

Abstract
Elastin-like polypeptides (ELPs) are a class of stimulus responsive biopolymers whose physicochemical properties and biocompatibility are particularly suitable for in vivo applications, such as drug delivery and tissue engineering. The lower critical solution temperature (LCST) behavior of ELPs allows them to be utilized as soluble macromolecules below their LCST, or as self-assembled nano-scale particles such as micelles, micron-scale coacervates, or viscous gels above their LCST, depending on the ELP architecture. As each ELP sequence is specified at its genetic level, functionalization of an ELP with peptides and proteins is simple to accomplish by the fusion of a gene encoding an ELP with that of the peptide or protein of interest. Protein ELP fusions, where the appended protein serves a therapeutic or targeting function, are suitable for applications in which the ELP can improve the systemic pharmacokinetics and biodistribution of the protein, or can be used as an injectable depot for sustained, local protein delivery. Here we describe considerations in the design of therapeutic protein ELP fusions and provide details of their gene design, expression, and purification.
1. Introduction
Elastin-like polypeptides (ELPs) are repetitive polypeptides inspired by the hydrophobic domain of elastin (Tatham and Shewry, 2000). The repeat unit in ELPs is a pentapeptide of (Val-Pro-Gly-X-Gly), where X is a ‘guest residue’ that can be any amino acid other than proline. These biopolymers exhibit lower critical solution temperature (LCST) behavior such that below a critical transition temperature (Tt) the ELP is a soluble unimer in aqueous solution, whereas above its Tt, the ELP undergoes a phase transition and aggregates into an insoluble coacervate (Urry, 1997). This phase transition is reversible, and the coacervated ELP can be redissolved when the temperature is lowered below the Tt. This stimulus response of ELPs can be carefully tuned to a desired temperature by manipulation of the ELP sequence by changing the guest residue or by manipulation of the ELP chain length by changing the number of pentapeptide repeats (Meyer and Chilkoti, 2004; Urry, 1997; Urry et al., 1991). The effect of guest residue composition is related to their hydropathy: hydrophobic guest residues lower the Tt while polar or charged residues raise it. Dependence of Tt on the ELP chain length follows an inverse relationship, such that longer ELP chains exhibit a lower Tt. The Tt of an ELP is also concentration dependent and generally shows an inverse-log dependence causing ELP in high concentration to transition at a lower Tt (Meyer and Chilkoti, 2004).
ELPs retain their thermal responsiveness when genetically fused to soluble proteins or peptides (Meyer and Chilkoti, 1999), but the Tt of the parent ELP is affected in what is described as the ‘fusion ΔTt effect’, defined as the difference between the Tt of the protein ELP fusion and the Tt of the free ELP. The fraction of hydrophobic surface area of the appended protein contributes to a negative ΔTt (Trabbic-Carlson et al., 2004b), while the fraction of charged surface area of the appended protein contributes to a positive ΔTt (unpublished). For proteins with a known atomic structure, these correlations allow in silico prediction of the ΔTt for a particular protein, and hence enable the rational choice of an ELP sequence and chain length to achieve a desired Tt for a specific protein ELP fusion.
The observation that ELPs retain their LCST behavior when fused to folded proteins has motivated their use as a purification tag. We have shown that protein ELP fusions can be expressed in E. coli and purified by utilizing their LCST behavior by a process termed inverse transition cycling (ITC) (Meyer and Chilkoti, 1999). After lysing cells that express a protein ELP fusion, the insoluble components of the cell lysate are precipitated by centrifugation. The supernatant, containing the protein ELP fusion is retained, and the phase transition of the protein ELP fusion is isothermally triggered by the addition of salt to lower the Tt below room temperature. The phase transition of the fusion protein results in the coacervation of micron-sized aggregates. After triggering the coacervation of the protein ELP fusion, the solution is centrifuged to form a pellet that contains the phase separated protein ELP fusions, thereby isolating them from the soluble contaminants in the supernatant; this step is termed a “hot spin”. The pellet is then resuspended in cold buffer (T<Tt) and the solution is centrifuged again to remove residual insoluble contaminants; this step is termed a “cold spin”. Together a hot and cold spin constitutes one round of ITC. With each round of ITC, the protein ELP fusion becomes purer as the coacervation step traps less contaminants during centrifugation.
The fact that ELP can be genetically appended to its protein or peptide partner with precise control over the gene design of ELP fusion (N-or C-terminal fusion), and that the thermal properties of the ELP tag can be easily tuned make ELP fusions attractive for recombinant protein purification (Ge et al., 2005; Meyer and Chilkoti, 1999; Trabbic-Carlson et al., 2004a), and for the design of biomacromolecular actuators (Kim and Chilkoti, 2008). Furthermore, their biodegradability, biocompatibility, and lack of toxicity make them attractive for in vivo applications, such as tissue engineering scaffolds (Nettles et al., 2010), and as vehicles for drug delivery (MacEwan and Chilkoti, 2010; MacKay et al., 2009; McDaniel et al., 2010a).
As carriers of an appended therapeutic protein, protein ELP fusions are capable of improving protein pharmacokinetics and biodistribution, thereby enhancing their accumulation at the disease site, and improving the protein's therapeutic efficacy. In the simplest form, a soluble ELP that is fused to a protein can be used as a soluble macromolecular carrier to improve the plasma half-life and prolong the circulation of systemically delivered proteins (Figure 1A). In a more sophisticated design, nano-scale assembly of ELP block copolymers into spherical micelles provides an additional means of improving pharmacokinetics and passively targeting their uptake to sites of disease such as tumors where the leaky vasculature permits accumulation of nanoparticles capable of traversing the vascular pores (MacKay et al., 2009). ELP micelles provide an additional advantage in that their coronas can be easily appended with proteins or peptides for therapeutic or targeting functions (Simnick et al., 2010) (Figure 1B). For local delivery of proteins fused to an ELP, the thermal responsiveness of protein ELP fusions can be tuned to achieve aggregation at physiological temperatures. For instance, the ELP sequence can be designed such that it is soluble at room temperature and undergoes aggregation at 37 °C to form a viscous coacervate (Figure 1C). This allows ease of injection at a desired site as a soluble material, while prolonging residence of the protein ELP fusion at the target by forming an insoluble depot (Liu et al., 2010).
Figure 1. Application of therapeutic protein ELP fusions.

The physiochemical properties and biocompatibility of ELPs make them a desirable material for physiological applications. With their fusion to pharmacological proteins ELPs can serve as therapeutic carriers in a variety of approaches to overcome challenges such as low solubility, fast clearance, and poor delivery that is typical to the application of protein drugs. The thermal properties of ELPs provide several means of improving the efficacy of its therapeutic protein cargo. Protein ELP fusions as soluble unimers provide improved solubility and prolonged circulation when systemically administered (A). ELP block copolymers, capable of forming spherical micelles, can enhance the avidity of interaction between proteins and their desired targets by the increased density of targeting peptides or proteins on the micelle corona (B). Alternatively, proteins can be sequestered and protected in a micelle core. By tuning of the ELP transition temperature, protein ELP fusions can be designed to aggregate at physiologic temperatures upon injection, and thereby provide an in situ depot to prolong the residence time of the therapeutic protein ELP fusion at the desired site of action (C) to act as a depot for loco-regional therapy or as a depot for the sustained release of the ELP fusion into systemic circulation.
2. Elastin-like polypeptide fusion protein design
A number of factors must be considered in the design of protein ELP fusions. Each protein and its fused ELP must be carefully tailored to the intended application and to the function of the therapeutic protein to ensure biological activity as a protein ELP fusion.
2.1 Therapeutic application considerations
The versatility of ELPs as stimulus responsive biomaterials has led to their use in a variety of physiological applications. The intended application of the protein ELP fusion dictates the ELP design. Considerations such as molecular weight, transition temperature, and self-assembly must be considered in choosing the number of pentapeptide repeats, the guest residue composition, and biopolymer architecture. Use of an ELP solely as a soluble macromolecular carrier provides advantages of improved pharmacokinetics and increased plasma half-life of the therapeutic fusion. For these ELPs, which are typically single segment polymers, the Tt should be designed to be sufficiently above body temperature to prevent aggregation at physiological conditions. Single segment ELP can also be used to provide a gel-like depot for prolonged local release of a therapeutic ELP fusion. Depot-forming ELPs require careful tuning of the Tt above the temperature at which they can be injected in their soluble form (for instance, at room temperature), and below the body temperature at which they should aggregate. ELPs can also be assembled into nano-scale structures by design of ELP block copolymers whose unique hydrophilic and hydrophobic domains maintain independent transition temperatures (Dreher et al., 2008; MacKay et al., 2009). Such ELP nanoparticles may improve systemic pharmacokinetics and enhance targeting to some disease sites such as solid tumors. The amino acid composition and molecular weight of hydrophobic and hydrophilic ELP domains will dictate the critical micelle temperature (CMT), influence micelle stability over the intended temperature range, and determine the micelle's hydrodynamic radius. By choice of ELP sequence and size, the CMT can be tuned well below body temperature, such that the ELP exists in a micellar form at all physiological thermal conditions. Alternatively, the CMT can be tuned to provide micelle assembly in response to a thermal trigger, such that heating of a disease target in vivo (at temperatures achieved by mild clinical hyperthermia, for instance) permits in situ self-assembly of ELP micelles only within the targeted tissue. This approach can be used, for example, to manipulate the density of peptide ligands presented on the micelle corona in order to improve local accumulation by high avidity interactions only within heated tissues (Simnick et al., 2010).
2.2 Expression and purification considerations
This protocol will describe the expression of protein ELP fusions in E. coli. Chilkoti and coworkers have previously expressed monomeric proteins ranging between 4-30 kDa and successfully used their ELP tag to obtain pure proteins. The properties of the protein and the ELP influence expression yields and thereby the chosen expression conditions. Herein, we discuss factors in the purification process and gene design that must be considered.
Protein properties
Chilkoti and coworkers have observed that proteins fused to an ELP affect the transition temperature of that ELP (Trabbic-Carlson et al., 2004b). A protein with an overall hydrophilic surface will result in an elevated transition temperature relative to the free ELP while a protein with a hydrophobic surface will result in a depressed transition temperature. This observation helps inform the choice of ELP and the choice of salt used in purification. If the target protein is extremely hydrophilic then a more hydrophobic ELP (e.g., with a valine guest residue) and a stronger salt (higher on the Hofmeister series) should be used for expression and purification. A more hydrophobic protein requires a more hydrophilic ELP (e.g. containing guest residues such as alanine and glycine) and a salt such as NaCl. Two ELP compositions have been typically used for purification: 1) 40 repeats with valine as guest residue. This ELP is used with hydrophilic proteins. 2) 90 repeats with valine, alanine, and glycine as guest residues at a ratio of 5:2:3. This ELP is the most commonly used ELP for purification. High yields have also been obtained for proteins (both hydrophilic and hydrophobic) that are fused to ELP block copolymers.
Protease site
If the target protein is to be cleaved from its ELP purification tag, a protease site must be engineered between the protein and ELP genes. Typically, protease sites for thrombin or tobacco etch virus (TEV) protease have been used. The choice of the protease depends on the desired amino acids to be left at the terminus of the protein after cleavage. Thrombin leaves behind a Leu-Val-Pro-Arg sequence if its substrate peptide (recognition sequence) is located on the C-terminus of the target protein and Gly-Ser if it is on the N-terminus of the target protein, while TEV leaves Glu-Asn-Leu-Tyr-Phe-Gln if its recognition sequence is located on the C-terminus and Gly if it is on the N-terminus of the protein.
Protein ELP fusion order
In a study performed by Chilkoti and coworkers, the fusion order of ELP to a protein was examined (Christensen et al., 2009). An ELP was fused to the N-terminus and C-terminus of various proteins and the expression yields and protein activity were determined. The expression yield as well as activity was generally higher for protein-ELP fusions in which the ELPs were fused at the C-terminus of the protein, though we note that exceptions to this rule have been observed (unpublished results). Therefore, it is recommended that expression vectors with a protein-ELP orientation be constructed and tested first and an ELP-protein construct be subsequently tested if yields are low or degradation of the protein is observed.
Choice of salt
Salts are known for their effect on the solubility of proteins. Anions have a stronger effect compared to cations. The order of the strength of these anions is described by the Hofmeister series (Hofmeister, 1888; Zhang and Cremer, 2006). Anions higher on the series than Cl- are referred to as kosmotropes and have a salting-out effect on proteins while anions lower on the series are referred to as chaotropes and have a salting-in effect.
For purification of protein ELP fusions, NaCl is the most commonly used salt. In the first ITC round, up to 3M NaCl is used to trigger isothermal transition of the protein ELP fusion. Subsequent rounds require less salt as the samples become more concentrated. However, extremely hydrophilic proteins or proteins with low yields may require a stronger kosmotrope to trigger the transition. The suggested salts are sodium citrate (0.15 M- 0.5M) and ammonium sulfate (0.4M-1M) in order of salting-out strength. Although the initial ITC round may require stronger salts, it is suggested that weaker salts be used in subsequent ITC rounds.
Choice of buffer
The choice of resuspension buffer depends on the specific characteristics of the target protein. For instance, hydrophobic proteins have been found to be more easily resuspended in water instead of buffer. HEPES buffer has been used for proteins that bind calcium ions since phosphate based buffers form precipitates in the presence of calcium ions. Purification can be carried out in any appropriate buffer and a final ITC round can then be performed to resuspend the protein ELP in a final storage buffer.
3. Recursive directional ligation by plasmid reconstruction
As ELPs consist of amino acids, the fusion of an ELP and a peptide or protein can be genetically encoded and synthesized by expression in a suitable heterologous host. There is, however, a significant challenge in the synthesis of the long repetitive genes that encode typical ELPs that we have used for purification of recombinant proteins as they range from 60-180 pentapeptides in length. Because chemically synthesized oligonucleotides –the starting point of gene synthesis– are typically available only up to approximately 150 bases, a “monomeric” gene that encodes a relatively short number of ELP repeats (~10 pentapeptides) is initially cloned into a vector from chemically synthesized oligonucleotides. This short sequence must then be oligomerized to provide the ELP gene of interest. Over the past decade, we have developed a number of methods to oligomerize genes that encode repetitive polypeptides such as ELPs. The first method that we developed is recursive directional ligation (RDL), which utilizes step-wise oligomerization (Meyer and Chilkoti, 2002). More recently, we have developed two new methods to improve upon RDL. The first method, recursive directional ligation by plasmid reconstruction (PRe-RDL) (McDaniel et al., 2010b), improves upon RDL by removing some of the sequence constraints imposed by RDL and enhancing the efficiency of the oligomerization process. The second method, overlap extension rolling circle amplification (OERCA) (Amiram et al., 2011), uses PCR methods to amplify repetitive sequences from a circular gene template. Herein, we will focus on PRe-RDL, a gene oligomerization method that provides both precise control of the ELP length and provides a modular cloning approach, which leads to the creation of libraries of ELP genes that encode a defined range of ELP molecular weights and allows ELP and protein or peptide genes to be easily swapped directly in and out of an expression vector. The following protocol for PRe-RDL was originally reported in detail in McDaniel et al., 2010.
3.1 Preparation of cloning vector for PRe-RDL
Prior to synthesis of the ELP gene, a pET-24a+ cloning vector must be modified to introduce endonuclease recognition sites for the type II enzymes used in PRe-RDL (AcuI and BseRI). In this step, a short oligonucleotide is ligated into the vector, which encodes the ribosome binding site, AcuI and BseRI recognition sites, as well as a leader and trailer sequence to precede and follow the ELP sequence, respectively. An example of oligonucleotides that encode this sequence is shown in Figure 2. In this example, methionine was chosen as the leader residue and tyrosine was chosen as the trailer residue followed by two stop codons. While initial modification of the cloning vector limits the number of amino acids that can be included in this rudimentary leader and trailer due to the base pair offset between the enzyme recognition site and cut site, later cloning steps can incorporate alternative leader and trailer sequences at the 5′- and 3′-end of the ELP, which are not restricted in length.
Figure 2. Modification of cloning vector for PRe-RDL.
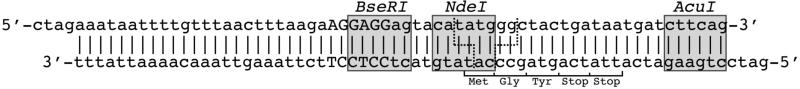
A short oligomer is inserted into the cloning vector, which has been digested with XbaI and BamHI. The oligomer design must include recognition sites for the PRe-RDL enzymes BseRI and AcuI (grey boxes), the ribosome binding site (capitalized), and short leader and trailer sequence (Met-Gly-Tyr-Stop-Stop). The cut site for NdeI (dotted line) and the overlapping cut site for BseRI and AcuI (dotted line) result in ta-5′ and gg-3′ overhangs respectively.
Modification of the pET-24a+ cloning vector is carried out as follows (Figure 3A):
Digest 1.5 μg of pET-24a+ DNA with 20 units XbaI and 20 units BamHI for 4 h at 37 °C.
Dephosphorylate the DNA product of the above reaction using 10 units of calf intestinal phosphatase (CIP) for 15 min at 37 °C and purify the product with a PCR purification kit (for example, Qiagen's PCR purification kit), and elute the DNA in 30 μl of H2O.
Anneal single stranded (ss) DNA for the sense and antisense strands that encode the PRe-RDL enzyme cleavage sites, ribosome binding site, and initial leader and trailer sequences – termed the PRe-RDL modifying insert. Heat 2 μM of each ssDNA that encodes for the sense and antisense strands of this insert in 50 μl of ligation buffer at 95 °C for 2 min, then allow to cool slowly to room temperature.
Ligate digested pET-24a+ with the annealed double stranded (ds) DNA product in a mixture that consists of 20 pmol of the annealed dsDNA, 0.1 pmol digested vector and 400 units T4 DNA ligase in ligation buffer at room temperature for 1 h.
Transform 5 μl of ligated DNA into competent cloning cells, then allow cells to recover at 37 °C in Super Optimal Broth with Catabolite Repression (SOC) for 1 h prior to plating on Terrific Broth (TB) plates containing 45 μg/ml kanamycin, and grow overnight at 37 °C.
Screen colonies for successful incorporation of the PRe-RDL insert by colony PCR or by a diagnostic restriction digest of purified DNA with XbaI and BamHI enzymes. Confirm successful insertion by DNA sequencing.
Figure 3. PRe-RDL cloning of protein ELP fusions.

Protein ELP fusions are designed and synthesized at the gene level from the repetitive cloning of an ELP motif, which is modified with the addition of leader or trailer peptide or protein sequences intended to precede or follow the ELP gene, respectively. Modification of the pET-24a+ cloning vector introduces the recognition sites necessary for PRe-RDL cloning (A). A short ELP oligomer is introduced into the BseRI cut site of the cloning vector and concatemerized to provide clones containing variable repeats of the ELP pentapeptide (B). The length of the ELP is further increased with PRe-RDL oligomerization, where ‘A cut’ and ‘B cut’ digestions of ELP are ligated to form an ELP gene with the sum of their lengths (C). This process may also be used to create block copolymer ELPs of various unique sequences. Lastly, protein sequences, optically active residues, or reactive amino acids may be appended at either terminus of the ELP by the addition of leader and trailer genes encoding these desired sequences (D).
3.2 ELP concatemerization
ELP genes of a desired length (typically encoding 60-180 pentapeptide repeats) require oligomerization of a synthetic gene that encodes a ‘monomer’ repeat, as typical chemical synthesis of DNA only yields ssDNA sequences of <150 bases in length. To speed the synthesis of long, repetitive genes initial oligomerization of several monomer gene repeats is achieved with concatemerization, in which multiple copies of small oligonucleotides can be inserted into the cloning vector in a single step. This initial insertion of multiple monomers of the repetitive gene provides little control over the length of ELP genes created and is a process typically done prior to gene oligomerization that is separate from PRe-RDL, which permits the creation of long ELP genes of precise length from the products of concatemerization. The monomer repeat on which the ELP sequence is based can be a single pentapeptide repeat or several pentapeptide repeats in length, depending on the desired length of the final ELP product (the total pentapeptide repeats must be a multiple of the repeats contained in the monomer unit) and the speed with which the gene encoding the final ELP product can be oligomerized by successive rounds of Pre-RDL (larger monomer repeats reduce the number of cloning steps toward a final ELP gene).
Preparation of the ELP-encoding oligonucleotides and their concatemerization in the modified pET-24a+ cloning vector is performed as follows (Figure 3B):
Digest 1.5 μg of modified pET-24a+ vector with 8 units of BseRI for 3 h at 37 °C.
Dephosphorylate the digested DNA with 10 units of CIP for 15 min at 37 °C and purify the vector DNA with a PCR purification kit, eluting the DNA in 30 μl of H2O.
Anneal complementary ssDNA encoding the desired ELP. The ELP cassette will contain a gg-3′ overhang on the sense strand and a cc-3′ overhang on the anti-sense strand that is complementary to the BseRI cleavage site in the cloning vector.
Ligate the digested vector and annealed ELP-encoding dsDNA in a mixture that consists of 0.1 pmol BseRI-digested vector, 20 pmol annealed ELP-encoding dsDNA, and 400 units of ligase in ligase buffer for 1 h at room temperature.
Transform the ligated DNA into competent cells, recover, and plate as described previously. Grow cells overnight at 37 °C.
Screen colonies for successful insertion of the ELP oligonucleotide by colony PCR or by a diagnostic restriction digest of purified plasmid DNA with XbaI and BamHI enzymes. Confirm the length and sequence of the insert by DNA sequencing.
3.3 ELP oligomerization
Following the initial insertion of the ‘monomer’ synthetic gene that encodes a defined number of ELP repeats obtained by concatemerization, PRe-RDL is used to oligomerize this DNA sequence further to attain the desired length of the final ELP. The length can be easily doubled in a single round of Pre-RDL by combining digestions of a single length of ELP, or can be tailored to any desired length by the combination of ELP genes whose lengths sum to the desired number of pentapeptide repeats. This step can also be utilized to synthesize genes encoding ELP block copolymers with different repeat motifs in each block.
The oligomerization of ELP genes and creation of ELP block copolymers is performed as follows (Figure 3C):
Create an ‘A cut’ of DNA of the ELP sequence intended for the N-terminus of the polypeptide product by digestion of 4 μg of DNA, 10 units AcuI, and 40 units BglI in appropriate buffer for 3 h at 37 °C.
Create a ‘B cut’ of DNA of the ELP sequence intended for the C-terminus of the polypeptide product by digestion of 4 μg DNA, 8 units of BseRI, and 40 units of BglI in appropriate buffer for 3 h at 37 °C.
Separate the ‘A cut’ and ‘B cut’ digestion products on a 1% low melting point agarose gel. The ‘A cut’ will create three fragments whose lengths in base pairs are 1586, 1821, and 1891 + the length of the ELP insert. The ‘B cut’ will create two bands whose lengths in base pairs are 1891 and 3407 + the length of the ELP insert. Excise the largest band from each digestion, purify the DNA with a gel extraction purification kit (for example, Qiagen's Gel Extraction Purification kit), and elute purified DNA in 30 μl H2O.
Ligate the purified ‘A cut’ and ‘B cut’ products in a mixture of each purified digestion product at 5 ng/μl and 400 units of ligase in 20 μl of ligase buffer for 1 h at room temperature.
Transform ligated DNA into competent cells, recover, and plate as described previously.
Screen colonies for successful oligomerization of the ELP by restriction digest of purified DNA with XbaI and BamHI enzymes. Confirm successful oligomerization of the ELP or assembly of the ELP block copolymer gene by DNA sequencing.
3.4 Modifying the N- and C-terminus of protein ELP fusions
The N- and C-terminus of the ELP can be further modified by the addition of leader or trailer sequences encoding amino acids, peptides, or proteins to precede or follow the ELP gene, respectively. For the purpose of creating protein ELP fusions, either the N- or C-terminus will contain the protein gene, while the other terminus may be used for the addition of functional residues for purposes such as protein quantification, fluorescent labeling, or drug conjugation. Leader and trailer sequences should be designed with complementary ssDNA such that the sense strand will contain a 5′-ta and gg-3′ overhang after annealing that is complementary to NdeI and BseRI cut sites.
Modification of the 5′- and 3′-end of the ELP gene is achieved as follows (Figure 3D):
- Digest 1.5 μg of the modified pET-24a+ cloning vector with 8 units of BseRI for 2 h at 37 °C in appropriate buffer. Add 60 units of NdeI and continue the digestion for 1 h at 37 °C. Purify the digestion product with a PCR purification kit and elute purified DNA in 30 μl H2O.This sequential digestion is necessary because the NdeI cut site lies between the BseRI recognition sequence and its respective cut site.
Anneal complementary ssDNA encoding the leader or trailer sequences as described previously. The leader or trailer sequence will contain a 5′-ta overhang and gg-3′ overhang on the sense strand, that are complementary to the NdeI and BseRI cut sites in the cloning vector.
Ligate digested pET-24a+ vector and annealed leader or trailer dsDNA and transform ligated DNA into competent cells, recover, and plate as described previously.
Screen colonies for successful insertion of leader or trailer sequences into the pET-24a+ vector by colony PCR or diagnostic digest with XbaI and BamHI. Confirm successful modification of the vector by DNA sequencing.
Digest the vector containing the leader sequence to form an ‘A cut’ and separately digest the vector containing the completed ELP sequence to form a ‘B cut’. Separate digestion products by gel electrophoresis, excise the largest DNA band from each digest, and purify DNA with a gel extraction purification kit. Ligate ‘A cut’ and ‘B cut’ digestion products as was performed in step 4 of the ELP oligomerization protocol and transform into competent cells, recover, and plate cells to grow overnight. Verify incorporation of the leader sequence to the 5′-end of the ELP gene by a diagnostic restriction digest with XbaI and BamHI and confirm by DNA sequencing.
Digest the vector containing the leader-ELP gene synthesized in the previous step to form an ‘A cut’ and separately digest the vector containing the trailer sequence to form a ‘B cut’. Separate the digestion products by gel electrophoresis, excise the largest DNA band from each lane, and purify the DNA with a gel extraction purification kit. Ligate the ‘A cut’ and ‘B cut’ digestion products as described in step 4 of the ELP oligomerization protocol. Transform competent cells suitable for recombinant protein expression with ligated DNA, allow the cells to recover, and plate the cells to grow overnight. Verify incorporation of the trailer sequence to the leader-ELP gene construct by a diagnostic restriction digest with XbaI and BamHI enzymes and confirm by DNA sequencing. The resulting leader-ELP-trailer gene construct is now ready for expression.
4. Expression and purification of protein ELP fusions
4.1 Expression and extraction of protein ELP fusions
This section will describe the conditions to grow and express protein ELP fusions.
- Transform expression plasmid with inserted gene of protein ELP fusion into an E. coli expression strain.BL21(DE3) is the most commonly used E. coli strain that is compatible with protein expression using the pET series of expression plasmid and other vectors which are under control of the T7 promoter. If a greater degree of control over protein expression is necessary, as may be the case for proteins that are toxic to E. coli (such as fusions of anti-microbial protein or peptides), BL21(DE3) pLysS cells may be appropriate to provide greater repression of baseline expression, prior to induction.
Grow single colony in 3 mL of autoclaved TB culture at 37 °C overnight while shaking at 225 rpm.
- Add 1 mL of overnight culture from step 2 to an autoclaved 1L TB culture in a 4L flask. Add appropriate volume of antibiotic to culture. Incubate at 37 °C while shaking at 225 rpm.The temperature and time of incubation vary depending on the optimum protein expression conditions. Proteins that do not fold properly at 37 °C, or are degraded, can be expressed at lower temperatures (as low as 16 °C) to obtain higher yields.Time of incubation can be 8 h or 24 h depending on whether the culture is induced or not. The T7 expression system is “leaky” resulting in continuous background expression of the target protein. Thus, cultures can be simply incubated for 24 h with no need to induce expression. However, if the protein is being degraded or is toxic to E. coli, growing the culture for shorter times with induction could improve yields. For expression by induction, a final concentration of 1 mM of IPTG should be added to the culture when the culture reaches an OD of 0.6. The culture should be grown for an additional 3-5 h before harvesting the cells.
Transfer culture into a 1 L bottle and centrifuge at 16000 xg for 15 min at 4 °C.
Decant supernatant and resuspend pellet in PBS to a volume of 45 mL and transfer to a 50 mL conical tube.
Sonicate cells while on ice at 85 W for 9 min in cycles of 10 sec on and 20 sec off.
- Add 2 mL of 10% (v/v) Polyethylimine (PEI) to the sonicated cell mixture.PEI is a positively charged polymer that will bind to negatively charged molecules in the cell lysate (including DNA), which assists in precipitating these molecules. If the protein being expressed is highly negatively charged, then proceed to step 8 without the addition of PEI.
- Centrifuge the mixture from step 7 at 16,000 ×g for 10 min at 4 °C.Typically, the cell lysate is divided equally between two 50 mL round bottomed centrifuge tubes.
- Transfer supernatant to a clean tube(s). Add salt to the supernatant step-wise until the solution turns turbid. The most common choice of salt is NaCl, which can be added at a concentration of up to 3 M.The choice of salt depends on the transition temperature of the ELP that the protein is fused to as well as the properties of the protein. If the ELP tag used is hydrophilic or short, then a stronger salt from the Hofmeister series is recommended. A protein with low yield (i.e. low concentration) also might require a stronger salt. Typically, sodium citrate (up to a concentration of 0.3 M) or ammonium sulfate (up to a concentration of 1 M) are used as the stronger salts. For further details refer to section 2.2.
- Centrifuge the sample at 16,000 ×g for 10 min at room temperature.A pellet (usually white) should form. The size of the pellet depends on the expression yield.
- Decant supernatant. Add 3-4 mL of PBS (or chosen purification buffer) to pellet and resuspend by pipetting solution.Cold PBS (4 °C) helps resuspend the pellet faster as the lower temperatures drive the resolubilization of the coacervated ELP. In addition, the pellets can be resuspended as they rotate at 4 °C in a refrigerator.
Once the pellet is completely resuspended, transfer solution into 1.5 mL centrifuge tubes.
4.2 Inverse transition cycling
This section will describe what is termed as an inverse transition cycle (ITC) (Figure 4). One round of ITC reduces the amount of soluble and insoluble contaminants. An ITC consists of one “cold spin” and one “hot spin”, which simply refer to the relative temperature at which the centrifugation steps are performed. The cold spin is carried out under conditions of salt and temperature at which the protein ELP fusion is soluble, and it removes contaminants that are insoluble from the solution. The hot spin, in contrast, is carried out under conditions of salt and temperature at which the ELP fusion protein is in its phase separated –coacervated– state. This step separates the coacervated protein ELP fusions from other soluble molecules in solution, thus leaving the soluble contaminants in the supernatant. Reduced amounts of salts should be added in each subsequent round of ITC as the protein ELP fusion becomes more concentrated (thus resulting in a lower transition temperature) and hence requires a lower concentration of salt to drive the ELP phase transition. Although salts such as NaCl have very poor ability to nonspecifically “salt-out” proteins, it is prudent to add the minimum amount of salt necessary to trigger the phase transition of the protein ELP fusion, as high salt concentrations increase the likelihood of coprecipitating contaminant proteins, especially for salts such as ammonium sulfate that are potent “salting-out” agents.
Figure 4. Purification of protein ELP fusions by inverse transition cycling (ITC).

The thermal properties of ELPs provide a means of fusion protein purification that is an excellent alternative to traditional purification methods such as chromatography. E. coli cells are collected from the culture media by centrifugation and lysed by sonication prior to purification by ITC (1). The ELP transition is triggered with the addition of heat or salt (2) and aggregated insoluble ELP is collected by centrifugation (3). The supernatant, containing soluble contaminants, is discarded while the pellet, containing insoluble ELP coacervate, is retained (4) and resolubilized in cold buffer (5). The solution is again centrifuged (6) and the pellet, containing insoluble contaminants, is discarded, while the supernatant, containing the soluble ELP, is retained (7). Steps 2 through 7 constitute one round of ITC, and should be repeated until desired purity is reached.
Note: We recommend saving 20 μL of the sample and supernatant at each step for subsequent analysis to help troubleshoot potential problems.
Add salt until the solution turns turbid. Centrifuge at 16,000 ×g for 10 min at room temperature.
- Remove the supernatant. Add ~750 μL of PBS (or chosen purification buffer) and resuspend pellet.In each subsequent round, smaller and smaller volumes of buffer can be added. The minimum amount of buffer that can be added depends on the level of expression (i.e. size of pellet).
- Centrifuge samples at 16,000 ×g for 10 min at 4 °C.A small pellet of insoluble contaminants should appear after centrifugation.
Transfer the supernatant to clean tubes without disrupting the pellet. If possible, combine samples into a smaller number of tubes.
- Repeat steps 1-5 until sample is pure. Typically, 3-5 rounds of ITC produce a pure sample.With each round of ITC, some protein ELP fusion is lost. Therefore, the number of rounds should be determined depending on the balance between the required purity and yield.
4.3 Evaluation and processing of purified protein ELP fusions
To verify the purity of the protein ELP fusion, the first step is to evaluate the purification product by SDS-PAGE analysis. Visualization of the protein band by SDS-PAGE verifies the presence or absence of contaminants as well as the molecular weight of the purified protein. SDS-PAGE should not, however, be relied upon to solely judge the molecular weight of the protein as ELPs can in some cases display anomalous migration in SDS-PAGE relative to protein standards (Meyer and Chilkoti, 2002). Other techniques such as mass spectrometry are more appropriate to precisely measure the mass of the protein.
- Samples from each step of purification by ITC should be incubated with an appropriate loading dye for 2 min at 100 °C.If native PAGE analysis is desired to visualize the undenatured protein, then an appropriate loading dye and buffer that does not contain SDS or β-mercaptoethanol should be used.
- Load the sample onto a pre-cast SDS-PAGE gel and run until the dye reaches the end of the gel.The degree of crosslinking of the polyacrylamide gel should be chosen based on the molecular weight of the protein.
- Stain the gel with a 0.5 M CuCl2 solution for 5 min or with a zinc solution according to the steps below:
- Incubate gel for 10-15 min in a 0.2 M imidazole, 0.1 % SDS (v/w) solution.
- Transfer the gel to a 0.3 M ZnSO4 solution and incubate for 30 sec (no longer than 45 sec).
- Rinse with water for 3 min.These chemicals work as negative stains; they will turn the gel white except for the protein bands. ELPs do not stain with Commassie blue and thus it is preferable to use a negative stain to ensure the visualization of any free ELP that might indicate the degradation or unspecific cleavage of the protein ELP fusion.
To cleave the ELP tag if desired, the corresponding protease cleavage kit should be purchased. It is highly recommended that a biotinylated protease (if available) be used in this step as subsequent removal of the protease from the mixture can be accomplished with streptavidin agarose beads. After cleavage of the protein from the ELP fusion, pure protein can be recovered as follows:
- Add the minimum amount of salt to transition the cleaved ELP.Adding a concentrated salt solution to the sample drop-wise prevents addition of excessive salt.
Centrifuge sample at 16,000 ×g for 10 min at room temperature. Save the supernatant.
To remove the excess salt, the sample can be either dialyzed using the appropriate molecular weight cut-off dialysis membrane or filtered through a gelfiltration column.
Repeat SDS-PAGE analysis to visualize the therapeutic protein and to ensure complete cleavage and purification from the protease and ELP tag.
For samples intended for in vivo applications, additional removal of endotoxin may be desired. Purified therapeutic protein can be passed through endotoxin removal columns (such as Pierce's Detoxi-gel). The presence of endotoxin can then be quantified by a gel clot assay (such as Lonza's Pyrogent assay).
Gel Analysis
In examining the SDS-PAGE results, a single band should be observed for a single sub-unit protein that corresponds to the correct molecular weight of the protein ELP fusion, as seen in Figure 5. Additional bands observed in that lane indicate contaminants that can be removed by additional rounds of ITC (step 4.2 (1-5)). If the lysed cell lane shows no overexpressed band at the molecular weight of the target protein ELP fusion, optimization of growth conditions (temperature, time, cell line and induction) is recommended. In addition, the choice of ELP guest residue composition or length should be reexamined.
Figure 5. SDS-PAGE analysis of protein ELP fusion purification by ITC.

A therapeutic interleukin-1 receptor antagonist ELP fusion, with applications for osteoarthritis, was examined throughout purification with two rounds of ITC. Each lane indicates the following purification steps: (1) cell lysate; (2) soluble cell lysate; (3) soluble lysate after addition of PEI; (4) insoluble pellet after addition of PEI; (5) supernatant after first cold spin; (6) supernatant after first hot spin; (7) supernatant after second cold spin. Note the presence of the overexpressed protein ELP fusion band in early stages of purification and the singular band of pure protein ELP fusion achieved with only two rounds of ITC. Figure adapted from (Shamji et al., 2007).
5. Physical characterization of protein ELP fusions
After cloning, expression, and purification of the protein ELP fusion, characterization of the thermal properties and higher order assembly are important to assess the applicability of the fusion to its intended application. Because the fusion of an ELP to a protein can alter the phase behavior, it is imperative that each fusion be evaluated with regard to its thermal behavior.
Evaluating the Tt of a protein ELP fusion provides the precise temperature at which ELP aggregation (or higher order self-assembly) will occur at a given concentration and buffer conditions. This thermal response can be measured by temperature-programmed turbidimetry where the light attenuation of the protein ELP fusion solution is monitored at 350 nm as the temperature is ramped, typically at a rate of 1 °C/min. The solubility of ELPs below their Tt leads to little light extinction. As the temperature is increased and approaches the Tt, the extinction of the solution will rapidly increase, indicating the coacervation of ELPs into micron size ELP-rich aggregates (Figure 6). The ELP Tt can be defined from this data as the temperature at which the first derivative of the absorbance reaches its maximum (otherwise defined as the inflection point of the absorbance curve). More subtle changes in the absorbance spectrum with respect to temperature can provide further information about nano-mesoscale assembly prior to the complete coacervation of the ELP. The assembly of ELP block copolymers into spherical micelles is one such example, where a region of increased absorbance occurs prior to aggregation in which the stable nano-scale ELP micelles contribute to the solution turbidity.
Figure 6. Thermal characterization of protein ELP fusion by temperature regulated turbidimetry.

The thermal properties of therapeutic fusions of interleukin-1 receptor antagonist (IL1Ra) with two unique ELP sequences were evaluated by temperature-programmed turbidimetry. Increasing the temperature (thick lines) leads to an increase in optical density at 350 nm and reveals transition temperatures of approximately 34 °C and 35-40 °C for an ELP fusion of 30 repeats with valine as the guest residue (ELP-(V)30-IL1Ra) and an ELP fusion of 90 repeats that contain valine, alanine, and glycine guest residues in a ratio of 5:2:3 (ELP-(V5A2G3)90-IL1Ra), respectively. Both ELP fusions demonstrate reversibility in their thermal transition as the optical density decreases as the temperature is reduced (thin lines). Figure adapted from (Shamji et al., 2007).
Additional information about the thermal response of protein ELP fusions can be inferred from temperature-programmed light scattering. While temperature-regulated turbidimetry can provide the temperature at which an ELP or its fusion protein self-assembles or phase separates into micron size aggregates, it cannot provide information about the size and shape of the assemblies nor is it sensitive to low levels of adventitious aggregation of a protein ELP fusion. Dynamic and static light scattering are two powerful techniques that can supply this information, providing additional evaluation of the size, molecular weight, and topology of such assemblies (Schartl, 2007).
6. Conclusions
As a stimulus responsive biopolymer, ELPs have unique potential as a polypeptide ”tag” for protein purification and as a carrier of therapeutic protein cargo. The genetic design of protein ELP fusions permits precise control of the ELP properties and affords complete control over the sequence of other peptides or proteins that can be appended at either end of the fusion. The biocompatibility of ELPs and the LCST behavior of protein ELP fusions make them suitable for a variety of physiological applications to improve the therapeutic efficacy of pharmaceutical protein ELP fusions by use of the ELP as a macromolecular carrier, depot-forming coacervate, or nano-scale assembly. ELPs thus provide the ability to enhance therapeutic action by improving the pharmacokinetics, prolonging the local residence, or targeting accumulation at a disease site by manipulation of their physicochemical properties via modulation of the ELP size, sequence, or architecture. The optimization of ELP design, genetically encoded synthesis, heterologous expression, and non-chromatographic purification attests to the simplicity with which new protein ELP fusions can be created and demonstrates the great utility that ELP fusions can have on the successful delivery of pharmaceutical proteins.
Table 1.
Timeline of protein ELP fusion cloning
| Step description | Step number | Time required | Notes |
|---|---|---|---|
| Modification of pET vector | 3.1-(1-6) | 3 days | Screen modified vectors by colony PCR |
| ELP concatemerization | 3.2-(1-6) | 3 days | Screen concatemerization products by colony PCR |
| ELP oligomerization by PRe-RDL | 3.3-(1-6) | 3 days | Screen oligomerization products by BamHI and XbaI diagnostic digest |
| Addition of leader and trailer sequences by PRe-RDL | 3.4-(1-6) | 9 days | Screen modified ELP sequences by BamHI and XbaI diagnostic digest |
Table 2.
Timeline of purification steps
| Step description | Step number | Time required | Notes |
|---|---|---|---|
| Transformation | 4.1-1 | 2 h | |
| Starter culture | 4.1-2 | Overnight | |
| Expression culture | 4.1-3 | 24 h | May vary depending on optimized growing conditions |
| Cell concentration and resuspension | 4.1-(4-6) | 15 min | Samples may be stored at -20 °C or -80 °C depending on cold denaturation point of therapeutic protein |
| First round of ITC | 4.1-(7-12) | 1 h | Samples may be stored at -20°C after this round is completed Note: Once step 4.1-6 is performed, the rest of the steps up to step 12 must be performed immediately |
| Successive round of ITC | 4.2-(1-5) | 30 min each | Samples may be stored at -20 °C after each round |
| Post-purification evaluation and processing | 4.3 | 1h-48h | Time varies depending on whether ELP tag is cleaved or not |
Acknowledgements
The work reported herein was supported by NIH grant R01 GM61232 to A.C.
References
- Amiram M, Quiroz FG, Callahan DJ, Chilkoti A. A highly parallel method for synthesizing DNA repeats enables the discovery of ‘smart’ protein polymers. Nat Mater. 2011;10:141–8. doi: 10.1038/nmat2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen T, Amiram M, Dagher S, Trabbic-Carlson K, Shamji M, Setton L, Chilkoti A. Fusion order controls expression level and activity of elastin-like polypeptide fusion proteins. Protein Science. 2009;18:1377–1387. doi: 10.1002/pro.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreher MR, Simnick AJ, Fischer K, Smith RJ, Patel A, Schmidt M, Chilkoti A. Temperature triggered self-assembly of polypeptides into multivalent spherical micelles. J Am Chem Soc. 2008;130:687–94. doi: 10.1021/ja0764862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge X, Yang DS, Trabbic-Carlson K, Kim B, Chilkoti A, Filipe CD. Self-cleavable stimulus responsive tags for protein purification without chromatography. J Am Chem Soc. 2005;127:11228–9. doi: 10.1021/ja0531125. [DOI] [PubMed] [Google Scholar]
- Hofmeister F. Zur Lehre von der Wirkung der Salze. Naunyn-Schmiedeberg's Archives of Pharmacology. 1888;24:247–260. [Google Scholar]
- Kim B, Chilkoti A. Allosteric actuation of inverse phase transition of a stimulus-responsive fusion polypeptide by ligand binding. J Am Chem Soc. 2008;130:17867–73. doi: 10.1021/ja8059057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, MacKay JA, Dreher MR, Chen M, McDaniel JR, Simnick AJ, Callahan DJ, Zalutsky MR, Chilkoti A. Injectable intratumoral depot of thermally responsive polypeptide-radionuclide conjugates delays tumor progression in a mouse model. J Control Release. 2010;144:2–9. doi: 10.1016/j.jconrel.2010.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacEwan SR, Chilkoti A. Elastin-like polypeptides: biomedical applications of tunable biopolymers. Biopolymers. 2010;94:60–77. doi: 10.1002/bip.21327. [DOI] [PubMed] [Google Scholar]
- MacKay JA, Chen M, McDaniel JR, Liu W, Simnick AJ, Chilkoti A. Self-assembling chimeric polypeptide-doxorubicin conjugate nanoparticles that abolish tumours after a single injection. Nat Mater. 2009;8:993–9. doi: 10.1038/nmat2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel JR, Callahan DJ, Chilkoti A. Drug delivery to solid tumors by elastin-like polypeptides. Adv Drug Deliv Rev. 2010a;62:1456–67. doi: 10.1016/j.addr.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel JR, Mackay JA, Quiroz FG, Chilkoti A. Recursive directional ligation by plasmid reconstruction allows rapid and seamless cloning of oligomeric genes. Biomacromolecules. 2010b;11:944–52. doi: 10.1021/bm901387t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer DE, Chilkoti A. Purification of recombinant proteins by fusion with thermally-responsive polypeptides. Nat Biotechnol. 1999;17:1112–5. doi: 10.1038/15100. [DOI] [PubMed] [Google Scholar]
- Meyer DE, Chilkoti A. Genetically encoded synthesis of protein-based polymers with precisely specified molecular weight and sequence by recursive directional ligation: examples from the elastin-like polypeptide system. Biomacromolecules. 2002;3:357–67. doi: 10.1021/bm015630n. [DOI] [PubMed] [Google Scholar]
- Meyer DE, Chilkoti A. Quantification of the effects of chain length and concentration on the thermal behavior of elastin-like polypeptides. Biomacromolecules. 2004;5:846–51. doi: 10.1021/bm034215n. [DOI] [PubMed] [Google Scholar]
- Nettles DL, Chilkoti A, Setton LA. Applications of elastin-like polypeptides in tissue engineering. Adv Drug Deliv Rev. 2010;62:1479–85. doi: 10.1016/j.addr.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schartl W. Light scattering from polymer solutions and nanoparticle dispersions. Springer; Berlin: 2007. [Google Scholar]
- Shamji MF, Betre H, Kraus VB, Chen J, Chilkoti A, Pichika R, Masuda K, Setton LA. Development and characterization of a fusion protein between thermally responsive elastin-like polypeptide and interleukin-1 receptor antagonist: sustained release of a local antiinflammatory therapeutic. Arthritis Rheum. 2007;56:3650–61. doi: 10.1002/art.22952. [DOI] [PubMed] [Google Scholar]
- Simnick AJ, Valencia CA, Liu R, Chilkoti A. Morphing low-affinity ligands into high-avidity nanoparticles by thermally triggered self-assembly of a genetically encoded polymer. ACS Nano. 2010;4:2217–27. doi: 10.1021/nn901732h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatham AS, Shewry PR. Elastomeric proteins: biological roles, structures and mechanisms. Trends Biochem Sci. 2000;25:567–71. doi: 10.1016/s0968-0004(00)01670-4. [DOI] [PubMed] [Google Scholar]
- Trabbic-Carlson K, Liu L, Kim B, Chilkoti A. Expression and purification of recombinant proteins from Escherichia coli: Comparison of an elastin-like polypeptide fusion with an oligohistidine fusion. Protein Sci. 2004a;13:3274–84. doi: 10.1110/ps.04931604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trabbic-Carlson K, Meyer DE, Liu L, Piervincenzi R, Nath N, LaBean T, Chilkoti A. Effect of protein fusion on the transition temperature of an environmentally responsive elastin-like polypeptide: a role for surface hydrophobicity? Protein Eng Des Sel. 2004b;17:57–66. doi: 10.1093/protein/gzh006. [DOI] [PubMed] [Google Scholar]
- Urry DW. Physical Chemistry of Biological Free Energy Transduction As Demonstrated by Elastin Protein-Based Polymers. J. Phys. Chem. B. 1997;101:11007–11028. [Google Scholar]
- Urry DW, Luan CH, Parker TM, Gowda DC, Prasad KU, Reid MC, Safavy A. Temperature of polypeptide inverse temperature transition depends on mean residue hydrophobicity. J. Am. Chem. Soc. 1991;113:4346–4348. [Google Scholar]
- Zhang YJ, Cremer PS. Interactions between macromolecules and ions: the Hofmeister series. Current Opinion in Chemical Biology. 2006;10:658–663. doi: 10.1016/j.cbpa.2006.09.020. [DOI] [PubMed] [Google Scholar]