

Abstract
CD40 is expressed on cells of the immune system and in some tissues that are targets for autoimmune-mediated damage. It is not known if CD40 expression in target tissues plays a role in the pathology of autoimmune diseases. This study shows that agonistic anti-CD40 induces strong and sustained proliferation of thyroid epithelial cells (TEC) or thyrocytes in IFN-γ−/− autoimmune-prone NOD and NOD.H-2h4 mice. TEC proliferation is accompanied by greatly increased expression of CD40 on TEC, development of fibrosis and hypothyroidism, and increased expression of proinflammatory molecules in thyroids. Bone marrow chimera experiments indicate that TEC expression of CD40 is required for anti-CD40-induced TEC proliferation, but lymphoid cells do not have to express CD40. TEC proliferation is reduced in WT mice given anti-CD40, presumably because they produce IFN-γ, which inhibits TEC proliferation. CD40 also increases on TEC during development of an autoimmune thyroid disease characterized by TEC hyperproliferation that develops spontaneously in IFN-γ−/− NOD.H-2h4 mice (TEC H/P). TEC H/P development is accelerated in mice given agonistic anti-CD40. These studies provide new information regarding the role of target tissue expression of CD40 in development of autoimmunity and suggest that use of agonistic anti-CD40 for tumor therapy could result in autoimmune disease.
Keywords: CD40, Thyroid, Hyperplasia
INTRODUCTION
Autoimmune thyroid diseases are the most common organ-specific autoimmune diseases in humans, and thyroid cancers account for >90% of all endocrine tumors (1). Thyroid nodules and thyroid hyperplasia are very common in humans and it is difficult to distinguish benign from neoplastic lesions (2–4). Since thyroid nodules and associated thyrocyte hyperplasia can be associated with an increased risk of developing thyroid cancer (1–3, 5), understanding the mechanisms underlying development of thyrocyte hyperplasia is very important.
Some strains of mice such as NOD and NOD.H-2h4 develop spontaneous autoimmune thyroiditis (SAT), i.e. without a requirement for immunization (6). NOD.H-2h4 mice that lack IFN-γ do not develop SAT, but they develop another autoimmune disease, thyroid epithelial cell hyperplasia/proliferation (TEC H/P) characterized by hyperplasia and extensive proliferation of thyroid epithelial cells, development of fibrosis, and loss of thyroid function (7, 8). TEC H/P is a chronic inflammatory autoimmune disease in which activated CD8+ T cells promote thyrocyte proliferation, at least in part, by producing TNF-α and TGF-β, which induce thyrocyte proliferation in vivo and in vitro (7, 9, 10). To further define the mechanisms by which CD8+ T cells promote thyrocyte hyperplasia, we generated CD4−/− IFN-γ−/− NOD.H-2h4 mice and showed that they developed TEC H/P comparable in severity to that of CD4+ IFN-γ−/− mice, but with a lower incidence. Splenocytes from CD4−/− donors were deficient in their ability to transfer TEC H/P to SCID recipients, suggesting that CD4+ T cells were needed to generate appropriately activated CD8+ T cells capable of transferring this disease (Yu et al., submitted). As part of these studies, we asked if an agonistic anti-CD40 antibody could be used to bypass the requirement for CD4+ T cells for activation of CD8+ cells as shown in other experimental models (11–14). Unexpectedly, the results indicated that agonistic anti-CD40 interacted with CD40 expressed on thyrocytes resulting in thyrocyte proliferation and greatly increased expression of CD40 by thyrocytes. These results provide new information as to how thyrocyte hyperplasia and autoimmune thyroid diseases can develop and also provide a cautionary note with respect to the use of anti-CD40 antibodies for tumor therapy.
CD40, a member of the tumor necrosis factor receptor (TNFR) superfamily, is expressed on cells of the immune system and also on some non-immune cells including microglia, epithelial cells, pancreatic beta cells and thyrocytes (15–19). CD40 signaling through its ligand, CD154, expressed on activated T cells is important for development of most immune responses (20), and expression of CD40 on non-immune cells or tissues may contribute to development of autoimmune diseases (17–19, 21, 22). Agonistic anti-CD40 antibodies can crosslink CD40 expressed on APC such as B cells, macrophages and dendritic cells leading to their activation, and can mimic the activity of CD154 by replacing the function of CD4+ T cells for activation of CD8+ T cells and antibody-producing B cells (11–14). In tumor bearing individuals, anti-CD40 can promote cytotoxic T cell responses, and ligation of CD40 on CD40 expressing tumor cells can have direct cytotoxic effects on some tumors (23–26). These multiple roles for CD40 have led to development of agonistic anti-CD40 antibodies used therapeutically for treatment of tumors (24, 25). However, because CD40 can be expressed in tissues known to be targets of self-reactive T cells that induce autoimmune diseases, it is important to know if CD40 expressing non-tumor cells might be targets for agonistic anti-CD40 antibodies and result in autoimmune diseases. The results of this study demonstrate profound and unexpected effects of agonistic anti-CD40 on thyroid epithelial cells (`thyrocytes') (TEC) of NOD and NOD.H-2h4 mice, with extensive proliferation of TEC, and earlier development and a greatly increased incidence and severity of the autoimmune disease TEC H/P in IFN-γ−/− NOD.H-2h4 mice.
MATERIALS AND METHODS
Mice
All NOD.H-2h4 and CBA/J, DBA/1 WT and DBA/1 IFN-γ−/− mice were generated in our breeding colonies at the University of Missouri. CD40−/− NOD mice were obtained from the colony of Drs. Diane Mathis and Christophe Benoist maintained at Jackson Laboratories. CD40−/− NOD mice were crossed with WT NOD.H-2h4 mice to generate CD40−/− NOD.H-2h4 mice. Mice were selected in the F2 generation for expression of the NOD.H-2h4 MHC by flow cytometry and for expression of the CD40−/− genotype by PCR analysis of tail DNA using the protocol and primer sequences provided by the Mathis/Benoist laboratory. CD40−/− IFN-γ−/− NOD.H-2h4 mice were obtained by crossing CD40−/− WT NOD.H-2h4 mice with IFN-γ−/− NOD.H-2h4 mice and selecting offspring for expression of the IFN-γ−/− and CD40−/− genotypes by PCR analysis of tail DNA. NOD, Fc null NOD, NOD.SCID, Balb/c and B6 Rag knockout mice were generously provided by Dr. Habib Zaghouani and were generated in his breeding colony at the University of Missouri. Fc null NOD mice were transferred to our colony through a Material Transfer agreement with RIKEN BRC who provided the original stock. Fc null NOD mice lacked Fc receptors both by flow cytometry and by PCR analysis of tail DNA using the primer sequences and protocol provided by RIKEN (27). IFN-γ−/− NOD mice were generated by crossing WT NOD mice with IFN-γ−/− NOD.H-2h4 mice and selecting for expression of the NOD MHC and IFN-γ genotypes by PCR analysis of tail DNA.
Anti-CD40
In most experiments, mice were given a single i.p. injection of 200 μg rat IgG2a anti-mouse CD40 antibody FGK45 (BioXCell, West Lebanon, NH). In preliminary experiments, 100–200μg anti-CD40 consistently induced strong thyrocyte proliferation 7 days later, whereas 10 μg gave less consistent results. Another rat IgG2a anti-CD40 mAb 1C10 (eBioscience, San Diego, CA) was used for some experiments, and had the same in vivo effects on the thyroid as FGK45. Control mice received 200 μg rat IgG2a isotype control (BioXCell).
Cell culture and transfer of TEC H/P to SCID recipients
Splenocytes from IFN-γ−/− or CD4−/− IFN-γ−/− donors with severe TEC H/P were pooled and cultured 72 hr as previously described in detail (7). Cells were harvested and 3×106 cells were transferred i.v. to IFN-γ−/− SCID recipients. Mice were given 0.08% NaI in their drinking water and thyroids were removed 4 or 8 wk later and evaluated for TEC H/P severity as described below.
Western blot
Protein was isolated from single frozen mouse thyroid lobes, quantitated, and 30 ug protein was added to a 10% SDS-PAGE gel as previously described (9). CD40 was quantified by Western blot using goat anti-mouse CD40 (1:500) (sc975; Santa Cruz Biotechnology, Santa Cruz, CA) followed by incubation with a secondary anti-rabbit peroxidase-conjugated antibody (Jackson ImmunoResearch). For normalization, membranes were stripped and reprobed with rabbit anti-actin primary antibody (sc 47778; Santa Cruz) and 1/3000 Horse Radish Peroxidase (HRP)-conjugated anti-rabbit IgG (Jackson ImmunoResearch) as described previously (9). Results are expressed as the ratio of CD40: β actin densitometric units ± SEM (× 100) and are representative of at least three independent experiments.
RT-PCR
Individual thyroid lobes were frozen in liquid nitrogen, homogenized in TRIzol (Life Technologies, Grand Island, NY), and RNA was extracted and reverse transcribed as described in detail previously (28). To compare relative levels of mRNA transcripts in different groups, samples were reverse transcribed and amplified at the same time using the same master mix. Densitometry analysis was done using an Alpha Imager imaging system (Alpha Innotech, Santa Clara, CA). Beta-actin was used as a housekeeping gene to control for differing RNA levels in individual thyroids. Densitometric units for each cytokine and β actin band were obtained, and results are expressed as the mean of the ratio of cytokine/β actin ± SEM.
Immunohistochemistry (IHC)
Frozen thyroids were sectioned and stained as previously described (7). Slides were blocked with 5% BSA/PBS for 60 min followed by 0.3% hydrogen peroxide for 30 min, then stained with primary antibody or isotype control at 4 C overnight. Primary antibody (anti-CD40 (1C10) (eBioscience, SanDiego, CA) was diluted in 1% BSA/PBS and used at a concentration of 5μg/ml. Biotinylated donkey anti-rat IgG (30 μg/ml in BSA/PBS) (Jackson ImmunoResearch, West Grove, PA) was added for 1 hr at room temperature followed by avidin-HRP using a Vectastain Elite kit (Vector Laboratories, Burlingame, CA) according to manufacturers instructions. Peroxidase activity was visualized with Nova Red substrate (Vector). A Nikon Eclipse 55i microscope (Nikon Corporation, Japan) with Nikon Plan 10×/0.25 and Nikon Plan 40×/0.65 objectives was used for imaging. Digital pictures of sections in Permount (Sigma) at 25 C were acquired with a Nikon DS Fi1 digital camera using NIS elements (version 3.22.00, build 710) imaging software.
Bone marrow chimeras
Bone marrow chimeras were generated as previously described (29). Briefly, recipient CD40−/− IFN-γ−/− or IFN-γ−/− NOD.H-2h4 mice 6–8 wk of age were irradiated (1000 Gy) and reconstituted with 5–10 × 106 bone marrow cells from CD40−/− IFN-γ−/− or CD40+ IFN-γ−/− donors. 6 wk later, when peripheral lymphocytes had reconstituted the hosts, mice were given 200 μg anti-CD40, and thyroids were removed 12–14 days later to assess thyrocyte proliferation. Chimerism of host mice was confirmed by flow cytometry.
Flow cytometry
Spleen cell suspensions were stained using antibodies specific for mouse CD4, CD8, CD40, CD19 and CD11b obtained from eBioscience or Biolegend (San Diego, CA). Samples were analyzed on a FACSCalibur (BD Biosciences) and analyzed using FlowJo (Treestar, Inc., Ashland, OR).
Primary cultures of thyrocytes/assessment of thyrocyte proliferation
Primary cultures of thyrocytes from naïve mice were generated as previously described (7, 9). After seeding in 8 well chamber slides, cells were maintained at 37 C with weekly changes of medium until they reached 70–80% confluence. Cells were then treated for 3 days with various concentrations of anti-CD40 (0.1–5 μg/ml) or isotype control IgG. Proliferation of thyrocytes was determined by IHC with the proliferation marker proliferating cell nuclear antigen (PCNA) (sc-7907, Santa Cruz Biotechnology, Santa Cruz, CA) as previously described (9). In some experiments, thyrocytes were cultured with 1 μg/ml anti-CD40 following a 4 hr preincubation with various concentrations (1–5 μg/ml) of 24G2 (Fc block) (eBioscience) to block interaction of anti-CD40 with Fc receptors on thyrocytes (30). To quantify proliferating cells, all cells in 5–6 randomly selected high power fields (× 400) were manually counted using image analysis software Metamorph version 6.3r6 (Molecular Devices) as previously described (9).
Evaluation of SAT and TEC H/P severity scores
When thyroids were removed, one lobe was fixed in formalin and stained using H&E). Thyroids were scored for severity of thyroid follicular cell (thyrocyte) hyperplasia/proliferation using a scale of 0–5+ as previously described (7, 8). Briefly, a score of 0-0+ indicates a normal thyroid with only very mild follicular changes and/or a few infiltrating inflammatory cells. A 1+ score indicates there are sufficient hyperplastic changes to cause replacement of several follicles, and a 2+ score indicates hyperplastic changes causing replacement or destruction of up to 1/4 of the gland. 3+ indicates that 1/4 to 1/2 of the gland is hyperplastic or destroyed, and 4+ indicates that greater than 1/2 of the normal thyroid follicles are proliferating and destroyed. Thyroids with a score of 5+ had few or no remaining normal follicles and extensive collagen deposition (fibrosis). Thyroids with 4–5+ severity scores always had widespread clusters of proliferating follicles, and the areas of proliferation were usually surrounded by collagen (8).
Serum T4 Assay
Serum thyroxine (T4) levels were determined using T4 ELISA kits (Leinco, Inc., St. Louis, MO) as previously described (8). Values for normal mouse serum ranged from 4–8 μg T4/dL of serum and values ≤ 3 μg T4/dL were considered to be low.
Statistical analysis
Mann-Whitney nonparametric analysis was used to analyze differences in TEC H/P severity scores between different groups of mice. Student's t test was used for all other analyses. The p values < 0.05 were considered significant.
RESULTS
NOD.H-2h4 SCID mice given anti-CD40 develop severe TEC H/P with fibrosis and loss of thyroid function
As mentioned in the Introduction, splenocytes from CD4−/− IFN-γ−/− NOD.H-2h4 mice with severe TEC H/P do not consistently transfer severe TEC H/P to SCID recipients (Yu et al., submitted), whereas highly purified CD8+ T cells transfer very severe TEC H/P (7). To test the hypothesis that agonistic anti-CD40 could provide a signal to activate splenocytes from CD4−/− mice to transfer severe TEC H/P, splenocytes from CD4−/− donors were cultured and transferred to SCID recipients as described in Methods. Recipients were given anti-CD40 or rat IgG isotype control the day of cell transfer, and thyroids were removed 4 wk later. All but one recipient given anti-CD40 developed very severe (5+) TEC H/P, whereas mice given isotype control had no TEC H/P (Figure. 1 A). Unexpectedly, however, SCID recipients given anti-CD40 but no splenocytes also had extensive thyrocyte proliferation (Fig. 1A). Grossly, thyroids of both groups of recipients were greatly enlarged (>10–20 fold) and white. Thyroid destruction was extensive as evidenced by collagen deposition (fibrosis) and hypothyroidism with low levels of serum T4 (Fig. 1A,B). Thyrocyte proliferation began as enlargement and hyperplasia of thyrocytes 3–4 days after injection of anti-CD40, and by 7 days, thyrocytes had proliferated extensively, forming large clusters that completely filled the follicular lumen (Fig. 1B). There was no evidence of resolution for at least 6 wk (Fig. 1 B; data not shown). There was mild infiltration of mononuclear cells in the livers of most SCID recipients 3–7 days after injection of anti-CD40, but this did not persist more than 10 days (Supplementary Fig. 1C and data not shown). Mononuclear cell infiltration was not seen in salivary glands, and was rarely seen in kidney or pancreas of SCID mice given anti-CD40 (data not shown). SCID mice given IgG2a isotype control had no changes in their thyroids or any other organs (Fig. 1B and not shown). Another rat IgG2a agonistic anti-CD40 mAb, 1C10, had similar effects on thyroids of SCID mice (data not shown). Therefore, agonistic anti-CD40 induces extensive proliferation of thyrocytes in NOD.H-2h4 SCID mice, and the primary target is the thyroid.
Figure 1.

Agonistic anti-CD40 induces thyrocyte hyperplasia in SCID mice. A. IFN-γ−/−NOD.H-2h4 SCID mice were given cultured splenocytes from CD4−/− IFN-γ−/− NOD.H-2h4 donors with severe (5+) TEC H/P or no cells. Recipients were given 200 μg anti-CD40 or isotype control the day of cell transfer. Thyroids were removed 4 wk later and serum T4 was determined by ELISA. Results for 2 experiments are shown, and are representative of 3 similar experiments. Each dot represents an individual mouse (Isotype, n = 5; cells anti-CD40, n = 9; no cells anti-CD40, n = 5). TEC H/P severity, * p < 0.01, Mann-Whitney. Serum T4 results were from some of the animals in the depicted experiment (Isotype, n = 3; cells/anti-CD40, n = 7; and no cells/anti-CD40, n = 3) and are expressed as the mean ± SEM; * p < 0.05, Student's t test. B. IFN-γ−/− NOD.H-2h4 SCID mice were given anti-CD40 or isotype control, and TEC H/P severity was determined 0.5, 1 or 4 wk later. Images are representative of 4 experiments, for isotype control, n = 10; days 3–4, n = 21; 1 wk, n = 25; and 4 wk, n = 27. (H&E stain, 100× bar is 0.1 mm, 400× bar is 0.05 mm.)
Effects of anti-CD40 in other strains of lymphopenic and nonlymphopenic mice
Because anti-CD40 had such unexpected effects on thyroids of NOD.H-2h4 SCID mice, it was important to determine if this was because of the lymphopenic SCID environment, or if anti-CD40 would induce thyrocyte proliferation in other lymphopenic and nonlymphopenic mice. To address this question, anti-CD40 was given to various strains of mice, and thyroids were examined after 1 wk (acute effects) and 3–8 wk (sustained effects). The results indicate that the NOD or NOD.H-2h4 genetic background and IFN-γ deficiency were the primary factors needed for induction of severe and sustained thyrocyte proliferation by anti-CD40 (Table I and Supplementary Fig. 1). Anti-CD40 induced strong and sustained thyrocyte proliferation in lymphopenic NOD.SCID and NOD.H-2h4 SCID mice, whereas it had no effect in lymphopenic B6 and Balb/c Rag−/−mice. Anti-CD40 induced strong and sustained thyrocyte proliferation in nonlymphopenic IFN-γ−/− NOD and IFN-γ−/− NOD.H-2h4 mice, whereas no changes were seen in thyroids of CD40−/− IFN-γ−/− NOD.H-2h4 mice given anti-CD40 (Table I; Supplementary Fig. 1A,B). Unexpectedly, anti-CD40 induced proliferation of thyrocytes of WT NOD.H-2h4 and WT NOD mice after 7 days, but the effects were more variable, and thyroids had essentially returned to normal by 21 days (Table I; Supplementary Fig. 1 A). Anti-CD40 induced no apparent changes in thyroids of CBA/J, WT DBA/1 or IFN-γ−/− DBA/1 mice, even though they have thyroiditis-susceptible MHC haplotypes (Table I). Excluding lymphoid organs, the major effects of agonistic anti-CD40 were always in the thyroid, although there was some mononuclear cell infiltration in the liver, and occasionally in the kidney and pancreas, 7 days after injection of anti-CD40 (Supplementary Fig. 1C and not shown). With the exception of the thyroid, mononuclear cell infiltrates in other organs did not persist longer than 10 days after injection of anti-CD40 (not shown).
Table I.
Effects of anti-CD40 on thyroids of different strains of mice
| Strain | 6–10 daysa | 3–8 wka | Serum T4 b |
|---|---|---|---|
| IFN-γ−/− NOD.H-2h4 SCID | 4–5+ (n = 25 ) | 4–5+ (n=13) | Low |
| IFN-γ−/− NOD.H-2h4 | 4–5+ (n=18) | 4–5+ (n=28) | Low |
| CD4−/− IFN-γ−/− NOD.H-2h4 | 4–5+ (n=20) | 4–5+(n=16) | Low |
| B cell −/− IFN-γ−/− NOD.H-2h4 | 4–5+ (n=5) | 4–5+ (n=9) | NDc |
| WT NOD.H-2h4 | 1–4+ (n=10) | 0–3+d (n=11) | Normal |
| CD40−/− NOD.H-2h4 | 0 (n=8) | ND | Normal |
| IFN-γ−/− NOD | 4–5+ (n=8) | 4–5+ (n=6) | Low |
| WT NOD | 0+−4+ (n=17) | 0+ (n=8) | Normal |
| Fc null NOD | 0+−4+ (n=14) | ND | ND |
| NOD. SCID | 4+ (n=9) | 4+ (n= 7) | Normal |
| IFN-γ−/− DBA/1 | 0 (n = 11) | 0 (n=6) | Normal |
| WT DBA/1 | 0 (n=3) | 0 (n=6) | Normal |
| CBA/J | 0 (n = 5) | ND | Normal |
| B6 Rag−/− | 0 (n = 3) | 0 (n = 3) | ND |
| Balb/c Rag−/− | 0 (n = 3) | ND | ND |
The indicated strains of mice were given 200 μg anti-CD40 and thyroids were removed after 6–10 days or 3–8 wk. TEC H/P severity scores are assigned as described in Methods. The range shown for each strain does not include an occasional mouse in the larger groups that failed to respond to anti-CD40, presumably because of an ineffective i.p. injection. Numbers of mice in each group are indicated in parentheses.
Serum T4 levels 6–10 days and 3–8 wk after anti-CD40. Normal serum T4 is defined as ≥ 3 μg/dL and mice with low serum T4 had T4 levels of ≤3 μg/dL. Mice with 5+ severity scores generally have low serum T4 levels whereas mice with 4+ or lower severity scores always have normal serum T4 levels.
ND = not determined.
WT NOD.H-2h4 mice given either isotype control or anti-CD40 had SAT but did not have TEC H/P 6–8 wk after receiving NaI in their water.
Changes in thyroids of all strains of mice that responded strongly to anti-CD40 were essentially identical to those described above for NOD.H-2h4 SCID mice (Fig. 1A, B). Thyrocyte proliferation was maintained for many months in all IFN-γ−/− mice, with deposition of collagen (fibrosis) that became more extensive over time (Supplementary Fig. 1 and not shown). Most SCID and IFN-γ−/− NOD.H-2h4 and IFN-γ−/− NOD mice had low serum T4 2 wk after anti-CD40 (Fig. 1A and Table I), whereas WT mice had little fibrosis and their serum T4 levels were normal (data not shown). The reason anti-CD40 did not induce persistent thyrocyte proliferation in WT thyroids is presumably because they produce IFN-γ which suppresses thyrocyte proliferation and survival in vitro and in vivo (9, 29, 31).
Thyroids express CD40 and agonistic anti-CD40 increases CD40 on thyrocytes
The effects of anti-CD40 on thyroids could be explained if thyrocytes expressed CD40 and if cross-linking of CD40 by agonistic anti-CD40 resulted in proliferation of thyrocytes. Both neoplastic and nonneoplastic human thyrocytes express CD40, and CD40 expression increases during inflammation (19, 22). To our knowledge, CD40 expression by murine thyrocytes during spontaneous development of autoimmune thyroid diseases has not been reported. To determine if thyrocytes express CD40 and if CD40 expression changes after administration of agonistic anti-CD40, IHC was used to examine protein expression of CD40 at various times after anti-CD40 administration. Thyrocytes of normal unmanipulated IFN-γ−/− NOD.H-2h4 SCID mice and mice given isotype control had no detectable CD40 protein by IHC (Fig. 2A), but low levels of CD40 were consistently detected in normal thyroids by Western blot (Fig. 2D). CD40 expression was variable and higher, 3–4 days after injection of anti-CD40 both by IHC and Western blot, further increased by day 7, was maximal by 21–28 days (Fig. 2A,E), and remained high for at least 6 wk (not shown). There were no histologic changes in thyroids of CD40−/− IFN-γ−/− mice given anti-CD40, and CD40 was undetectable in CD40−/− thyroids by IHC or Western blot even after longer exposure of the gels (Fig. 2 A, D). CD40 protein also increased 7 days after anti-CD40 in thyroids of nonlymphopenic WT and IFN-γ−/− NOD.H-2h4 mice (Fig. 2 B,C,F) as well as NOD.SCID and WT and IFN-γ−/− NOD mice (not shown). CD40 protein expression remained high in IFN-γ−/− thyroids for many weeks (Fig. 2 B,F and data not shown), but decreased in WT thyroids 3 wk after anti-CD40 (Fig. 2C and data not shown). CD40 was barely detectable in thyroids of IFN-γ−/− DBA/1 and WT DBA/1 and CBA/J mice given anti-CD40 even after exposure of gels for 1 hr (data not shown). It is not known why thyroids of mice that do not develop spontaneous autoimmunity, in particular IFN-γ−/− DBA/1, had no apparent response to agonistic anti-CD40. Splenic B cells from DBA/1 mice express as much CD40 as B cells from NOD.H-2h4 mice, and anti-CD40 induced comparable expansion of splenic B cells in both strains (data not shown). As will be shown below, CD40 is also upregulated on IFN-γ−/− NOD.H-2h4 thyrocytes during spontaneous development of TEC H/P without administration of anti-CD40. This suggests that upregulation of CD40 on thyrocytes is a normal consequence of the autoimmune inflammatory response that leads to spontaneous development of TEC H/P in IFN-γ−/− NOD.H-2h4 mice, and this does not occur in strains of mice that do not spontaneously develop autoimmune thyroid disease.
Figure 2.

Anti-CD40 induces upregulation of CD40 by thyrocytes. A. Frozen sections of thyroids from IFN-γ−/− NOD.H-2h4 SCID or CD40−/− IFN-γ−/− NOD.H-2h4 SCID mice given anti-CD40 as indicated were examined for expression of CD40 by IHC. (100× bar is 0.1 mm and 400× bar is 0.05 mm) Images are representative of 2 experiments, n = 6. B. CD40 expression in thyroids of IFN-γ−/− NOD.H-2h4 mice 1 and 4 weeks after anti-CD40. Images are representative of 2 experiments, n = 6. C. CD40 expression in thyroids of wild type NOD.H-2h4 mice 1 and 4 weeks after anti-CD40. Images are representative of 2 experiments, n = 6. D, E, F. CD40 protein expression in thyroids determined by Western Blot. D, thyroids of CD40−/− IFN-γ−/− NOD.H-2h4 SCID mice given anti-CD40 1 wk earlier, and CD40+ IFN-γ−/− NOD.H-2h4 SCID mice given anti-CD40 or isotype control 4 days earlier. Films were developed for 10 minutes. E, thyroids of IFN-γ−/− NOD.H-2h4 SCID mice 0.5, 1 and 4 weeks after anti-CD40. F, thyroids of WT and IFN-γ−/− NOD.H-2h4 mice 1 wk after injection of anti-CD40 or isotype control. Gels in E and F were developed for 30 sec. Bar graphs are means of the ratio of CD40 /actin ± SEM for the bands shown on the left of each graph. * p < 0.05, Student's t test. Results are representative of 5–6 separate samples per group.
Anti-CD40 induces thyrocyte proliferation in the absence of CD40 expression by lymphoid cells
Agonistic anti-CD40 antibody activates and induces proliferation of CD40 expressing lymphoid cells, especially B cells, dendritic cells and macrophages (13, 24, 25). In these studies, splenic dendritic cells and macrophages expanded in all SCID mice 3–7 days after injection of anti-CD40, and anti-CD40 induced expansion of splenic B cells in nonlymphopenic mice, which was maximal 4–7 days after injection of anti-CD40. NOD and NOD.H-2h4 mice also have CD40-expressing T cells (15), but anti-CD40 did not expand CD40-expressing T cells (data not shown). Because agonistic anti-CD40 induced changes both in thyroids and lymphoid cells of NOD.H-2h4 mice, it was important to determine if the lymphoid or nonlymphoid compartment was most important for proliferation of thyrocytes in mice given anti-CD40. To address this question, bone marrow chimeras were generated in which thyroid (nonlymphoid) or lymphoid compartments were derived either from CD40+ or CD40−/− IFN-γ−/− NOD.H-2h4 mice. Six weeks after irradiation and bone marrow reconstitution, all mice were given anti-CD40 and thyroids were removed 12–14 days later. The results clearly show that CD40 expression by non-lymphoid cells (presumably the thyroid) was necessary and sufficient for anti-CD40 to induce thyrocyte proliferation (Fig. 3A). When lymphocytes were CD40-negative and thyrocytes and other nonlymphoid cells were CD40+, thyrocyte proliferation tended to be slightly greater than in mice whose thyroids and lymphocytes were both CD40+. Conversely, when thyroids and other nonlymphoid cells were CD40-negative, anti-CD40 did not induce thyrocyte proliferation even if lymphoid cells were CD40+. Essentially all splenic T cells, B cells, macrophages and dendritic cells were derived from the bone marrow donors as shown by flow cytometry (Fig. 3B). It is not known why anti-CD40-mediated proliferation of CD40+ thyroids tended to be greater when lymphoid cells were CD40-negative, but this trend was seen in all experiments. One possibility is that more anti-CD40 was available to stimulate the thyroids when there were no CD40+ cells for anti-CD40 to react with in peripheral lymphoid organs. Clearly, the same amount of antibody had to react with many more cells in mice given CD40+ lymphocytes.
Figure 3.

CD40 expression by thyroids/ nonlymphoid cells is required for anti-CD40 to induce thyrocyte proliferation, but lymphoid cells do not have to express CD40. A. Mice expressing CD40 either on lymphoid or non-lymphoid cells were generated by creating bone marrow chimeras in which recipient nonlymphoid cells were CD40+ or CD40-negative and donor lymphoid cells were CD40+ or CD40-negative. All donors and recipients were IFN-γ−/−. 6 wk after irradiation and bone marrow reconstitution, all mice were given 200 μg anti-CD40. Thyroids were removed 12 d later and scored for severity of thyrocyte proliferation. * p < 0.01, Mann-Whitney. Data represents 2 of 4 total experiments. n = 14, 14, 14, 11. Error bars are ± SEM. B. Splenocytes from bone marrow chimeras were examined for expression of CD8 and CD40 or CD19 andCD40 by flow cytometry. * p < 0.05, Student's t test. Data are the percentages of the indicated cell types; 2 experiments, n = 10 for each group. Graphs on the right are representative flow cytometry plots for each group.
Anti-CD40 induces thyrocyte proliferation in vitro
Anti-CD40 induces proliferation of thyrocytes of NOD and NOD.H-2h4 mice in vivo via an apparently direct effect of anti-CD40 on the thyroid. To determine if anti-CD40 can induce proliferation of thyrocytes vitro in the absence of any other cells, thyrocyte cultures were generated as previously described (7, 9). After they reached 60–70% confluence, anti-CD40 or isotype control IgG was added, thyrocytes were harvested and proliferation was evaluated by immunostaining for PCNA. Anti-CD40 induced a concentration-dependent increase in PCNA+ cells in CD40+ thyrocytes, but had no effect on CD40−/− thyrocytes (Fig. 4A). These results are also consistent with the results shown in Fig. 3, both approaches demonstrating that anti-CD40 can induce thyrocyte proliferation independently of any CD40 contribution by lymphoid cells.
Figure 4.

Anti-CD40 promotes proliferation of thyrocytes in vitro and in vivo independent of FcγRIIB. A. 60–70 % confluent primary cultures of TEC from CD40+ IFN-γ−/− or CD40−/− IFN-γ−/− NOD.H-2h4 mice were incubated with 0.2 to 5 μg/ml anti-CD40 or isotype control for 3 days as indicated. Thyrocyte proliferation was determined by immunostaining with anti-PCNA. Data was quantified by counting 5–6 fields for expression of PCNA+ cells (red). Results are the mean ± SEM of 5–6 fields. * p < 0.05, Student's t-test. Data is representative of 3 separate experiments, n = 3. 400× bar is 0.05 mm. B. Primary cultures of 60–70% confluent TEC from NOD and FcγR null NOD mice were incubated with 1 μg/ml anti-CD40 or isotype control for 3 days. Thyrocyte proliferation was determined as in A. Data is representative of 2 experiments, n = 2. (400× bar is 0.05 mm). Data was quantified as in A. C. Primary cultures of thyrocytes from CD40 + IFN-γ−/− NOD.H-2h4 mice were cultured 3 days with 1 μg/ml anti-CD40 in the presence or absence of 1 μg/ml Fc block (24G2). Thyrocyte proliferation was determined as in A. Data is representative of 2 experiments. D. Severity of thyrocyte proliferation in wild type and FcγR null NOD mice given anti-CD40 or isotype control 7 days earlier. Data represents mean severity scores in 2 of 4 experiments; n = 5, 6. E, Representative H&E stained sections of thyroids in D. 100× bar is 0.1 mm, 400× bar is 0.05 mm.
The effects of anti-CD40 are independent of FcγRIIB
The in vivo and in vitro effects of agonistic anti-CD40 antibodies were recently shown to require engagement of anti-CD40 by FcγRIIB (30, 32). Human thyrocytes can express FcγRIIB (33), so it was of interest to determine if coengagement of CD40 and FcγRIIB was required for anti-CD40 to induce thyrocyte proliferation. Others showed that effects of anti-CD40 could be blocked by coadministration of a blocking FcγR antibody (24G2) (30), but 24G2 had no effect on the ability of anti-CD40 to induce thyrocyte proliferation in vitro (Figure 4C) or in vivo (data not shown). To directly address a role for FcγRIIB in thyrocyte proliferation, we used NOD and Fc null NOD mice. Anti-CD40 induced comparable proliferation of NOD and Fc null NOD thyrocytes in vitro (Fig. 4B), and had equivalent effects on thyrocytes of NOD and Fc null NOD mice in vivo (Figure 4D, E). These results indicate that coengagement of FcγRIIB and anti-CD40 was apparently not required for anti-CD40 to induce thyrocyte proliferation.
Anti-CD40 leads to increased expression of cytokines and chemokines in thyroids of SCID mice
To determine if agonistic anti-CD40 induced expression of proinflammatory cytokines or chemokines in the thyroid, IFN-γ−/− NOD.H-2h4 SCID mice were given anti-CD40 and thyroids were removed after various intervals. RNA was isolated from single thyroid lobes and expression of proinflammatory molecules was examined by RT-PCR. The results indicate that expression of mRNA for several proinflammatory molecules involved in innate immunity, e.g. IL-6, IL-1 and IL-12, increased significantly 4 days after injection of anti-CD40 (Figure 5). IL-6 and IL-1 mRNA remained elevated for the next 3 wk, while IL-12 mRNA declined after day 8. Expression of MCP-1, a chemokine upregulated by anti-CD40 in other studies (13, 21), was also increased. CD40 mRNA expression was relatively high in thyroids of mice given isotype control (in contrast to CD40 protein expression which was very low; see above) and CD40 mRNA remained elevated after injection of anti-CD40. Expression of thyroglobulin mRNA declined, probably because thyroids that proliferate in response to anti-CD40 have abnormal thyroid follicles that have almost no colloid, the source of thyroglobulin in normal thyroid follicles. It is not known if the proinflammatory molecules in thyroids of SCID mice given anti-CD40 are produced by thryocytes or by macrophages and dendritic cells recruited to the thyroid after injection of anti-CD40, although it is known that thyrocytes can produce both IL-6 and MCP-1 in response to agonistic anti-CD40 (21).
Figure 5.

Anti-CD40 induces expression of proinflammatory cytokines and chemokines in thyroids of SCID mice. SCID mice were given isotype control or anti-CD40 and thyroids were removed 4, 8, 10 or 21 days later as indicated. RNA was isolated from individual thyroid lobes, and cDNA was amplified using primers specific for the indicated molecules. Results are expressed as the mean ratio of the indicated molecule/β actin ± SEM for 4–5 individual samples from each group. * p<0.05 compared to isotype control group, Student's t test.
Anti-CD40 promotes development of severe thyrocyte hyperplasia (TEC H/P) and fibrosis in IFN-γ−/− NOD.H-2h4 mice
Agonistic anti-CD40 increases CD40 expression and induces proliferation of NOD and NOD.H-2h4 thyrocytes, and its effects are most evident and sustained when IFN-γ is absent. IFN-γ−/− NOD.H-2h4 mice develop an autoimmune disease characterized by extensive proliferation of thyrocytes, thyroid fibrosis and hypothyroidism (7, 8). Severe TEC H/P develops 6–7 mo after administration of NaI in the drinking water with an incidence of 60–70% (6, 8). If upregulation of CD40 on thyrocytes is important for development of thyroid autoimmunity, it should also occur during spontaneous development of TEC H/P, and agonistic anti-CD40 should induce earlier development of and/or a greater incidence of severe TEC H/P. To test this hypothesis, IFN-γ−/− NOD.H-2h4 mice were given NaI in their drinking water, and anti-CD40 or isotype control, and thyroids were removed after various intervals. At each time interval, most anti-CD40 treated mice had severe thyrocyte hyperplasia, while control mice given rat IgG had very mild or no TEC H/P unless they were given NaI water for > 6 mo (Fig. 6A). Splenocytes or purified CD8+ splenic T cells from mice with severe TEC H/P transfer severe TEC H/P to SCID recipients (7), and splenocytes from IFN-γ−/− mice given anti-CD40 2 months previously transfer severe TEC H/P to SCID recipients (data not shown). Therefore agonistic anti-CD40 leads to increased expression of CD40 on thyrocytes (see above), and promotes earlier development and a greatly increased incidence of severe TEC H/P in IFN-γ−/− NOD.H-2h4 mice.
Figure 6.

Anti-CD40 results in early development of TEC H/P in IFN-γ−/− NOD.H-2h4 mice. A. All mice were given NaI in their drinking water. At various times after injection of anti-CD40 or isotype control, thyroids were removed and scored for severity of thyrocyte proliferation. * p < 0.01, Mann Whitney. Error bars are ± SEM, and represent 4 experiments. For isotype, n = 10; anti-CD40, 1 wk, n = 15; 4 wk, n = 15; 8 wk, n = 18, and isotype 6–7 mo, n = 10. B. CD40 expression by thyrocytes of IFN-γ−/− NOD.H-2h4 mice with TEC H/P. Images are representative of 2 experiments, n = 6. 100× magnification bar is 0.01 mm, 400× magnification bar is 0.05 mm. C. Splenocytes from IFN-γ−/− NOD.H-2h4 mice with severe TEC H/P were cultured and transferred to IFN-γ−/− SCID NOD.H-2h4 mice as described in Methods. CD40 expression was examined 4 and 8 wk later in mice with varying TEC H/P severity scores. Images are representative of 2 experiments, n= 6. D. CD40 protein expression in thyroids of IFN-γ−/− mice with 0+ or 5+ TEC H/P severity scores or IFN-γ−/− mice given anti-CD40 1 wk earlier. Data was quantified by comparing the ratio of CD40 to actin as in Figure 3. Results are representative of 2 experiments, n=6. Error bars are ± SEM; * p<0.05, Student's t test.
To determine if CD40 increases on thyrocytes as a normal consequence of development of TEC H/P, CD40 protein was determined in thyroids of IFN-γ−/− NOD.H-2h4 mice that did or did not have TEC H/P. CD40 was highly expressed in thyroids of all mice with severe TEC H/P, whereas CD40 was undetectable by IHC in thyroids of mice that did not develop TEC H/P (Fig. 6B). CD40 was also highly expressed and sustained in thyroids of SCID recipients of splenocytes from IFN-γ−/− donors when they developed severe TEC H/P (4–5+ severity scores), but thyroids of mice with 1–2+ TEC H/P severity scores had lower expression of CD40 (Fig. 6C and not shown). CD40 expression in thyroids of mice developing severe TEC H/P was comparable to that in mice given agonistic anti-CD40 (Fig. 6D). Therefore, CD40 is upregulated on thyrocytes during spontaneous development of TEC H/P, an autoimmune thyroid disease characterized by hyperproliferation of thyrocytes. Agonistic anti-CD40 mimics the events that occur spontaneously, but greatly accelerates development of TEC H/P in IFN-γ−/− NOD.H-2h4 mice. These results therefore establish a previously unrecognized mechanism by which autoimmune thyroid diseases, particularly those associated with thyrocyte hyperplasia, can develop.
DISCUSSION
The initial goal of this study was to determine if agonistic anti-CD40 could provide a signal for activation of T cells from CD4−/− donors to transfer severe TEC H/P to SCID recipients. However, agonistic anti-CD40 had unexpected and very profound effects on thyroids of NOD.H-2h4 SCID recipients (Fig. 1), leading to extensive proliferation of thyrocytes in NOD.H-2h4 SCID mice, with fibrosis and sufficient loss of normal thyroid follicles to result in hypothyroidism (Fig. 1). Anti-CD40 had similar effects on TEC of all IFN-γ−/− NOD and IFN-γ−/− NOD.H-2h4 mice (Supplementary Fig. 1 and Table I). The ability of anti-CD40 to induce thyrocyte proliferation was accompanied by increased CD40 protein expression on thyrocytes (Fig. 2), and increased mRNA expression of proinflammatory cytokines and chemokines in thyroids (Fig. 5). In the absence of T and B cells (SCID mice), anti-CD40 induced expression of mRNA for molecules usually produced by APC during inflammation, e.g. IL-1, IL-6 and IL-12. Similar proinflammatory molecules were produced in thyroids of mice with transgenic overexpression of CD40 in thyroids (21), in guts of mice given anti-CD40 to induce colitis (13), and by thyrocytes in response to anti-CD40 in vitro (21).
Human thyrocytes express CD40 (19, 22) and transgenic overexpression of CD40 in the thyroid promotes cytokine and autoantibody production, and results in more severe thyroid lesions in a mouse model of Graves' disease (21). To our knowledge, the current study is the first to demonstrate that agonistic anti-CD40 increases CD40 expression on thyrocytes, and to show that CD40 expression increases on thyrocytes during spontaneous development of the autoimmune thyroid disease TEC H/P. CD40 cross-linking leads to binding of TNFR associated factors (TRAFs) to CD40, resulting in activation of MAP kinases (17). Although the precise mechanism by which agonistic anti-CD40 induces thyrocyte proliferation is unknown, MAP kinases contribute to cell cycle regulation induced by cAMP in FRTL-5 thyroid cells (34), and it is reasonable to suggest that anti-CD40 might promote thyrocyte proliferation through activation of this pathway.
Anti-CD40 induced thyrocyte proliferation in thyroids of WT NOD and WT NOD.H-2h4 mice after 7–10 days, but proliferation was not sustained, and WT thyroids expressed less CD40 than IFN-γ−/− thyroids (Fig. 2). Because IFN-γ inhibits thyrocyte proliferation and survival in vitro and in vivo (9, 29, 31, 35), the early proliferation and upregulation of thyrocyte CD40 in WT mice probably occurs before significant amounts of IFN-γ are produced. After T cells are activated, they migrate to the thyroid, and produce IFN-γ to inhibit thyrocyte proliferation (9, 29).
Importantly, our results demonstrate that agonistic anti-CD40 greatly potentiates early development of severe TEC H/P, an autoimmune disease that normally requires an induction period of > 6 months (6, 8). When IFN-γ−/− NOD.H-2h4 mice are given a single injection of anti-CD40 at 2–3 mo of age, most mice have extensive thyrocyte proliferation 1 week later which persists for many months. In contrast, thyrocyte proliferation is not seen in most age-matched mice given isotype control unless they are given iodine in their water for >6 months (Fig. 6). Thyroids of IFN-γ−/− mice given anti-CD40 8 wk earlier have T cell infiltrates similar to those of mice that develop TEC H/P after 6–7 mo, and their splenocytes transfer severe TEC H/P to SCID recipients (data not shown). Thyroids with severe thyrocyte proliferation 1–2 wk after injection of anti-CD40 have relatively few infiltrating T cells, and their splenocytes do not transfer severe TEC H/P to SCID recipients (our unpublished results). Therefore, the early thyrocyte proliferation that occurs in response to agonistic anti-CD40 is simply thyrocyte proliferation that can be severe enough to result in hypothyroidism. In contrast, TEC H/P is an autoimmune disease that is transferrable to SCID recipients by T cells present in spleens of mice with TEC H/P. Agonistic anti-CD40 promotes earlier development of TEC H/P, probably due to its ability to activate APC and promote T cell activation.
CD40 is upregulated on thyrocytes as a natural consequence of developing the spontaneous autoimmune disease TEC H/P, and CD40 expression levels generally correlate with TEC H/P severity scores (Fig. 6). Based on these results, we hypothesize that agonistic anti-CD40 facilitates a process that occurs spontaneously in IFN-γ−/− NOD.H-2h4 mice, resulting in a greater incidence and earlier development of severe TEC H/P as shown in Fig. 7. The results of this study suggest that upregulation of CD40 by thyrocytes is critical for development of thyroid autoimmunity and if CD40 is absent or signals leading to its upregulation are absent, thyroid autoimmunity will not develop. This is consistent with our finding that CD40−/− IFN-γ−/− NOD.H-2h4 mice are resistant to TEC H/P (our unpublished results), and that susceptibility to Graves' hyperthyroidism is associated with expression of a specific single nucleotide polymorphism in the CD40 gene (21, 36). These results suggest a previously unrecognized mechanism for development of autoimmune thyroid diseases, in particular those associated with hyperplasia/proliferation of thyrocytes. Thyrocyte hyperplasia in humans is very common, and can be associated with an increased risk of thyroid cancer (1, 3–5). Because many thyroid tumors express CD40 (19), these results could have important implications for understanding the mechanisms underlying development of thyroid cancer. In addition, when agonistic anti-CD40 antibodies are used for tumor therapy (23, 25, 26), autoimmunity or damage to CD40-expressing tissues could be an undesirable consequence of such treatment.
Figure 7.
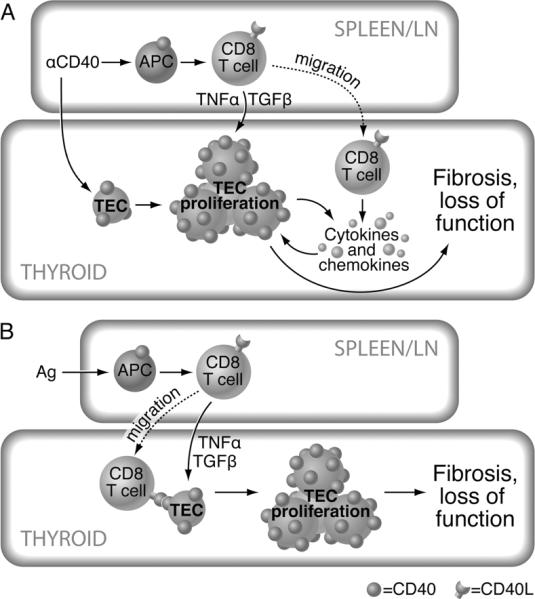
A. Proposed mechanism by which agonistic anti-CD40 induces thyrocyte proliferation. Anti-CD40 crosslinks CD40 on thyroid epithelial cells (TEC) (thyrocytes) leading to increased CD40 expression on TEC, proliferation of TEC and production of proinflammatory cytokines by TEC and/or monocytes or dendritic cells in the thyroid. Anti-CD40 also activates APC in peripheral lymphoid organs. In nonlymphopenic mice, the activated APC facilitate activation of CD8+ T cells which traffic to the thyroid where they produce cytokines such as TNF-α and TGF-β (7, 9) that promote thyrocyte (TEC) proliferation, resulting in TEC H/P. B. Proposed mechanism for spontaneous development of TEC H/P. Antigen is presented to T cells by APC, leading to upregulation of CD40L, activation of APC and activation of CD8+ T cells. This process may be facilitated by increased environmental iodine. Activated T cells traffic to the thyroid where they interact with CD40 on TEC and produce cytokines leading to increased expression of CD40 on TEC, TEC proliferation, further cytokine production and recruitment of additional cells to the thyroid. Note that T cells in the figure are depicted as CD8+ T cells because CD8+ T cells have been shown to be the major effectors in TEC H/P (7). However, CD4+ T cells are also activated and migrate to the thyroid, and are known to play a role during initial activation of the CD8+ T cells.
Our results also have implications for other autoimmune diseases in which anti-CD40 might lead to increased expresson of CD40 in target organs and result in autoimmunity (17). In the model studied here, the major target of agonistic anti-CD40 is the thyroid itself as shown in Fig. 7A. While B cells and other APC are also targets of anti-CD40, their response is not needed for development of the proliferative changes in the thyroid (Fig. 3). Expression of CD40 in tissues that are targets of autoimmune inflammation is important for development of EAE (18, 37) and transgenic overexpression of CD40 in the thyroid results in more severe Graves' disease in mice (21). In other models, agonistic anti-CD40 led to increased tissue destruction and proliferation of self-reactive CD8+ T cells (38), promoted development of CD8+ memory T cells in diabetes (14) and induced increases in inflammatory cytokines and intestinal inflammation in mice (13).
Because the effects of agonistic anti-CD40 on thyrocytes were greatest in mice lacking IFN-γ, promotion of autoimmunity by agonistic anti-CD40 might be relatively rare in patients receiving agonistic anti-CD40 for therapy of tumors. However, drugs or radiation treatments given to cancer patients generally lead to immunosuppression, including suppression of cells that produce IFN-γ. Moreover, when tumors are actively growing, tumor-reactive cytotoxic T cells that potentially produce IFN-γ would likely be inhibited, and would not be present in large numbers at the tumor site. Therefore, while humans are not IFN-γ-deficient in the same sense that our mice are, there are clinical situations where IFN-γ is likely to be low.
In summary, the results of this study demonstrate a previously unrecognized effect of agonistic anti-CD40 on thyroids of two autoimmune-prone strains of mice, NOD and NOD.H-2h4. We show for the first time that cross-linking CD40 on thyrocytes leads to greatly increased expression of CD40 and extensive thyrocyte proliferation. Thyrocyte proliferation can occur in the absence of T and B lymphocytes, e.g. in SCID mice, but when T cells are present, anti-CD40 greatly accelerates development of the autoimmune disease TEC H/P in IFN-γ−/− NOD.H-2h4 mice. TEC/HP is chronic and results in thyroid fibrosis and low serum T4. Most importantly, although the process is much slower in the absence of anti-CD40, the outcome and the underlying mechanisms by which TEC H/P develops are the same, i.e. T cell activation, upregulation of CD40 on thyrocytes, chronic and severe thyrocyte proliferation, fibrosis and low serum T4. These experiments define a previously unrecognized role of CD40 in development of autoimmunity and suggest that increased CD40 expression on thyrocytes could play a role in development of thyrocyte hyperplasia and thyroid cancer.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr. Habib Zaghouani and members of his laboratory for generously providing some of the mice used in this study.
2. Abbreviations used in this article
- TEC
thyroid epithelial cells or thyrocytes
- TEC H/P
thryoid epithelial cell hyperplasia and proliferation
- T4
thyroxine
- HRP
horse radish peroxidase
- PCNA
proliferating cell nuclear antigen
Footnotes
This work was supported by National Institutes of Health Grant RO1 AI 074857 and by the Lottie Caroline Hardy Trust.
References
- 1.Kobawala TP, Patel GH, Gajjar DR, Patel KN, Thakor PB, Parekh UB, Patel KM, Shukla SN, Shah PM. Clinical utility of serum interleukin-8 and interferon-alpha in thyroid diseases. J Thyroid Res. 2011;2011:270149. doi: 10.4061/2011/270149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Asa SL. My approach to oncocytic tumours of the thyroid. J Clin Pathol. 2004;57:225–232. doi: 10.1136/jcp.2003.008474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DeMay RM. Follicular lesions of the thyroid. W(h)ither follicular carcinoma? Am J Clin Pathol. 2000;114:681–683. doi: 10.1309/BUL2-7V05-DGXU-HU6E. [DOI] [PubMed] [Google Scholar]
- 4.Derwahl M, Studer H. Hyperplasia versus adenoma in endocrine tissues: are they different? Trends Endocrinol Metab. 2002;13:23–28. doi: 10.1016/s1043-2760(01)00519-7. [DOI] [PubMed] [Google Scholar]
- 5.Powell DJ, Jr., Russell JP, Li G, Kuo BA, Fidanza V, Huebner K, Rothstein JL. Altered gene expression in immunogenic poorly differentiated thyroid carcinomas from RET/PTC3p53−/− mice. Oncogene. 2001;20:3235–3246. doi: 10.1038/sj.onc.1204425. [DOI] [PubMed] [Google Scholar]
- 6.Yu S, Sharp GC, Braley-Mullen H. Dual roles for IFN-gamma, but not for IL-4, in spontaneous autoimmune thyroiditis in NOD.H-2h4 mice. J Immunol. 2002;169:3999–4007. doi: 10.4049/jimmunol.169.7.3999. [DOI] [PubMed] [Google Scholar]
- 7.Yu S, Fang Y, Sharav T, Sharp GC, Braley-Mullen H. CD8+ T cells induce thyroid epithelial cell hyperplasia and fibrosis. J Immunol. 2011;186:2655–2662. doi: 10.4049/jimmunol.1002884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu S, Sharp GC, Braley-Mullen H. Thyroid epithelial cell hyperplasia in IFN-gamma deficient NOD.H-2h4 mice. Clin Immunol. 2006;118:92–100. doi: 10.1016/j.clim.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 9.Fang Y, Yu S, Braley-Mullen H. TGF-beta promotes proliferation of thyroid epithelial cells in IFN-gamma(−/−) mice by down-regulation of p21 and p27 via AKT pathway. Am J Pathol. 2012;180:650–660. doi: 10.1016/j.ajpath.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu S, Sharp GC, Braley-Mullen H. TGF-beta promotes thyroid epithelial cell hyperplasia and fibrosis in IFN-gamma-deficient NOD.H-2h4 mice. J Immunol. 2008;181:2238–2245. doi: 10.4049/jimmunol.181.3.2238. [DOI] [PubMed] [Google Scholar]
- 11.Bennett SR, Carbone FR, Karamalis F, Flavell RA, Miller JF, Heath WR. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. 1998;393:478–480. doi: 10.1038/30996. [DOI] [PubMed] [Google Scholar]
- 12.French RR, Chan HT, Tutt AL, Glennie MJ. CD40 antibody evokes a cytotoxic T-cell response that eradicates lymphoma and bypasses T-cell help. Nat Med. 1999;5:548–553. doi: 10.1038/8426. [DOI] [PubMed] [Google Scholar]
- 13.Uhlig HH, McKenzie BS, Hue S, Thompson C, Joyce-Shaikh B, Stepankova R, Robinson N, Buonocore S, Tlaskalova-Hogenova H, Cua DJ, Powrie F. Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity. 2006;25:309–318. doi: 10.1016/j.immuni.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 14.Shameli A, Clemente-Casares X, Wang J, Santamaria P. Development of memory-like autoregulatory CD8+ T cells is CD4+ T cell dependent. J Immunol. 2011;187:2859–2866. doi: 10.4049/jimmunol.1101117. [DOI] [PubMed] [Google Scholar]
- 15.Baker RL, Wagner DH, Jr., Haskins K. CD40 on NOD CD4 T cells contributes to their activation and pathogenicity. J Autoimmun. 2008;31:385–392. doi: 10.1016/j.jaut.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 16.Barbe-Tuana FM, Klein D, Ichii H, Berman DM, Coffey L, Kenyon NS, Ricordi C, Pastori RL. CD40-CD40 ligand interaction activates proinflammatory pathways in pancreatic islets. Diabetes. 2006;55:2437–2445. doi: 10.2337/db05-1673. [DOI] [PubMed] [Google Scholar]
- 17.Peters AL, Stunz LL, Bishop GA. CD40 and autoimmunity: the dark side of a great activator. Semin Immunol. 2009;21:293–300. doi: 10.1016/j.smim.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ponomarev ED, Shriver LP, Dittel BN. CD40 expression by microglial cells is required for their completion of a two-step activation process during central nervous system autoimmune inflammation. J Immunol. 2006;176:1402–1410. doi: 10.4049/jimmunol.176.3.1402. [DOI] [PubMed] [Google Scholar]
- 19.Smith TJ, Sciaky D, Phipps RP, Jennings TA. CD40 expression in human thyroid tissue: evidence for involvement of multiple cell types in autoimmune and neoplastic diseases. Thyroid. 1999;9:749–755. doi: 10.1089/thy.1999.9.749. [DOI] [PubMed] [Google Scholar]
- 20.Grewal IS, Flavell RA. CD40 and CD154 in cell-mediated immunity. Annu Rev Immunol. 1998;16:111–135. doi: 10.1146/annurev.immunol.16.1.111. [DOI] [PubMed] [Google Scholar]
- 21.Huber AK, Finkelman FD, Li CW, Concepcion E, Smith E, Jacobson E, Latif R, Keddache M, Zhang W, Tomer Y. Genetically Driven Target Tissue Overexpression of CD40: A Novel Mechanism in Autoimmune Disease. J Immunol. 2012;189:3043–3053. doi: 10.4049/jimmunol.1200311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Metcalfe RA, McIntosh RS, Marelli-Berg F, Lombardi G, Lechler R, Weetman AP. Detection of CD40 on human thyroid follicular cells: analysis of expression and function. J Clin Endocrinol Metab. 1998;83:1268–1274. doi: 10.1210/jcem.83.4.4732. [DOI] [PubMed] [Google Scholar]
- 23.Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, Torigian DA, O'Dwyer PJ, Vonderheide RH. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–1616. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khalil M, Vonderheide RH. Anti-CD40 agonist antibodies: preclinical and clinical experience. Update Cancer Ther. 2007;2:61–65. doi: 10.1016/j.uct.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khong A, Nelson DJ, Nowak AK, Lake RA, Robinson BW. The use of agonistic anti-CD40 therapy in treatments for cancer. Int Rev Immunol. 2012;31:246–266. doi: 10.3109/08830185.2012.698338. [DOI] [PubMed] [Google Scholar]
- 26.Murugaiyan G, Martin S, Saha B. CD40-induced countercurrent conduits for tumor escape or elimination? Trends Immunol. 2007;28:467–473. doi: 10.1016/j.it.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 27.Inoue Y, Kaifu T, Sugahara-Tobinai A, Nakamura A, Miyazaki J, Takai T. Activating Fc gamma receptors participate in the development of autoimmune diabetes in NOD mice. J Immunol. 2007;179:764–774. doi: 10.4049/jimmunol.179.2.764. [DOI] [PubMed] [Google Scholar]
- 28.Tang H, Sharp GC, Chen K, Braley-Mullen H. The kinetics of cytokine gene expression in the thyroids of mice developing granulomatous experimental autoimmune thyroiditis. J Autoimmun. 1998;11:581–589. doi: 10.1006/jaut.1998.0247. [DOI] [PubMed] [Google Scholar]
- 29.Yu S, Sharp GC, Braley-Mullen H. Thyrocytes responding to IFN-gamma are essential for development of lymphocytic spontaneous autoimmune thyroiditis and inhibition of thyrocyte hyperplasia. J Immunol. 2006;176:1259–1265. doi: 10.4049/jimmunol.176.2.1259. [DOI] [PubMed] [Google Scholar]
- 30.Li F, Ravetch JV. Inhibitory Fcγ receptor engagement drives adjuvant and anti-tumor activities of agonistic CD40 antibodies. Science. 2011;333:1030–1034. doi: 10.1126/science.1206954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yip I, Pang XP, Berg L, Hershman JM. Antitumor actions of interferon-gamma and interleukin-1 beta on human papillary thyroid carcinoma cell lines. J Clin Endocrinol Metab. 1995;80:1664–1669. doi: 10.1210/jcem.80.5.7745015. [DOI] [PubMed] [Google Scholar]
- 32.White AL, Chan HT, Roghanian A, French RR, Mockridge CI, Tutt AL, Dixon SV, Ajona D, Verbeek JS, Al-Shamkhani A, Cragg MS, Beers SA, Glennie MJ. Interaction with FcγRIIB is critical for the agonistic activity of anti-CD40 monoclonal antibody. J Immunol. 2011;187:1754–1763. doi: 10.4049/jimmunol.1101135. [DOI] [PubMed] [Google Scholar]
- 33.Estienne V, Duthoit C, Reichert M, Praetor A, Carayon P, Hunziker W, Ruf J. Androgen-dependent expression of FcγRIIB2 by thyrocytes from patients with autoimmune Graves' disease: a possible molecular clue for sex dependence of autoimmune disease. FASEB J. 2002;16:1087–1092. doi: 10.1096/fj.01-0998hyp. [DOI] [PubMed] [Google Scholar]
- 34.Correze C, Blondeau JP, Pomerance M. p38 mitogen-activated protein kinase contributes to cell cycle regulation by cAMP in FRTL-5 thyroid cells. Eur J Endocrinol. 2005;153:123–133. doi: 10.1530/eje.1.01942. [DOI] [PubMed] [Google Scholar]
- 35.McLachlan SM, Taverne J, Atherton MC, Cooke A, Middleton S, Pegg CA, Clark F, Rees Smith B. Cytokines, thyroid autoantibody synthesis and thyroid cell survival in culture. Clin Exp Immunol. 1990;79:175–181. doi: 10.1111/j.1365-2249.1990.tb05175.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tomer Y, Concepcion E, Greenberg DA. A C/T single-nucleotide polymorphism in the region of the CD40 gene is associated with Graves' disease. Thyroid. 2002;12:1129–1135. doi: 10.1089/105072502321085234. [DOI] [PubMed] [Google Scholar]
- 37.Becher B, Durell BG, Miga AV, Hickey WF, Noelle RJ. The clinical course of experimental autoimmune encephalomyelitis and inflammation is controlled by the expression of CD40 within the central nervous system. J Exp Med. 2001;193:967–974. doi: 10.1084/jem.193.8.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roth E, Schwartzkopff J, Pircher H. CD40 ligation in the presence of self-reactive CD8 T cells leads to severe immunopathology. J Immunol. 2002;168:5124–5129. doi: 10.4049/jimmunol.168.10.5124. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.