

Abstract
Sleep deprivation is a common feature in modern society, and one of the consequences of sleep loss is the impairment of cognitive function. Although it has been widely accepted that sleep deprivation affects learning and memory, only recently has research begun to address which molecular signalling pathways are altered by sleep loss and, more importantly, which pathways can be targeted to reverse the memory impairments resulting from sleep deprivation. In this review, we discuss the different methods used to sleep deprive animals and the effects of different durations of sleep deprivation on learning and memory with an emphasis on hippocampus-dependent memory. We then review the molecular signalling pathways that are sensitive to sleep loss, with a focus on those thought to play a critical role in the memory and synaptic plasticity deficits observed after sleep deprivation. Finally, we highlight several recent attempts to reverse the effects of sleep deprivation on memory and synaptic plasticity. Future research building on these studies promises to contribute to the development of novel strategies to ameliorate the effects of sleep loss on cognition.
Keywords: sleep loss, learning, hippocampus, astrocytes, cAMP, transcription
1. Introduction
Millions of people worldwide experience sleep deprivation on a daily basis [1]. The pressure to stay up longer in our modern 24/7 society impacts a growing percentage of the population [2, 3]. A population-based study indicated that, over the past 50 years, sleep duration in adult and adolescent Americans has decreased by 1.5–2 hours per night in adults and adolescents, with 30% reporting sleep of 6 hours per night or less[4].
One of the first indications that sleep might be beneficial for the formation of memories came from a study by Jenkins and Dallenbach [5] that showed that sleep attenuated the rate of forgetting. In the 1960’s, Morris and colleagues found that sleep deprivation impaired memory processing [6]. In the decades thereafter, it became apparent in both humans and animal models that specific forms of memory are affected by sleep deprivation [7–10]. To combat the effects of sleep deprivation, it is critical to understand the molecular and cellular mechanisms by which sleep deprivation leads to cognitive deficits. Here, we review current knowledge of the intracellular signalling pathways that are affected by sleep deprivation, with an emphasis on the impact of sleep deprivation on hippocampal function In addition, we discuss the different approaches that have been developed to reverse memory and plasticity deficits induced by sleep deprivation.
2. Methods for sleep deprivation in rodents: advantages and drawbacks
To elucidate which cellular and molecular effects of sleep deprivation lead to memory impairments, many research laboratories have utilized rodents as study objects. Three primary techniques have been used to deprive laboratory rodents of sleep. Each of these methods has particular advantages and drawbacks, as discussed below.
The first is the platform-over-water, pedestal, or “flower pot” method, which is the best method to selectively deprive animals of rapid eye movement (REM) sleep for one or multiple days with only intermittent monitoring by the researcher [11]. Animals are placed in a chamber with one or multiple small platforms surrounded by water. When the animals enter REM sleep, their muscle tone diminishes and the animals fall into the water, waking them up and preventing them from going into REM sleep. For control animals, the small platforms are replaced with larger ones, allowing them to enter REM sleep without falling into the water [12]. The concern with this particular method is that large-platform control animals, which obtain normal amounts of REM sleep, also show some alterations in neuronal function (see section 4 of this review). This suggests that aspects of the method other than the loss of REM sleep are responsible for some of the phenotypes observed after using the platform-over-water method [13].
The second method of sleep deprivation utilizes forced locomotion, in which the animal is placed in a chamber with a revolving floor or rotating drum that forces the animal to reposition itself with each revolution [14, 15]. This method can be tailored to achieve total deprivation or selective deprivation of a particular sleep stage. Control animals can be manipulated to move just as much as deprived animals, but with longer periods of rest in between, thereby preventing excessive sleep loss. For example, the schedule for rotation of the chamber for a sleep-deprived animal might be 10 seconds on, 30 seconds off, whereas the schedule for a control animal might be 10 minutes on, 30 minutes off. Thus, the control animal has repeated periods of 30 minutes during which it can sleep undisturbed, but the sleep-deprived animal has a maximum of 30 seconds to rest before it is forced to move again (see [16]). This technique is often used to model sleep fragmentation, such as might occur due to sleep apnea, caring for an infant throughout the night, or otherwise fitful sleep. The interpretation of the effect of sleep deprivation using this technique can be challenging and depends on using the correct control groups, because locomotor activity or stress evoked using this technique may mask or reverse the effects the effects of sleep deprivation.
The third sleep deprivation method is based on gentle handling or mild stimulation. In this technique, researchers make mild noises, gently jostle the animal’s home cage, disturb the animal’s nesting material, and in some cases stroke the animal [17–19]. Gentle handling is very effective at inducing total sleep deprivation as determined by electroencephalography [20], and seems to be a strong model of typical sleep deprivation in humans. One downside of the gentle handling technique is that it requires constant vigilance by the researcher, with the result that gentle handling is rarely carried out for longer than 12 hours.
Because the gentle handling method involves direct contact between the researcher and the cage or the animal itself, a concern is whether the memory deficits resulting from sleep deprivation by gentle handling are due to sleep loss or non-specific side effects of the technique itself. To test this issue, Hagewoud and colleagues [21] trained animals at the beginning of the resting phase (in rodents, this is the light phase) and recorded the amount of stimulation needed to keep them awake during the following 6 hours. Next, the authors trained a new cohort of rats at the end of the resting phase and determined whether giving the animals the same amount of stimulation during the first 6 hours of the active phase (in rodents, this is the dark phase, in which they spend only a short time sleeping) would also induce memory deficits. They found that giving the same amount of stimulation during the first 6 hours of the active phase, in contrast to the resting phase, did not induce a memory deficit. These findings indicated that the memory deficits observed after sleep deprivation in the light phase were not likely a consequence of the gentle handling method used to sleep deprive animals but rather a consequence of sleep loss.
In some other sleep deprivation studies, the introduction of novel objects or new nesting material are used to keep animals awake, or animals are allowed to explore novel environments [22, 23]. Although these techniques have successfully kept animals awake without elevating plasma corticosterone levels [23], they may be problematic when the goal is to study the effects of sleep deprivation on memory or synaptic plasticity. For example, it has been reported that the exploration of new environments and new objects facilitates hippocampal LTD and occludes LTP in vivo [24, 25], increases the phosphorylation of NMDA and AMPA receptor subunits, and activates ERK1/2 signalling [26]. Furthermore, nest building behaviour has been reported to accelerate REM sleep generation [27]. It would be useful, as suggested by Hagewoud and colleagues [21], to separate the effects of exposure to a novel environment, novel objects, or new nesting material from the effects of prolonged wakefulness on plasticity and memory, perhaps by applying these methods of sleep deprivation during the animals’ active phase, in which they naturally sleep much less.
A recurring matter of debate in the field of sleep research is how many of the molecular and cellular changes observed after sleep deprivation can be attributed to stress, rather than to sleep loss itself. We believe that the role of stress as the major factor in the memory and plasticity deficits observed particularly after brief sleep deprivation is overstated. Two recent studies in which the activation of the stress signalling pathway is temporarily or permanently disrupted have shown that sleep deprivation leads to cognitive impairments even in the absence of stress-mediated signalling [28, 29]. Also, the changes in plasma corticosterone levels during brief sleep deprivation by gentle handling in many cases do not reach levels beyond those observed during the stress hormone’s natural circadian oscillation [30], during exploration of a novel environment containing novel objects [31], or due to exposure to a conspecific of the opposite sex [32]. In some cases, the mildly increased plasma corticosterone levels observed after sleep deprivation do not even reach statistical significance [18, 19, 21, 33]. Further if brief sleep deprivation acts as a mild stressor, one might expect that this would enhance memory, as mild increases in glucocorticoids have been shown to facilitate rather than impair memory and plasticity formation [34]. Thus, although mild stress is inherent to most forms of sleep deprivation in the laboratory as in the real world, it is unlikely that this stress component can explain the effects of sleep deprivation on the molecular signalling pathways that result in impairments in memory and synaptic plasticity.
In summary, the sleep deprivation techniques described above all have particular advantages, but each also has its limitations. Because of the different characteristics of each of these sleep deprivation methods, it is important to consider the technique and duration of sleep deprivation used in each of the studies described below.
3. Sleep deprivation and memory
Using the platform-over-water method, Fishbein and colleagues [35] tested the effects of 24 hours of REM sleep deprivation immediately prior to training in a classical conditioning task called inhibitory avoidance. Although 24 hours of REM sleep deprivation had no effect on acquisition and short-term memory, long-term memory (tested 1–7 days after training) was significantly impaired [35, 36]. Follow-up studies applying REM sleep deprivation directly after conditioning also attenuated memory formation [37–39], indicating that REM sleep deprivation was particularly detrimental for memory consolidation (reviewed in [40]).
A series of experiments conducted years later showed that memory consolidation requiring the hippocampus is particularly sensitive to sleep deprivation. This was first shown using the Morris water maze, a task that can be designed to assess learning and memory formation that are either hippocampus-dependent (the hidden platform version of the task), or hippocampus-independent (the visible platform version of the task) [41]. Twelve hours of REM sleep deprivation directly after training in the hippocampus-dependent hidden platform version of the Morris water maze task impaired long-term memory formation for platform location, tested 24 hours after training. In contrast, the 12 hours of REM sleep deprivation did not affect performance or memory in the hippocampus-independent visible platform version of the same task. REM sleep deprivation from 13–24 hours after training did not affect memory formation for either task, suggesting that the consolidation period following training is specifically sensitive to sleep loss [42]. The same laboratory then elaborated on this finding by using shorter periods of deprivation, and found that REM sleep deprivation during the first 4 hours directly after training in the water maze had a negative effect on hippocampus-dependent spatial learning and memory formation, but not on hippocampus-independent learning and memory formation [42]. Although some laboratories have confirmed the finding that REM sleep deprivation impairs hippocampus-dependent learning in the Morris water maze task [43–46], others have challenged the idea that REM sleep is essential for spatial learning in the Morris water maze [47, 48].
Like the Morris water maze task, the fear conditioning task is a learning paradigm that enables researchers to examine both hippocampus-dependent and hippocampus-independent learning processes. The ability to associate a foot shock with the specific environment, or context, in which the shock was administered is a process dependent on the hippocampal formation [49, 50]. In contrast, learning to associate a shock with an explicitly paired cue, such as a tone, does not require the hippocampus (for review, see [49, 50]). Five hours of total sleep deprivation using the gentle handling method immediately following training caused a selective impairment in contextual, but not cued memory [51]. The finding that consolidation of contextual fear memories is sensitive to sleep deprivation has been confirmed in many other studies [21, 30, 52, 53].
More recently, Hagewoud and colleagues [33] determined whether hippocampus-dependent working memory was affected by 6–12 hours of total sleep deprivation using the gentle handling method. Mice were deprived of sleep for 6 or 12 hours using the gentle handling method, and immediately at the end of the sleep deprivation period were allowed to explore 2 out of 3 accessible arms in a symmetrical Y maze for 10 minutes. After the training session in the novel arm recognition task, mice remained in their home cage for 2 minutes followed by a 5 minute test session in which all arms were accessible. Mice deprived of sleep for 12 hours spent a similar time exploring the novel and familiar arm. This indicated that the sleep-deprived mice failed to discriminate between the familiar versus novel arms of the maze, suggesting that hippocampus-dependent working memory was impaired. In summary, the studies described above indicate that hippocampus-dependent memory formation and working memory are particularly sensitive to (brief) sleep deprivation.
Because the effects of sleep deprivation can be brain region-specific, the negative effects of sleep deprivation on memory can be subtle and sometimes difficult to assess due to compensation by other less-impacted brain regions. For example, Hagewoud and colleagues [18], showed that five hours of sleep deprivation immediately after each daily training session did not affect directly the rate at which mice learned the location of a food-reward in a two-arm reference task. However, sleep deprivation did induce a shift in the behavioral strategy used to locate the food reward: whereas non-sleep-deprived mice favoured to use a hippocampus-dependent strategy, mice subjected to sleep deprivation switched from using a hippocampus-dependent spatial strategy to using a hippocampus-independent strategy necessitating the striatum. Because this strategy could also be used to locate the food-reward, the effect of sleep deprivation did not become apparent during training. Surprisingly, although sleep deprivation during training did not directly affect performance during the course of training, it did impair performance during consecutive reversal training (in which the food reward was relocated to the previously non-baited arm). The authors argued that the delayed effect of sleep deprivation on performance was due to the shift from using a hippocampus-dependent to a striatum-dependent strategy during the initial training. Because the striatal system is less flexible, it was more difficult for sleep-deprived mice to adapt the previously formed but now incorrect memory for the location of the food reward [18]. Thus, this study concluded that sleep deprivation can lead to the use of alternative learning mechanisms and brain regions. However, there can be consequences of using these alternative suboptimal mechanisms, such as a greater difficulty with updating outdated memories.
Cortical regions important for memory formation are affected by sleep loss as well [22, 54]. For example, sleep deprivation affects the consolidation of memory for objects (referred to as recognition memory) [31, 55]. Object recognition is a process that, depending on the training conditions, does not critically rely on the hippocampus [56, 57], but rather requires extra-hippocampal cortical regions including the perirhinal cortex [57] and insular cortex [58]. In line with these observations in rodents, studies in humans have shown that sleep deprivation affects cortical systems involved in a variety of functions, including executive attention, working memory, and higher cognitive abilities (reviewed in [59]).
An important finding in behavioural studies of the effects of sleep deprivation is that there are particular periods following learning during which sleep deprivation impacts memory consolidation. The timing of these windows differs depending on the nature of the training [42, 60], but most studies have found that sleep deprivation has the greatest detrimental effect when applied within 5–6 hours after training in the light period [21, 30, 31, 51, 55, 61–64].
In the case of contextual fear conditioning, 5 hours of sleep deprivation at the beginning of the resting phase (light phase in rodents) impaired memory consolidation when applied directly after training, but when the period of sleep deprivation was delayed by 5 hours, memory for contextual fear was not affected [51]. This critical time window coincides with the consolidation period following learning when transcription, translation, and cAMP-PKA signalling are required for memory consolidation in this task (reviewed in [65]), which has suggested that sleep deprivation might disrupt these crucial molecular processes. To address this possibility, many laboratories have examined the effects of sleep deprivation on hippocampal synaptic plasticity to determine which molecular mechanisms that are critical for memory consolidation are affected by (brief) sleep deprivation.
4. Sleep deprivation and hippocampal synaptic plasticity
As described above, behavioural studies looking at the effect of sleep deprivation on memory indicated that the hippocampus is particularly vulnerable to sleep loss. Therefore, electrophysiological studies were conducted to determine the effect of sleep deprivation on various forms of hippocampal synaptic plasticity. One prominent form of hippocampal synaptic plasticity is long-term potentiation (LTP), a long-lasting change in the strength of synaptic connections that is a frequently used model to study the mechanisms underlying learning and memory. Despite using different methods to study hippocampal synaptic plasticity, it is generally agreed that sleep deprivation attenuates LTP in the hippocampus. In rats, the use of forced locomotion to induce total sleep deprivation for 12 hours [15], or sleep fragmentation for 24 hours [66], impaired hippocampal LTP recorded from Schaffer collateral CA1 synapses. Similar LTP deficits were also produced by REM sleep deprivation using the platform-over-water technique for 24 to 75 hrs hours in area CA1 both in vitro and in vivo [52, 67–70]. Shorter durations of REM deprivation for 3, 6, or 9 hours also impaired LTP in vivo in a “dose”-dependent fashion when recordings were performed immediately after the deprivation period [13]. Schaffer collateral LTP at CA1 synapses was also attenuated by shorter periods (~4–6 hours) of total sleep deprivation induced by either exposing mice to a novel environment [23] or gentle handling [30]. In all of the LTP studies described thus far, sleep deprivation was performed prior to the induction of plasticity. Two in vivo studies using recordings from the dentate gyrus in non-anesthetized rats found that REM-specific sleep deprivation by gentle handling performed after the induction of LTP caused significant deficits in LTP maintenance [71, 72]. Importantly, when the same method was used to specifically deprive animals of non-REM sleep, no LTP deficits in the dentate gyrus were observed [72], suggesting that REM sleep plays a critical role in the maintenance of LTP in the dentate gyrus.
In contrast to the abundance of studies looking at the effects of sleep deprivation on LTP, there are only three studies to our knowledge that examine the effect of sleep deprivation on LTD. Tadavarty et al. [73] deprived rats of sleep for 12 hours using the gentle handling method and then induced LTD in rat CA1 slices with 30 seconds of 20 Hz stimulation to the Schaffer collaterals. They found that LTD was enhanced in hippocampal slices from sleep-deprived rats. In a follow-up study, the authors showed that the enhancement of LTD was accompanied by elevated expression and heterodimerization of γ-aminobutyric acid (GABA)B and metabotropic glutamate 1α receptors in the hippocampus [74].
In line with these observations, Kopp and colleagues [23] reported that 4 hours of sleep deprivation attenuated LTP induced by 2 × 100-Hz trains, while it facilitated LTD induced by stimulations in the theta frequency range (5 Hz). No effect of sleep deprivation was observed on LTD induced by 1-Hz stimulation [23]. Together, these studies examining the effects of sleep deprivation on synaptic plasticity suggest that sleep deprivation inhibits synaptic potentiation in the hippocampus (but see [22]), while it facilitates certain forms of synaptic depression. In the paragraphs below, we will discuss the role of specific signalling pathways that may contribute to the plasticity changes observed after sleep deprivation.
While many studies have assessed the effect of sleep deprivation on synaptic plasticity, only a few studies have attempted to identify which specific molecular signalling pathways mediate the changes in synaptic efficacy observed after sleep deprivation. Two studies from the same laboratory suggested that 24 hours of REM sleep deprivation in mice disrupted the function of N-methyl-D-Aspartate receptors (NMDAr) in the dentate gyrus [75] and 72 hours of REM deprivation in rats disrupted NMDAr function in CA1 [68]. Specifically, both reports found decreased NMDAr/2-amino-3-(5-methyl-3-oxo-1,2- oxazol-4-yl) propanoic acid receptor AMPAr current ratios that were attributed to decreased surface expression of NR1 subunits. Reduced hippocampal NR1 expression was also reported in a later study in which rats were deprived of REM sleep for 75 hours [67]. The most intriguing finding in the experiments by McDermott et al. [68] was that application of glycine, which enhances NMDAr function, reversed the effects of 24–48 hours of sleep deprivation on LTP in area CA1. Thus, attenuated NMDAr function appears to be a functional contributor to the hippocampal plasticity deficits observed after longer periods of sleep deprivation [68].
As mentioned above, partial LTP deficits have been observed in large-platform control animals, suggesting that aspects of the treatment other than sleep deprivation may have been somewhat responsible for the impairment of LTP [13]. This effect could potentially also be mediated by the strong stress hormone induction that builds over successive days of platform-over-water treatment in both control and sleep-deprived animals (for a more in-depth discussion of the role of stress in long-term sleep deprivation experiments, see [76]).
Although multiple studies have described changes in NMDAr subunit composition and function after long-term sleep deprivation, the debate remains whether brief sleep deprivation induces similar changes in NMDAr composition and function, and whether these changes underlie the deficits in plasticity and memory that have been reported after brief periods of sleep deprivation. A study by Kopp et al. [23] described changes in NMDAr composition and function after only 4 hours of total sleep deprivation. In contrast with this study, a recent report by Vecsey et al. [30] that applied 5 hours of sleep deprivation did not observe any changes in NMDAr function in area CA1. It is important to note that these studies used different methods of sleep deprivation, which may explain this discrepancy. Kopp and colleagues [23] used exposure to a novel environment and ad libitum access to nesting material to keep mice awake, whereas Vecsey et al. [30] used the gentle handling method.
NMDA receptors are only required during LTP induction, but not during the maintenance of LTP [77] (reviewed in [78]). As described above, changes in NMDAr composition and function, affecting the induction of LTP, have been particularly prominent in studies applying long-term sleep deprivation [52, 68, 75]. To determine whether sleep deprivation impairs the maintenance of LTP in the dentate gyrus, Ishikawa et al. [72] deprived rats of REM sleep for 24–48 hours, in this case by stroking with a brush starting directly after LTP induction. They found that LTP in the dentate gyrus was still impaired, suggesting that the molecular processes underlying the maintenance of LTP are sensitive to sleep loss. In line with the latter in vivo study, several sleep deprivation studies using hippocampal slices have shown that 4–5 hours of sleep deprivation can impair the maintenance phase of LTP without affecting its induction [30, 71, 79]. In conclusion, although modulation of NMDAr function has been found to be modulated by sleep deprivation, additional cellular signalling mechanisms are likely to be altered by sleep loss, particularly in the case of brief periods of sleep deprivation, as described below.
5. Sleep deprivation, cholinergic, and GABAergic signalling
The cholinergic system plays a critical role in memory formation (reviewed in [80, 81] and is a major modulator of neuronal activity (reviewed in [82]). Ninety-six hours of REM sleep deprivation increases acetylcholinesterase (the enzyme that breaks down acetylcholine) in the pons, thalamus, and medulla oblongata, but not in other brain regions including the hippocampus [83]. It is important to note that the pons contains cholinergic cells involved in the generation of REM, while the thalamus and medulla oblongata receive cholinergic input from the pons. The higher levels of acetylcholinesterase suggest that there is a higher turnover of acetylcholine in these regions as a consequence of sleep deprivation. Receptor expression studies have indicated that REM sleep deprivation reduces muscarinic M2 cholinergic receptors in the pons and hippocampus [84].
There have been efforts to boost cholinergic activity with the aim to reverse the effect of sleep deprivation on memory formation. Activation of nicotinic acetylcholine receptors, in particular the α7 nACh receptor, through systemic application of nicotine, was found to rescue the memory deficits in the hippocampus-dependent radial arm maze task observed after 24 and 48 hours of REM sleep deprivation using the platform-over-water method [85, 86]. In addition to rescuing hippocampus-dependent memory consolidation, nicotine treatment during REM sleep deprivation reversed the impairment in hippocampal LTP in vivo in area CA1 and in the dentate gyrus [86]. The authors offered two hypotheses for how nicotine treatment might be reversing the effects of sleep deprivation on memory and plasticity: 1) nicotine treatment activates pre-synaptic nicotinic receptors, leading to elevated glutamate release from pre-synaptic terminals [87], thereby increasing the activity of excitatory neurons, and 2) nicotine treatment could facilitate activity of excitatory neurons through desensitization of α7 nACh receptors in GABAergic neurons reducing the release of GABA [88]. Finally, Aleisa et al. [89] showed that chronic nicotine treatment reversed stress-induced reductions in protein levels of the brain-derived neurotrophic factor (BDNF), a key protein in hippocampal synaptic plasticity [90]. Like stress, sleep loss can lead to reductions in hippocampal BDNF protein levels in the hippocampus [91] and more specifically in the dentate gyrus [92]. However, to our knowledge, no experiments have been conducted to assay whether nicotine treatment reverses the sleep deprivation-induced decrease in BDNF protein levels, or whether changes in BDNF content play a critical role in the rescue of sleep deprivation-mediated memory impairments. In conclusion, although the rescue of sleep deprivation-induced memory deficiencies by nicotine is exciting, much work remains to ascertain the molecular mechanisms by which nicotine treatment ameliorates sleep loss-induced cognitive deficits.
Fast synaptic inhibition in the adult brain is primarily mediated by γ-aminobutyric acid receptors (GABArs). Regulation of GABAAr surface expression at synapses - a process that is critical for maintaining the correct level of synaptic inhibition - is important for memory consolidation [93]. Wang and Li [94] reported higher GABA levels in cortex, hypothalamus, and brain stem after 72 hours of sleep deprivation in mice. This suggested that sleep deprivation might increase GABA tone, leading to increased GABAergic signalling and a suppression of activity of excitatory neurons. Modirrousta et al. [95] showed that expression of the GABAr β2–3 subunit is enhanced in cholinergic cells in the basal forebrain after sleep deprivation, suggesting that one way through which prolonged wake reduces cholinergic activity is through higher GABAergic activity. Increases in GABABr receptor protein levels in hippocampal lysates after 12 hours of sleep deprivation using the gentle handling method have also been reported by others [74]. It remains to be elucidated whether other neurotransmitter systems besides the cholinergic system are also inhibited after sleep deprivation through elevation of GABAergic activity.
6. Sleep deprivation and cAMP signalling
As mentioned previously, sleep deprivation can impair the maintenance of LTP without affecting LTP induction [30, 71, 72, 79]. These observations suggest that specific intracellular signalling pathways important for the maintenance of LTP are affected by brief sleep deprivation. To determine which pathways are impacted, the Abel laboratory conducted a set of electrophysiological experiments with differing molecular requirements [30]. The authors found that 5 hours of sleep deprivation impaired spaced four-train LTP, theta-burst LTP, and forskolin-induced potentiation [30]. These forms of LTP have the common feature that they are all dependent on the cAMP-PKA pathway, a pathway generally acknowledged to play a critical role in memory consolidation [96] (reviewed in [97]). Importantly, one-train LTP and massed 4-train LTP, both PKA-independent forms of plasticity, were not affected by 5 hours of sleep deprivation. Biochemical analysis of hippocampal CA1 mini slices indicated that both basal and forskolin-induced cAMP levels were reduced after 5 hours of sleep deprivation. Thus, in contrast to studies, using longer periods of sleep deprivation allowing exploration of novel environments and ad libitum access to nesting material to achieve sleep deprivation in which changes in NMDA receptor function were observed, Vecsey et al. [30] showed that brief 5 hour sleep deprivation using the gentle handling method caused specific disruptions of the cAMP-PKA pathway in the hippocampus, without affecting NMDA receptor function in area CA1 as determined using whole-cell recordings.
Other studies looking at downstream targets of the cAMP-PKA pathway have also suggested that this pathway is sensitive to sleep deprivation. Although longer periods of sleep deprivation were applied, both Ravassard et al. [67] and Hagewoud et al. [33] found that AMPA receptor GluR1 (or GluA1) serine 845 phosphorylation, a target substrate of the cAMP-PKA pathway that controls AMPA receptor function [98], was reduced after 75 hours of REM sleep deprivation using the platform-over-water method and 12 hours of total sleep deprivation respectively. However, Vyazovskiy et al. described opposite results [22]. They found that 4 hours of spontaneous or enforced wakefulness led to elevated levels of AMPA receptor GluR1 S845 phosphorylation in both hippocampus and cortex. These different observations may be due to the length of the sleep deprivation period.
A second PKA target that has been studied in relation to the effects of sleep deprivation on plasticity and memory is the cAMP response element binding protein (CREB), a transcription factor playing a crucial role in memory and plasticity (for review see [99]). Vecsey et al. [30] showed that brief sleep deprivation reduced hippocampal CREB phosphorylation of the serine 133 site in area CA1 and the dentate gyrus of the hippocampus. Other studies have also found decreased hippocampal CREB phosphorylation at serine 133, although longer periods of sleep deprivation were applied [70, 100]. A recent study by Hagewoud et al. [18] looked at the effect of daily sleep deprivation for 5 hours on learning-induced changes in hippocampal and striatal phosphorylation of CREB at serine 133. The authors found that performance in a Y-maze reference memory task was not affected by 5 hours of sleep deprivation after each daily training session. However, sleep-deprived mice avoided using a hippocampus-dependent strategy and preferred to use a striatum-dependent response strategy to locate the food reward. In line with the shift in behavioural strategy used to locate the reward, the training-induced increase in CREB serine 133 phosphorylation shifted from hippocampus to striatum indicating that daily brief sleep deprivation attenuated CREB function in the hippocampus and shifted it to the striatum. A question that remains to be answered is whether sleep deprivation induces a shift in the behavioural strategy used, thus leading to a change in CREB function, or alternatively, whether sleep deprivation directly limits CREB signalling leading to the change in behavioural strategy used.
It should be noted that these reductions in CREB serine 133 phosphorylation are not necessarily uniform for the entire brain. Cirelli and Tononi [54] reported that keeping animals awake for 1–9 hours through the introduction of novel objects in their recording cage elevated CREB phosphorylation in the cortex. Furthermore, Vecsey et al. [30] reported that CREB phosphorylation in the amygdala was not affected by 5 hours of sleep deprivation using the gentle handling method.
Both the cAMP-PKA and the extracellular signal-regulated kinase (ERK, also known as MAPK) pathway critically regulate changes in synaptic efficacy important for memory formation [101–103], and crosstalk between both pathways has been described [104] through the exchange protein activated by cAMP (Epac) and Ras [105]. Because sleep deprivation attenuates hippocampal cAMP levels [30], one would hypothesize that sleep deprivation would also indirectly affect the ERK pathway. Indeed, Ravassard et al. [67] showed that 75 hours of sleep deprivation using the platform-over-water method reduced ERK P44/P42 phosphorylation in area CA1. Likewise, in an earlier study, Guan et al. [44] found reductions in ERK P44/P42 phosphorylation after 6 hrs of total sleep deprivation.
Because the sole means to inactivate cAMP is through degradation by cAMP phosphodiesterases (PDEs) (reviewed in [106, 107]), Vecsey et al. [30] determined whether the impairments in hippocampal Schaffer collateral LTP observed after 5 hours of sleep deprivation could be reversed by bath application of the general PDE inhibitor IBMX or the PDE4-specific inhibitor rolipram. Indeed, blocking PDE4 activity rescued the LTP deficits induced by sleep deprivation. To determine whether PDE4-mediated degradation of cAMP induced by sleep deprivation was also the cause of contextual fear memory deficits, the authors repeated the contextual fear conditioning experiment with systemic administration of the PDE4 inhibitor rolipram during the 5 hours of sleep deprivation. In line with the LTP findings, rolipram treatment rescued the sleep deprivation-induced memory deficit for contextual fear.
The rescue of plasticity and behaviour in sleep-deprived animals with PDE inhibitors suggested that elevated degradation of cAMP might be responsible for the effects of SD on cAMP signalling. Indeed, biochemical analysis of hippocampal tissue indicated that 5 hours of sleep deprivation led to the upregulation of both PDE4 enzymatic activity and the protein levels of one specific PDE4 isoform, PDE4A5, rather than an upregulation of all of the 20 described PDE4 isoforms [30]. Each PDE4 isoform has a unique N-terminal region resulting in distinct intracellular localization and compartment-specific local degradation of cAMP (reviewed in [106, 107]). Therefore, the finding that sleep deprivation specifically increases protein levels of the PDE4A5 isoform may indicate that sleep deprivation reduces cAMP in specific PDE4A5-containing intracellular compartments rather than degrading cAMP in a global fashion. The next challenge will be to determine the molecular consequences of sleep deprivation-mediated changes in PDE4A5 activity in the hippocampus. A first step will be to determine the activity of proteins known to interact with PDE4A5, which will help identify which PDE4A5-containing signalling complexes and pathways are altered by sleep loss. These proteins include MK2 (also known as MAPK-activated protein kinase 2), which phosphorylates PDE4A5 thereby reducing its activity [108], caspase-3, which causes N-terminal cleavage of PDE4A5 [109], and the immunophilin XAP2, which inhibits PDE4A5 activity [110]. Another challenge will be to develop pharmacological agents that specifically target PDE4A5 (or its human ortholog PDE4A4 [106]) and determine whether the cognitive impairments induced by sleep loss can be reversed by targeting this particular PDE4 isoform without affecting the other PDE4 isoforms in both mice and humans. This approach of developing drugs that target specific PDE4 isoforms is currently underway with the hope of providing novel therapeutic strategies for the treatment of inflammatory diseases including asthma, chronic obstructive pulmonary disease, psoriasis, and depression [111].
Treatment with caffeine has also been reported to reverse the effects of sleep deprivation on hippocampal synaptic plasticity and memory [70, 92]. It is important to note that one of the many biological actions of caffeine is the inhibition of several PDE families (e.g. PDE1, PDE4 and PDE5) (reviewed in [112]). Thus, one potential mechanism through which caffeine treatment could reverse the effect of sleep deprivation is through inhibition of PDE4 activity, thereby facilitating cAMP-PKA signalling [30]. Another hint that caffeine may indeed act on the cAMP-PKA pathway came from recent work in flies [113], which found that caffeine reduced and fragmented sleep in flies, and that those effects were mitigated in flies with reduced PKA activity. Caffeine could also prevent the effects of sleep deprivation on memory and plasticity by antagonizing adenosine receptors (reviewed in [112]), which inhibit cAMP signalling through coupling to Gi proteins [114, 115]. This is a particularly interesting possibility, given the role of adenosine signalling in the impairments in plasticity and memory induced by sleep deprivation as discussed in detail below.
7. Sleep deprivation, adenosine, and astrocytes
Adenosine, a degradation product of ATP whose extracellular levels increase with brain metabolism, plays a critical role in sleep regulation through the modulation of slow wave activity [116–118]. In rats, extracellular adenosine levels have been reported to be higher during the circadian active period (the dark phase) than during the resting period (the light phase) in both the hippocampus and neostriatum [119]. Adenosine levels have also been reported to decline during sleep [120]. A direct link between increased adenosine levels and elevated sleep drive came from a study by Porkka-Heiskanen et al. [120], which showed that sleep deprivation or pharmacologically elevating adenosine levels, both led to similar changes in sleep/wake patterns: a reduction in wakefulness and an increase in slow wave sleep. With prolonged wakefulness, elevation of extracellular adenosine levels leads to activation of the adenosine A1 receptor, which in turn attenuates transmitter release of surrounding excitatory presynaptic terminals by increasing potassium currents [121]. Conditional deletion of the adenosine A1 receptor from forebrain neurons reduces the rebound in slow wave activity observed in wild-type animals [122]. The adenosine A1 receptor primarily couples to Gi proteins, which inhibit the production of cAMP [114, 115]. Therefore higher levels of adenosine, as a consequence of sleep loss, may lead to increased Gi signalling, thereby attenuating cAMP levels. In addition to affecting the cAMP pathway, changes in adenosine levels may also alter the activity of the cholinergic neurons in the basal forebrain, which appear to be central in the regulation of recovery sleep after sleep deprivation [120, 123, 124].
One intriguing topic that has only recently received attention is to identify which brain cells are responsible for the build up of adenosine after sleep deprivation. In addition to neurons, astrocytes have been shown to release transmitter molecules such as glutamate, ATP, and D-serine, thereby altering neuronal function [125–128]. These have been termed ‘gliotransmitters’, although their role in vivo remains a topic of debate [129]. Transmitter release from astrocytes is triggered by elevation of intracellular calcium levels, and is suggested to require the formation of a soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex between the target membrane and vesicles [130]. Pascual et al. [128] overexpressed the cytosolic portion of the SNARE domain of synaptobrevin 2 (DN-SNARE) selectively in astrocytes, thereby preventing release of gliotransmitters from astrocytes. Using the same mouse model, Halassa et al. [55] determined whether attenuating release of gliotransmitters prevented the effect of brief sleep deprivation on memory formation. They found that mice expressing the DN-SNARE protein did not have any rebound sleep after brief sleep deprivation and also showed no memory deficits as a consequence of sleep deprivation immediately following training in a hippocampus-independent learning task. These findings indicate that astrocytes are potentially implicated in the build-up of adenosine after prolonged wakefulness. The authors hypothesized that the crucial gliotransmitter mediating the effects of sleep deprivation on memory was ATP, which is quickly converted to adenosine after release [131] and acts through the adenosine A1 receptor. Chronic infusion of the adenosine A1 receptor antagonist (8-cyclopentyl-1,3-dimethylxanthine (CPT) into the brain prevented sleep deprivation-induced deficits in hippocampus-independent memory formation. Subsequent research has demonstrated that sleep deprivation-induced impairments in hippocampal LTP and memory are also rescued by DN-SNARE expression or by targeted CPT infusion into the hippocampus [79]. Therefore, astrocyte-derived adenosine induced by sleep deprivation may impact synaptic plasticity underlying memory formation in multiple brain areas. Because the DN-SNARE mutant mice exhibit reductions in sleep rebound and a resistance to the cognitive impact of sleep deprivation, these studies raise the intriguing question of what the role of recovery sleep is and how it might impact cognitive function.
Together, the studies using CPT and mice expressing the DN-SNARE protein have suggested that the build-up of extracellular adenosine through gliotransmitter release from astrocytes plays a key role in the plasticity and memory deficits observed after brief sleep deprivation, and suggest that the A1 receptor may be a potential therapeutic target to combat the cognitive deficits observed after sleep deprivation. An important question that remains to be answered is how overstimulation of the adenosine A1 receptor - as observed after sleep deprivation - leads to memory impairments. Based on the known signalling pathways downstream of the A1 receptor, Florian and colleagues [79] have hypothesized that sleep deprivation-induced elevations in adenosine levels may activate presynaptic A1 receptors, inhibiting adenylyl cyclase activity and resulting in reductions in cAMP levels and ERK pathway signalling. Although the studies by Florian et al. [79] and Halassa et al. [55] indicate that sleep loss may result in memory deficits through stimulation of the adenosine A1 receptor, potentially attenuating the production of cAMP, it does not explain how sleep loss leads to elevated hippocampal PDE4A5 protein levels and increased PDE4 activity as described by Vecsey et al. [30]. Thus, it may very well be that multiple signalling pathways in parallel attenuate the cAMP-PKA pathway, leading to plasticity and memory impairments as a consequence of sleep loss.
8. Sleep deprivation, gene transcription, and translation
In addition to looking at specific signalling pathways to identify the mechanisms underlying the memory deficits caused by sleep loss, many laboratories have used gene expression studies to identify the molecular targets of sleep deprivation. In particular, microarray studies allowing for the simultaneous analysis of thousands of transcripts have led to the identification of many genes whose expression change after sleep deprivation (for in depth review of the gene expression studies see [132–136]. Based on a computational analysis of multiple previously published gene expression studies assessing the effect short-term sleep deprivation on gene expression, Wang et al. [135] concluded that almost all genes affected by sleep deprivation are either directly or indirectly regulated by the cAMP-responsive element. In addition, they found that across studies, expression levels of several immediate early genes were elevated with sleep deprivation, including the Egr family (Egr1, Egr2, Egr3), Homer1a, Nr4a1, and Arc. Consistently down-regulated genes include cold-induced RNA binding proteins such as Rbm3 and Cirbp, which are involved in RNA/lipid metabolic processing.
Gene expression studies are well-suited to determine which gene families, gene clusters, and molecular pathways are affected by manipulations such as sleep deprivation. However, one caveat is that meta-analysis has shown that changes in gene expression after sleep deprivation are not uniform throughout the brain. For example, Arc mRNA expression was elevated in most brain subregions, but was reduced in the lateral hypothalamus (in the vicinity of hypocretin neurons and in the suprachiasmatic nucleus) [135]. Thus, gene expression alterations in one region of cortex may not be representative of the changes taking place in another area. This may be an interesting phenomenon to probe further, by comparing the effects of sleep deprivation across multiple brain regions.
Another important issue with gene expression studies is that a change in mRNA expression is not necessarily indicative of functional changes at the protein level. An example of differently regulated transcription and translation came from a study of genes in the Egr family by Cheval and colleagues [137], which determined the effect of LTP induction on changes in gene expression and protein levels for the Egr family. Although LTP induction led to an increase in Egr1, Egr2 and Egr3 mRNA expression, protein levels were enhanced only for Egr1 but not for Egr2 and Egr3. Unfortunately, none of the microarray studies looking at the effect of sleep deprivation on gene transcription also examined protein levels in parallel. Several studies have suggested that sleep promotes translation [138–140], and microarray analysis has shown that gene clusters important for protein synthesis are altered by sleep deprivation [133, 141]. Therefore, future studies are needed to determine whether sleep deprivation-induced changes in gene expression are paralleled by altered protein levels.
In summary, gene transcription studies have significantly advanced the insight into the effects of sleep deprivation on intracellular signalling pathways. However, it should be taken into account that changes at the RNA level do not automatically mean that protein levels and function are affected in parallel or that these changes are uniform throughout the brain. Thus, in order to fully understand how sleep deprivation affects both neuronal and glial function, future research should compare effects of sleep deprivation on gene expression in specific brain regions and determine whether these changes reflect functional alterations at the protein level. Such studies may be useful to determine why particular brain functions such as memory are disrupted by sleep loss.
9. Conclusions and future directions
One of the hallmarks of our modern society is to work longer each day and for more days each year, usually at the expense of sleep time, which can lead to cognitive impairments. Over the last few decades, significant advances have been made in unravelling the mechanisms underlying the memory and plasticity deficits observed after both brief and longer periods of sleep deprivation. The role of specific genes and signalling pathways responsible for sleep deprivation-induced deficits have been elucidated and efforts have been undertaken to reverse the effects of sleep deprivation on memory and plasticity by targeting these specific molecular substrates. Still, many important questions remain unanswered and methodological issues have not been addressed. As described in this review, depending on the method used for sleep deprivation, opposite effects of sleep deprivation on specific signalling pathways have been described. Thus, it remains to be determined which sleep deprivation method is best suited to study the effects of sleep deprivation on cognitive performance as observed in humans. One exciting approach, which may avoid many of the potential side effects of the currently used sleep deprivation methods, would be to use optogenetic strategies - the use of light-sensitive receptors to modulate the activity of genetically defined cells - to manipulate sleep. A recent set of studies showed that this may very well be feasible in the near future. Adamantidis et al. [142] developed an optogenetic approach to increase the occurrence of transitions from a sleep state to wakefulness in freely moving mice, and a follow-up study showed that an optogenetic approach could be successfully used to disrupt sleep, which led to impaired memory consolidation [143].
One interesting but yet unanswered question is why certain brain regions are more vulnerable to sleep deprivation than others. As described above, mice that were sleep deprived directly after training for 6 hours per day shifted from using a hippocampus-dependent learning strategy to a strategy that requires the striatum [18], suggesting that the hippocampus in comparison with the striatum is more sensitive to sleep loss. In addition, although most studies have reported that sleep deprivation hampers hippocampal function, some evidence suggests that sleep loss facilitates plasticity in the medial prefrontal cortex [71]. Gene expression studies showing varied effects of sleep deprivation also emphasize that sleep deprivation does not impact all brain regions identically. Future studies will have to determine the molecular mechanisms that underlie the different sensitivity to sleep loss.
Another challenge for the field of memory and sleep research is to determine whether the molecular signalling mechanisms underlying memory consolidation are different during the animals’ resting phase and active phase. Many studies have shown that 5 to 6 hours of sleep deprivation directly following training in the animals resting phase leads to long-term memory deficits. This sleep deprivation-sensitive period coincides with the time-window in which pharmacological intervention of specific molecular processes including PKA signalling and protein synthesis impairs memory consolidation (reviewed in [65]). In contrast to the resting phase, 6 hours of sleep deprivation during the animals’ active phase is not sufficient to induce consolidation deficits [21, 55]. The memory impairment was only seen when animals were sleep deprived for 12 hours during their active phase [21]. Because rodents sleep 60–80% of the time in the resting phase (the light period) in contrast to 20–35% in the active phase (the dark period) [144–146], the amount of sleep lost during 6 hours of sleep deprivation in the resting phase was equivalent to the amount of sleep lost during 12 hours of sleep deprivation during the active phase [21]. That is, both the timing and amount of sleep loss are crucial to determine whether memory is disrupted (for further discussion see [147]). This finding raises several important questions: Does the consolidation window in the active period (with relatively little sleep) extend beyond the 5–6 hours that has been reported when learning occurs during the resting phase (with much sleep)? In other words, does sleep accelerate the molecular processes underlying memory consolidation? Future studies will have to elucidate whether this is indeed the case.
In summary, our knowledge of the molecular mechanisms underlying the cognitive and plasticity deficits observed after sleep deprivation has greatly advanced in recent years. A handful of signalling pathways susceptible to sleep loss have been identified and several in vivo studies have demonstrated that modulation of the nicotine, adenosine, or cAMP signalling pathways can ameliorate sleep loss-induced cognitive impairments. It remains unclear whether these signalling pathways affected by sleep deprivation are interconnected, or if they are independent from each other. Further research on these issues may ultimately lead to novel and improved therapeutic and pharmacological approaches to reverse the deleterious effects of sleep loss.
Figure 1.
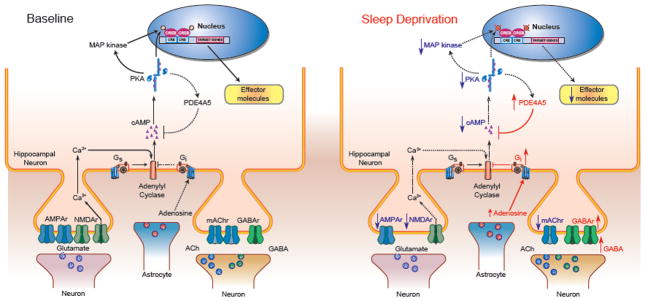
A schematic overview of hippocampal signalling pathways whose modulation by sleep deprivation may contribute to the effects of sleep deprivation on memory formation. (A) Signalling pathways under baseline conditions. (B) Sleep deprivation has been reported to reduce glutamatergic signalling and cholinergic signalling while increasing adenosine levels and GABAergic signalling. Sleep deprivation also attenuates cAMP signalling and CREB-mediated gene transcription. All of these molecular events are shown in a single connected pathway in order to demonstrate how the effects of sleep deprivation could potentially interact to impact learning and memory. Dashed black lines and black arrows pointing down indicate attenuation of the signalling pathway. Red lines and upward pointing arrows indicate an increase of the signalling pathway.
Highlights.
Hippocampal function is particularly sensitive to sleep loss
Sleep deprivation alters hippocampal glutamate, acetylcholine, and GABA systems
Sleep deprivation attenuates hippocampal cAMP signalling
Rescuing cAMP signalling prevents effects of sleep deprivation on the hippocampus
Astrocytes contribute to the effects of sleep deprivation on memory and plasticity
Acknowledgments
We thank Mathieu Wimmer, Dr. Jennifer H.K. Choi, and Dr. Sara J. Aton for input on a previous version of the manuscript and Paul Schiffmacher for help with the illustration. This research was supported by the Netherlands Organization for Scientific Research (NWO-Rubicon grant 825.07.029 to RH), NIA (5P01AG017628-09 to TA; Principal Investigator Allan Pack), NIMH (RO1 MH086415-01 to T.A.), and F32 post-doctoral NRSA, MH090711NRSA (to C.G.V).
Abbreviations
- AMPAr
2-amino-3-(5-methyl-3-oxo-1,2- oxazol-4-yl) propanoic acid receptors
- CREB
cAMP Response element binding protein
- ERK
extracellular signal-regulated kinase
- GABAr
gamma-aminobutyric acid receptor
- LTP
long-term potentiation
- mAChr
muscarinic cholinergic receptor
- nAChr
nicotinic acetylcholine receptor
- NMDAr
N-Methyl-D-aspartate receptor
- PDE
phosphodiesterase
- PDE4
cyclic AMP specific phosphodiesterase-4
- PKA
cyclic AMP-dependent protein kinase A
- REM sleep
rapid eye movement sleep
- Rolipram
4-[3-(cyclopentyloxy)-4-methoxyphenyl]-2-pyrrolidinone
- SNARE
N-ethylmaleimide-sensitive factor attachment protein receptor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hublin C, Kaprio J, Partinen M, Koskenvuo M. Sleep. 2001;24:392–400. doi: 10.1093/sleep/24.4.392. [DOI] [PubMed] [Google Scholar]
- 2.Bonnet MH, Arand DL. Sleep. 1995;18:908–911. doi: 10.1093/sleep/18.10.908. [DOI] [PubMed] [Google Scholar]
- 3.Rajaratnam SM, Arendt J. Lancet. 2001;358:999–1005. doi: 10.1016/S0140-6736(01)06108-6. [DOI] [PubMed] [Google Scholar]
- 4.National Health Inverview Survey. Quickstats: Percentage of Adults Who Reported an Average of <6 Hours of Sleep per 24-Hour Period bSaAG-US. MMWR Morb Mortal Wkly Rep. 2005;54:933. [Google Scholar]
- 5.Jenkins JG, Dallenbach KM. American Journal of Psychology. 1924;35:605–612. [Google Scholar]
- 6.Morris GO, Williams HL, Lubin A. Archives of General Psychiatry. 1960;2:247–254. [Google Scholar]
- 7.Walker MP. Ann N Y Acad Sci. 2009;1156:168–197. doi: 10.1111/j.1749-6632.2009.04416.x. [DOI] [PubMed] [Google Scholar]
- 8.Stickgold R, Walker MP. Sleep Med. 2007;8:331–343. doi: 10.1016/j.sleep.2007.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Whitney P, Hinson JM. Prog Brain Res. 2010;185:37–48. doi: 10.1016/B978-0-444-53702-7.00003-8. [DOI] [PubMed] [Google Scholar]
- 10.Axmacher N, Draguhn A, Elger CE, Fell J. Cell Mol Life Sci. 2009;66:2285–2297. doi: 10.1007/s00018-009-0019-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jouvet D, Vimont P, Delorme F. J Physiol (Paris) 1964;56:381. [PubMed] [Google Scholar]
- 12.Kim EY, Mahmoud GS, Grover LM. Neurosci Lett. 2005;388:163–167. doi: 10.1016/j.neulet.2005.06.057. [DOI] [PubMed] [Google Scholar]
- 13.Marks CA, Wayner MJ. Brain Res Bull. 2005;66:114–119. doi: 10.1016/j.brainresbull.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 14.Friedman L, Bergmann BM, Rechtschaffen A. Sleep. 1979;1:369–391. doi: 10.1093/sleep/1.4.369. [DOI] [PubMed] [Google Scholar]
- 15.Campbell IG, Guinan MJ, Horowitz JM. J Neurophysiol. 2002;88:1073–1076. doi: 10.1152/jn.2002.88.2.1073. [DOI] [PubMed] [Google Scholar]
- 16.Roman V, Walstra I, Luiten PG, Meerlo P. Sleep. 2005;28:1505–1510. [PubMed] [Google Scholar]
- 17.Ledoux L, Sastre JP, Buda C, Luppi PH, Jouvet M. Brain Res. 1996;735:108–118. doi: 10.1016/0006-8993(96)00599-9. [DOI] [PubMed] [Google Scholar]
- 18.Hagewoud R, Havekes R, Tiba PA, Novati A, Hogenelst K, Weinreder P, Van der Zee EA, Meerlo P. Sleep. 2010;33:1465–1473. doi: 10.1093/sleep/33.11.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van der Borght K, Ferrari F, Klauke K, Roman V, Havekes R, Sgoifo A, van der Zee EA, Meerlo P. Behav Brain Res. 2006;167:36–41. doi: 10.1016/j.bbr.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 20.Meerlo P, de Bruin EA, Strijkstra AM, Daan S. Physiol Behav. 2001;73:331–335. doi: 10.1016/s0031-9384(01)00451-6. [DOI] [PubMed] [Google Scholar]
- 21.Hagewoud R, Whitcomb SN, Heeringa AN, Havekes R, Koolhaas JM, Meerlo P. Sleep. 2010;33:1315–1322. doi: 10.1093/sleep/33.10.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vyazovskiy VV, Cirelli C, Pfister-Genskow M, Faraguna U, Tononi G. Nat Neurosci. 2008;11:200–208. doi: 10.1038/nn2035. [DOI] [PubMed] [Google Scholar]
- 23.Kopp C, Longordo F, Nicholson JR, Luthi A. J Neurosci. 2006;26:12456–12465. doi: 10.1523/JNEUROSCI.2702-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kemp A, Manahan-Vaughan D. Proc Natl Acad Sci U S A. 2004;101:8192–8197. doi: 10.1073/pnas.0402650101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu L, Anwyl R, Rowan MJ. Nature. 1997;387:497–500. doi: 10.1038/387497a0. [DOI] [PubMed] [Google Scholar]
- 26.Sarantis K, Antoniou K, Matsokis N, Angelatou F. Neurochem Int. 2011 doi: 10.1016/j.neuint.2011.10.018. [DOI] [PubMed] [Google Scholar]
- 27.Isokawa M, Shimokochi M. Physiol Behav. 1983;30:689–695. doi: 10.1016/0031-9384(83)90164-6. [DOI] [PubMed] [Google Scholar]
- 28.Tiba PA, Oliveira MG, Rossi VC, Tufik S, Suchecki D. Sleep. 2008;31:505–515. doi: 10.1093/sleep/31.4.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruskin DN, Dunn KE, Billiot I, Bazan NG, LaHoste GJ. Life Sci. 2006;78:2833–2838. doi: 10.1016/j.lfs.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 30.Vecsey CG, Baillie GS, Jaganath D, Havekes R, Daniels A, Wimmer M, Huang T, Brown KM, Li XY, Descalzi G, Kim SS, Chen T, Shang YZ, Zhuo M, Houslay MD, Abel T. Nature. 2009;461:1122–1125. doi: 10.1038/nature08488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palchykova S, Winsky-Sommerer R, Meerlo P, Durr R, Tobler I. Neurobiol Learn Mem. 2006;85:263–271. doi: 10.1016/j.nlm.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 32.Meerlo P, Turek FW. Brain Res. 2001;907:84–92. doi: 10.1016/s0006-8993(01)02603-8. [DOI] [PubMed] [Google Scholar]
- 33.Hagewoud R, Havekes R, Novati A, Keijser JN, EA VDZ, Meerlo P. J Sleep Res. 2009 doi: 10.1111/j.1365-2869.2009.00799.x. [DOI] [PubMed] [Google Scholar]
- 34.Roozendaal B. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:1213–1223. doi: 10.1016/j.pnpbp.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 35.Fishbein W. communications in Behavioral Biology (A) 1970;5:171–175. [Google Scholar]
- 36.Sagales T, Domino EF. Psychopharmacologia. 1973;29:307–315. doi: 10.1007/BF00429278. [DOI] [PubMed] [Google Scholar]
- 37.Fishbein W. Physiol Behav. 1971;6:279–282. doi: 10.1016/0031-9384(71)90155-7. [DOI] [PubMed] [Google Scholar]
- 38.Wolfowitz BE, Holdstock TL. Communications in Behavioral Biology. 1971;6:281–284. [Google Scholar]
- 39.Linden ER, Bern D, Fishbein W. Physiol Behav. 1975;14:409–412. doi: 10.1016/0031-9384(75)90004-9. [DOI] [PubMed] [Google Scholar]
- 40.Fishbein W, Gutwein BM. Behav Biol. 1977;19:425–464. doi: 10.1016/s0091-6773(77)91903-4. [DOI] [PubMed] [Google Scholar]
- 41.Morris RG, Garrud P, Rawlins JN, O’Keefe J. Nature. 1982;297:681–683. doi: 10.1038/297681a0. [DOI] [PubMed] [Google Scholar]
- 42.Smith C, Rose GM. Physiol Behav. 1996;59:93–97. doi: 10.1016/0031-9384(95)02054-3. [DOI] [PubMed] [Google Scholar]
- 43.Youngblood BD, Zhou J, Smagin GN, Ryan DH, Harris RB. Physiol Behav. 1997;61:249–256. doi: 10.1016/s0031-9384(96)00363-0. [DOI] [PubMed] [Google Scholar]
- 44.Guan Z, Peng X, Fang J. Brain Res. 2004;1018:38–47. doi: 10.1016/j.brainres.2004.05.032. [DOI] [PubMed] [Google Scholar]
- 45.Yang RH, Hu SJ, Wang Y, Zhang WB, Luo WJ, Chen JY. Brain Res. 2008;1230:224–232. doi: 10.1016/j.brainres.2008.07.033. [DOI] [PubMed] [Google Scholar]
- 46.Li S, Tian Y, Ding Y, Jin X, Yan C, Shen X. Learn Behav. 2009;37:246–253. doi: 10.3758/LB.37.3.246. [DOI] [PubMed] [Google Scholar]
- 47.Wang GP, Huang LQ, Wu HJ, Zhang L, You ZD, Zhao ZX. Neuroreport. 2009;20:1172–1176. doi: 10.1097/WNR.0b013e32832f0772. [DOI] [PubMed] [Google Scholar]
- 48.Walsh CM, Booth V, Poe GR. Learn Mem. 2011;18:422–434. doi: 10.1101/lm.2099011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.LeDoux JE. Annu Rev Neurosci. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- 50.Maren S. Annu Rev Neurosci. 2001;24:897–931. doi: 10.1146/annurev.neuro.24.1.897. [DOI] [PubMed] [Google Scholar]
- 51.Graves LA, Heller EA, Pack AI, Abel T. Learn Mem. 2003;10:168–176. doi: 10.1101/lm.48803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McDermott CM, LaHoste GJ, Chen C, Musto A, Bazan NG, Magee JC. J Neurosci. 2003;23:9687–9695. doi: 10.1523/JNEUROSCI.23-29-09687.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ruskin DN, Liu C, Dunn KE, Bazan NG, LaHoste GJ. Eur J Neurosci. 2004;19:3121–3124. doi: 10.1111/j.0953-816X.2004.03426.x. [DOI] [PubMed] [Google Scholar]
- 54.Cirelli C, Tononi G. J Neurosci. 2000;20:9187–9194. doi: 10.1523/JNEUROSCI.20-24-09187.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Halassa MM, Florian C, Fellin T, Munoz JR, Lee SY, Abel T, Haydon PG, Frank MG. Neuron. 2009;61:213–219. doi: 10.1016/j.neuron.2008.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oliveira AM, Hawk JD, Abel T, Havekes R. Learn Mem. 2010;17:155–160. doi: 10.1101/lm.1625310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Winters BD, Forwood SE, Cowell RA, Saksida LM, Bussey TJ. J Neurosci. 2004;24:5901–5908. doi: 10.1523/JNEUROSCI.1346-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Roozendaal B, Hernandez A, Cabrera SM, Hagewoud R, Malvaez M, Stefanko DP, Haettig J, Wood MA. J Neurosci. 2010;30:5037–5046. doi: 10.1523/JNEUROSCI.5717-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Goel N, Rao H, Durmer JS, Dinges DF. Semin Neurol. 2009;29:320–339. doi: 10.1055/s-0029-1237117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smith C, Butler S. Physiol Behav. 1982;29:469–473. doi: 10.1016/0031-9384(82)90268-2. [DOI] [PubMed] [Google Scholar]
- 61.Pearlman C. Physiol Behav. 1973;11:233–237. doi: 10.1016/0031-9384(73)90355-7. [DOI] [PubMed] [Google Scholar]
- 62.Smith C, Rose GM. Behav Neurosci. 1997;111:1197–1204. doi: 10.1037//0735-7044.111.6.1197. [DOI] [PubMed] [Google Scholar]
- 63.Smith CT, Conway JM, Rose GM. Neurobiol Learn Mem. 1998;69:211–217. doi: 10.1006/nlme.1997.3809. [DOI] [PubMed] [Google Scholar]
- 64.Bjorness TE, Riley BT, Tysor MK, Poe GR. Learn Mem. 2005;12:352–359. doi: 10.1101/lm.84805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Graves L, Pack A, Abel T. Trends Neurosci. 2001;24:237–243. doi: 10.1016/s0166-2236(00)01744-6. [DOI] [PubMed] [Google Scholar]
- 66.Tartar JL, Ward CP, McKenna JT, Thakkar M, Arrigoni E, McCarley RW, Brown RE, Strecker RE. Eur J Neurosci. 2006;23:2739–2748. doi: 10.1111/j.1460-9568.2006.04808.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ravassard P, Pachoud B, Comte JC, Mejia-Perez C, Scote-Blachon C, Gay N, Claustrat B, Touret M, Luppi PH, Salin PA. Sleep. 2009;32:227–240. doi: 10.1093/sleep/32.2.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McDermott CM, Hardy MN, Bazan NG, Magee JC. J Physiol. 2006;570:553–565. doi: 10.1113/jphysiol.2005.093781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Davis CJ, Harding JW, Wright JW. Brain Res. 2003;973:293–297. doi: 10.1016/s0006-8993(03)02508-3. [DOI] [PubMed] [Google Scholar]
- 70.Alhaider IA, Aleisa AM, Tran TT, Alkadhi KA. Mol Cell Neurosci. 2011;46:742–751. doi: 10.1016/j.mcn.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 71.Romcy-Pereira R, Pavlides C. Eur J Neurosci. 2004;20:3453–3462. doi: 10.1111/j.1460-9568.2004.03808.x. [DOI] [PubMed] [Google Scholar]
- 72.Ishikawa A, Kanayama Y, Matsumura H, Tsuchimochi H, Ishida Y, Nakamura S. Eur J Neurosci. 2006;24:243–248. doi: 10.1111/j.1460-9568.2006.04874.x. [DOI] [PubMed] [Google Scholar]
- 73.Tadavarty R, Kaan TK, Sastry BR. Exp Neurol. 2009;216:239–242. doi: 10.1016/j.expneurol.2008.11.012. [DOI] [PubMed] [Google Scholar]
- 74.Tadavarty R, Rajput PS, Wong JM, Kumar U, Sastry BR. PLoS One. 2011;6:e24933. doi: 10.1371/journal.pone.0024933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen C, Hardy M, Zhang J, LaHoste GJ, Bazan NG. Biochem Biophys Res Commun. 2006;340:435–440. doi: 10.1016/j.bbrc.2005.12.021. [DOI] [PubMed] [Google Scholar]
- 76.Meerlo P, Sgoifo A, Suchecki D. Sleep Med Rev. 2008;12:197–210. doi: 10.1016/j.smrv.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 77.Collingridge GL, Kehl SJ, McLennan H. J Physiol. 1983;334:33–46. doi: 10.1113/jphysiol.1983.sp014478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bennett MR. Prog Neurobiol. 2000;60:109–137. doi: 10.1016/s0301-0082(99)00006-4. [DOI] [PubMed] [Google Scholar]
- 79.Florian C, Vecsey CG, Halassa MM, Haydon PG, Abel T. J Neurosci. 2011;31:6956–6962. doi: 10.1523/JNEUROSCI.5761-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Havekes R, Abel T, Van der Zee EA. Behav Brain Res. 2011;221:412–423. doi: 10.1016/j.bbr.2010.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Deiana S, Platt B, Riedel G. Behav Brain Res. 2011;221:389–411. doi: 10.1016/j.bbr.2010.11.036. [DOI] [PubMed] [Google Scholar]
- 82.Boonstra TW, Stins JF, Daffertshofer A, Beek PJ. Cell Mol Life Sci. 2007;64:934–946. doi: 10.1007/s00018-007-6457-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Benedito MA, Camarini R. Braz J Med Biol Res. 2001;34:103–109. doi: 10.1590/s0100-879x2001000100012. [DOI] [PubMed] [Google Scholar]
- 84.Salin-Pascual RJ, Diaz-Munoz M, Rivera-Valerdi L, Ortiz-Lopez L, Blanco-Centurion C. Sleep Res Online. 1998;1:19–23. [PubMed] [Google Scholar]
- 85.Aleisa AM, Alzoubi KH, Alkadhi KA. Neurosci Lett. 2011;499:28–31. doi: 10.1016/j.neulet.2011.05.025. [DOI] [PubMed] [Google Scholar]
- 86.Aleisa AM, Helal G, Alhaider IA, Alzoubi KH, Srivareerat M, Tran TT, Al-Rejaie SS, Alkadhi KA. Hippocampus. 2010;21:899–909. doi: 10.1002/hipo.20806. [DOI] [PubMed] [Google Scholar]
- 87.Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA. Nature. 1996;383:713–716. doi: 10.1038/383713a0. [DOI] [PubMed] [Google Scholar]
- 88.Alkondon M, Braga MF, Pereira EF, Maelicke A, Albuquerque EX. Eur J Pharmacol. 2000;393:59–67. doi: 10.1016/s0014-2999(00)00006-6. [DOI] [PubMed] [Google Scholar]
- 89.Aleisa AM, Alzoubi KH, Gerges NZ, Alkadhi KA. Neurobiol Dis. 2006;22:453–462. doi: 10.1016/j.nbd.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 90.Lu Y, Christian K, Lu B. Neurobiol Learn Mem. 2008;89:312–323. doi: 10.1016/j.nlm.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Guzman-Marin R, Ying Z, Suntsova N, Methippara M, Bashir T, Szymusiak R, Gomez-Pinilla F, McGinty D. J Physiol. 2006;575:807–819. doi: 10.1113/jphysiol.2006.115287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Alhaider IA, Aleisa AM, Tran TT, Alkadhi KA. Eur J Neurosci. 2010;31:1368–1376. doi: 10.1111/j.1460-9568.2010.07175.x. [DOI] [PubMed] [Google Scholar]
- 93.Tretter V, Revilla-Sanchez R, Houston C, Terunuma M, Havekes R, Florian C, Jurd R, Vithlani M, Michels G, Couve A, Sieghart W, Brandon N, Abel T, Smart TG, Moss SJ. Proc Natl Acad Sci U S A. 2009;106:20039–20044. doi: 10.1073/pnas.0908840106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang SX, Li QS. Di Yi Jun Yi Da Xue Xue Bao. 2002;22:888–890. [PubMed] [Google Scholar]
- 95.Modirrousta M, Mainville L, Jones BE. BMC Neurosci. 2007;8:15. doi: 10.1186/1471-2202-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Abel T, Nguyen PV, Barad M, Deuel TA, Kandel ER, Bourtchouladze R. Cell. 1997;88:615–626. doi: 10.1016/s0092-8674(00)81904-2. [DOI] [PubMed] [Google Scholar]
- 97.Havekes R, Abel T. Adv Genet. 2009;65:1–38. doi: 10.1016/S0065-2660(09)65001-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Roche KW, O’Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Neuron. 1996;16:1179–1188. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- 99.Abel T, Kandel E. Brain Res Brain Res Rev. 1998;26:360–378. doi: 10.1016/s0165-0173(97)00050-7. [DOI] [PubMed] [Google Scholar]
- 100.Zhao Z, Huang L, Wu H, Li Y, Zhang L, Yin Y, Xiang Z. Neuroreport. 2010;21:623–628. doi: 10.1097/WNR.0b013e328339b5f9. [DOI] [PubMed] [Google Scholar]
- 101.Impey S, Obrietan K, Storm DR. Neuron. 1999;23:11–14. doi: 10.1016/s0896-6273(00)80747-3. [DOI] [PubMed] [Google Scholar]
- 102.Mazzucchelli C, Brambilla R. Cell Mol Life Sci. 2000;57:604–611. doi: 10.1007/PL00000722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sweatt JD. Curr Opin Neurobiol. 2004;14:311–317. doi: 10.1016/j.conb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 104.Sindreu CB, Scheiner ZS, Storm DR. Neuron. 2007;53:79–89. doi: 10.1016/j.neuron.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Roth TL, Sweatt JD. Nat Neurosci. 2008;11:993–994. doi: 10.1038/nn0908-993. [DOI] [PubMed] [Google Scholar]
- 106.Houslay MD. Trends Biochem Sci. 2009;35:91–100. doi: 10.1016/j.tibs.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 107.Houslay MD, Adams DR. Biochem J. 2003;370:1–18. doi: 10.1042/BJ20021698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.MacKenzie KF, Wallace DA, Hill EV, Anthony DF, Henderson DJ, Houslay DM, Arthur JS, Baillie GS, Houslay MD. Biochem J. 2011;435:755–769. doi: 10.1042/BJ20101184. [DOI] [PubMed] [Google Scholar]
- 109.Huston E, Beard M, McCallum F, Pyne NJ, Vandenabeele P, Scotland G, Houslay MD. J Biol Chem. 2000;275:28063–28074. doi: 10.1074/jbc.M906144199. [DOI] [PubMed] [Google Scholar]
- 110.Bolger GB, Peden AH, Steele MR, MacKenzie C, McEwan DG, Wallace DA, Huston E, Baillie GS, Houslay MD. J Biol Chem. 2003;278:33351–33363. doi: 10.1074/jbc.M303269200. [DOI] [PubMed] [Google Scholar]
- 111.Houslay MD, Schafer P, Zhang KY. Drug Discov Today. 2005;10:1503–1519. doi: 10.1016/S1359-6446(05)03622-6. [DOI] [PubMed] [Google Scholar]
- 112.Ribeiro JA, Sebastiao AM. J Alzheimers Dis. 2010;20(Suppl 1):S3–15. doi: 10.3233/JAD-2010-1379. [DOI] [PubMed] [Google Scholar]
- 113.Wu MN, Ho K, Crocker A, Yue Z, Koh K, Sehgal A. J Neurosci. 2009;29:11029–11037. doi: 10.1523/JNEUROSCI.1653-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Libert F, Van Sande J, Lefort A, Czernilofsky A, Dumont JE, Vassart G, Ensinger HA, Mendla KD. Biochem Biophys Res Commun. 1992;187:919–926. doi: 10.1016/0006-291x(92)91285-x. [DOI] [PubMed] [Google Scholar]
- 115.Olah ME, Stiles GL. Annu Rev Pharmacol Toxicol. 1995;35:581–606. doi: 10.1146/annurev.pa.35.040195.003053. [DOI] [PubMed] [Google Scholar]
- 116.Radulovacki M. Rev Clin Basic Pharm. 1985;5:327–339. [PubMed] [Google Scholar]
- 117.Benington JH, Heller HC. Prog Neurobiol. 1995;45:347–360. doi: 10.1016/0301-0082(94)00057-o. [DOI] [PubMed] [Google Scholar]
- 118.Scharf MT, Naidoo N, Zimmerman JE, Pack AI. Prog Neurobiol. 2008;86:264–280. doi: 10.1016/j.pneurobio.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Huston JP, Haas HL, Boix F, Pfister M, Decking U, Schrader J, Schwarting RK. Neuroscience. 1996;73:99–107. doi: 10.1016/0306-4522(96)00021-8. [DOI] [PubMed] [Google Scholar]
- 120.Porkka-Heiskanen T, Strecker RE, Thakkar M, Bjorkum AA, Greene RW, McCarley RW. Science. 1997;276:1265–1268. doi: 10.1126/science.276.5316.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Newman EA. J Neurosci. 2003;23:1659–1666. doi: 10.1523/JNEUROSCI.23-05-01659.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bjorness TE, Kelly CL, Gao T, Poffenberger V, Greene RW. J Neurosci. 2009;29:1267–1276. doi: 10.1523/JNEUROSCI.2942-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rainnie DG, Grunze HC, McCarley RW, Greene RW. Science. 1994;263:689–692. doi: 10.1126/science.8303279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Porkka-Heiskanen T, Kalinchuk AV. Sleep Med Rev. 2011;15:123–135. doi: 10.1016/j.smrv.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 125.Parpura V, Zorec R. Brain Res Rev. 2010;63:83–92. doi: 10.1016/j.brainresrev.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhang Q, Haydon PG. J Neural Transm. 2005;112:121–125. doi: 10.1007/s00702-004-0119-x. [DOI] [PubMed] [Google Scholar]
- 127.Jourdain P, Bergersen LH, Bhaukaurally K, Bezzi P, Santello M, Domercq M, Matute C, Tonello F, Gundersen V, Volterra A. Nat Neurosci. 2007;10:331–339. doi: 10.1038/nn1849. [DOI] [PubMed] [Google Scholar]
- 128.Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, Takano H, Moss SJ, McCarthy K, Haydon PG. Science. 2005;310:113–116. doi: 10.1126/science.1116916. [DOI] [PubMed] [Google Scholar]
- 129.Hamilton NB, Attwell D. Nat Rev Neurosci. 2010;11:227–238. doi: 10.1038/nrn2803. [DOI] [PubMed] [Google Scholar]
- 130.Scales SJ, Chen YA, Yoo BY, Patel SM, Doung YC, Scheller RH. Neuron. 2000;26:457–464. doi: 10.1016/s0896-6273(00)81177-0. [DOI] [PubMed] [Google Scholar]
- 131.Dunwiddie TV, Diao L, Proctor WR. J Neurosci. 1997;17:7673–7682. doi: 10.1523/JNEUROSCI.17-20-07673.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mackiewicz M, Zimmerman JE, Shockley KR, Churchill GA, Pack AI. Trends Mol Med. 2009;15:79–87. doi: 10.1016/j.molmed.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cirelli C. J Appl Physiol. 2002;92:394–400. doi: 10.1152/jappl.2002.92.1.394. [DOI] [PubMed] [Google Scholar]
- 134.Cirelli C, Faraguna U, Tononi G. J Neurochem. 2006;98:1632–1645. doi: 10.1111/j.1471-4159.2006.04058.x. [DOI] [PubMed] [Google Scholar]
- 135.Wang H, Liu Y, Briesemann M, Yan J. Physiol Genomics. 2010;42:427–436. doi: 10.1152/physiolgenomics.00205.2009. [DOI] [PubMed] [Google Scholar]
- 136.Tononi G, Cirelli C. Neuropsychopharmacology. 2001;25:S28–35. doi: 10.1016/S0893-133X(01)00322-0. [DOI] [PubMed] [Google Scholar]
- 137.Cheval H, Chagneau C, Levasseur G, Veyrac A, Faucon-Biguet N, Laroche S, Davis S. Hippocampus. 2011 doi: 10.1002/hipo.20926. [DOI] [PubMed] [Google Scholar]
- 138.Naidoo N, Giang W, Galante RJ, Pack AI. J Neurochem. 2005;92:1150–1157. doi: 10.1111/j.1471-4159.2004.02952.x. [DOI] [PubMed] [Google Scholar]
- 139.Nakanishi H, Sun Y, Nakamura RK, Mori K, Ito M, Suda S, Namba H, Storch FI, Dang TP, Mendelson W, Mishkin M, Kennedy C, Gillin JC, Smith CB, Sokoloff L. Eur J Neurosci. 1997;9:271–279. doi: 10.1111/j.1460-9568.1997.tb01397.x. [DOI] [PubMed] [Google Scholar]
- 140.Ramm P, Smith CT. Physiol Behav. 1990;48:749–753. doi: 10.1016/0031-9384(90)90220-x. [DOI] [PubMed] [Google Scholar]
- 141.Cirelli C, Gutierrez CM, Tononi G. Neuron. 2004;41:35–43. doi: 10.1016/s0896-6273(03)00814-6. [DOI] [PubMed] [Google Scholar]
- 142.Adamantidis AR, Zhang F, Aravanis AM, Deisseroth K, de Lecea L. Nature. 2007;450:420–424. doi: 10.1038/nature06310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Rolls A, Colas D, Adamantidis A, Carter M, Lanre-Amos T, Heller HC, de Lecea L. Proc Natl Acad Sci U S A. 2011;108:13305–13310. doi: 10.1073/pnas.1015633108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lancel M, Kerkhof GA. Physiol Behav. 1989;45:289–297. doi: 10.1016/0031-9384(89)90130-3. [DOI] [PubMed] [Google Scholar]
- 145.Franken P, Dijk DJ, Tobler I, Borbely AA. Am J Physiol. 1991;261:R198–208. doi: 10.1152/ajpregu.1991.261.1.R198. [DOI] [PubMed] [Google Scholar]
- 146.Tang X, Yang L, Sanford LD. Behav Brain Res. 2007;180:62–68. doi: 10.1016/j.bbr.2007.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Poe GR, Walsh CM, Bjorness TE. Sleep. 2010;33:1277–1278. doi: 10.1093/sleep/33.10.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]