

Abstract
Despite expanded definition of the leukocyte adhesion cascade and mechanisms underlying individual steps, very little is known about regulatory mechanisms controlling sequential shifts between steps. We tested the hypothesis that metalloproteinases provide a mechanism to rapidly transition monocytes between different steps. Our study identifies diapedesis as a step targeted by metalloproteinase activity. Time-lapse video microscopy shows that the presence of a metalloproteinase inhibitor results in a doubling of the time required for human monocytes to complete diapedesis on unactivated or inflamed human endothelium, under both static and physiological-flow conditions. Thus, diapedesis is promoted by metalloproteinase activity. In contrast, neither adhesion of monocytes nor their locomotion over the endothelium is altered by metalloproteinase inhibition. We further demonstrate that metalloproteinase inhibition significantly elevates monocyte cell-surface levels of integrins CD11b/CD18 (Mac-1) specifically during transendothelial migration. Interestingly, such alterations are not detected for other endothelial- and monocyte-adhesion molecules that are presumed metalloproteinase substrates. Two major transmembrane metalloproteinases, ADAM17 and ADAM10, are identified as enzymes that control constitutive cleavage of Mac-1. We further establish that knockdown of monocyte ADAM17, but not endothelial ADAM10 or ADAM17, or monocyte ADAM10, reproduces the diapedesis delay observed with metalloproteinase inhibition. Therefore, we conclude that monocyte ADAM17 facilitates the completion of transendothelial migration by accelerating the rate of diapedesis. We propose that the progression of diapedesis may be regulated by spatial and temporal cleavage of Mac-1, which is triggered upon interaction with endothelium.
Keywords: human monocytes, adhesion molecules, cell trafficking, inflammation
Introduction
Inflammation is a key process in disease pathogenesis, and leukocyte transendothelial migration into inflamed tissues controls both initiation and progression of acute and chronic inflammatory diseases. Transendothelial migration is a sequential, multi-step process that consists of leukocyte rolling, capture and firm adhesion to endothelium, followed by locomotion across the endothelium and diapedesis through the intact vessel into the tissue, usually at endothelial junctions (1–3). Engagement of key ligands and their counter receptors expressed on both leukocytes and endothelial cells have been shown to be responsible for each step of transendothelial migration (1–3). However, very little is known about regulatory mechanisms underlying the progression through each step and transitions between steps.
A molecular mechanism capable of rapidly reducing adhesion molecule interactions is their proteolytic cleavage or “ectodomain shedding” (4). In fact, soluble forms of multiple adhesion molecules involved in each step of leukocyte transendothelial migration are proteolytically shed, and elevated levels are detected in physiological fluids from various inflammatory diseases (4, 5). Additionally, recent studies suggest roles for ectodomain shedding in leukocyte trafficking. For example, shedding of Mac-1 (CD11b/CD18), a major integrin dimer on leukocytes, has been reported to accelerate macrophage efflux from an inflammatory site (6) and regulate neutrophil detachment in vitro (7). However, specific steps in leukocyte transendothelial migration modulated by ectodomain shedding have not been determined.
Proteases responsible for shedding of a large number of cell-surface proteins involved in leukocyte transendothelial migration on both leukocytes and endothelial cells are zinc-dependent endopeptidases comprised of a family of matrix metalloproteinases (MMPs)1, that include secreted and membrane types (MT)-MMPs, and the ADAM (A Disintegrin And Metalloproteinase) family of transmembrane metalloproteinases (4, 5). Analyses of specific enzymes have identified some key proteolytic cleavage events, but most studies have focused on regulation of neutrophil and lymphocyte transendothelial migration. For example, knockdown of ADAM10 on T cells, HUVECs, or both impairs T cell transendothelial migration in vitro (8). Although VE-cadherin was identified as a possible endothelial target of ADAM10, the relative role of VE-cadherin cleavage was not determined (8). Neutrophil transendothelial migration is inhibited by soluble forms of junctional adhesion molecule (JAM)-A, which is shed from endothelium by ADAM10 and ADAM17 (9). Recently, ADAM17 shedding of L-selectin was shown to limit neutrophil recruitment, but monocyte emigration is independent of L-selectin shedding (10). A more limited number of studies have examined monocyte transendothelial migration. An antibody to MT1-MMP impairs transendothelial migration of human monocytes, but only when HUVECs are preactivated and monocyte chemotactic protein (MCP)-1 is present (11). Although ICAM-1 has been proposed as a possible substrate (12), the step(s) targeted by MT1-MMP is still unclear. Thus, there is a need to evaluate particular steps targeted by regulated proteolysis during transendothelial migration, and to better define the enzymes and substrates involved.
In this report, we identify the metalloproteinase-regulated mechanisms underlying transendothelial migration utilized by monocytes, cells of central importance in the outcome of acute and chronic inflammatory disease pathogenesis. In the presence of GM6001, which blocks metalloproteinases expressed on both monocytes and endothelial cells, we show that diapedesis is significantly delayed without any marked effect on adhesion or locomotion across the endothelial surface. To interrogate possible substrates whose impaired cleavage contributes to the delay of diapedesis, we separately screened monocytes and endothelial cells for adhesion molecules whose cell-surface expression was elevated by GM6001 following their co-incubation, which mimics transendothelial migration. Despite the presence of multiple substrates on both cells, only monocyte surface levels of Mac-1 increase upon co-incubation with GM6001. We further show that ADAM10 and ADAM17 are responsible for metalloproteinase-dependent shedding of Mac-1, and that absence of monocyte ADAM17 leads to a significant prolongation of diapedesis. In contrast, depletion of monocyte ADAM10, or endothelial ADAM10 or ADAM17, does not affect diapedesis. Therefore, we demonstrate that monocyte ADAM17 promotes diapedesis and suggest that ADAM17 cleavage of Mac-1 may serve as a regulatory mechanism.
Materials and methods
Human endothelial cell culture and monocyte isolation
HUVECs (Cascade Biologics-Invitrogen) were cultured in gelatin-coated flasks using M199 supplemented with 20% FCS, EGM-2 SingleQuots™ (Lonza) and antibiotics, and used up to passage 3. Human PBMCs were freshly isolated from citrate anti-coagulated whole blood of healthy donors by Ficoll-Paque Plus (Amersham Biosciences) separation and subjected to enrichment for monocytes by negative selection using the Monocyte Isolation Kit II (Miltenyi Biotech), plus biotinylated antibodies against CD42b (GeneTex) in some experiments to remove platelets (13). Enriched monocytes (>90% estimated by FACS analysis) were resuspended in assay medium (phenol-red-free M199 with 20% FCS, 20 mM HEPES, pH 8.0) and kept on ice unless specified. Metalloproteinase activity was blocked by a 30 min preincubation with 50 μM GM6001 (Elastin Products) and 0.1% DMSO as a control, unless specified otherwise. Since GM6001 blockade is reversible, all subsequent experiments also included inhibitor or control. The University of Washington Human Subjects Review Committee has approved all protocols.
Transwell assays
HUVECs were seeded at 3.0 × 104 cells/well onto Costar Transwell filters (3-μm pore, 6.5-mm diameter) previously coated with 5 μg/ml human fibronectin and grown to confluence. PBMCs (3.0 × 104 cells/100 μl) were labeled with Calcein-AM, and migrated leukocytes in the lower well and bottom filter were counted using ImageJ software (NIH).
Static assay of monocyte transendothelial migration
The in vitro system previously reported to model monocyte transendothelial migration under static conditions using confluent HUVEC monolayers grown on type-I collagen gels (14) was modified to enable simultaneous recording of multiple conditions. Briefly, flat collagen gels (~0.12-mm thickness) were prepared in 4-well silicon chamber (9-mm diameter, ~3-mm depth), which is made in a 40-mm glass-bottom dish (Willco Wells) in combination with Secure-Seal™ Adhesive Spacer and Press-to-Seal™ Silicon Isolator (Molecular Probes). Detailed methodology is available upon request. The collagen gels were coated with human plasma fibronectin, and HUVECs were plated and grown to confluence. After 30-min equilibration at 37° C and 30-min pretreatment with DMSO or GM6001, both in assay medium, experiments were started by addition of a monocyte suspension (50 μl droplet/well, 3–4 ×104 cells) to the HUVECs (100 μl/well). Multiple fields were randomly chosen so that at least 25 diapedesis events per condition could be monitored. The recording was started after 20 min for unactivated HUVECs and 10 min for TNF-activated HUVECs.
Time-lapse video microscopy was performed with a 40x differential interface contrast (DIC) objective lens, unless specified otherwise, on an inverted microscope (Zeiss Axiovert 200M) equipped with a charge-coupled device camera (Zeiss AxioCam MRm) and controlled by Axiovision software (Zeiss V. 4.5). The culture dish was maintained at 37°C in a humidified 5% CO2 chamber mounted on the stage. Image acquisition was done sequentially on the defined fields to be monitored in a single time point (35–100 s intervals).
Flow assay of monocyte transendothelial migration
Analysis of transendothelial migration under shear flow was performed as previously described(15). Briefly, HUVEC were grown to confluence on 25-mm diameter fibronectin-coated glass coverslips and preactivated for 4 h with 10 ng/ml TNF-α followed by preincubation with DMSO (0.1%) or 50 mM GM6001 for 30 min at 37°C. The prepared coverslip was inserted into the in vitro flow device as described. Platelet-free monocytes were kept at 8°C and used within 6 h of preparation. Pre-incubation of monocytes with DMSO or GM6001 was at 37°C for 5 min to minimize the loss of CD62L, which is essential for adhesion under flow conditions. The monocytes were then introduced into the flow chamber as a bolus (106 cells/100 μl), followed by drawing them across the HUVEC monolayer with a flow rate estimated to be 0.5 dyn/cm2 for 30 min in flow medium (PBS with 0.75 mM Ca2+ and Mg2+, 0.125% human serum albumin) containing DMSO or GM6001. The degree of shear stress employed induces maximal adhesion of monocytes at the beginning of the assay. Acquisition of time-lapse 20x DIC images was started when ~10 monocytes were adherent in the monitored field, and images were collected every 10 s thereafter.
Analysis or monocyte migration captured by time-lapse video microscopy
Monocyte migration on endothelium was analyzed in a blinded manner by two separate investigators using ImageJ with MTrackJ plug-in software (Erik Meijering, PhD, Biomedical Imaging Group Rotterdam of the Erasmus MC-University Medical Center Rotterdam, Netherlands). Monocyte cell bodies were traced on the time-lapse images by moving MTrackJ pointer when they displaced more than their radius. Analyses were done only on monocytes that ultimately underwent diapedesis, and monocytes undergoing transmigration at impaired endothelial junctions were excluded.
Flow cytometry-based screening of metalloproteinase substrates on coincubated endothelial cells and monocytes
HUVECs were grown to confluence in human plasma fibronectin (5 μg/ml)-coated 10-cm dishes and stained for 10 min with 1 μM CellTracker Green CMFDA (Molecular Probes) 16 hr prior to use. Monolayers were stimulated for 4 h with TNF-α at 10 ng/ml, and subjected to preincubation with DMSO or GM6001 for 20 min. Freshly prepared PBMCs (107 per condition) were similarly preincubated, and then co-incubated with the HUVEC monolayers in the presence of DMSO or GM6001 for 2 h. The co-cultures were washed three times to remove non-adherent cells and collected with ice-cold PBS supplemented with 0.1% BSA and 10 mM EDTA (1 ml/dish) using cell lifters. Following incubation with FcR blocking reagent (Miltenyi Biotech), cell suspensions were stained at 4° C for 30 min with anti-CD14 and saturating amounts of PE-labeled antibodies for candidate molecules (Table SI). Simultaneously, cells incubated separately for 2 h were collected and stained as described above as cell suspensions with the same ratio of endothelial cells and monocytes. All stained cells were analyzed on FACScan (BD Biosciences) and flow data were analyzed using FlowJo 8.4 software (TreeStar).
Knockdown of ADAM10 and ADAM17 in primary monocytes
Two reported siRNA sequences for knockdown of ADAM10 and ADAM17 were synthesized by Ambion, and those with the best downregulation of target ADAMs were employed for the experiments shown: ADAM10-construct, 5′-AGA CAU UAU GAA GGA UUA U 3′ (16); ADAM17-construct, 5′-GCU UGA UUC UUU GCU CUC A 3′ (17). A non-targeting siRNA (Silencer Negative Control #1, Ambion) was used as control.
Platelet-free primary monocytes (5×106) were transfected with siRNA (300 nM siRNA per reaction) using Nucleofector II and Human Monocyte Nucleofector Kit (Amaxa). To achieve maximal knockdown, monocytes were allowed to recover at 37° C for 16 h in IMDM (Lonza) containing 20% human plasma-derived serum (or FCS to evaluate of soluble CD18 dimers). Monocytes to be used for transmigration were incubated in a 50-ml conical tube on an orbital shaker to give an estimated shear of 7.5 dyn/cm2 to maintain monocyte migratory properties as determined in pilot experiments. Monocytes were subjected to Annexin-V-based Dead Cell Removal Kit (Miltenyi Biotech) to enrich for healthy cells (~95%), and then applied to the static assay of transendothelial migration.
Statistical analysis
For analysis of 2 groups within a single experiment of time-lapse microscopy using monocytes from a single donor, unpaired t tests were performed after data were confirmed to fulfill the criteria. Otherwise, Mann-Whitney U tests were applied. In comparison of means or medians obtained from 3 or 4 experiments, paired t tests were performed. To validate significance for fold-differences, paired t test were performed after log transformation. All statistical analyses were performed 2-sided using InStat (GraphPad), and p values less than 0.05 were considered significant.
Results
A broad-spectrum metalloproteinase inhibitor impairs transendothelial migration of monocytes
We put forward the hypothesis that proteolytic shedding of cell surface proteins provides a mechanism to aid in the rapid transition of cells between different steps and coordinate the complex, multi-step process of leukocyte recruitment to inflammatory sites (4). To test this hypothesis, we examined the impact of a broad-spectrum, zinc-dependent metalloproteinase inhibitor, GM6001, on net migration of human PBMCs across HUVEC monolayers using a Transwell assay. As shown in Figure 1A, GM6001 impairs net migration of leukocytes across unactivated endothelium to 40% of vehicle control. When endothelial cells are preactivated by the inflammatory stimulant TNF-α (0.1 ng/ml for 4–5 h), GM6001 reduces net leukocyte migration to a similar extent (Figure 1B). We confirmed that monocytes are the major cell type that migrates across endothelial monolayers under all conditions tested (>85% as estimated by FACS analysis, data not shown), and quantification of net monocyte migration shows that GM6001 impairs migration by 70% (Figure 1C).
Figure 1. Net migration of mononuclear cells across endothelial monolayers is reduced by the broad-spectrum metalloproteinase inhibitor GM6001.
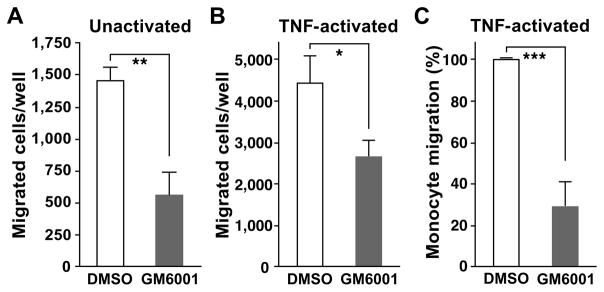
A and B. Peripheral blood mononuclear cells were co-incubated for 4 h with HUVECs grown to confluence on fibronectin-coated porous transwell inserts to assess migration in the presence of 50 μM GM6001 or 0.1% DMSO (vehicle control). Before co-incubation, the HUVEC monolayers and leukocytes were separately pre-incubated for 30 min with vehicle or GM6001. HUVECs were unactivated (A) or pre-activated with 0.1 ng/ml TNFα for 4 h (B). Representative data from at least 3 different experiments are shown, *p <0.02, **p<0.002 (unpaired t test). C. Monocyte migration across pre-activated HUVEC monolayers (10 ng/ml TNF-α) was determined by FACS analysis for mononuclear cell transendothelial migration as shown in B. Monocyte numbers in the presence of GM6001 are expressed as ratios relative to those of DMSO. Means ± SD of 6 experiments using different mononuclear cell donors are shown, ***p <0.001 (paired t test).
Metalloproteinase blockade delays completion of monocyte transendothelial migration under both static and flow conditions
Time-lapse video microscopy was then employed to monitor the interactions of purified monocytes with endothelial cells to dissect at which step(s) GM6001 is acting in transendothelial migration. To assess migration under static conditions, unactivated endothelial monolayers on collagen gels were co-incubated with primary monocytes (18), an in vitro assay in which key mediators of transmigration have subsequently been confirmed in vivo (19–23) and almost all leukocytes cross the endothelial cells at the cell borders, undergoing paracellular migration (24). As shown in Figure 2A, GM6001 postpones the frequency of monocyte completion of transendothelial migration, the point at which remnants of monocyte cell body disappear from the apical surface of endothelial monolayers in the process of diapedesis. Since the total frequency of transendothelial migration in the presence of GM6001 is comparable to vehicle control (Figure 2A), GM6001 does not inhibit transendothelial migration. Rather, GM6001 delays monocyte transendothelial migration under static conditions.
Figure 2. Metalloproteinase blockade by GM6001 delays monocyte transendothelial migration under static and flow conditions.

A. Human monocytes were added to unactivated HUVEC monolayers in the presence of DMSO or GM6001 under static conditions and observed using time-lapse video microscopy every 37.8 s. Individual monocytes migrating on the apical surface of the monolayers were tracked until they complete migration across the monolayers. Monocytes that complete their transendothelial migration up to each time point are expressed as % of total monocytes in the field. In one experiment, an average of 50 monocytes were evaluated for DMSO and 46 for GM6001. B. Monocyte transendothelial migration under flow conditions in the presence of DMSO (Video 1) or GM6001 (Video 2) was analyzed as described in A. Monocytes were drawn across pre-activated HUVEC monolayers (10 ng/ml TNF-α for 4–6 h) at 0.5 dyn/cm2 in flow medium containing DMSO or GM6001. Pretreatment with DMSO or GM6001 was 30 min for HUVECs and 5 min for monocytes. Time-lapse video microscopy was performed every 10 s for 30 min. Means ± SD of 3 different experiments are shown, *p < 0.05, **p <0.01, and ***p <0.005 (paired t test).
The effects of GM6001 on monocyte transendothelial migration were also tested under flow conditions that more closely model physiological conditions. Monocytes were drawn across a TNF-α-activated endothelial layer for 30 min in medium including vehicle or GM6001 at an estimated flow rate of 0.5 dyn/cm2, conditions that induce maximal initial adhesion to allow evaluation of post-adhesion events (15). In vehicle control (Video 1), monocytes rapidly adhere, spread on the activated endothelium followed by locomotion for a relatively short distance to reach endothelial junctions. Monocytes then undergo diapedesis, thereby finishing their paracellular transendothelial migration, which is particularly easy to distinguish under flow conditions. It takes monocytes ~10 min to complete all of these steps (data not shown). However, in the presence of GM6001 (Video 2), monocyte tails remain on the apical surface of endothelial monolayers for longer durations until they complete transendothelial migration (Figure 2B), and GM6001 delays the completion of transendothelial migration under flow conditions as observed under static conditions (Figure 2A). Together our data indicate that metalloproteinase activity is rate limiting and is required for optimal monocyte transendothelial migration.
Metalloproteinase activity facilitates progression of diapedesis but not other steps of monocyte transendothelial migration
Since the process of monocyte transendothelial migration involves multiple steps, each was quantified to identify the metalloproteinase-targeted step(s). As summarized in Table I, GM6001 does not affect the initial adhesion or locomotion across the endothelium. We also verified that GM6001 does not alter endothelial junctions during the period of the migration assay as compared with vehicle control (data not shown). To analyze diapedesis, a frame-by-frame analysis of vehicle control and GM6001 conditions was performed on unactivated endothelial monolayers under static conditions (Figure 3A). In vehicle control (Figure 3A, upper panel; Video 3) monocytes first squeeze a membrane protrusion, and then their main cell body, between the endothelial junctions, and within a short time their tails retract from the apical surface into the junctions. In contrast, monocytes in the presence of GM6001 (Figure 3A, lower panel; Video 4) take a longer time to squeeze their cell body between endothelial junctions and their remnant (tail) remains on the apical surface for extended periods while the main cell body passes through the endothelial monolayer. We quantified the duration of diapedesis as the time interval between the arrival of monocytes at the site of diapedesis and their completion of diapedesis as assessed from the apical surface. The distribution of diapedesis duration shows that under control conditions approximately 90% of monocytes complete diapedesis within 20 min, whereas only 40% do in the presence of GM6001 (Figure 3B). Comparison of diapedesis duration for monocytes prepared from different donors reveals that GM6001 prolongs diapedesis 2-fold as compared with control conditions (Figure 3C, left). When the endothelium was pre-activated with TNF-α, similar results were obtained with a 1.6-fold increase in the duration of diapedesis by GM6001 (Figure 3C, right). Under physiological flow conditions, GM6001 results in 2-fold increase even though the diapedesis duration in vehicle control is much shorter than observed under static conditions (Figure 3D). Together, these data demonstrate that prolongation of diapedesis by metalloproteinase blockade is responsible for the delay in completion of monocyte transendothelial migration.
Table 1. GM6001 does not alter initial adhesion or locomotion of monocytes under static conditions.
Adhesion and locomotion were measured as described in footnotes. Values are means ± SD of representative data from at least 3 experiments.
| Condition |
|
|
|---|---|---|
| Steps of transendothelial migration
| ||
| Index of initial adhesion a | Locomotion speed (μm/min) b | |
| Unstimulated HUVECs: | ||
| DMSO | 0.139 ± 0.012 | 3.01 ± 1.42 |
| GM6001 | 0.147 ± 0.011 | 3.82 ± 3.35 |
| TNF-activated HUVECs: | ||
| DMSO | 0.455 ± 0.021 | 3.63 ± 1.91 |
| GM6001 | 0.464 ± 0.047 | 3.22 ± 2.03 |
Calcein AM-labeled PBMCs (3×105/well) were co-incubated for 20 min with unstimulated or TNF-activated HUVEC monolayers in a 96-well plate. The index of adhesion is the ratio of fluorescent intensities of adhering cells after washing over the input intensity.
The locomotion step of individual monocytes was tracked on time-lapse movies until the frame where the membrane protrusion of the cells reaches the site of diapedesis. To estimate speeds of locomotion, time-course plots of inclusive migration distance for individual monocytes were subjected to the linear least-square method. The slopes of resultant approximated lines were referred to as locomotion speeds.
Figure 3. Monocyte diapedesis is prolonged by metalloproteinase inhibition under both static and flow conditions.

A. Sequential time-lapse images from the beginning to the end of diapedesis in the presence of DMSO (top, Video 3) and GM6001 (bottom, Video 4) are shown every 75.6 s. The beginning of diapedesis (Start) was defined as the image frame where the cell body or monocyte membrane protrusion reached the site of diapedesis (asterisk). Completion of diapedesis (End) was denoted as the image frame where the remnant of the monocyte cell body disappeared from the apical surface of HUVEC monolayer. Arrows indicate the direction of monocyte migration. B. Histograms of the diapedesis duration in DMSO and GM6001 are shown for unactivated HUVEC monolayers. Diapedesis duration is defined as the interval between the start and the end frames of diapedesis, as defined in A. A representative data set from 3 different experiments is shown. Medians for this data sets are 8.8 min, n=47 monocytes for DMSO; 21.7 min, n=26 monocytes for GM6001, p <0.0001 (Mann-Whitney test). C. Means ± SD of diapedesis duration from different experiments on unactivated (n=3) and TNF-activated HUVEC (0.1 ng/ml for 4 h, n=4) are shown for monocytes prepared from different donors. * p < 0.05; ** p < 0.02 (paired t test). D. Duration of diapedesis under flow conditions in the presence of DMSO or GM6001 was evaluated as described above. Means ± SD of medians obtained from 3 different experiments are shown, ***p <0.005 (paired t test). In a single experiment, an average of 45 monocytes were evaluated for DMSO and 37 for GM6001.
Monocytes shed CD18 integrin dimers in a metalloproteinase-dependent manner upon co-incubation with endothelial cells and CD11b/CD18 is cleaved by transmembrane proteases ADAM17 and ADAM10
Diapedesis is dependent upon both endothelial and monocyte adhesion molecules, several of which have been shown to be shed by metalloproteinases, including leukocyte-specific integrin CD18 (6, 25) and its partner alpha integrins (CD11a, CD11b (6), CD11c and CD11d), intercellular adhesion molecule (ICAM)-1 (12, 26), activated leukocyte cell adhesion molecule (ALCAM) (27), platelet/endothelial cell adhesion molecule (PECAM)-1 (28), and JAM-A (9). To identify specific molecules(s) involved in metalloproteinase-mediated regulation of diapedesis, we initially screened these adhesion molecules by flow cytometry to determine whether monocyte surface levels were increased in the presence of GM6001 under the same conditions used for evaluation of transendothelial migration (Figure 4A and B, Table SII). CD14+ monocytes were evaluated following PBMC incubation with or without TNF-activated HUVEC monolayers in the presence of vehicle and GM6001 for 2 h, a time period during which the majority of monocytes complete transendothelial migration (Figure 2A). As highlighted in Figure 4A and B, only surface levels of CD11b are upregulated (~1.4-fold) following co-incubation with HUVECs in the presence of GM6001 (Figure 4B). ICAM-1 levels were also increased by GM6001 treatment, but both with and without HUVEC co-incubation (Table SII). Monocyte ICAM-1 does not seem relevant to transendothelial migration since monocyte-targeted, functional blockade of ICAM-1 did not significantly affect net Transwell migration of mononuclear cells (data not shown). CD11a, ALCAM-1, PECAM-1, and JAM-A levels were not altered by GM6001 (Figure 4A, Table SII). Together these data demonstrate a metalloproteinase-dependent and selective increase in CD11b/CD18 levels on monocytes upon co-incubation with endothelial cells.
Figure 4. CD18 integrins are shed from human monocytes in a metalloproteinase-dependent manner both constitutively and inducibly upon interaction with endothelial cells.

A and B. Cell-surface expression of CD11a (A), and CD11b (B) on monocytes was determined by FACS analysis after co-incubation with TNF-activated HUVEC monolayers (EC+PBMC) or separate incubation (PBMC alone) for 2 h in the presence of DMSO or GM6001. For analysis of cell surface expression on monocytes, first green-fluorescence endothelial cells were gated out and then CD14+ positive cells were analyzed further. At least 5,000 events were collected. The expression levels are presented as ratios relative to separate incubation with DMSO. Each column represents mean ± SD from 3 different experiments for CD11a, and 5 for CD11b, *p <0.05, **p <0.01 (paired t test). C. Platelet-free monocytes were incubated in Opti-MEM at 106/ml in the presence of DMSO (−) or GM6001 (+) for 16 h at 37°C. Cells were lysed on ice with NP-40 buffer supplemented with proteinase inhibitors and conditioned media (CMs) were concentrated using YM-30 membrane (Millipore) following centrifugation. CMs (40x concentrated, 40 μl) and cell lysates (4 μg) were resolved by 7.5% SDS-PAGE, and evaluated by Western blotting with antibody to the ectodomain of human CD18. Arrows indicate full-length CD18 in the lysates and shed forms (soluble) of CD18 in CM. D. CD18 integrin complexes shed in 16-h CM were quantitated by sandwich ELISA using antibodies for CD11a and CD11b for capture and CD18 for detection. See Table SI for antibodies used.
To confirm that CD18 is shed from primary monocytes in a metalloproteinase-dependent manner, we analyzed cell lysates and conditioned media (CMs) prepared from monocytes treated with GM6001 for 16 h. Soluble integrin CD18 is decreased in 16-h CM with GM6001 treatment as determined by Western analysis (Figure 4C), and this is associated with increased integrin CD18 in cell lysates. The modest difference in molecular weight of soluble and cellular integrin CD18 is similar to previous reports (6, 7, 29), indicating that metalloproteinase cleavage within the ectodomain close to the cell membrane results in loss of the transmembrane and the small cytoplasmic domains. The relatively small increase in cellular CD18 with GM6001 is consistent with significant intracellular stores of CD18 (30). Further analysis of monocyte CMs demonstrates that both CD11b/CD18 (Mac-1) and CD11a/CD18 (LFA-1) are shed by metalloproteinases, but the amount of shed Mac-1 was ~7.8 times higher than that of LFA-1, suggesting a difference in the extent of constitutive shedding (Figure 4D).
Since shedding of a number of cell-surface adhesion molecules can be mediated by the constitutively expressed transmembrane proteases ADAM10 and ADAM 17 (4, 5), we tested whether one or both of these enzymes contribute to cleavage of LFA-1 and Mac-1. Primary monocytes were transfected with siRNAs to deplete ADAM17 or ADAM10 using the Nucleofector system, and maximal knockdown was achieved 16 h after transfection (Figure 5A) with a transfection efficiency >95% (data not shown) and comparable viabilities (~75%, data not shown). While levels of soluble LFA-1 in CM collected from post-transfection culture (16 h) are not altered by knockdown of ADAM10 or ADAM17, soluble Mac-1 levels are reduced 20% and 30% by ADAM10 and ADAM17 siRNA, respectively, as compared with control-transfected cells (Figure 5B). Thus, both monocyte ADAM17 and ADAM10 can contribute to ectodomain shedding of Mac-1.
Figure 5. Depletion of ADAM17 and ADAM10 from primary monocytes decreases shedding of CD11b/CD18.

Human primary monocytes were transfected with siRNAs for ADAM17, ADAM10 and a non-specific control sequence. A. Knockdown of ADAM17 and ADAM10 was evaluated by immunoblotting of cell lysates 16 h post-transfection. Cells were lysed with NP-40 lysis buffer and 10 μg/lane were separated by 7.5% SDS-PAGE under reducing conditions and probed with rabbit polyclonal antibodies against the cytoplasmic domains of ADAM10 and ADAM17 or Pan-actin antibody (see Table SI for antibodies). Arrows indicate specific bands for each protein. B–C. Soluble CD11a/CD18 (B) and CD11b/CD18 (C) integrins was evaluated by sandwich ELISA of 16-h CMs prepared from siRNA-transfected monocytes as described in Figure 4D. Representative data are shown from 2 independent experiments. Means±SD are calculated from measurements of triplicate wells, *p< 0.005, and **p<0.0001 (unpaired t test). For comparison between control and ADAM10 in B, p< 0.05 (unpaired t test).
Candidate endothelial substrates are not altered by metalloproteinase blockade and neither endothelial ADAM10 nor ADAM17 promotes monocyte diapedesis
Since endothelial junctions may serve as gatekeepers for leukocyte transmigration (31), we also assessed changes in surface levels of endothelial candidate substrates during monocyte transendothelial migration in the presence of GM6001 (Table SIII) as shown in the analysis of monocyte surface adhesion molecules (Table SII). Four candidate substrates were screened, including vascular cell adhesion molecule (VCAM)-1 (32), ICAM-1 (12, 26), ALCAM (27), and JAM-A (9). However, GM6001 treatment has no effect on surface levels of any of the 4 candidate substrates, with or without co-incubation (Table SIII).
Diapedesis duration was also assessed following separate knockdown of endothelial ADAM10 and ADAM17 from HUVECs using siRNA (Figure S1). Depletion of ADAM10 consistently leads to a significant decrease in endothelial-junctional permeability (Figure S1C), which agrees with a previous report (8). However, neither ADAM10 nor ADAM17 depletion from HUVECs reproduces GM6001-mediated prolongation of monocyte diapedesis (Figures S1A–B). Taken together, our data demonstrates that cleavage of metalloproteinase substrates on endothelial cells and endothelial ADAM10 and ADAM17 proteolytic activity are not major contributors to metalloproteinase-mediated facilitation of monocyte diapedesis.
ADAM17 expressed on monocytes, but not ADAM10, promotes their diapedesis
To determine the monocyte metalloproteinases contribute to regulation of diapedesis, we first analyzed MT1-MMP previously reported to promote monocyte transmigration on activated endothelium based on partial inhibition by an MT1-MMP antibody, but only if MCP-1 was present as a chemoattractant (11). Using the same antibody, we observed no effect on monocyte transmigration under our conditions lacking MCP-1 (data not shown). Thus, MT1-MMP does not appear to be a major regulatory mechanism for diapedesis. We then used siRNA knockdown to evaluate whether monocyte ADAM17 and/or ADAM10 are involved in metalloproteinase-mediated regulation of diapedesis. GM6001-dependent prolongation of diapedesis was verified for control-transfected monocytes (Figure 6A), and a similar extent of inhibition (1.5-fold delay) to that with freshly prepared monocytes (compare with Figure 2C) was observed. In contrast, depletion of ADAM17 (Video 6), but not ADAM10 (Figure 6B), extends the duration of monocyte diapedesis compared with control-transfected monocytes (Video 5), although the extent of inhibition (1.3-fold) was less than that observed with GM6001 (Figure 6A). Together, these data demonstrate that monocyte ADAM17, but not monocyte ADAM10, makes a significantly contribution to metalloproteinase-dependent promotion of diapedesis.
Figure 6. Depletion of ADAM17, but not ADAM10, from monocytes prolongs the diapedesis step.
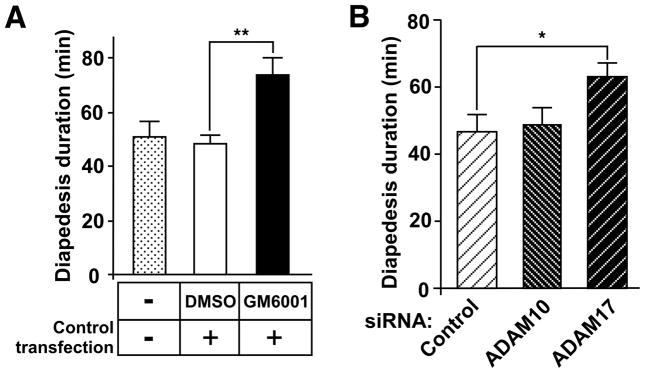
A. Untransfected monocytes and monocytes transfected with control siRNA were generated as described in the Materials and Methods, and subjected to time-lapse video analysis on TNF-activated endothelium in the presence of DMSO or GM6001. Representative data from 2 independent experiments are expressed as means ± SEM, **p <0.0005 (unpaired t test with Welch correction; n=19, 37, and 27 monocytes for untransfected, DMSO, and GM6001, respectively). B. Duration of monocyte diapedesis was determined for monocytes transfected with control, ADAM10 or ADAM17 siRNA. Means ± SD from 3 different experiments are shown, *p <0.01 (paired t test). All monocyte preparations show compatible viability (>95% as determined by trypan blue exclusion).
Discussion
Although metalloproteinase-mediated “ectodomain shedding” has been implicated in the regulation of leukocyte trafficking (4–9), the steps targeted, enzymes responsible, and key substrates are still unclear, particularly for monocytes. The present study shifts the paradigms regarding metalloproteinase regulation of monocyte transendothelial cell migration at several levels. First, we demonstrate for the first time that metalloproteinases promote monocyte transendothelial migration through facilitation of the diapedesis step. Second, our studies reveal that blockade of metalloproteinases elevates monocyte surface Mac-1 (CD11b/CD18) levels specifically upon co-incubation with activated endothelial cells, conditions that mimic transendothelial cell migration. Further, despite previous documentation of metalloproteinase-mediated shedding of multiple other adhesion molecules on both monocytes and endothelial cells, none of these show elevated surface levels dependent upon monocyte-endothelial co-incubation and metalloproteinases. Third, we show two enzymes, ADAM10 and ADAM17, contribute to metalloproteinase-dependent, constitutive shedding of Mac-1. Finally, depletion of monocyte ADAM17 significantly impairs the diapedesis step, while neither monocyte ADAM10 nor endothelial ADAM10 or ADAM17 are involved in diapedesis regulation.
Our study highlights the role of metalloproteinases, specifically monocyte ADAM17, in regulating the rate of diapedesis. The delay in diapedesis by metalloproteinase blockade is a form of regulation distinct from total blockade of the process as previously shown with blocking antibodies to PECAM-1(18) and CD99 (21). As our time-lapse imaging shows, the main cell body of the monocyte migrates into the endothelial junction, even in the presence of GM6001. Thus, blocking metalloproteinase-mediated cleavage does not impair initiation of diapedesis and functions related to movement of the cell body into the junction between endothelial cells. The marked effect of GM6001 treatment is that monocyte tails are persistent on the apical surface of endothelium, even after the main cell body is spreading in the subendothelial space. Therefore, we suggest that metalloproteinase inhibition impairs organized retraction of the monocyte cell body from the endothelial apical surface and thus diapedesis. The greater inhibitory efficacy of GM6001 shown in the transwell assay as compared with apical video microscopy (Figures 1 and 3) further suggests that post-diapedesis mechanisms may additionally be targeted by metalloproteinases. Interesting possibilities for future studies include new steps that have been recently defined beneath the endothelium for leukocyte emigration into tissues in vivo in which uropod release from the endothelial basal surface (33) is followed by cell crawling and penetration of vascular basement membranes and pericyte sheaths (34).
A surprising finding in our study is the failure to observe any increase in endothelial surface expression of previously demonstrated substrates of metalloproteinases, including those of ADAM10 and ADAM17, upon co-incubation of monocytes and endothelial cells in the presence of GM6001. Co-incubation was limited to 2 h to focus on cleavage events during transendothelial migration. Using a similar detection system, Koenen et al. showed that enhanced shedding of endothelial JAM-A upon 2-h co-incubation with neutrophils is associated with significant decrease in their endothelial surface levels (9). Consistent with our inability to detect surface changes in endothelial ADAM substrates, which could contribute to the regulation of diapedesis, endothelial knockdown of ADAM10 or ADAM17 had no effect on the rate of diapedesis. These data are in contrast to previous reports that endothelial ADAM10 regulates T cell transendothelial migration, although the specific step targeted by ADAM10 was not determined (8). Endothelial ADAM10 and ADAM17 also control neutrophil transendothelial migration through generation of soluble JAM-A (9). These data strongly suggest that transendothelial migration of different leukocytes may be regulated by distinct mechanisms.
In contrast, analysis of expression levels of candidate monocyte substrates under conditions comparable to transendothelial migration identified a selective increase in surface levels of the integrin dimer Mac-1 (CD11b/CD18). Mac-1 has been shown to involved in diapedesis (35), as well as other steps of leukocyte transendothelial migration (1). We were able to confirm that the broad inhibitor GM6001, as well as knockdown of ADAM17 and ADAM10, decreased shedding of soluble Mac-1. To our knowledge, this is the first report of ADAM-mediated cleavage of leukocyte integrins.
Our data further suggest that ADAM17 cleavage of Mac-1 may play a role in facilitation of monocyte diapedesis since knockdown of monocyte ADAM17 impairs diapedesis (Figure 6B). Under normal conditions, soluble Mac-1 may also contribute to the facilitation of diapedesis since it retains its ability to bind to its endothelial ligand, ICAM-1 (6, 7). Interaction of leukocytes and endothelial cells triggers multiple signaling cascades and activation of leukocyte integrins (36). Since we have shown that integrin activation is not sufficient to induce cleavage of Mac-1 (6), we speculate that monocyte-endothelial contact may activate monocyte ADAM17 or promote its binding to Mac-1, and thus enhance temporally and spatially regulated cleavage. The dissociation of Mac-1/ICAM-1 interactions could then facilitate monocyte retraction from the apical side of endothelium, thereby achieving rapid progression of diapedesis (22). However, since Mac-1 functional blocking antibodies impair locomotion and thus prevent paracellular diapedesis (22), it is not possible to use this approach to define the contribution of Mac-1 cleavage to progression of monocyte diapedesis. Alternate strategies, such as uncleavable mutants, will be needed to eliminate the possible contribution of other effects of ADAM17 knockdown and to provide a direct link between ADAM17 cleavage of Mac-1 and facilitation of diapedesis. It also remains possible that additional metalloproteinases may be involved since GM6001 resulted in a greater suppression of diapedesis and soluble integrin CD18 dimer release than those following siRNA knockdown of ADAM17. Shedding of additional substrates not included in our screen may also contribute to the regulation of monocyte diapedesis.
Interestingly, depletion of monocyte ADAM10 did not alter diapedesis, suggesting that ADAM10-mediated cleavage of Mac-1 is not enhanced by interaction with endothelial cells. Indeed, it is known that constitutive shedding by ADAM17 and ADAM10 can be enhanced through distinct stimuli and signaling pathways (37, 38). Previous experiments that defined the monocyte locomotion step also demonstrated a requirement for both monocyte CD11a/CD18 and CD11b/CD18 interaction with endothelial ICAM-1 and ICAM-2 (22). Since endothelial ICAM-1 has been identified as a substrate of ADAM17 (26) and MT1-MMP (12), inhibition of ICAM-1 cleavage has been suggested as a mechanism to regulate its interaction with CD18 integrins capable of altering the locomotion step. However, our inability to detect any effect of metalloproteinase blockade on the locomotion step, in spite of demonstrating metalloproteinase-dependent cleavage of both CD11a/CD18 and CD11b/CD18, suggests that other mechanisms are to modulate their ICAM interactions, such as alteration of integrin affinity (39), may be more important for transitioning between locomotion and diapedesis.
Potential in vivo relevance for our prolongation of in vitro monocyte diapedesis is suggested by intriguing similarities with data from a recent study of in vivo neutrophil diapedesis. Woodfin et al (40) described two modes of abnormal neutrophil diapedesis in ischemia-reperfusion stimulated cremaster venules: hesitant diapedesis and reverse diapedesis. In hesitant diapedesis, neutrophils move back and forth at endothelial junctions before finally completing migration into the subendothelial space as we observed with monocyte diapedesis in the presence of GM6001, most clearly seen under flow conditions (Video 2). In vivo, neutrophils show delayed diapedesis similar to our studies. The delayed diapedesis of neutrophils is caused by relocalization of JAM-C to the venular surface from endothelial junctions (40), which is triggered by ischemia-reperfusion. JAM-C has been reported to mediate monocyte diapedesis in kidney models of ischemia reperfusion (41), and to maintain monocyte unidirectional diapedesis in vitro (42), suggesting that monocytes may show delayed diapedesis in specific organs and conditions. Interestingly, JAM-C controls diapedesis thorough interaction with Mac-1 (35, 43, 44). Thus, it is possible that metalloproteinase cleavage of Mac-1 may be involved in JAM-C regulated unidirectional diapedesis. Although we observed no delay in vivo in monocyte accumulation in the peritoneal cavity 24 hrs after injection of the sterile irritant thioglycollate in mice lacking ADAM17 in circulating cells (10), the potential role of Mac-1 and JAM-C has not been investigated in a 30% decrease in monocyte accumulation 48 hrs after thioglycollate seen in the same mice (Tang et al., unpublished observations). However, as discussed above, alternate approaches such as generation of uncleavable mutants are needed to address this question.
In conclusion, this investigation demonstrates that metalloproteinase activity promotes the diapedesis step of monocyte transendothelial migration under both static and physiological flow conditions in vitro by increasing the rate of diapedesis. During transendothelial migration, metalloproteinase activity leads to cleavage of monocyte Mac-1, but not other reported adhesion substrates expressed on monocytes and endothelial cells. Knockdown of monocyte ADAM17 impairs Mac-1 shedding and diapedesis, suggesting that the metalloproteinase-mediated promotion of diapedesis may involve monocyte ADAM17 cleavage of Mac-1. Since diapedesis is a central component of leukocyte recruitment, our study establishes cell-type specific regulatory mechanisms of diapedesis, which are critical for the development of cell-targeted therapy in chronic inflammatory diseases such as atherosclerosis.
Supplementary Material
Acknowledgments
We gratefully acknowledge the assistance of University of Washington colleagues Brian Fish (Nutrition Obesity Research Center), Roderick Theobald and Chris Jordan-Squire (Department of Statistics) and Nancy Temkin (Department of Biostatistics) with statistical evaluation of our experiments. We thank Li-Chuan Huang for excellent technical assistance. We also acknowledge Francis W. Luscinskas for help to establish the flow assays and insightful discussions; and José López and Jummei Chen (Puget Sound Blood Center) for their assistance with the flow assays. John M. Harlan, Carole L. Wilson and Jingjing Tang also provided helpful comments and suggestions during the preparation of the manuscript.
This work was partially supported by grants from the National Institutes of Health (HL 067267 and HL018645 to E.W.R.), Developmental Biology Training grant (NIH-5732-HD07183 to P.W.L.T.), and P30 DK035816 (University of Washington, Nutrition Obesity Research Center) for statistical analyses.
Footnotes
Abbreviations used in this paper: MMP, matrix metalloproteinase; MT1-MMP, membrane-type-1 MMP; ADAM, a disintegrin and metalloproteinase; JAM, junctional adhesion molecule; CM, conditioned media; MCP-1, monocyte chemotactic protein-1.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 2.Petri B, Phillipson M, Kubes P. The physiology of leukocyte recruitment: an in vivo perspective. J Immunol. 2008;180:6439–6446. doi: 10.4049/jimmunol.180.10.6439. [DOI] [PubMed] [Google Scholar]
- 3.Muller WA. Mechanisms of leukocyte transendothelial migration. Annu Rev Pathol. 2011;6:323–344. doi: 10.1146/annurev-pathol-011110-130224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garton KJ, Gough PJ, Raines EW. Emerging roles for ectodomain shedding in the regulation of inflammatory responses. J Leukoc Biol. 2006;79:1105–1116. doi: 10.1189/jlb.0106038. [DOI] [PubMed] [Google Scholar]
- 5.Reiss K, Saftig P. The “a disintegrin and metalloprotease” (ADAM) family of sheddases: physiological and cellular functions. Semin Cell Dev Biol. 2009;20:126–137. doi: 10.1016/j.semcdb.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 6.Gomez IG, Tang J, Wilson CL, Yan W, Heinecke JW, Harlan JM, Raines EW. Metalloproteinase-mediated Shedding of Integrin beta2 promotes macrophage efflux from inflammatory sites. J Biol Chem. 2012;287:4581–4589. doi: 10.1074/jbc.M111.321182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zen K, Guo YL, Li LM, Bian Z, Zhang CY, Liu Y. Cleavage of the CD11b extracellular domain by the leukocyte serprocidins is critical for neutrophil detachment during chemotaxis. Blood. 2011;117:4885–4894. doi: 10.1182/blood-2010-05-287722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schulz B, Pruessmeyer J, Maretzky T, Ludwig A, Blobel CP, Saftig P, Reiss K. ADAM10 regulates endothelial permeability and T-Cell transmigration by proteolysis of vascular endothelial cadherin. Circ Res. 2008;102:1192–1201. doi: 10.1161/CIRCRESAHA.107.169805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koenen RR, Pruessmeyer J, Soehnlein O, Fraemohs L, Zernecke A, Schwarz N, Reiss K, Sarabi A, Lindbom L, Hackeng TM, Weber C, Ludwig A. Regulated release and functional modulation of junctional adhesion molecule A by disintegrin metalloproteinases. Blood. 2009;113:4799–4809. doi: 10.1182/blood-2008-04-152330. [DOI] [PubMed] [Google Scholar]
- 10.Tang J, Zarbock A, Gomez I, Wilson CL, Lefort CT, Stadtmann A, Bell B, Huang LC, Ley K, Raines EW. Adam17-dependent shedding limits early neutrophil influx but does not alter early monocyte recruitment to inflammatory sites. Blood. 2011;118:786–794. doi: 10.1182/blood-2010-11-321406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matias-Roman S, Galvez BG, Genis L, Yanez-Mo M, de la Rosa G, Sanchez-Mateos P, Sanchez-Madrid F, Arroyo AG. Membrane type 1-matrix metalloproteinase is involved in migration of human monocytes and is regulated through their interaction with fibronectin or endothelium. Blood. 2005;105:3956–3964. doi: 10.1182/blood-2004-06-2382. [DOI] [PubMed] [Google Scholar]
- 12.Sithu SD, English WR, Olson P, Krubasik D, Baker AH, Murphy G, D’Souza SE. Membrane-type 1-matrix metalloproteinase regulates intracellular adhesion molecule-1 (ICAM-1)-mediated monocyte transmigration. J Biol Chem. 2007;282:25010–25019. doi: 10.1074/jbc.M611273200. [DOI] [PubMed] [Google Scholar]
- 13.Williams MR, Sakurai Y, Zughaier SM, Eskin SG, McIntire LV. Transmigration across activated endothelium induces transcriptional changes, inhibits apoptosis, and decreases antimicrobial protein expression in human monocytes. J Leukoc Biol. 2009;86:1331–1343. doi: 10.1189/jlb.0209062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muller WA, Weigl SA. Monocyte-selective transendothelial migration: dissection of the binding and transmigration phases by an in vitro assay. J Exp Med. 1992;176:819–828. doi: 10.1084/jem.176.3.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luscinskas FW, Kansas GS, Ding H, Pizcueta P, Schleiffenbaum BE, Tedder TF, Gimbrone MA., Jr Monocyte rolling, arrest and spreading on IL-4-activated vascular endothelium under flow is mediated via sequential action of L-selectin, beta 1-integrins, and beta 2-integrins. J Cell Biol. 1994;125:1417–1427. doi: 10.1083/jcb.125.6.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murai T, Miyazaki Y, Nishinakamura H, Sugahara KN, Miyauchi T, Sako Y, Yanagida T, Miyasaka M. Engagement of CD44 promotes Rac activation and CD44 cleavage during tumor cell migration. J Biol Chem. 2004;279:4541–4550. doi: 10.1074/jbc.M307356200. [DOI] [PubMed] [Google Scholar]
- 17.Tanida S, Joh T, Itoh K, Kataoka H, Sasaki M, Ohara H, Nakazawa T, Nomura T, Kinugasa Y, Ohmoto H, Ishiguro H, Yoshino K, Higashiyama S, Itoh M. The mechanism of cleavage of EGFR ligands induced by inflammatory cytokines in gastric cancer cells. Gastroenterology. 2004;127:559–569. doi: 10.1053/j.gastro.2004.05.017. [DOI] [PubMed] [Google Scholar]
- 18.Muller WA, Weigl SA, Deng X, Phillips DM. PECAM-1 is required for transendothelial migration of leukocytes. J Exp Med. 1993;178:449–460. doi: 10.1084/jem.178.2.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bogen S, Pak J, Garifallou M, Deng X, Muller WA. Monoclonal antibody to murine PECAM-1 (CD31) blocks acute inflammation in vivo. J Exp Med. 1994;179:1059–1064. doi: 10.1084/jem.179.3.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liao F, Ali J, Greene T, Muller WA. Soluble domain 1 of platelet-endothelial cell adhesion molecule (PECAM) is sufficient to block transendothelial migration in vitro and in vivo. J Exp Med. 1997;185:1349–1357. doi: 10.1084/jem.185.7.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schenkel AR, Mamdouh Z, Chen X, Liebman RM, Muller WA. CD99 plays a major role in the migration of monocytes through endothelial junctions. Nat Immunol. 2002;3:143–150. doi: 10.1038/ni749. [DOI] [PubMed] [Google Scholar]
- 22.Schenkel AR, Mamdouh Z, Muller WA. Locomotion of monocytes on endothelium is a critical step during extravasation. Nat Immunol. 2004;5:393–400. doi: 10.1038/ni1051. [DOI] [PubMed] [Google Scholar]
- 23.Dufour EM, Deroche A, Bae Y, Muller WA. CD99 is essential for leukocyte diapedesis in vivo. Cell Commun Adhes. 2008;15:351–363. doi: 10.1080/15419060802442191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muller WA. Leukocyte-endothelial cell interactions in the inflammatory response. Lab Invest. 2002;82:521–533. doi: 10.1038/labinvest.3780446. [DOI] [PubMed] [Google Scholar]
- 25.Vaisar T, Kassim SY, Gomez IG, Green PS, Hargarten S, Gough PJ, Parks WC, Wilson CL, Raines EW, Heinecke JW. MMP-9 sheds the beta2 integrin subunit (CD18) from macrophages. Mol Cell Proteomics. 2009;8:1044–1060. doi: 10.1074/mcp.M800449-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsakadze NL, Sithu SD, Sen U, English WR, Murphy G, D’Souza SE. Tumor necrosis factor-alpha-converting enzyme (TACE/ADAM-17) mediates the ectodomain cleavage of intercellular adhesion molecule-1 (ICAM-1) J Biol Chem. 2006;281:3157–3164. doi: 10.1074/jbc.M510797200. [DOI] [PubMed] [Google Scholar]
- 27.Bech-Serra JJ, Santiago-Josefat B, Esselens C, Saftig P, Baselga J, Arribas J, Canals F. Proteomic identification of desmoglein 2 and activated leukocyte cell adhesion molecule as substrates of ADAM17 and ADAM10 by difference gel electrophoresis. Mol Cell Biol. 2006;26:5086–5095. doi: 10.1128/MCB.02380-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weskamp G, Mendelson K, Swendeman S, Le Gall S, Ma Y, Lyman S, Hinoki A, Eguchi S, Guaiquil V, Horiuchi K, Blobel CP. Pathological neovascularization is reduced by inactivation of ADAM17 in endothelial cells but not in pericytes. Circ Res. 2010;106:932–940. doi: 10.1161/CIRCRESAHA.109.207415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Evans BJ, McDowall A, Taylor PC, Hogg N, Haskard DO, Landis RC. Shedding of lymphocyte function-associated antigen-1 (LFA-1) in a human inflammatory response. Blood. 2006;107:3593–3599. doi: 10.1182/blood-2005-09-3695. [DOI] [PubMed] [Google Scholar]
- 30.Miller LJ, Bainton DF, Borregaard N, Springer TA. Stimulated mobilization of monocyte Mac-1 and p150,95 adhesion proteins from an intracellular vesicular compartment to the cell surface. J Clin Invest. 1987;80:535–544. doi: 10.1172/JCI113102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alcaide P, Auerbach S, Luscinskas FW. Neutrophil recruitment under shear flow: it’s all about endothelial cell rings and gaps. Microcirculation. 2009;16:43–57. doi: 10.1080/10739680802273892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garton KJ, Gough PJ, Philalay J, Wille PT, Blobel CP, Whitehead RH, Dempsey PJ, Raines EW. Stimulated shedding of vascular cell adhesion molecule 1 (VCAM-1) is mediated by tumor necrosis factor-alpha-converting enzyme (ADAM 17) J Biol Chem. 2003;278:37459–37464. doi: 10.1074/jbc.M305877200. [DOI] [PubMed] [Google Scholar]
- 33.Hyun YM, Sumagin R, Sarangi PP, Lomakina E, Overstreet MG, Baker CM, Fowell DJ, Waugh RE, Sarelius IH, Kim M. Uropod elongation is a common final step in leukocyte extravasation through inflamed vessels. J Exp Med. 2012;209:1349–1362. doi: 10.1084/jem.20111426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Proebstl D, Voisin MB, Woodfin A, Whiteford J, D’Acquisto F, Jones GE, Rowe D, Nourshargh S. Pericytes support neutrophil subendothelial cell crawling and breaching of venular walls in vivo. J Exp Med. 2012;209:1219–1234. doi: 10.1084/jem.20111622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chavakis T, Keiper T, Matz-Westphal R, Hersemeyer K, Sachs UJ, Nawroth PP, Preissner KT, Santoso S. The junctional adhesion molecule-C promotes neutrophil transendothelial migration in vitro and in vivo. J Biol Chem. 2004;279:55602–55608. doi: 10.1074/jbc.M404676200. [DOI] [PubMed] [Google Scholar]
- 36.Dixit N, Simon SI. Chemokines, selectins and intracellular calcium flux: temporal and spatial cues for leukocyte arrest. Front Immunol. 2012;3:188. doi: 10.3389/fimmu.2012.00188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Le Gall SM, Maretzky T, Issuree PD, Niu XD, Reiss K, Saftig P, Khokha R, Lundell D, Blobel CP. ADAM17 is regulated by a rapid and reversible mechanism that controls access to its catalytic site. J Cell Sci. 2010;123:3913–3922. doi: 10.1242/jcs.069997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Le Gall SM, Bobe P, Reiss K, Horiuchi K, Niu XD, Lundell D, Gibb DR, Conrad D, Saftig P, Blobel CP. ADAMs 10 and 17 represent differentially regulated components of a general shedding machinery for membrane proteins such as transforming growth factor alpha, L-selectin, and tumor necrosis factor alpha. Mol Biol Cell. 2009;20:1785–1794. doi: 10.1091/mbc.E08-11-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Semmrich M, Smith A, Feterowski C, Beer S, Engelhardt B, Busch D, Bartsch B, Laschinger M, Hogg N, Pfeffer K, Holzmann B. Importance of integrin LFA-1 deactivation for the generation of immune responses. J Exp Med. 2005;201:1987–1998. doi: 10.1084/jem.20041850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Woodfin A, Voisin MB, Beyrau M, Colom B, Caille D, Diapouli FM, Nash GB, Chavakis T, Albelda SM, Rainger GE, Meda P, Imhof BA, Nourshargh S. The junctional adhesion molecule JAM-C regulates polarized transendothelial migration of neutrophils in vivo. Nat Immunol. 2011;12:761–769. doi: 10.1038/ni.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scheiermann C, Colom B, Meda P, Patel NS, Voisin MB, Marrelli A, Woodfin A, Pitzalis C, Thiemermann C, Aurrand-Lions M, Imhof BA, Nourshargh S. Junctional adhesion molecule-C mediates leukocyte infiltration in response to ischemia reperfusion injury. Arterioscler Thromb Vasc Biol. 2009;29:1509–1515. doi: 10.1161/ATVBAHA.109.187559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bradfield PF, Scheiermann C, Nourshargh S, Ody C, Luscinskas FW, Rainger GE, Nash GB, Miljkovic-Licina M, Aurrand-Lions M, Imhof BA. JAM-C regulates unidirectional monocyte transendothelial migration in inflammation. Blood. 2007;110:2545–2555. doi: 10.1182/blood-2007-03-078733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Santoso S, Sachs UJ, Kroll H, Linder M, Ruf A, Preissner KT, Chavakis T. The junctional adhesion molecule 3 (JAM-3) on human platelets is a counterreceptor for the leukocyte integrin Mac-1. J Exp Med. 2002;196:679–691. doi: 10.1084/jem.20020267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lamagna C, Meda P, Mandicourt G, Brown J, Gilbert RJ, Jones EY, Kiefer F, Ruga P, Imhof BA, Aurrand-Lions M. Dual interaction of JAM-C with JAMB and alpha(M)beta2 integrin: function in junctional complexes and leukocyte adhesion. Mol Biol Cell. 2005;16:4992–5003. doi: 10.1091/mbc.E05-04-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.