

Abstract
Rationale
During the transition from compensated hypertrophy to heart failure, the signaling between L-type Ca2+ channels (LCCs) in the cell membrane/T-tubules (TTs) and ryanodine receptors (RyRs) in the sarcoplasmic reticulum (SR) becomes defective, partially due to the decreased expression of a TT-SR anchoring protein, junctophilin-2 (JP2). MiR-24, a JP2 suppressing microRNA, is up-regulated in hypertrophied and failing cardiomyocytes.
Objective
To test whether miR-24 suppression can protect the structural and functional integrity of LCC-RyR signaling in hypertrophied cardiomyocytes.
Methods and Results
In vivo silencing of miR-24 by a specific antagomir in an aorta-constricted mouse model effectively prevented the degradation of heart contraction but not ventricular hypertrophy. Electrophysiology and confocal imaging studies showed that antagomir treatment prevented the decreases in LCC-RyR signaling fidelity/efficiency and whole-cell Ca2+ transients. Further studies showed that antagomir treatment stabilized JP2 expression and protected the ultrastructure of TT-SR junctions from disruption.
Conclusions
MiR-24 suppression prevented the transition from compensated hypertrophy to decompensated hypertrophy, providing a potential strategy for early treatment against heart failure.
Keywords: Hypertrophy, remodeling heart failure, myocardial contraction, Ca2+ signaling, hypertrophic cardiomyopathy
INTRODUCTION
Transition from compensated hypertrophy to decompensated hypertrophy represents a key step in the development of heart failure.1,2 One of the hallmarks of this transition is the decreased strength of cardiac contraction.1,3 In heart cells, the contraction is initiated by periodic transient increases in intracellular Ca2+. During each Ca2+ transient, the Ca2+ influx through L-type Ca2+ channels (LCCs) in the cell membrane and transverse tubules (TTs) triggers Ca2+ release from ryanodine receptors (RyRs) in the sarcoplasmic reticulum (SR).4–7 The structural integrity of the LCC-RyR signaling apparatus relies on a TT-SR linker protein, known as junctophilin-2 (JP2),8–10 which is down-regulated in all tested animal models and human specimens of decompensated hypertrophy and heart failure.10–14 Recently, we found that miR-24, a microRNA that suppresses JP2 expression, is up-regulated in hypertrophy/heart failure.15 Since over-expression of miR-24 suppresses both JP2 expression and E-C coupling efficiency,15 we hypothesized that miR-24 up-regulation is a key factor in the transition from compensated hypertrophy to heart failure.
In the present study, we tested this hypothesis by treating aorta-constricted mouse models of hypertrophy with a specific antagomir16 against miR-24. We found that in vivo silencing of miR-24 indeed protected the E-C coupling from structural and functional remodeling, preventing the transition from compensated hypertrophy to decompensated hypertrophy.
METHODS
We created a chronic mouse model of pressure-overload hypertrophy by transverse aortic constriction (TAC) surgery as described.17 In one of the TAC groups, we suppressed the expression of miR-24 by periodic injection (Online Figure I) of a chemically modified antisense oligonucleotide antagomir16 specific for miR-24. An oligonucleotide with mismatches to miR-24 was injected into another TAC group for negative control (NC). Single cardiomyocytes were isolated around 30 weeks after surgery for structural and functional analysis using electron microscopy,10 electrophysiology12 and confocal Ca2+ imaging12 as described. The methods are detailed in the online supplemental materials.
RESULTS
MiR-24 suppression prevented decompensation but not hypertrophy
Compared with that in the sham-operated group, the miR-24 level in isolated ventricular myocytes exhibited a ~2.5-fold increase in the NC group, but not in the antagomir group (Fig. 1A), indicating that the up-regulation of miR-24 associated with TAC-induced hypertrophy was successfully suppressed by the antagomir treatment.
Figure 1. In vivo miR-24 silencing in mouse hypertrophy models.
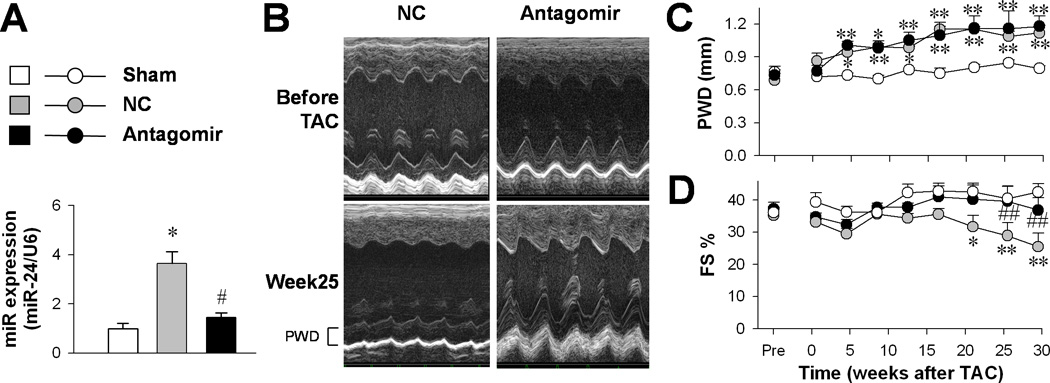
A, Real-time PCR assay of miR-24 expression in sham (n = 4), NC (n = 3) and antagomir (n = 3) groups. B, Representative echocardiograms before and 25 weeks after TAC surgery in NC and antagomir groups. C, Left ventricle wall thickness (PWD, upper) and, D, fractional shortening (FS, lower) measured by echocardiography. *P <0.05 and **P <0.01 vs. sham; #P <0.05 and ##P <0.01 vs. NC.
Echocardiographic measurements (Fig. 1B) showed that left ventricle hypertrophy developed 4 weeks after TAC surgery in our models (Fig. 1C). Around 15 weeks later, the fractional shortening became decreased (Fig. 1D), indicating a transition from compensated to decompensated hypertrophy. Notably, although in vivo antagomir treatment did not interfere with the development of hypertrophy (Fig. 1C), it did prevent the reduction of fractional shortening (Fig. 1D), indicating that the transition toward decompensated hypertrophy was effectively prevented by miR-24 suppression.
In vivo miR-24 suppression protected E-C coupling in cardiomyocytes
To examine whether miR-24 suppression protected E-C coupling at the cellular level, we recorded the Ca2+ transient evoked by whole-cell LCC Ca2+ current (ICa) (Fig. 2A) under a condition (resting cardiomyocytes equilibrated in 2 mM extracellular Ca2+) where the SR Ca2+ load was comparable among all groups (Online Figure II). In the NC group, TAC induced a significant reduction in Ca2+ transient amplitude without altering ICa density (Fig. 2B), leading to a decreased gain of E-C coupling (Fig. 2C) and reduced fraction of cell contraction (Fig. 2D). In contrast, the Ca2+ transient amplitude (Fig. 2B), the E-C coupling gain (Fig. 2C) and the fractional shortening (Fig. 2D) were well maintained after TAC in the antagomir group, indicating that miR-24 suppression protected the integrity of E-C coupling in hypertrophied cardiomyocytes.
Figure 2. The effect of miR-24 silencing on E-C coupling.
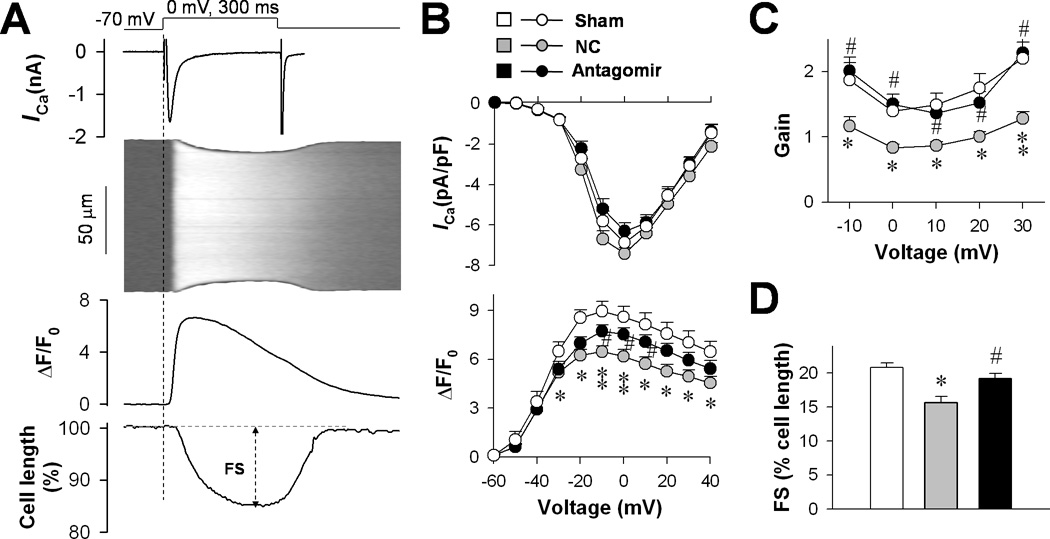
A, Whole-cell patch-clamp and confocal imaging were used to measure ICa density (upper), Ca2+ transients (middle) and cell shortening (lower). B, ICa density and amplitude of Ca2+ transients were compared among sham (14 cells), NC (19 cells) and antagomir (18 cells) groups. C, Gain of E-C coupling calculated as the amplitude of Ca2+ transient per unit ICa density. D, Fractional shortening of cardiomyocytes measured by cell edge-detection of Ca2+ transients at 0 mV. *P <0.05 and **P <0.01 vs. sham; #P <0.05 vs. NC.
Ca2+ transients are composed of numerous Ca2+ sparks evoked by LCC openings. Using unique loose-patch confocal imaging technology,7,12 we investigated the effect of the antagomir on LCC-RyR intermolecular Ca2+ signaling. To visualize single LCC activity, in the form of Ca2+ sparklets,7 we included in the pipette solution 20 mM Ca2+ and 10 µM FPL64176, an LCC agonist. Depolarization of on-cell patches evoked two distinct populations of local Ca2+ events (Fig. 3A): steep, ryanodine-sensitive Ca2+ sparks from RyRs; and flat, ryanodine-resistant but nifedipine-sensitive Ca2+ sparklets from individual LCCs.7 With comparable Ca2+ release duration (time-to-peak), the amplitude of Ca2+ sparks was significantly lower in the NC group but not in the TAC antagomir group (Fig. 3B), indicating that the TAC-induced decrease of local Ca2+ release flux was prevented by antagomir treatment. To quantify the fidelity of LCC-RyR coupling, we measured the percentage of the first detectable Ca2+ sparklets that successfully triggered Ca2+ sparks during the depolarization. The fidelity was decreased significantly in the NC group but unchanged in the antagomir group (Fig. 3C, upper). Also, the percentage of depolarization pulses that failed to trigger a Ca2+ spark (“miss index”) was increased in the NC group but not in the antagomir group (Fig. 3C, lower). We also quantified LCC-RyR coupling kinetics by the latency from the onset of a Ca2+ sparklet to the takeoff of a triggered Ca2+ spark (Fig. 3D). Exponential fitting of the coupling latency (Fig. 3E) showed that the time constant for LCC-RyR coupling was prolonged in the NC group but unchanged in the antagomir group (Fig. 3F). These results indicated that miR-24 suppression effectively prevented the decreased efficiency and slowed kinetics of LCC-RyR signaling in failing heart cells12,18.
Figure 3. The effect of miR-24 silencing on LCC-RyR communications.
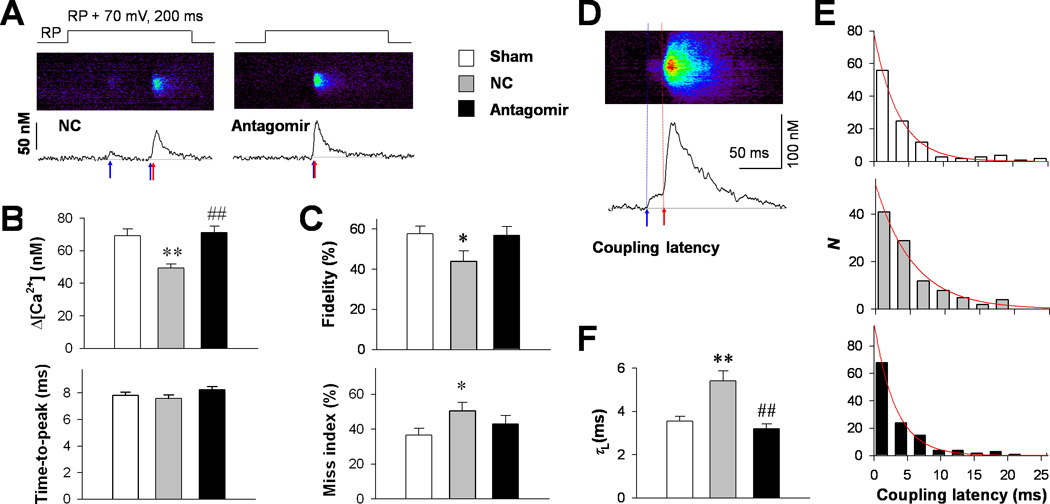
A, Representative loose-patch confocal images (middle) and their time profiles (lower) in NC and antagomir groups, showing that LCC Ca2+ sparklets (blue arrows) triggered RyR Ca2+ sparks (red arrows) in a probabilistic manner during 70-mV depolarizations from resting potential (RP+70, upper). B, Amplitude (upper) and time-to-peak (lower) of triggered Ca2+ sparks in sham (187 events), NC (150 events) and antagomir (185 events) groups. C, LCC-RyR coupling fidelity (upper) was indexed by the percentage of the first apparent Ca2+ sparklet that successfully activated a Ca2+ spark during a patch depolarization. The miss index (lower) was defined as the percentage of depolarizing pulses that failed to trigger any Ca2+ spark. The percentages were first determined for each cell, and then averaged in the sham (59 cells), NC (52 cells) and antagomir (62 cells) groups. D, Example of a confocal image (upper) and its time profile (lower) from the antagomir group, illustrating the measurement of LCC-RyR coupling latency from the onset of a Ca2+ sparklet (blue arrow) to the takeoff of the triggered Ca2+ spark (red arrow). E, The distributions (bars) and their exponential fits (curves) of coupling latency in sham (109 events), NC (105 events) and antagomir (123 events) groups. F, Comparison of time constants (τL) of the LCC-RyR coupling latency among groups. *P <0.05 and **P <0.01 vs. sham; ##P <0.01 vs. NC.
MiR-24 suppression prevented structural remodeling of E-C coupling apparatus
Next, we checked the ultrastructural basis of LCC-RyR communication using transmission electron microscopy. Stereological analysis (Online Figure III) showed that the volume density and the surface area of TTs apparently coupled to SRs were dramatically decreased in the NC group but not in the antagomir group (Fig. 4A). The increase of bald TTs and decrease of junctional SRs were also suppressed by the antagomir. In failing heart cells, TT-SR junctions were displaced from the Z-line area, exhibiting increased junction-Z distance (Fig. 4B and C).10 The increased junction-Z distance was not observed in the antagomir group (Fig. 4C). The spatial span of individual TT-SR junctions is one of the determinants of LCC-RyR signaling efficiency.10 We found that the antagomir prevented the shrinkage of individual junction size (Fig. 4D). These data indicated that the defects of TT-SR junctions in failing cardiomyocytes were prevented by miR-24 suppression.
Figure 4. Effect of miR-24 silencing on the structure of TT-SR junctions.
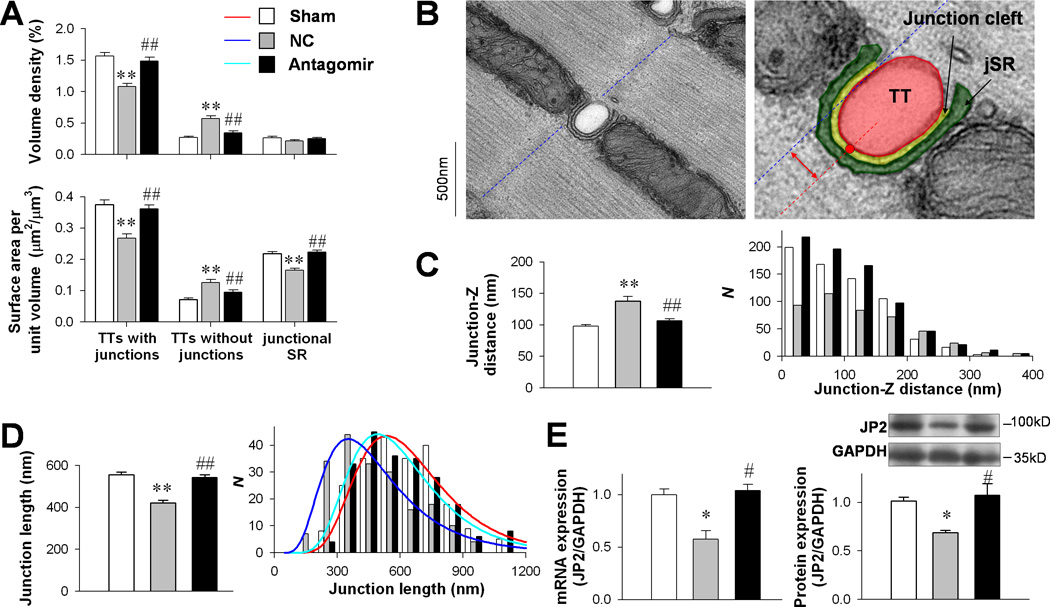
A, Results of stereological analysis of volume density (upper) and surface area per unit volume (lower) of TTs coupled with SRs, bald TTs and junctional SRs (JSRs) in sham (183 images), NC (154 images) and antagomir (169 images) groups. B, Typical images showing the measurement of junction-Z distance (red double arrow) between the center of a junction cleft (red line) and its adjacent Z-line (blue line). C, Comparison of junction-Z distance (left) and its distribution (right) among sham (183 images), NC (154 images) and antagomir (169 images) groups. D, TT-SR junction length was measured as the curvilinear length of the junctional cleft (marked in yellow in B). E,Comparison of JP2 mRNA (left) and protein (right) expression levels among sham (n = 4), NC (n = 3) and antagomir (n = 3) groups. *P < 0.05 and ** P < 0.01 vs. sham; #P <0.05 and ##P <0.01 vs. NC.
JP2 is a structural protein maintaining the morphology of TT-SR junctions and efficiency of LCC-RyR signaling.8–10 We found that the levels of both JP2 mRNA and protein, which were significantly decreased in the NC group, were unchanged in the antagomir group (Fig. 4E).
DISCUSSION
E-C coupling becomes defective during the chronic transition from compensated hypertrophy to heart failure.12,20 In the present study, we show that in vivo silencing of miR-24 in an aortic-constricted mouse model effectively protects cardiomyocytes from structural/functional disruption of E-C coupling and prevents the transition toward decompensated hypertrophy.
MiR-24 is expressed in cardiomyocytes and many other cell types and regulates multiple target proteins.19–22 We have recently shown that over-expression of miR-24, as observed in heart failure/hypertrophy models, suppresses JP2 expression and leads to defective E-C coupling in carrdiomyocytes.15 In the present study, we show that the JP2 down-regulation is prevented by the miR-24 antagomir in TAC mice. As our bioinformatic analysis was not able to identify other miR-24 targets with known function related to E-C coupling, the stabilization of JP2 at least partially explains the protective effects of miR-24 suppression on TT-SR junctions and E-C coupling. Besides E-C coupling, whether other histological/molecular hallmarks of decompensation, such as fibrosis, are altered by miR-24 modulation still needs further in-depth studies.
The pathogenesis of hypertrophy and heart failure involves a variety of intracellular signaling cascades, including the calcineurin-nuclear factor of activated T-cells (NFAT) pathway, the calmodulin-dependent protein kinase pathway, and pathways involving other protein kinases.23,24 The calcineurin-NFATc3 pathway controls the microRNA cluster miR-23a~27a~24-2, which is up-regulated in hypertrophy.21,22,25 In this cluster, miR-23, but not miR-24 and miR-27, is found essential in the isoproterenol/aldosterone-induced cardiomyocyte hypertrophy.25 Agreeing with this report, our present study shows that miR-24 suppression in vivo does not prevent TAC-induced hypertrophy. Excitingly, miR-24 suppression does prevent the structural and functional degradation of E-C coupling, indicating that miR-24 up-regulation is important in the transition from compensated hypertrophy to heart failure.
Supplementary Material
Novelty and Significance.
What Is Known?
Cardiac excitation-contraction (E-C) coupling becomes defective during the transition from compensated hypertrophy to heart failure.
The defective E-C coupling in cardiac myocytes of failing hearts could be partially attributed to the physical uncoupling between T-tubules and sarcoplasmic reticulum (SR) associated with the down-regulation of junctophilin-2 (JP2).
MiR-24, a microRNA that suppresses JP2 expression, is up-regulated in hypertrophied/failing cardiomyocytes.
What New Information Does This Article Contribute?
In vivo suppression of miR-24 does not interfere with transverse aortic constriction (TAC)-induced hypertrophy, but prevents the progressive decrease in the contraction of the left ventricule.
MiR-24 suppression protects cardiomyocytes from TAC-induced defects in L-type calcium channel- ryanodine receptor Ca2+ signaling.
Suppression of miR-24 prevents TAC-induced de-stabilization of TT-SR junctions in cardiac myocytes, presumably by maintaining JP2 levels.
During the transition from compensated hypertrophy to heart failure, cardiac E-C coupling becomes defective, partially due to the down-regulation of T-tubule SR anchoring protein - JP2. Because miR-24, which suppresses JP2, is up-regulated in failing cardiomyocytes, we tested whether suppression of miR-24 protects the integrity of E-C coupling. We found that in vivo silencing of miR-24 blocks the transition to decompensate hypertrophy while allowing compensated hypertrophy to persist in mice subjected to TAC. Cellular studies showed that miR-24 antagomir treatment protects cardiac myocytes from structural and functional remodeling of E-C coupling apparatus. These findings suggest that miR-24 may be a potential target in the treatment of heart failure.
ACKNOWLEDGEMENTS
We thank Drs. Xue-Mei Hao and Ying-Chun Hu for professional technical support.
SOURCES OF FUNDING
This study was supported by the 973 Program of China (2011CB809101), the National Natural Science Foundation of China (81070196, 81030001, 81121061), the Strategic Priority Research Program of Chinese Academy of Sciences (XDA01020105), the Program for New Century Excellent Talents in University, the Beijing Talents Foundation, and the NIH, USA (R01 TW007269).
Non-standard Abbreviations
- E-C
excitation-contraction
- ICa
whole-cell Ca2+ current through L-type Ca2+ channels
- JP2
junctophilin-2
- LCC
L-type Ca2+ channel
- NC
negative control
- NFAT
nuclear factor of activated T-cells
- RyR
ryanodine receptor
- SR
sarcoplasmic reticulum.
- TAC
transverse aortic constriction
- TT
transverse tubule
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES
None
REFERENCES
- 1.Braunwald E, Bristow MR. Congestive heart failure: fifty years of progress. Circulation. 2000;102:IV14–IV23. doi: 10.1161/01.cir.102.suppl_4.iv-14. [DOI] [PubMed] [Google Scholar]
- 2.Hunter JJ, Chien KR. Signaling pathways for cardiac hypertrophy and failure. N Engl J Med. 1999;341:1276–1283. doi: 10.1056/NEJM199910213411706. [DOI] [PubMed] [Google Scholar]
- 3.Houser SR, Margulies KB. Is depressed myocyte contractility centrally involved in heart failure? Circ Res. 2003;92:350–358. doi: 10.1161/01.RES.0000060027.40275.A6. [DOI] [PubMed] [Google Scholar]
- 4.Fabiato A. Time and calcium dependence of activation and inactivation of calcium-induced release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. J Gen Physiol. 1985;85:247–289. doi: 10.1085/jgp.85.2.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lopez-Lopez JR, Shacklock PS, Balke CW, Wier WG. Local calcium transients triggered by single L-type calcium channel currents in cardiac cells. Science. 1995;268:1042–1045. doi: 10.1126/science.7754383. [DOI] [PubMed] [Google Scholar]
- 6.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 7.Wang SQ, Song LS, Lakatta EG, Cheng H. Ca2+ signalling between single L-type Ca2+ channels and ryanodine receptors in heart cells. Nature. 2001;410:592–596. doi: 10.1038/35069083. [DOI] [PubMed] [Google Scholar]
- 8.Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K. Junctophilins: a novel family of junctional membrane complex proteins. Mol Cell. 2000;6:11–22. doi: 10.1016/s1097-2765(00)00003-4. [DOI] [PubMed] [Google Scholar]
- 9.van Oort RJ, Garbino A, Wang W, Dixit SS, Landstrom AP, Gaur N, De Almeida AC, Skapura DG, Rudy Y, Burns AR, Ackerman MJ, Wehrens XH. Disrupted junctional membrane complexes and hyperactive ryanodine receptors after acute junctophilin knockdown in mice. Circulation. 2011;123:979–988. doi: 10.1161/CIRCULATIONAHA.110.006437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu HD, Xu M, Li RC, Guo L, Lai YS, Xu SM, Li SF, Lu QL, Li LL, Zhang HB, Zhang YY, Zhang CM, Wang SQ. Ultrastructural remodelling of Ca2+ signalling apparatus in failing heart cells. Cardiovasc Res. 2012;95:430–438. doi: 10.1093/cvr/cvs195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Minamisawa S, Oshikawa J, Takeshima H, Hoshijima M, Wang Y, Chien KR, Ishikawa Y, Matsuoka R. Junctophilin type 2 is associated with caveolin-3 and is down-regulated in the hypertrophic and dilated cardiomyopathies. Biochem Biophys Res Commun. 2004;325:852–856. doi: 10.1016/j.bbrc.2004.10.107. [DOI] [PubMed] [Google Scholar]
- 12.Xu M, Zhou P, Xu SM, Liu Y, Feng X, Bai SH, Bai Y, Hao XM, Han Q, Zhang Y, Wang SQ. Intermolecular failure of L-type Ca2+ channel and ryanodine receptor signaling in hypertrophy. PLoS Biol. 2007;5:e21. doi: 10.1371/journal.pbio.0050021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wei S, Guo A, Chen B, Kutschke W, Xie YP, Zimmerman K, Weiss RM, Anderson ME, Cheng H, Song LS. T-tubule remodeling during transition from hypertrophy to heart failure. Circ Res. 2010;107:520–531. doi: 10.1161/CIRCRESAHA.109.212324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wagner E, Lauterbach MA, Kohl T, Westphal V, Williams GS, Steinbrecher JH, Streich JH, Korff B, Tuan HT, Hagen B, Luther S, Hasenfuss G, Parlitz U, Jafri MS, Hell SW, Lederer WJ, Lehnart SE. Stimulated emission depletion live-cell super-resolution imaging shows proliferative remodeling of T-tubule membrane structures after myocardial infarction. Circ Res. 2012;111:402–414. doi: 10.1161/CIRCRESAHA.112.274530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu M, Wu HD, Li RC, Zhang HB, Wang M, Tao J, Feng XH, Guo YB, Li SF, Lai ST, Zhou P, Li LL, Yang HQ, Luo GZ, Bai Y, Xi JJ, Gao W, Han QD, Zhang YY, Wang XJ, Meng X, Wang SQ. Mir-24 regulates junctophilin-2 expression in cardiomyocytes. Circ Res. 2012;111:837–841. doi: 10.1161/CIRCRESAHA.112.277418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with 'antagomirs'. Nature. 2005;438:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 17.Xiao H, Ma X, Feng W, Fu Y, Lu Z, Xu M, Shen Q, Zhu Y, Zhang Y. Metformin attenuates cardiac fibrosis by inhibiting the TGFbeta1-Smad3 signalling pathway. Cardiovasc Res. 2010;87:504–513. doi: 10.1093/cvr/cvq066. [DOI] [PubMed] [Google Scholar]
- 18.Gomez AM, Valdivia HH, Cheng H, Lederer MR, Santana LF, Cannell MB, McCune SA, Altschuld RA, Lederer WJ. Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science. 1997;276:800–806. doi: 10.1126/science.276.5313.800. [DOI] [PubMed] [Google Scholar]
- 19.Qian L, Van Laake LW, Huang Y, Liu S, Wendland MF, Srivastava D. miR-24 inhibits apoptosis and represses Bim in mouse cardiomyocytes. J Exp Med. 2011;208:549–560. doi: 10.1084/jem.20101547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fiedler J, Jazbutyte V, Kirchmaier BC, Gupta SK, Lorenzen J, Hartmann D, Galuppo P, Kneitz S, Pena JT, Sohn-Lee C, Loyer X, Soutschek J, Brand T, Tuschl T, Heineke J, Martin U, Schulte-Merker S, Ertl G, Engelhardt S, Bauersachs J, Thum T. MicroRNA-24 regulates vascularity after myocardial infarction. Circulation. 2011;124:720–730. doi: 10.1161/CIRCULATIONAHA.111.039008. [DOI] [PubMed] [Google Scholar]
- 21.Sayed D, Hong C, Chen IY, Lypowy J, Abdellatif M. MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ Res. 2007;100:416–424. doi: 10.1161/01.RES.0000257913.42552.23. [DOI] [PubMed] [Google Scholar]
- 22.van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, Richardson JA, Olson EN. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci U S A. 2006;103:18255–18260. doi: 10.1073/pnas.0608791103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shah AM, Mann DL. In search of new therapeutic targets and strategies for heart failure: recent advances in basic science. Lancet. 2011;378:704–712. doi: 10.1016/S0140-6736(11)60894-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anderson ME, Brown JH, Bers DM. CaMKII in myocardial hypertrophy and heart failure. J Mol Cell Cardiol. 2011;51:468–473. doi: 10.1016/j.yjmcc.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin Z, Murtaza I, Wang K, Jiao J, Gao J, Li PF. miR-23a functions downstream of NFATc3 to regulate cardiac hypertrophy. Proc Natl Acad Sci U S A. 2009;106:12103–12108. doi: 10.1073/pnas.0811371106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.