informed consent was obtained from all participants.
Bacteria, cells and culture
Virulent Mtb (H37Rv) was grown to mid-log phase in Middlebrook 7H9 broth (BD Biosciences) with BBL Middlebrook OADC Enrichment (Becton Dickenson) and 0.05% Tween 80 (Difco). Aggregation was prevented by sonication for 10s. CD11b+ peritoneal macrophages were harvested after being elicited with 3% thioglycollate followed by CD11b positive selection using magnetic activated cell sorting (MACS) columns. Purified cells were over 95% F4/80+ CD11b+, as determined by flow cytometry. Mϕ (1 × 105 cells per well) were seeded in a 96-well culture plate in complete RPMI 1640 medium (Invitrogen Life Technologies) supplemented with 10% fetal bovine serum (hyclone), 1 mM pyruvate, 1% nonessential amino acids, 1% minimal essential amino acids, 2mM L-glutamine, 7 mM NaOH, 20 mM HEPES (all from Gibco). Cells were allowed to adhere for 2–24 h prior to in vitro infection with Mtb.
In vitro infections and co-cultures
Peritoneal Mϕ were infected with H37Rv at a multiplicity of infection (MOI) = 10 as described previously (22). In brief, Mtb were opsonized for 5 min using RPMI 1640 medium supplemented with 2% human serum/10% FBS/0.05% Tween 80, washed twice with complete medium without antibiotics. Bacteria were passed through a 5 μm syringe filter (Millipore), counted in a Petroff-Hausser chamber and added to Mϕ at the MOI indicated. The length of infection was 2 h for all experiments (unless otherwise indicated). Infected Mϕ were cultured overnight before the addition of cytokines, Tim3-Ig and other conditions. At days 1 and 5 post infection, cells were lysed with 1% TritonX-100 for 5 min and mycobacteria enumerated by plating serial dilutions of cell lysates on Middlebrook 7H10 agar plates and culture at 37°C. Colonies were counted after 21 d. In all the culture conditions mentioned below, CFU was enumerated at day 1 (unless otherwise indicated) in untreated infected Mϕ to determine initial inoculum and at day 5 post infection to determine growth of intracellular Mtb in untreated infected Mϕ and relative suppression/killing mediated by various treatments. The “experimental” MOI is calculated by dividing the CFU recovered on d1 by the number of macrophages per well. For example, recovery of 20,000 CFU in a well containing 100,000 Mϕ indicates an experimental MOI of 0.2 (Fig 3D). % Growth Inhibition of Mtb was calculated as follows: 100 × [CFUd5; conditions − CFUd1; media]/[CFUd5; media − CFUd1; media].
Figure 3. Tim3 and IL-1β mediated control of Mtb replication requires TNF.

(A) Mtb infected WT Mϕ were cultured either alone or in the presence of 10 μg/mL Tim3-Ig fusion protein with and without 25 μg/mL anti-TNF neutralizing antibody (αTNF). (B) WT and TNF−/−Mϕ were infected with Mtb in parallel. On d1, 10 μg/mL of Tim3-Ig or Hu IgG (control) was added to the Mϕ. (C) Mtb infected WT Mϕ were cultured alone or with 10 ng/mL of IL-1β. 24 h later, culture supernatants from triplicate wells were assayed for TNF. Open bars indicate uninfected Mϕ and closed bars indicate Mtb infected Mϕ. (D) Mtb infected murine peritoneal Mϕ were cultured either alone or in the presence of 10 ng/mL IL-1β or 10ng/mL TNF with and without 25 μg/mL anti-murine TNF (αTNF) or anti-murine IL-1β (αIL-1β) neutralizing antibody. (E) Percent inhibition in CFU in WT, IL-1R−/− and MyD88−/− Mϕ treated with and without 10 ng/mL of IL-1β or TNF. (F) Mtb infected human MDM were cultured either alone or in the presence of 10 ng/mL IL-1β with and without 25 μg/mL anti-murine TNF neutralizing antibody (αTNF). CFU in A, B, D, F (left panel) was determined on d1 and d4 or d5 post infection. Percent inhibition in E, F (right panel) is CFU growth in test conditions normalized to untreated Mϕ alone. Data is representative of 2 (A, B), 3 (C, F left panel) or 4 (D) independent experiments. Pooled data from 3 independent experiments is shown in E and F, right panel. *p<0.05, **p<0.01, ***p<0.001, one-way anova compared to control conditions: (A) isotype control, (B) Hu IgG (D) isotype control (E) WT Mϕ (F) isotype control. IC, Isotype control. Bars represent mean ± SEM.
Human monocyte derived macrophage isolation and infection
Human peripheral monocyte derived macrophages (MDM) were from heparinized blood from healthy donors by centrifugation using Ficoll-Paque PLUS gradient. Monocytes from peripheral blood mononuclear cells (PBMC) were enriched through CD14 positive selection using MACS columns. Purified cells were over 90% CD14+, as determined by flow cytometry. CD14+ monocytes were plated at 1 × 105 cells/well in 96-well plates supplemented with RPMI 1640 medium (Invitrogen Life Technologies) supplemented with 2mM L-glutamine, streptomycin, penicillin (all from Gibco), and 10% human serum. After 7-day incubation, non-adherent cells were washed off with sterile PBS and the medium was replenished; viable cells were considered monocyte derived macrophages (MDM) based on their adherence to tissue culture plates and expression profile of CD14 and CD68. Human MDM were infected with H37Rv according to methods mentioned above for peritoneal Mϕ.
Tim3-Ig fusion protein, IL-1β, TNF treatment and TNF, IL-1β, caspase inhibition studies
Tim3-immunoglobulin (Tim3-Ig) constructed as human IgG1 Fc tail fusion protein, is available as full length (FL) or soluble (s) Tim3-Ig based on the domains included in the fusion protein construct (23, 24). Fusion proteins contained <0.1EU/μg of LPS (Chimerigen Laboratories, Allston, MA). In all in vitro infections, both flTim3-Ig and sTim3-Ig were used and data obtained were identical. However, for simplicity, data from either fusion protein is included in the figures. FlTim3-Ig, sTim3-Ig and human IgG1 (Hu IgG; control) were added to Mtb infected Mϕ at concentrations indicated in the figure legends. Following 20 min incubation, goat F(ab′)2 anti-human Ig at a final concentration of 2.5 μg/mL was added to cross-link the fusion proteins or Hu IgG. To test control of Mtb by IL-1α, IL-1β in murine peritoneal Mϕ, cytokines at final concentrations of 10, 1 and 0.1 ng/mL were added directly to media containing infected Mϕ. In human MDM, IL-1β at final concentrations of 10, 5, 2.5, 1.25 and 0.625 ng/mL were added directly to media containing infected Mϕ. For all cytokine blockade studies using cytokine neutralizing antibodies (anti-IL-1β and anti-TNF), 25 μg/mL of neutralizing antibody was added either separately to infected Mϕ or along with treatment (Tim3-Ig, TNF or IL-1β) as indicated on the figure legend. For example, to determine whether TNF was required for Tim3-Ig mediated Mtb killing, anti-TNF (25 μg/mL; final concentration) was added separately to infected Mϕ along with 10 μg/mL Tim3-Ig. To account for any cells dying following Tim3-Ig treatment and releasing mycobacteria into cell culture supernatant leading to underestimation of total Mtb in infected Mϕ, we measured CFU following (1) removal of supernatant, lysis of Mϕ in 1% triton X-100 and plating the supernatant and the Mϕ lysate or (2) lysing Mϕ without the removal of cell culture supernatant by adding 10% triton X-100 at 1/10th the cell culture volume. Under the standard in vitro infection conditions mentioned above, we detect <10% of the total CFU in the cell culture supernatant. The CFU present in the supernatant was not statistically significant between Mϕ treated with media, Tim3-Ig, Hu IgG, IL-1β or TNF. To determine whether caspase-3, -8, and -9 was involved in Tim3-Ig and IL-1β mediated control, caspase-3 (Z-DEVD-Fmk); caspase-8 (Z-IETD-Fmk) or caspase-9 (Z-LEHD-Fmk) peptide inhibitors at 10 μM final concentration was added to the culture media 30 min prior to Tim3-Ig or Il-1β treatment. Negative control peptide (Z-FA-Fmk) was added at 10 μM final concentration. As a control for cytotoxicity, we treated Mϕ with caspase-3, -8 or -9 inhibitors and the negative control peptide in the absence of Tim3-Ig or IL-1β. Our ability to recover similar levels of intracellular mycobacteria in caspase-3, -8, -9/negative peptide inhibitor treated Mϕ and untreated Mϕ indicated that there were minimal cytotoxicity associated with these inhibitors at the concentrations used.
Cytokine detection
Culture supernatants from uninfected and infected Mϕ following IL-1β treatment was filtered through 0.2 μM filter to remove any bacteria. Supernatants were assayed for TNF by sandwich ELISA performed in accordance with the manufacturer’s instructions (Biolegend) and absorbance recorded at 405 nm on SoftMax Pro ELISA analysis software (Molecular Devices). IL-1β in culture supernatants was quantified by comparison with the appropriate recombinant standard (purchased from Biolegend).
Flow Cytometry
Uninfected and Mtb infected Mϕ following 24 hr Tim3-Ig, IL-1β or TNF treatment were stained for 30 min at 4°C with 25 μg/mL of anti-CD120b-PE, anti-CD120a-APC, anti-CD11b-APC-Cy7 and anti-F4/80-Pacific Blue antibodies. To inhibit nonspecific staining, murine FC receptors were blocked with 25 μg/ml of Fc-block for 20 min at 4 °C prior to staining with fluorochrome-conjugated antibodies and appropriate isotype controls. To determine apoptosis following 24 hr Tim3-Ig or IL-1β treatment, uninfected and Mtb infected Mϕ were stained for activated caspase-3 using (a) FLICA reagent FAM-DEVD-FMK, 1 hr at 37°C (Immunochemistry Technologies; 1:10; optimal excitation range = 490 – 495 nm, and emission range = 515 – 525 nm) and (b) Alexa Fluor 647 rabbit anti-active caspase-3, 30 min at 4°C (BD560626; 1:25). Activated caspase-8 and -9 were also detected using FLICA reagents FAM-LETD-FMK and FAM-LEHD-FMK respectively. FLICA reagent FAM-DEVD-FMK, FAM-LETD-FMK and FAM-LEHD-FMK are a cell-permeable non-toxic fluorescently conjugated caspase-3, -8 and -9 peptide that irreversibly binds to activated caspases with a preference for its target peptide sequence (FAM-DEVD-Fmk, FAM-LETD-FMK and FAM-LEHD-FMK highly specific for caspase-3/7, -8 and -9 respectively). Because the FLICA reagent becomes covalently coupled to the active caspase enzymes, it is retained within the cell during wash steps, while any unbound FLICA reagent diffuses out of the cell and is washed away. The remaining green fluorescent signal is a direct measure of the amount of caspase-3, -8 or -9 activity present in the cell at the time the reagent was added. Alexa Fluor 647 rabbit anti-active caspase-3 preferentially binds activated caspase-3 and was added to Mϕ following fixation and permeabilization performed according to manufacturer’s instructions (BD Biosciences). Both staining strategies (Caspase-3 FLICA for 1 hr and Alexa Fluor 647 rabbit anti-active caspase-3 for 30 min) yielded similar results. The cells were co-stained with Live/Dead viability dye (Life Technologies). Data were collected using a FACS Canto II (BD Biosciences) and analyzed with FlowJo (Tree Star).
In vitro assays of necrosis and apoptosis
Necrosis/pyroptosis in Mϕ was evaluated through the release of intracellular enzyme, Lactate dehydrogenase (LDH) in cell culture supernatants. The in vitro infections performed were done at a low (actual) MOI resulting in 1–2% of cells being infected. Under these standard in vitro infection conditions, we did not detect LDH release. To improve the sensitivity of the LDH assay we sought to increase the frequency of infected cells by infecting the Mϕ overnight as opposed to 2 hr. At the times indicated after infection, the LDH activity of the culture supernatants of infected cells was measured by using a cytotoxicity detection kit according to the manufacturer’s protocol. % LDH release was calculated according to the formula: (LDH activitytest sample, SN − LDH activityuntreated uninfected Mϕ, SN)/(Maximal releasable LDHSN+lys − LDH activityuntreated uninfected Mϕ, SN), where SN is the amount in the supernatant and Lys is the amount in the lysates. Apoptosis and necrosis were measured by enzyme-linked immunosorbent assay cell (Cell Death Detection ELISAPLUS; 11 920685 001; Roche Applied Science) for quantification of cytoplasmic (apoptosis) and extracellular (necrosis) histone-associated DNA fragments according to the specifications of the manufacturer. The relative amount of necrosis or apoptosis was calculated as a ratio of the absorbance of infected macrophages to that of uninfected control Mϕ.
Statistical analysis
One-way ANOVA was used to analyze the in vitro macrophage infections and Dunnett’s post-test was used to compare appropriate control conditions as indicated in figure legends. Analysis was performed using Prism 5.0 software (GraphPad Software, Inc., San Diego, CA).
RESULTS
IL-1 restricts intracellular bacterial replication in Mtb infected murine Mϕ
We previously showed that Tim3 binding to Galectin-9 expressed by Mtb infected Mϕ led to Mϕ activation and IL-1β dependent Mtb control in both murine peritoneal and alveolar Mϕ (9). To determine the full potential of IL-1β to mediate antimicrobial activity in murine Mϕ, we treated Mtb infected Mϕ with recombinant IL-1β. IL-1β inhibited Mtb replication in thioglycolate elicited peritoneal Mϕ in a dose dependent manner (Fig 1A). At a dose of 10 ng/mL, bacterial replication was inhibited by 80–90% (p<0.05, 1-way ANOVA). In addition to IL-1β, IL-1α also binds specifically to the IL-1 receptor (IL-1R). However, IL-1α suppressed bacterial growth less potently (Fig 1A). The greater activity of IL-1β is consistent with its greater affinity for the IL-1R. Suppression of Mtb replication was a specific action of IL-1β, since neither Tim3-Ig nor IL-1β were able to inhibit bacterial growth in infected Mϕ lacking IL-1R (Fig 1B). Finally, the antimicrobial function of both Tim3-Ig and IL-1β were dependent on MyD88, as neither molecule was able to control Mtb growth in MyD88−/− Mϕ (Fig. 1B). These data show that the antimicrobial action of IL-1β requires signaling via the IL-1R and the adaptor molecule MyD88.
Figure 1. IL-1 restricts intracellular bacterial replication in Mtb infected murine Mϕ.
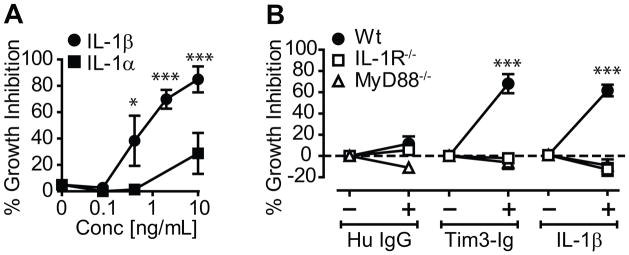
(A) Mtb infected thioglycolate-elicited peritoneal Mϕ were cultured alone or in the presence of increasing amounts of IL-1α or IL-1β. Bacterial growth was determined on d4. Percent Mtb inhibition was calculated as described in the methods section (B) Percent inhibition of Mtb growth in WT, IL-1R−/− and MyD88−/− Mϕ cultured in the absence or presence of Tim3-Ig or Hu IgG (10 μg/mL), or 10 ng/mL of IL-1β. Data is representative of 4 independent experiments in (A) or pooled data from 3 independent experiments is shown in (B). Data points in each graph represents mean ± S.E.M. *p<0.05, ***p<0.001, one-way anova compared to control conditions: (A) untreated Mϕ alone [0 ng/mL] (B) WT Mϕ.
IL-1β suppresses intracellular mycobacterial growth in human Mϕ
An essential question is whether IL-1β induced similar anti-mycobacterial activity in human Mϕ. Peripheral monocytes were isolated from healthy donors and allowed to differentiate into Mϕ (MDM). Treatment with recombinant human IL-1β led to a significant reduction in intracellular mycobacterial growth in human MDM (Fig 2A). MDM from six anonymous donors from the U.S. and Mexico were infected in vitro with Mtb and treated with hu IL-1β. IL-1β inhibited intracellular growth of Mtb in all donors. A significant CFU reduction was seen at a dose of 1.25 ng/mL hu IL-1β and 10 ng/mL completely suppressed Mtb replication (Fig 2B). These results show that IL-1β signaling activates human Mϕ to suppress intracellular growth of mycobacteria.
Figure 2. IL-1β restricts intracellular bacterial replication in Mtb infected human Mϕ.

(A) Peripheral blood monocyte-derived human Mϕ (MDM) from a single donor was infected in vitro with Mtb and were cultured alone or in the presence of 10 ng/mL IL-1β. Six replicate cultures per condition were performed. (B) Mtb infected MDMs from 6 healthy donors were cultured alone or in the presence of increasing concentrations of IL-1β. The effect of IL-1β on bacterial growth was calculated as % inhibition, as described in the methods. *p<0.05, **p<0.01, ***p<0.001, one-way anova compared to control conditions: (A) d6 media (B) untreated Mϕ alone [0 ng/mL].
Tim3/Gal9 and IL-1β induced Mtb control in Mϕ requires TNF
We previously observed that Tim3-Ig treatment of Mtb infected Mϕ not only induced IL-1α and IL-1β, but also stimulated significant production of IL-6, TNF, MIP1α, MIP1β, and G-CSF compared to untreated infected or uninfected Mϕ (9). Our previous observation that induction of these cytokines by Tim3-Ig was dependent on caspase-1 suggested that Tim3-induced IL-1β acts in an autocrine manner to stimulate the production of additional cytokines. Given the importance of TNF in antimicrobial immunity, we hypothesized that IL-1β induction of TNF is important in restricting bacterial growth. Indeed, neutralizing antibodies to TNF inhibited Tim3-Ig mediated suppression of Mtb growth (Fig 3A) and Tim3-Ig could not induce antimicrobial activity in TNF deficient Mϕ (Fig 3B). Addition of TNF to infected Mϕ also suppressed Mtb growth (Fig 3A).
Since both IL-1β and TNF were essential for Tim3-Ig mediated control, we wished to determine whether IL-1β and TNF act in parallel or in series, and possibly regulate each other. IL-1β treatment of uninfected and Mtb infected Mϕ increased TNF transcription (data not shown) and TNF secretion (Fig 3C). In contrast, TNF did not induce IL-1β transcription or secretion (Supplemental Fig. 1). Furthermore, TNF did not induce the secretion of other NF-kB dependent cytokines such as IL-1α or IL-6 (Supplemental Fig. 1). These data indicate that IL-1β induces TNF production by Mtb infected Mϕ. To determine whether TNF contributed to the antimicrobial activity of IL-1β, Mtb infected Mϕ were treated with IL-1β in the presence of neutralizing antibody to TNF. Treatment with anti-TNF but not an isotype control, abrogated the antimicrobial effect of IL-1β (Fig 3D). In contrast, the antimicrobial effect of TNF was not dependent on IL-1β, since anti-IL-1β neutralizing antibody had no intrinsic effect on TNF-mediated Mtb control (Fig 3D). Neither anti-TNF nor anti-IL-1β mAb by itself has any effect on Mtb growth. As expected, the antimicrobial effect of TNF was independent of IL-1R and MyD88 (Fig 3E).
To determine whether IL-1β stimulated TNF production led to control of intracellular Mtb growth in human Mϕ, infected MDM were treated with IL-1β in the presence of TNF blocking antibodies. IL-1β efficiently suppressed Mtb growth by ~90%. As predicted based on our murine data, addition of anti-human TNF mAb abrogated the antimicrobial activity of IL-1β (Fig 3F). Collectively, these data show that IL-1β signaling induces TNF, which in turn activates downstream antimicrobial activity in both murine and human Mϕ.
IL-1β modulates TNFR1 expression to mediate Mtb control
Soluble and membrane bound TNF signals via two distinct receptors, TNFR1 and TNFR2 (25). On average, 80–95% of thioglycolate-elicited peritoneal Mϕ express cell surface TNFR2 but only a small fraction (4–5%) of these Mϕ express cell surface TNFR1 (Fig 4A). The expression of TNFR1 and TNFR2 were not significantly altered following Mtb infection. However, IL-1β treatment of Mtb-infected Mϕ, but not uninfected Mϕ, upregulated cell surface levels of TNFR1 but had little effect on TNFR2. A similar effect was observed when the Mϕ were activated by TNF. In the absence of IL-1R, IL-1β had no effect on TNFR1 expression in Mtb-infected Mϕ (Fig 4B). To determine which TNFR was required for Tim-3 and IL-1β mediated Mtb control, we treated TNFR1−/− and TNFR2−/− Mϕ with Tim3-Ig, IL-1β and TNF. TNFR1 was essential while TNFR2 was dispensable for soluble TNF mediated Mtb control confirming previously established TNF-signaling pathways for TNF receptors. Tim3-Ig and IL-1β mediated Mtb control was dependent on TNFR1 and largely independent of TNFR2 signaling. (Fig 4C). These data indicate that TNFR1 is required for antimicrobial activities of Tim3-Ig and IL-1β.
Figure 4. Regulation of TNF receptor expression and its requirement for IL-1β mediated Mtb control.

(A) Uninfected and Mtb infected (MOI 10) peritoneal Mϕ were cultured alone or in the presence of IL-1β or TNF (10 ng/mL). Cell surface expression of TNFR1 and TNFR2 of CD11b+ F4/80+ Mϕ was assessed by flow cytometry after 24 h. (B) Cell surface expression of TNFR1 is shown for uninfected and Mtb infected (MOI 10) WT and IL-1R−/− Mϕ. (C) Mtb infected WT, TNFR1−/− and TNFR2−/− Mϕ were treated with 10 μg/mL of Tim3-Ig or Hu IgG (control), 10 ng/mL IL-1β or TNF. Data is representative of 2 (A, B) independent experiments. Pooled data from 2 independent experiments is shown in (C). **p<0.01, ***p<0.001, one-way anova compared to control conditions: (C) WT Mϕ.
Tim3/Gal9 binding and IL-1β both induce caspase 3 activation in Mtb infected macrophages
As Tim3-Ig and IL-1β treatment promotes control of intracellular bacterial replication by infected Mϕ in a TNF dependent manner, we hypothesized that IL-1β was modulating the death modality of infected Mϕ via induction of extrinsic death ligands such as TNFR1. We measured the release of the intracellular enzyme lactate dehydrogenase (LDH) into cell culture supernatant, as a measure of plasma cell membrane disruption. Using our standard infection protocol, no necrosis was observed. Since the actual multiplicity of infection (MOI) is very low with <10% of Mϕ being infected, we increased the length of infection from 2 hours to overnight and varied the MOI to increase the percentage of Mϕ that were infected. Under these conditions, necrosis was detected 3 days after infection and correlated with increasing MOI; however, neither IL-1β nor Tim3-Ig caused an increase in LDH release (data not shown). These data are consistent with our prior data that Tim3-Ig doesn’t induce necrosis (9).
We next asked whether Tim3-Ig or IL-1β induced apoptosis of Mtb infected macrophages. Using an ELISA that detects intracellular and extracellular DNA-histone complexes as a measure of apoptosis and necrosis, respectively, we previously did not find any evidence that Tim-Ig induces apoptosis. However, the ELISA is of unknown sensitivity, and because only 1–10% of the macrophages are infected, we sought to use an assay that can detect apoptosis at the single cell level. Macrophages infected with MCherry-Mtb were treated with Tim3-Ig or IL-1β, and after fixation, a TUNEL assay was performed. Despite trying multiple MOIs and time points, a consistent increase in TUNEL+ cells was not detected after Tim3-Ig or IL-1β treatment (data not shown). In parallel, to determine whether Tim3-Ig or IL-1β leads to the activation of caspase-3, the executioner caspase that has a pivotal role in apoptosis, we measured caspase-3 activation in Mtb infected Mϕ using the fluorescently conjugated caspase-3 substrate Z-DEVD-Fmk. Caspase-3 is activated both by extrinsic (death ligand) and intrinsic (mitochondrial) pathways via caspase-8 and capase-9 initiator caspases. On average, 5–8% of uninfected thioglycolate-elicited peritoneal Mϕ express active caspase-3. The number of Mϕ expressing active caspase-3 increased two-fold following Mtb infection (Fig 5A). Tim3-Ig treated Mtb-infected Mϕ, but not uninfected Mϕ or control treated Mtb-infected Mϕ had increased caspase-3 activity (Fig 5A, B). A similar effect was observed when murine Mϕ were treated with IL-1β (Fig 5C). The action of IL-1β was specific since IL-1β treatment of Mtb infected IL-1R−/− Mϕ did not increase active caspase-3 (Fig 5D). As predicted based on our murine data, IL-1β also induced caspase-3 activation in Mtb infected human Mϕ (Fig 5E). To further confirm TNF was responsible for this caspase-3 activation, infected MDM were treated with IL-1β in the presence of TNF blocking antibodies. The addition of anti-human TNF mAb diminished the increased frequency of active caspase-3 in HMDM to baseline levels in untreated media alone conditions (Fig 5E, F). Similar results were obtained when intracellular staining with a mAb specific for active caspase 3 was used (data not shown). In addition, IL-1β treatment of Mtb infected HMDM led to TNF-dependent activation of caspase-8 and -9 (see Supplemental Figure 2). We conclude that in Mtb-infected Mϕ, stimulation of IL-1β by Tim3/Gal9 binding, or addition of exogenous IL-1β, leads to activation of caspase-3, the key executioner caspase that mediates apoptosis.
Figure 5. Tim3 and IL-1β induce caspase-3 activation.

(A) Mtb infected and uninfected thioglycolate-elicited Mϕ were cultured either alone or in the presence of 10 μg/mL Tim3-Ig or Hu IgG (Control). 24 h later, intracellular expression of active caspase-3 (aCaspase-3) in CD11b+ F4/80+ Mϕ was assessed by flow cytometry. (B) Fold induction of active caspase-3 compared to untreated uninfected Mϕ is graphically plotted. Stauro, staurosporine [1 μM], positive control. (C) Mtb infected and uninfected thioglycolate-elicited Mϕ were cultured either alone or in the presence of 10 ng/mL IL-1β. 24 h later, intracellular expression of active caspase-3 in CD11b+ F4/80+ Mϕ was assessed by flow cytometry. (D) Fold induction of active caspase-3 over untreated uninfected Mϕ is graphically plotted for uninfected and Mtb infected WT and IL-1R−/− Mϕ. (E) Mtb infected and uninfected human MDM were cultured either alone or in the presence of 10 ng/mL IL-1β with and without 25 μg/mL anti-TNF neutralizing antibody (αTNF). 24 h later, intracellular expression of active caspase-3 in CD14+ Mϕ was assessed by flow cytometry. (F) Fold induction of active caspase-3 compared to untreated uninfected Mϕ is graphically plotted. Data is representative of 2 (A, B, E, F) and 3 (C) independent experiments. Pooled data from 3 independent experiments is shown in (D). *p<0.05, **p<0.01, ***p<0.001, one-way anova compared to conditions: (B) Tim3-Ig and Staurosporine, (D) IL-1β treated WT Mϕ or (F) IL-1β treated human MDM. Bars indicate mean ± SEM from 3 replicate cultures.
The antimicrobial effect of IL-1β requires efferocytosis
We next evaluated whether active caspase-3, -8 and -9 was required for Tim3-Ig and IL-1β mediated Mtb control in murine peritoneal macrophages and HMDM. Mtb infected Mϕ were treated with Tim3-Ig or IL-1β in the presence of Z-DEVD-Fmk, Z-IETD–Fmk, Z-LEHD–Fmk, peptide inhibitors of caspase-3, -8 and -9 activity respectively. Z-DEVD–Fmk, Z-IETD–Fmk, Z-LEHD–Fmk abrogated Tim3-Ig and IL-1β mediated control of Mtb infection (Fig 6A, B), showing that caspase-3, -8 and -9 are required for Tim3-Ig and IL-1β mediated Mtb control in murine and human macrophages. In the absence of Tim3-Ig or IL-1β, Z-DEVD-Fmk, Z-IETD–Fmk, Z-LEHD–Fmk had no effect on bacterial replication (data not shown). To rule out any possible toxic effects of caspase inhibitors, we monitored the secretion of TNF, IL-1α and IL-6 in the presence of Tim3-Ig and IL-1β treatment (Supplemental Fig. 3). While we saw reduction in the levels of IL-6 following addition of peptide inhibitors, we did not observe any reduction in the levels of soluble TNF and IL-1α following addition of caspase-3 peptide inhibitor suggesting that loss of CFU control in IL-1β treatment conditions was not due to caspase-3 mediated inhibition of TNF.
Figure 6. Active caspase-3 and efferocytosis is required for Mtb control.

(A) H37Rv infected WT Mϕ was co-cultured with Tim3-Ig or IL-1β in the presence or absence of Z-DEVD-Fmk, Z-IETD–Fmk, Z-LEHD–Fmk, peptide inhibitors of caspase-3, -8 and -9 activity. (B) H37Rv infected human MDM was co-cultured with 10 ng/mL IL-1β in the presence or absence of Z-DEVD-Fmk, Z-IETD Fmk, Z-LEHD–Fmk, peptide inhibitors of caspase-3, -8 and -9 activity. Control peptide is Z-FA-Fmk. (C) H37Rv infected WT Mϕ were cultured either alone or in the presence of 10 ng/mL IL-1β with and without 25 μg/mL blocking antibody to PS receptor (αPS-R). Data is representative of 4 (A) or 2 (B) independent experiments. *p<0.05, **p<0.01, ***p<0.001, one-way anova compared to conditions: (A) Tim3-Ig or IL-1β treated WT Mϕ (B) Isotype control, IC. Bars indicate mean ± SEM from 3 replicate cultures.
We have recently shown that the viability of Mtb is reduced when Mtb sequestered within apoptotic vesicles is rapidly taken up by uninfected bystander Mϕ by a constitutive process called efferocytosis (26). In contrast, necrosis of Mtb infected Mϕ leads to bacterial dispersal and ongoing growth. Uninfected bystander Mϕ recognize dying apoptotic cells through “find me” and “eat me” signals such as phosphatidylserine (PS). To determine whether IL-1β mediated program of antimicrobial activity requires efferocytic uptake of apoptotic Mϕ, Mtb infected Mϕ were treated with blocking antibody to the PS receptor (Tim-4) in the presence of IL-1β (27, 28). Blocking antibody to the PS receptor but not the control antibody completely abolished IL-1β mediated Mtb control (Fig 6C). We found that IL-1β did not enhance efferocytosis of apoptotic cells (data not shown). These data support the hypothesis that IL-1β induces apoptosis in Mtb infected Mϕ that then are engulfed by the process of efferocytosis which is required to restrict Mtb replication.
DISCUSSION
The inflammasome complex, IL-1β and downstream innate adaptor, MyD88 are all important in modulating host susceptibility to Mtb (16). Mice deficient in either IL-1R or IL-1β are acutely susceptible to Mtb (14, 16). Although it has been recently described that inflammatory monocytes and dendritic cells (DC), distinguished by their cell surface expression of CD11c and Ly6C, are the primary sources of IL-1β production in the lungs of Mtb infected mice, why IL-1β is essential for host resistance to Mtb is unknown (29). Here, we provide the first evidence that IL-1β directly activates innate antimicrobial activity in murine Mϕ and human monocyte-derived Mϕ leading to restriction of Mtb replication. Using in vitro models of Mtb infection in Mϕ, we were able to ascertain for the first time the antimicrobial molecules activated following IL-1R signaling that contribute to IL-1β-mediated Mtb control.
We previously reported that the caspase-1–processed cytokine IL-1β promotes innate antimicrobial immunity in Mtb infected Mϕ following interaction between Tim3 expressed by T cells and Gal9 expressed by Mϕ (9). We found that in Mtb infected Mϕ, Tim3 activates a program of antimicrobial immunity that involves the initial secretion of IL-1β followed by IL-1β mediated recruitment of other antimicrobial effectors including IL-6, TNF, MIP1α, MIP1β, and G-CSF. Our previous finding that genetic ablation of caspase-1 in Mtb infected Mϕ suppressed TNF production strongly indicated that IL-1β was acting in an autocrine manner to promote production of additional antimicrobial effectors (9). Here we show that IL-1β directly increased TNF transcription and secretion, an activity that was augmented in Mtb infected Mϕ. We do not believe that autocrine action of IL-1β on macrophages is the sole source of TNF in vivo. Indeed, IL-1R−/− and IL-1β−/− exhibit higher levels of TNF in lungs of infected mice than WT controls (16, 29). This indicates that mechanisms other than IL-1β activation of macrophages are important for TNF production during Mtb infection. Finally, we found that IL-1β potentially augments TNF signal transduction by upregulating TNFR1 cell surface expression on infected Mϕ. The antimicrobial action of both Tim3-Ig and IL-1β were largely dependent on TNFR1 and partially dependent on TNFR2. TNFR1 can be activated by both soluble (sTNF) and membrane (mTNF) while TNFR2 have some exclusivity for membrane TNF (mTNF). The nature of the TNF ligand produced following IL-1β signaling is unknown. However, one explanation for our finding is the ‘ligand-passing’ mechanism by which TNFR2 holds ligand (TNF), enhances the local TNF concentration in the vicinity of TNFR1 which in turn accepts TNF from TNFR2 leading to TNFR1 signaling (25). While TNFR1 is indispensable for IL-1β mediated Mtb control, TNFR2 appears to play a minor role. Thus, the ability of Tim3-Ig and IL-1β to stimulate TNF production by Mtb infected Mϕ is required for their antimicrobial activity.
The ability of the Tim3/Gal9 pathway to restrict intracellular Mtb replication is independent of IFNγ and nitric oxide (9), two of the chief mediators that lead to bacterial control. Therefore, we considered whether Tim3/Gal9 signaling altered the death modality of infected macrophages. Induction of Mϕ necrosis, which is the cellular outcome more frequently associated with infection by virulent Mtb, leads to enhanced bacterial growth in vitro. In contrast, apoptosis of Mtb-infected Mϕ is associated with the control of intracellular bacterial growth (30). Apoptosis, a tightly regulated complex process initiated by extrinsic (caspase-8 dependent) or intrinsic signals (caspase-9 dependent) and culminates in the activation of executioner caspase-3 or caspase-7. TNF is a pleiotropic molecule that plays crucial roles in cellular stress, inflammation during infection, tissue damage and TB granuloma formation in the lung (6, 31–34). TNF signaling via its receptors TNFR1 and TNFR2 influences cell fate and can lead to three distinct outcomes: (a) necrosis, (b) apoptosis through the death domain TRADD, and (c) cellular survival via NF-kB activation (25). Mtb can evade TNF-mediated apoptosis by stimulating human Mϕ to secrete sTNFR2, which serves as an antagonist of TNFR1 signaling (35). Virulent mycobacteria can also evade TNF-mediated apoptosis by inducing necrosis (30, 36, 37).
Tim3-Ig did not induce more necrosis or apoptosis of infected macrophages (9). However, the characterization of cell death during Mtb infection suffers from difficulties including differing death modalities, lack of synchronization, secondary necrosis, varying kinetics and poor performance of some experimental assays following fixation. In particular, we were concerned that the DNA-histone ELISA may lack the sensitivity to detect rare events, particularly in our low MOI culture system. We next turned to a microscopy-based assay using MCherry-expressing H37Rv and TUNEL. While sensitive, this assay gave inconsistent results. However, other data suggested that Tim3-Ig was modulating cell death. After treatment with Tim3-Ig, more Mtb infected macrophages expressed activate capsase-3. Although the frequency of caspase-3+ cells was higher than expected based on our TUNEL staining, these results were confirmed by showing that CFU control was dependent on caspase-3, suggesting that Tim3-Ig induced greater apoptosis. In parallel, CFU control was inhibited by caspase-8 and -9 inhibitors. Such co-dependence on both the intrinsic and extrinsic pathways is observed when initial precursor pool of caspase-8 is not sufficient to drive subsequent caspase-3 activation. In certain cell types (M2 macrophages) the initial precursor pool of active caspase-8 leads to truncation of Bid, which translocates to the mitochondria that serves as a platform for caspase-9 activation and subsequent caspase-3 activation and apoptosis (38). We cannot completely rule out that the effect of these peptide inhibitors is due to inhibition of serine proteases other than caspase-3, -8, and -9. However, the known enzymes that are cross-inhibited are other caspases or cathepsins, which also have a role in cell death.
We have recently described that macrophage engulfment of apoptotic infected cells leads to destruction of intracellular Mtb (26). This process, commonly referred to efferocytosis, occurs when macrophages recognize and engulf apoptotic cells. Apoptosis is not inherently bactericidal; rather the efferocytic uptake of dying apoptotic macrophages by uninfected bystander macrophages restricts Mtb viability. We have further shown the mechanism of Mtb control to the confinement of Mtb within the efferocytic phagosome in the eating macrophages and subsequent phagosome maturation (26). The recognition of phosphatidylserine (PS) by a variety of PS receptors is critical for efferocytosis. Although several PS receptors have been identified, the dominant one used by peritoneal macrophages is Tim4. Our data that anti-Tim4 mAb blocks bacterial control induced by IL-1β and Tim3-Ig implicates efferocytosis of apoptotic cells as part of the mechanism.
Our finding that IL-1β requires active caspase-8, -9 and -3 for its antimicrobial activity suggests that IL-1R signaling leads to TNF/TNFR1-mediated activation of the extrinsic caspase-8 pathway that feeds into the intrinsic caspase-9 pathway resulting in caspase-3 mediated apoptosis of Mtb infected Mϕ and control of intracellular bacterial replication. Phagocytic cells clear apoptotic cells by efferocytosis, a highly conserved process that is important in preventing autoimmunity and inappropriate inflammation. Defective efferocytosis often seen in atherosclerotic plaques is a failure to clear lipid laden apoptotic Mϕ (also called as foamy Mϕ) that leads to post-apoptotic necrosis (secondary necrosis) (39). Foamy Mϕ are also characteristic of caseating necrotic lesions in tuberculous granuloma (40). Could the presence of foamy Mϕ in TB granulomas therefore be indicative of defective efferocytosis where lipid laden Mϕ are not efficiently cleared by bystander Mϕ? We find that IL-1β (a) induces apoptosis in Mtb infected Mϕ, (b) requires efferocytosis for Mtb control, but, (c) does not by itself regulate efferocytosis. These data suggest a model in which IL-1β production by infected Mϕ or neighboring cells stimulates TNF production by Mtb infected Mϕ, leading to their apoptotic cell death. These dying cells are engulfed by recruited Mϕ (efferocytosis), which we believe represents a common final pathway that restricts Mtb replication.
The extensive pulmonary necrosis seen in infected Mtb IL-1R−/− mice may reflect the role that IL-1β plays in modulating cell death and host resistance to Mtb (16). Given the high levels of IL-1R antagonist in lungs of infected WT mice that can block IL-1R signaling, we speculate that inhibited IL-1R signaling could impair control of bacterial growth and contribute to recrudescence and lung pathology (caseating necrosis) in Mtb infected WT mice (16). The functional overlap between IL-1R signaling and TNFR1/TNFR2 signaling has been previously illustrated in lung models of airway inflammation. IL-1β augments TNF-mediated secretion of chemokines such as MIP-2 and KC, key chemoattractants for neutrophils and Mϕ, thereby altering the microenvironment of the lung (41–43). While the outcome of enhanced Mϕ and neutrophil recruitment on host resistance is unpredictable, it is likely that tight regulation of the IL-1β/TNF signaling axis prevents over-exuberant inflammation and tissue damage, possibly at the cost of optimal bacterial control (44).
In summary, our work provides the first evidence that IL-1β directly activates antimicrobial effector pathways in Mtb-infected human and murine Mϕ. We find that IL-1β acts in an autocrine fashion to modulate TNFR signaling through increased TNF and TNFR1 expression in Mϕ. This leads to caspase-3 activation and apoptosis, which restricts Mtb replication by efferocytosis.
Supplementary Material
Acknowledgments
The authors thank K. Kobayashi (Harvard Medical School) for MyD88−/− mice.
Abbreviations used
- −/−
knockout
- APC
antigen presenting cells
- CFU
colony forming units
- Gal9
Galectin-9
- IFNγ
interferon-γ
- iNOS
inducible nitric oxide synthase
- Hu IgG
human immunoglobulin-gamma
- IL-1α
interleukin-1 alpha
- IL-1β
interleukin-1 beta
- IL-1R
interleukin-1 receptor
- LDH
lactate dehydrogenase
- Mϕ
macrophage
- Mtb
Mycobacterium tuberculosis
- NOS2
nitric oxide synthase-2
- ROS/RNS
reactive oxygen & nitrogen species
- TIM-3
T cell immunoglobulin and mucin domain-3
- Tim3-Ig
Tim3-Immunoglobulin fusion protein
- TNF
tumor necrosis factor
- TNFR1
TNF-receptor 1
- TNFR2
TNF-receptor 2
- TLR
Toll-like receptor
- Type I and II IFN
type I and II interferon
- WT
wild type
Footnotes
This work was supported by the National Institutes of Health grants R01 AI085669 and AI098637 to S.M. Behar, R01 AI072143 and R01 AI 073774 to H. G. Remold, TIM program project grant P01 AI 073748 to V. K. Kuchroo, the American Lung Association postdoctoral research training fellowship (RT-123085-N) and Harvard University Center for AIDS Research (CFAR) Scholar Award to P. Jayaraman. The HU CFAR Scholar award to P. Jayaraman is an NIH funded program (P30 AI060354), which is supported by the following NIH Co-Funding and Participating Institutes and Centers: NIAID, NCI, NICHD, NHLBI, NIDA, NIMH, NIA, NCCAM, FIC, and OAR.
The authors have no competing financial interests.
AUTHOR CONTRIBUTIONS
P.J. and S.M.B conceived of and designed the experiments, analyzed the data and wrote the paper; P.J., I.S.O., and T.N. did the experiments; A.C.A., V.K.K. and H.G.R. provided reagents and intellectual input.
References
- 1.Kim BH, Shenoy AR, Kumar P, Das R, Tiwari S, MacMicking JD. A family of IFN-gamma-inducible 65-kD GTPases protects against bacterial infection. Science. 2011;332:717–721. doi: 10.1126/science.1201711. [DOI] [PubMed] [Google Scholar]
- 2.MacMicking JD, Taylor GA, McKinney JD. Immune control of tuberculosis by IFN-gamma-inducible LRG-47. Science. 2003;302:654–659. doi: 10.1126/science.1088063. [DOI] [PubMed] [Google Scholar]
- 3.Cooper AM, Dalton DK, Stewart TA, Griffin JP, Russell DG, Orme IM. Disseminated tuberculosis in interferon gamma gene-disrupted mice. J Exp Med. 1993;178:2243–2247. doi: 10.1084/jem.178.6.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, Bloom BR. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J Exp Med. 1993;178:2249–2254. doi: 10.1084/jem.178.6.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.MacMicking JD, North RJ, LaCourse R, Mudgett JS, Shah SK, Nathan CF. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc Natl Acad Sci U S A. 1997;94:5243–5248. doi: 10.1073/pnas.94.10.5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bean AG, Roach DR, Briscoe H, France MP, Korner H, Sedgwick JD, Britton WJ. Structural deficiencies in granuloma formation in TNF gene-targeted mice underlie the heightened susceptibility to aerosol Mycobacterium tuberculosis infection, which is not compensated for by lymphotoxin. J Immunol. 1999;162:3504–3511. [PubMed] [Google Scholar]
- 7.Flynn JL, Goldstein MM, Chan J, Triebold KJ, Pfeffer K, Lowenstein CJ, Schrelber R, Mak TW, Bloom BR. Tumor necrosis factor-[alpha] is required in the protective immune response against mycobacterium tuberculosis in mice. Immunity. 1995;2:561–572. doi: 10.1016/1074-7613(95)90001-2. [DOI] [PubMed] [Google Scholar]
- 8.Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, Manning S, Greenfield EA, Coyle AJ, Sobel RA, Freeman GJ, Kuchroo VK. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. 2002;415:536–541. doi: 10.1038/415536a. [DOI] [PubMed] [Google Scholar]
- 9.Jayaraman P, Sada-Ovalle I, Beladi S, Anderson AC, Dardalhon V, Hotta C, Kuchroo VK, Behar SM. Tim3 binding to galectin-9 stimulates antimicrobial immunity. J Exp Med. 2010;207:2343–2354. doi: 10.1084/jem.20100687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Imaizumi T, Kumagai M, Sasaki N, Kurotaki H, Mori F, Seki M, Nishi N, Fujimoto K, Tanji K, Shibata T, Tamo W, Matsumiya T, Yoshida H, Cui XF, Takanashi S, Hanada K, Okumura K, Yagihashi S, Wakabayashi K, Nakamura T, Hirashima M, Satoh K. Interferon-gamma stimulates the expression of galectin-9 in cultured human endothelial cells. J Leukoc Biol. 2002;72:486–491. [PubMed] [Google Scholar]
- 11.Kashio Y, Nakamura K, Abedin MJ, Seki M, Nishi N, Yoshida N, Nakamura T, Hirashima M. Galectin-9 induces apoptosis through the calcium-calpain-caspase-1 pathway. J Immunol. 2003;170:3631–3636. doi: 10.4049/jimmunol.170.7.3631. [DOI] [PubMed] [Google Scholar]
- 12.Yoshida H, Imaizumi T, Kumagai M, Kimura K, Satoh C, Hanada N, Fujimoto K, Nishi N, Tanji K, Matsumiya T, Mori F, Cui XF, Tamo W, Shibata T, Takanashi S, Okumura K, Nakamura T, Wakabayashi K, Hirashima M, Sato Y, Satoh K. Interleukin-1beta stimulates galectin-9 expression in human astrocytes. Neuroreport. 2001;12:3755–3758. doi: 10.1097/00001756-200112040-00030. [DOI] [PubMed] [Google Scholar]
- 13.Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, Zheng XX, Strom TB, Kuchroo VK. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005;6:1245–1252. doi: 10.1038/ni1271. [DOI] [PubMed] [Google Scholar]
- 14.Fremond CM, Togbe D, Doz E, Rose S, Vasseur V, Maillet I, Jacobs M, Ryffel B, Quesniaux VF. IL-1 receptor-mediated signal is an essential component of MyD88-dependent innate response to Mycobacterium tuberculosis infection. J Immunol. 2007;179:1178–1189. doi: 10.4049/jimmunol.179.2.1178. [DOI] [PubMed] [Google Scholar]
- 15.Juffermans NP, Florquin S, Camoglio L, Verbon A, Kolk AH, Speelman P, van Deventer SJ, van Der Poll T. Interleukin-1 signaling is essential for host defense during murine pulmonary tuberculosis. J Infect Dis. 2000;182:902–908. doi: 10.1086/315771. [DOI] [PubMed] [Google Scholar]
- 16.Mayer-Barber KD, Barber DL, Shenderov K, White SD, Wilson MS, Cheever A, Kugler D, Hieny S, Caspar P, Nunez G, Schlueter D, Flavell RA, Sutterwala FS, Sher A. Caspase-1 independent IL-1beta production is critical for host resistance to mycobacterium tuberculosis and does not require TLR signaling in vivo. J Immunol. 2010;184:3326–3330. doi: 10.4049/jimmunol.0904189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sugawara I, Yamada H, Hua S, Mizuno S. Role of interleukin (IL)-1 type 1 receptor in mycobacterial infection. Microbiology and immunology. 2001;45:743–750. doi: 10.1111/j.1348-0421.2001.tb01310.x. [DOI] [PubMed] [Google Scholar]
- 18.Yamada H, Mizumo S, Horai R, Iwakura Y, Sugawara I. Protective role of interleukin-1 in mycobacterial infection in IL-1 alpha/beta double-knockout mice. Lab Invest. 2000;80:759–767. doi: 10.1038/labinvest.3780079. [DOI] [PubMed] [Google Scholar]
- 19.Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–550. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 20.Guma M, Ronacher L, Liu-Bryan R, Takai S, Karin M, Corr M. Caspase 1-independent activation of interleukin-1beta in neutrophil-predominant inflammation. Arthritis Rheum. 2009;60:3642–3650. doi: 10.1002/art.24959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, Akira S. Targeted disruption of the MyD88 gene results in loss of IL-1-and IL-18-mediated function. Immunity. 1998;9:143–150. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- 22.Sada-Ovalle I, Chiba A, Gonzales A, Brenner MB, Behar SM. Innate invariant NKT cells recognize Mycobacterium tuberculosis-infected macrophages, produce interferon-gamma, and kill intracellular bacteria. PLoS Pathog. 2008;4:e1000239. doi: 10.1371/journal.ppat.1000239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sabatos CA, Chakravarti S, Cha E, Schubart A, Sanchez-Fueyo A, Zheng XX, Coyle AJ, Strom TB, Freeman GJ, Kuchroo VK. Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat Immunol. 2003;4:1102–1110. doi: 10.1038/ni988. [DOI] [PubMed] [Google Scholar]
- 24.Sanchez-Fueyo A, Tian J, Picarella D, Domenig C, Zheng XX, Sabatos CA, Manlongat N, Bender O, Kamradt T, Kuchroo VK, Gutierrez-Ramos JC, Coyle AJ, Strom TB. Tim-3 inhibits T helper type 1-mediated auto-and alloimmune responses and promotes immunological tolerance. Nat Immunol. 2003;4:1093–1101. doi: 10.1038/ni987. [DOI] [PubMed] [Google Scholar]
- 25.MacEwan DJ. TNF ligands and receptors--a matter of life and death. Br J Pharmacol. 2002;135:855–875. doi: 10.1038/sj.bjp.0704549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martin CJ, Booty MG, Rosebrock TR, Nunes-Alves C, Desjardins DM, Keren I, Fortune SM, Remold HG, Behar SM. Efferocytosis is an innate antibacterial mechanism. Cell Host Microbe. 2012;12:289–300. doi: 10.1016/j.chom.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a phosphatidylserine receptor. Nature. 2007;450:435–439. doi: 10.1038/nature06307. [DOI] [PubMed] [Google Scholar]
- 28.Rodriguez-Manzanet R, Sanjuan MA, Wu HY, Quintana FJ, Xiao S, Anderson AC, Weiner HL, Green DR, Kuchroo VK. T and B cell hyperactivity and autoimmunity associated with niche-specific defects in apoptotic body clearance in TIM-4-deficient mice. Proc Natl Acad Sci U S A. 2010;107:8706–8711. doi: 10.1073/pnas.0910359107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mayer-Barber KD, Andrade BB, Barber DL, Hieny S, Feng CG, Caspar P, Oland S, Gordon S, Sher A. Innate and Adaptive Interferons Suppress IL-1alpha and IL-1beta Production by Distinct Pulmonary Myeloid Subsets during Mycobacterium tuberculosis Infection. Immunity. 2011;35:1023–1034. doi: 10.1016/j.immuni.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen M, Divangahi M, Gan H, Shin DS, Hong S, Lee DM, Serhan CN, Behar SM, Remold HG. Lipid mediators in innate immunity against tuberculosis: opposing roles of PGE2 and LXA4 in the induction of macrophage death. J Exp Med. 2008;205:2791–2801. doi: 10.1084/jem.20080767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clay H, Volkman HE, Ramakrishnan L. Tumor necrosis factor signaling mediates resistance to mycobacteria by inhibiting bacterial growth and macrophage death. Immunity. 2008;29:283–294. doi: 10.1016/j.immuni.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chakravarty SD, Zhu G, Tsai MC, Mohan VP, Marino S, Kirschner DE, Huang L, Flynn J, Chan J. Tumor necrosis factor blockade in chronic murine tuberculosis enhances granulomatous inflammation and disorganizes granulomas in the lungs. Infect Immun. 2008;76:916–926. doi: 10.1128/IAI.01011-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Algood HM, Lin PL, Flynn JL. Tumor necrosis factor and chemokine interactions in the formation and maintenance of granulomas in tuberculosis. Clin Infect Dis. 2005;41(Suppl 3):S189–193. doi: 10.1086/429994. [DOI] [PubMed] [Google Scholar]
- 34.Flynn JL, Goldstein MM, Chan J, Triebold KJ, Pfeffer K, Lowenstein CJ, Schreiber R, Mak TW, Bloom BR. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity. 1995;2:561–572. doi: 10.1016/1074-7613(95)90001-2. [DOI] [PubMed] [Google Scholar]
- 35.Balcewicz-Sablinska MK, Keane J, Kornfeld H, Remold HG. Pathogenic Mycobacterium tuberculosis evades apoptosis of host macrophages by release of TNF-R2, resulting in inactivation of TNF-alpha. J Immunol. 1998;161:2636–2641. [PubMed] [Google Scholar]
- 36.Keane J, Remold HG, Kornfeld H. Virulent Mycobacterium tuberculosis strains evade apoptosis of infected alveolar macrophages. J Immunol. 2000;164:2016–2020. doi: 10.4049/jimmunol.164.4.2016. [DOI] [PubMed] [Google Scholar]
- 37.Gan H, Lee J, Ren F, Chen M, Kornfeld H, Remold HG. Mycobacterium tuberculosis blocks crosslinking of annexin-1 and apoptotic envelope formation on infected macrophages to maintain virulence. Nat Immunol. 2008;9:1189–1197. doi: 10.1038/ni.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scaffidi C, Schmitz I, Zha J, Korsmeyer SJ, Krammer PH, Peter ME. Differential modulation of apoptosis sensitivity in CD95 type I and type II cells. The Journal of biological chemistry. 1999;274:22532–22538. doi: 10.1074/jbc.274.32.22532. [DOI] [PubMed] [Google Scholar]
- 39.Thorp E, Subramanian M, Tabas I. The role of macrophages and dendritic cells in the clearance of apoptotic cells in advanced atherosclerosis. Eur J Immunol. 2011;41:2515–2518. doi: 10.1002/eji.201141719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hunter RL, Jagannath C, Actor JK. Pathology of postprimary tuberculosis in humans and mice: contradiction of long-held beliefs. Tuberculosis (Edinb) 2007;87:267–278. doi: 10.1016/j.tube.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 41.Cardell LO, Uddman R, Zhang Y, Adner M. Interleukin-1beta up-regulates tumor necrosis factor receptors in the mouse airways. Pulm Pharmacol Ther. 2008;21:675–681. doi: 10.1016/j.pupt.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 42.Saperstein S, Chen L, Oakes D, Pryhuber G, Finkelstein J. IL-1beta augments TNF-alpha-mediated inflammatory responses from lung epithelial cells. J Interferon Cytokine Res. 2009;29:273–284. doi: 10.1089/jir.2008.0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saperstein S, Huyck H, Kimball E, Johnston C, Finkelstein J, Pryhuber G. The effects of interleukin-1beta in tumor necrosis factor-alpha-induced acute pulmonary inflammation in mice. Mediators Inflamm. 2009;2009:958658. doi: 10.1155/2009/958658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nandi B, Behar SM. Regulation of neutrophils by interferon-{gamma} limits lung inflammation during tuberculosis infection. J Exp Med. 2011;208:2251–2262. doi: 10.1084/jem.20110919. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.