

Abstract
Several studies suggest a role for the amyloid precursor protein (APP) in neurite outgrowth and synaptogenesis, but the downstream interactions that mediate the function of APP during neuron development are unknown. By introducing interaction-deficient FE65 into cultured hippocampal neurons using adenovirus, we show that a complex including APP, FE65 and an additional protein is involved in neurite outgrowth at early stages of neuronal development. Both FE65 that is unable to interact with APP (PID2 mutants) or a WW mutant increased axon branching. Although the FE65 mutants did not affect total neurite output, both mutants decreased axon segment length, consistent with an overall slowing of axonal growth cones. FE65 mutants did not alter the localization of either APP or FE65 in axonal growth cones, suggesting that the effects on neurite outgrowth are achieved by alterations in local complex formation within the axonal growth cone.
Keywords: Alzheimer’s disease, APP, FE65, Mena, Neurite outgrowth, Branching
Introduction
The normal function of the Alzheimer’s disease (AD) amyloid precursor protein (APP) has been the subject of intense investigation. Several lines of evidence suggest that APP regulates neurite outgrowth. APP synthesis and axonal transport coincide with periods of axon elongation and synapse formation (Hung et al., 1992; Moya et al., 1994). Secreted fragments of APP and over-expression of transmembrane APP increase neurite outgrowth (Milward et al., 1992; Jin et al., 1994; Small et al., 1994; Perez et al., 1997). When APP expression is decreased, process outgrowth is altered (LeBlanc et al., 1992; Majocha et al., 1994; Allinquant et al., 1995; Perez et al., 1997). In Drosophila APP promotes post-developmental neurite arborization and is involved in PNS development (Merdes et al., 2004; Leyssen et al., 2005). Despite this evidence that APP regulates neurite outgrowth, the molecular mechanisms of this regulation remain unknown.
FE65 binds the cytoplasmic domain of APP and alters APP processing and trafficking (Sabo et al., 1999). FE65 contains several protein–protein interaction domains including one WW domain and two PI domains (PID). FE65 interacts with APP through its carboxy-terminal PI domain (PID2) (Borg et al., 1996; Guenette et al., 1999). We have shown that FE65, in a complex with APP, can regulate cell motility (Sabo et al., 2001). APP and FE65 colocalize in growth cones of developing neurons (Sabo et al., 2003), raising the hypothesis that APP regulates growth cone motility and neurite growth via its interaction with FE65. However, a function for FE65 in growth cone behavior and neurite growth has yet to be demonstrated.
It is likely that FE65 acts by targeting some effector molecule to APP through one of its other protein-interaction domains. One candidate for this role is Mena, which interacts with the WW domain of FE65 (Ermekova et al., 1997) and colocalizes with APP and FE65 in growth cones (Sabo et al., 2003). Mena regulates growth cone dynamics and is required for normal neural development (Gertler et al., 1990, 1995; Lanier, 1999). Moreover, the phenotypic abnormalities arising from disruption of two members of the FE65 family (Guenette et al., 2006) or deletion of all three members of the APP family (Herms et al., 2004) resemble those observed in Mena-deficient mice (Lanier, 1999; Goh et al., 2002), suggesting a functional interaction between these three protein families during neuronal development in vivo. Together, these data suggest that the APP/FE65 complex may regulate neuronal development through its interaction with Mena.
Here we show that disrupting the interaction between APP and FE65 by introducing interaction-deficient FE65 mutants into cultured hippocampal neurons increases neurite branching without affecting total neurite output. Furthermore, neurite branching similarly increases when the WW domain of FE65 is mutated to disrupt the interaction between FE65 and a WW domain-binding protein, which may be Mena or a related protein. These data provide the first evidence that APP regulates neurite branching through a complex with FE65.
Results
Expression of FE65 that cannot interact with APP increases neurite branching
APP, FE65 and Mena form a tripartite complex in cultured cells and neuronal growth cones and regulate motility in non-neuronal cells (Sabo et al., 2001; Sabo et al., 2003). To test whether a similarly functional complex exists in growth cones as well we introduced interaction-deficient FE65 into cultured neurons using adenovirus. The following constructs were used (depicted in Fig. 1A): 1) wild type FE65 (WT); 2) FE65 with a point mutation in the second PI domain (C602V; mPID2), which has previously been shown to prevents it from binding to APP (Borg et al., 1996); and 3) FE65 in which PID2 was deleted in its entirety (ΔPID2). When the PID2 mutants are over-expressed, they function as dominant-negatives by sequestering the third protein normally associated with the complex away from APP thus preventing formation of a complete tripartite complex. FE65 PID2 mutants can still interact with the third partner normally associated with the complex (e.g. Mena or a similar protein), but cannot interact with APP. Because the mutant FE65 is over-expressed, most of the third protein should be bound to mutant FE65 and unavailable for interaction with endogenous FE65. Therefore, although endogenous APP and FE65 can still interact with each other, the endogenous tripartite complex is left incomplete. Virus expressing GFP alone was used as control (CONT). Hippocampal neurons were infected with the various viruses and fixed at 24 h in vitro. Infected neurons were identified by the presence of GFP, which is co-expressed with FE65 in the same virus. Number and length of neurites from at least 30 neurons per treatment were counted and measured.
Fig. 1.
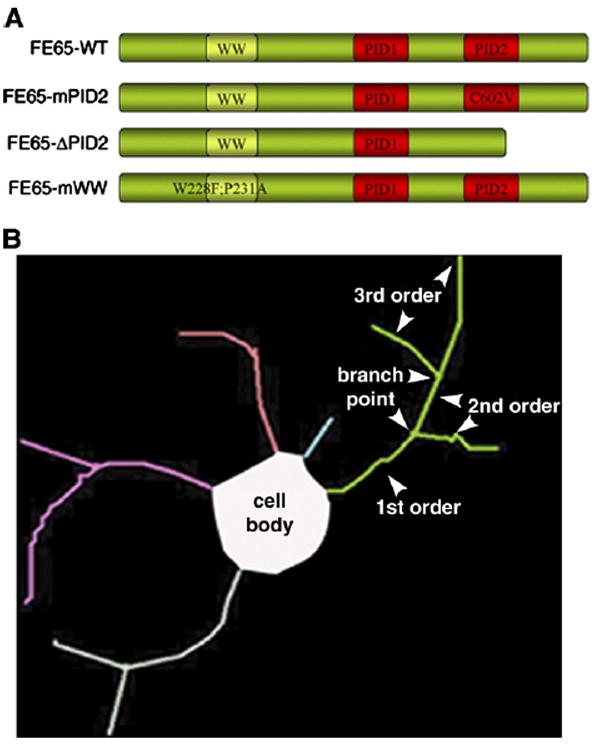
Scheme of viruses and tracing method used. (A) Schematic of the FE65 dominant-negative viral constructs used. FE65-ΔPID2 lacks the entire PID2 domain. FE65-mPID2 and FE65-mWW contain the indicated point mutations, which disrupt the interaction of FE65 with APP and Mena, respectively. (B) Example of a Neurolucida trace depicting the terms used (i.e. neurite order, branch points, etc.).
Disrupting the interaction between APP and FE65 caused a significant increase in neurite branching compared to control virus (Fig. 2A). Neurites expressing PID2 mutants had 2–3-fold more branches than control neurons (mean second order neurites: CONT= 3.67 ± 1.2; mPID2 = 6.8 ± 1.5; ΔPID2 = 6.3 ± 1.0; mean third order neurites: CONT = 0.7 ± 0.4; mPID2 = 3.2 ± 1.2; ΔPID2=2.1±0.9; p<0.05; see illustration in Fig. 1B for explanation of terms). Similar results were obtained when treated cells were fixed after 48 h in vitro (not shown; see Fig. 2C for neuron images). There were no differences between control virus- and FE65-WT virus-infected cells. This is most likely due to the abundance of endogenous FE65 in neurons.
Fig. 2.
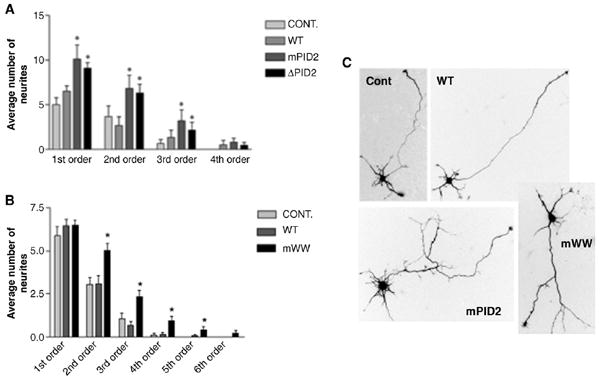
FE65 negatively regulates neurite branching as part of a macromolecular complex. (A, B) Expression of the FE65 PID2 (A) and WW (B) mutants illustrated in Fig. 1A increase neurite branching. Neurons were infected with adenovirus expressing wild type FE65 (WT), FE65-ΔPID2 (ΔPID2), FE65-mPID2 (mPID2) or GFP virus (CONT) 3h after plating. Neurites of infected 1 DIV (A) or 2 DIV (B) neurons were traced, counted and plotted as a function of neurite order number. 1st order neurites are defined as the segments extending directly from the cell soma. Branches correspond to 2nd order and higher neurites (see illustration in Fig. 1B). Branches correspond to 2nd order and higher neurites (n=30 neurons per treatment; *p<0.05). The data shown represent the mean ± S.E.M. (C) Examples of hippocampal neurons expressing GFP (Cont), wild type FE65 (WT), FE65-mPID2 (mPID2) and FE65-mWW (mWW). Scale bar equals 10 μm.
The FE65 mutants could affect neurite branching by altering the localization of the APP/FE65 complex within growth cones. To test this we transfected 1-day-old hippocampal cultures with GFP-fusion proteins of FE65-WT and FE65-mPID2. Cells were fixed and stained 1 day later. We detected no differences in the localization of either APP or GFP-FE65 between the WT- and mPID2-transfected neurons (Figs. 3A, B, D). Both constructs still showed growth cone enrichment and co-localized with APP. FE65 was found consistently in growth cones extending from the neurite shaft as well (Fig. 3E). This suggests that the effect of the FE65 mutants is on complex formation within the growth cone rather than on trafficking to or from the growth cone.
Fig. 3.

Expression of the FE65 mutants does not alter the localization of either FE65 or APP. Representative images of growth cones expressing GFP-fusion constructs (green) of WT-FE65 (A), FE65-mPID2 (B) and FE65-mWW (C) labeled for βIII-tubulin (blue) and actin (red) or APP (D) TF: transfection. (E) FE65 (green) is consistently found in growth cones extending from the neurite shaft. Scale bar equals 10 μm.
The APP/FE65 complex must interact with a third protein to inhibit branching
The results described above suggest that the APP/FE65 complex modulates neurite branching. If the PID2 mutation increases branch formation by sequestering an additional protein away from the APP/FE65-containing tripartite complex, then preventing the third protein in the complex from interacting with FE65 should have a similar effect on neurite branching. To test the possibility that this effect may be mediated through formation of a tripartite complex with a WW domain-interacting protein, we infected neurons with adenovirus expressing FE65 with two point mutations in the WW domain (W228F and P231A; mWW; Fig. 1A), which were previously shown to prevent the association between FE65 and Mena (Ermekova et al., 1997), and fixed at 48 h in vitro. As shown in Fig. 2B, this WW mutation caused a similar increase in neurite branching to that obtained by preventing FE65 from binding to APP (~2-fold increase in branching for WW mutants as compared to control; mean second order neurites: CONT=3.0±0.5; mWW=5.0±0.4; mean third order neurites: CONT=1.0±0.3; mWW=2.3±0.4; p<0.05; see illustration in Fig. 1B; also compare neuron images in Fig. 2C). Once again there was no difference in the localization of APP and FE65 upon transfection with FE65-mWW as compared to FE65-WT-transfected neurons (Figs. 3A, C, D) suggesting that the mWW mutant caused the WW domain-binding protein to dissociate from the complex with APP and FE65 without altering the localization of either FE65 or APP.
Axon, but not dendrite, branching is regulated by APP/FE65 complex early in neurite development
To determine whether the observed branching effect was on the axon, dendrites or both, infected neurons were fixed at 48 h in vitro and cells in which an axon was clearly distinguishable were analyzed. There was a significant increase in axon branching in the presence of the PID2 and WW mutants as assessed by the number of branch points per axon (WT=2.1± 0.4, mPID2=3.7±0.3, mWW=5.0±0.5; n=30; p<0.05 for mPID2; p<0.001 for mWW; Fig. 4A). Although a similar trend was seen in the dendrites, the differences failed to reach significance (p=0.12; Fig. 4B). It should be noted that in these cultures dendrites do not mature until 4–5 days in vitro. Therefore it is likely that the APP/FE65-containing tripartite complex would become involved in dendrite branching at later developmental stages.
Fig. 4.
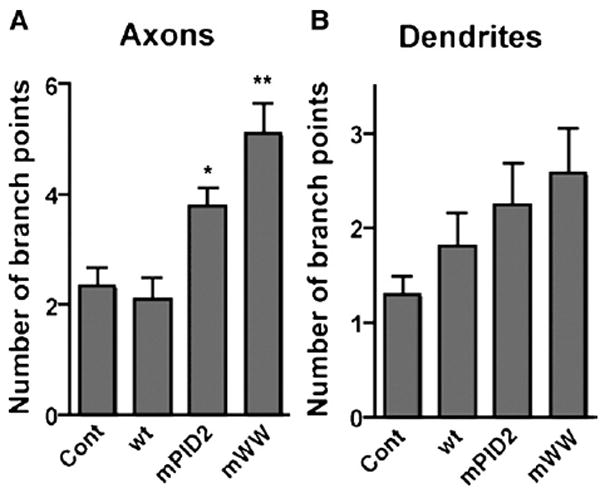
Interaction-deficient FE65 increases axonal but not dendritic branching early in neuronal development. (A) FE65 mutants that cannot interact with APP (mPID2) or its WW domain-interacting partner (e.g. Mena or a related protein; mWW) increase axon branching. Neurites of 2 DIV neurons with defined axons were traced and number of branch points per axon counted (n=30; *p<0.05, **p<0.01). (B) FE65 mutants have no significant affect on dendrite branching in 2 DIV neurons. The number of branch points per dendrite was counted. The means±S.E.M. are indicated in each plot.
Axonal branches form in vitro at sites where growth cones pause for an extended time period. At this point the growth cone enlarges and rearranges (Szebenyi et al., 1998). If either the duration or frequency of pausing were increased, we would expect the average length of axonal segments between branch points to be shorter. Consistent with this, the axon segments were significantly shorter in the presence of either the PID2 mutant or the WW mutant (Fig. 5A; p<0.05 for mPID2; p<0.001 for mWW). FE65 mutants had no effect on total neurite output (expressed as total neurite length; p=0.265; Fig. 5B) or total axon length (including all branches; p=0.28; Fig. 5C), suggesting that FE65 mutants affect branching by altering the frequency or duration of growth cone pausing but not the overall rate of neurite extension. The data are therefore consistent with APP/FE65 complex being most important for neurite branching rather than elongation. We did not observe significant differences in growth cone sizes in our cultures between neurons expressing wild type or mutant FE65, probably because growth cone enlargements are transient and may not be readily detectable in fixed cultures. Nevertheless, the data demonstrate that expression of the PID2 or WW mutant significantly increases axon branching but not overall neurite growth.
Fig. 5.

Interaction-deficient FE65 increases average axon segment length but not total neurite or axon length. (A) FE65 mutants that cannot interact with either APP [FE65-mPID2 (mPID2)] or Mena [FE65-mWW (mWW)] (Fig. 1A) decrease the average axon segment length. Axon segment length is assessed by dividing the total axon length (shown in C) by the number of segments per axon (n=30; *p<0.05, **p<0.001). The data shown represent the mean±S.E.M. The data are representative of several such experiments with similar results. (B) The FE65 PID2 mutants illustrated in Fig. 1A do not alter total neurite length. Neurons were infected with adenovirus expressing wild type FE65 (WT), FE65-ΔPID2 (ΔPID2), FE65-mPID2 (mPID2) or GFP virus (CONT) 3 h after plating. Neurites of infected 1 DIV were traced and measured. Total neurite length corresponds to the sum of all segments per process, including the segment extending directly from the soma (see illustration in Fig. 1B). The data are representative of several such experiments with similar results. (C) FE65 mutants do not affect total axon length. Total axon length, the sum of the lengths of all axon segments, was measured in neurons expressing wild type FE65 (WT), FE65-mPID2 (mPID2), FE65-mWW (mWW) or empty virus (CONT).
Discussion
In this study we provide evidence that a macromolecular complex containing APP and FE65 negatively regulates axon branching early in neuronal development. We show that disrupting the interaction between APP and FE65 in cultured hippocampal neurons caused a significant increase in axonal branching without affecting the total axon length. Inhibiting the interaction between FE65 and a third protein that binds to the WW domain of FE65 produced a similar effect on neurite branching. The increase in axonal branching appeared to be a result of altered growth cone pausing behavior since both the PID2 and WW mutants significantly reduced axon segment length without affecting total neurite output. The localizations of APP and FE65 were unaltered upon expression of the FE65 mutants, suggesting that the effect on branching is through variations in complex formation within the growth cone rather than aberrant targeting of either protein to the growth cone.
Our data are consistent with the FE65 mutants either increasing the amount of time the growth cone pauses leading to branch formation or the probability that pausing would result in branching. Since axonal branches form at choice points where the primary growth cone pauses for an extended time period (Szebenyi et al., 1998), our results support a model in which the complex of APP, FE65 and an additional protein normally results in a fast-moving growth cone that pauses less often or for a shorter time period and hence reduces branch formation. When this complex is disrupted either by sequestering APP away from the third protein in the presence of the WW mutant or by sequestering the third protein away from APP in the presence of the PID2 mutant, the growth cone slows down and pauses more often or longer resulting in increased branch formation. While it is possible that the dominant-negative FE65 mutants may disrupt other APP-containing macromolecular complexes that do not contain FE65, the fact that both the PID2 and the WW mutants had the same effect on branching with the same magnitude supports the idea that the two mutants are disrupting the same complex.
FE65 was previously shown to bind to the growth cone enriched protein Mena, and the two point mutations in the FE65 WW domain used here were shown to disrupt this interaction (Ermekova et al., 1997). This makes Mena and the Ena/VASP family of proteins likely candidates for the third protein in the APP/FE65-containing tripartite complex. Strong support for a role of APP/FE65/Mena in early brain development is provided by the study of FE65 and FE65L1 double knockout mice. These mice are impaired in pial basement membrane integrity, neural positioning and the establishment of normal axonal projections during cortical development (Guenette et al., 2006). Remarkably, the phenotype of the FE65/FE65L1 null mice is very similar to that of mutant mice lacking all three members of the APP family (Herms et al., 2004) as well as to Mena null mice (Lanier et al., 1999).
Although the FE65 WW domain may interact with proteins other than Mena, several additional observations support the hypothesis that Mena is likely to be the third partner in an APP/FE65 tripartite complex that regulates neurite branching. First, FE65 can interact with APP and Mena simultaneously, and all three proteins co-localize within axonal growth cones (Sabo et al., 2001, 2003). In addition, previous reports support a role for Mena in growth cone pausing and branching. For example, when Mena was depleted from the growth cone as a result of expressing a dominant-negative form of the ADP-ribosylation factor (ARF) nucleotide-binding site opener (ARNO) or dominant negative ARF6, neurite branching significantly increased (Hernandez-Deviez et al., 2004). Increasing the expression of Mena at the growth cone membrane also caused an increase in axon branching (Lebrand et al., 2004). Finally, early studies in Drosophila showed that growth cones of Ena mutants bypass choice points where they would normally pause (Gertler et al., 1995) suggesting that Mena is required for pausing behavior (Lanier and Gertler, 2000). In the current study, we have not changed the localization or expression of Mena directly, but rather its ability to interact with local macro-molecular complexes containing FE65. It is therefore likely that Mena may have differing effects on axon branching and extension depending on which protein complex it associates with.
A recent study in Drosophila demonstrated that both the Drosophila APP homologue APPL and exogenously expressed human APP induce axonal arborization post-developmentally in adult fly brains (Leyssen et al., 2005). This effect was dependent on the interaction between APP and the Abelson tyrosine kinase (Abl) through the adaptor protein Dab, which binds to the same site in APP as FE65. Dab may substitute here for FE65 since there does not appear to be an FE65 homologue in Drosophila. Nevertheless these findings are consistent with the idea that APP-containing complexes play an important role in axon branching and arborization.
Axonal loss, synaptic disconnection and aberrant neuritic sprouting correlate with dementia in Alzheimer’s disease (AD) suggesting that neurite sprouting in AD may be associated with disrupted axonal and dendritic remodeling and synapse loss (Geddes et al., 1986). The interaction of APP with the FE65-containing complex and their role in neurite growth may contribute to this AD pathology. First, the increased APP cleavage associated with AD will disrupt the APP/FE65/Mena complex, resulting in increased axonal branching or sprouting. In addition, the reduced neuronal FE65 expression seen in the frontal and temporal cortices in AD brains (Hu et al., 2000) would further disrupt the complex, exacerbating axonal sprouting. Thus, if the APP/FE65 complex retains its role in axon branching into adulthood, disruption of this complex might contribute to the progression of cognitive decline seen in AD.
Interestingly, FE65 modulates APP processing, increasing production of β-amyloid (Aβ) (Sabo et al., 1999), which is the major constituent of the plaques found in AD brains. It has been postulated that insoluble Aβ deposits may cause sprouting by transforming normal neuronal cytoskeletal proteins (Dickson et al., 1999). In future studies, it would be interesting to determine whether the APP/FE65/Mena complex influences branching by regulating the proteolytic processing of APP or whether these two functions of the APP/FE65 interaction are independent.
In summary, we propose a mechanism by which FE65 in complex with APP and Mena and/or another member of the Ena/VASP family may play a role in early neuronal development as well as contribute to the neurodegenerative pathology in Alzheimer’s disease. This is the first study demonstrating a role for FE65 in neurite branching through its interaction with APP.
Experimental methods
Cell cultures
All media and supplements were purchased from Invitrogen. Poly-d-lysine was purchased from Sigma. Primary hippocampal and glial cultures were prepared from E16 and P0 Swiss Webster mice, respectively as described for E18 and P0 rat (Banker, 1998). Neuronal cells were plated on poly-d-lysine-coated 15 mm diameter coverslips at 50,000 cells/coverslip in plating medium (MEM with 10% horse serum, 3% glucose, 1 mM pyruvate, 10 mM Hepes). After 3 h, the medium was changed to neurobasal medium supplemented with B27 supplement, which had been pre-conditioned for 2 days by astroglial cultures. At this point the cells were also infected with the various viruses.
Adenovirus construction and infection
Site-directed mutagenesis of FE65 was performed by 2-step PCR using wild type FE65 in pcDNA3 (Sabo et al., 1999) as a template. Viral constructs were prepared using a kit from Q-biogene (AdEasy™) according to the manufacturer’s instructions. The adenovirus shuttle vector pAdTrack-CMV was kindly provided by Dr. Bert Vogelstein (HHMI, Johns Hopkins University). The FE65 genes were excised from pcDNA3 and ligated into the shuttle vector, which was then used to form the virus by recombination with the viral backbone vector (pAdEasy-1). Neurons were infected at an MOI of 20. All viruses express EGFP under the control of a separate CMV promoter to allow detection of infected cells.
Neurite outgrowth assessment
Infected neurons were fixed 24 or 48 h after plating in 4% para-formaldehyde in PHEM buffer (60 mM PIPES (pH 7), 25 mM HEPES (pH 7), 10 mM EGTA, 2 mM MgCl2, 4% sucrose) for 30 min at 37 °C. Cells were permeabilized and stained for βIII-tubulin to distinguish neurons from glia. Infected cells were visualized using a Zeiss Axiophot Photomicroscope fluorescence microscope (Carl Zeiss, Thornwood, NY), attached to a CCD camera (Diagnostic Instruments). All images were captured using the same microscope/camera setup. Images were collected using a 40×Plan-Neofluar oil objective and analyzed using Neurolucida (Microbrightfield). The term “neurite” here refers to the branch segments including the one extending directly from the cell body (the 1st order neurite). The average neurite length, number of neurites, branch number etc. were analysed using Neuroexplorer software (MBF Bioscience) and subsequently ANOVA. At least 30 neurons from each treatment were traced and analysed. Each experiment was conducted at least 3 times with similar results.
Transfection of neurons
For transfection with FE65-GFP, FE65-mPID2-GFP and FE65-mWW-GFP constructs, neurons were dissociated from P1 rat hippocampi and plated on astrocyte monolayers. In this case, neurons were maintained in MEM with N2 supplement, glucose, 1% ovalbumin, 1 mM pyruvate, and 10 mM Hepes, as described (Banker, 1998). Neurons were transfected 24 h after plating using calcium phosphate. 48 h after transfection, neurons were fixed, permeabilized and labelled with anti-GFP (chicken, 1:2500; Chemicon), βIII-tubulin (mouse (TUJ-1), 1:1000; Abcam)) and APP (rabbit, 369, 1:200) antibodies. Secondary antibodies were goat anti-chicken Alexa-488, goat anti-rabbit Alexa-405, and goat anti-mouse Alexa-647 (1:400; Molecular Probes). Images were collected with a Zeiss LSM 510 confocal system using a 40× (1.3 NA) objective. Lasers were Diode 405, Ar 488, HeNe 633.
Acknowledgments
This work was supported by a grant from the American Health Assistance Foundation to AFI.
References
- Allinquant B, Hantraye P, Mailleux P, Moya K, Bouillot C, Prochiantz A. Downregulation of amyloid precursor protein inhibits neurite outgrowth in vitro. J Cell Biol. 1995;128:919–927. doi: 10.1083/jcb.128.5.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banker G, Goslin K. Culturing Nerve Cells. Massachusetts Institute of Technology (2) 1998 [Google Scholar]
- Borg JP, Ooi J, Levy E, Margolis B. The phosphotyrosine interaction domains of X11 and FE65 bind to distinct sites on the YENPTY motif of amyloid precursor protein. Mol Cell Biol. 1996;16:6229–6241. doi: 10.1128/mcb.16.11.6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson TC, King CE, McCormack GH, Vickers JC. Neurochemical diversity of dystrophic neurites in the early and late stages of Alzheimer’s disease. Exp Neurol. 1999;156:100–110. doi: 10.1006/exnr.1998.7010. [DOI] [PubMed] [Google Scholar]
- Ermekova KS, Zambrano N, Linn H, Minopoli G, Gertler F, Russo T, Sudol The WW domain of neural protein FE65 interacts with proline-rich motifs in Mena, the mammalian homolog of Drosophila enabled. J Biol Chem. 1997;272:32869–32877. doi: 10.1074/jbc.272.52.32869. [DOI] [PubMed] [Google Scholar]
- Geddes JW, Anderson KJ, Cotman CW. Senile plaques as aberrant sprout-stimulating structures. Exp Neurol. 1986;94:767–776. doi: 10.1016/0014-4886(86)90254-2. [DOI] [PubMed] [Google Scholar]
- Gertler FB, Doctor JS, Hoffmann FM. Genetic suppression of mutations in the Drosophila Abl proto-oncogene homolog. Science. 1990;248:857–860. doi: 10.1126/science.2188361. [DOI] [PubMed] [Google Scholar]
- Gertler FB, Comer AR, Juang JL, Ahern SM, Clark MJ, Liebl EC, Hoffmann FM. Enabled, a dosage-sensitive suppressor of mutations in the Drosophila Abl tyrosine kinase, encodes an Abl substrate with SH3 domain-binding properties. Genes Dev. 1995;9:521–533. doi: 10.1101/gad.9.5.521. [DOI] [PubMed] [Google Scholar]
- Goh KL, Cai L, Cepko CL. Ena/VASP proteins regulate cortical neuronal positioning. Curr Biol. 2002;12:565–569. doi: 10.1016/s0960-9822(02)00725-x. [DOI] [PubMed] [Google Scholar]
- Guenette SY, Chen J, Ferland A, Haass C, Capell A, Tanzi RE. hFE65L influences amyloid precursor protein maturation and secretion. J Neurochem. 1999;73:985–993. doi: 10.1046/j.1471-4159.1999.0730985.x. [DOI] [PubMed] [Google Scholar]
- Guenette S, Chang Y, Hiesberger T, Richardson JA, Eckman CB, Eckman EA, Hammer RE, Herz J. Essential roles for the FE65 amyloid precursor protein-interacting proteins in brain development. EMBO J. 2006;25:420–431. doi: 10.1038/sj.emboj.7600926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herms J, Anliker B, Heber S, Ring S, Fuhrmann M, Kretzschmar H, Sisodia S. Cortical dysplasia resembling human type 2 lissencephaly in mice lacking all three APP family members. EMBO J. 2004;23:4106–4115. doi: 10.1038/sj.emboj.7600390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Deviez DJ, Roth MG, Casanova JE. ARNO and ARF6 regulate axonal elongation and branching through downstream activation of phosphatidylinositol 4-phosphate 5-kinase alpha. Mol Biol Cell. 2004;15:111–120. doi: 10.1091/mbc.E03-06-0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q, Jin LW, Starbuck MY, Martin MY. Broadly altered expression of the mRNA isoforms of FE65, a facilitator of beta amyloidogenesis, in Alzheimer cerebellum and other brain regions. J Neurosci Res. 2000;60:73–86. doi: 10.1002/(SICI)1097-4547(20000401)60:1<73::AID-JNR8>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Hung AY, Koo EH, Haass C, Selkoe DJ. Increased expression of beta-amyloid precursor protein during neuronal differentiation is not accompanied by secretory cleavage. Proc Natl Acad Sci U S A. 1992;89:9439–9443. doi: 10.1073/pnas.89.20.9439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin LW, Ninomiya H, Roch JM, Schubert D, Masliah E, Otero DA. Peptides containing the RERMS sequence of amyloid beta/A4 protein precursor bind cell surface and promote neurite extension. J Neurosci. 1994;14:5461–5470. doi: 10.1523/JNEUROSCI.14-09-05461.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier LM, Gertler FB. From Abl to actin: Abl tyrosine kinase and associated proteins in growth cone motility. Curr Opin Neurobiol. 2000;10:80–87. doi: 10.1016/s0959-4388(99)00058-6. [DOI] [PubMed] [Google Scholar]
- Lanier LM, Gates MA, Witke W, Menzies AS, Wehman AM, Macklis JD, Kwiatkowski D, Soriano P, Gertler FB. Mena is required for neurulation and commissure formation. Neuron. 1999;22:313–325. doi: 10.1016/s0896-6273(00)81092-2. [DOI] [PubMed] [Google Scholar]
- LeBlanc AC, Kovacs DM, Chen HY, Villare F, Tykocinski M, Autilio-Gambetti L, Gambetti P. Role of amyloid precursor protein (APP): study with antisense transfection of human neuroblastoma cells. J Neurosci Res. 1992;31:635–645. doi: 10.1002/jnr.490310407. [DOI] [PubMed] [Google Scholar]
- Lebrand C, Dent EW, Strasser GA, Lanier LM, Krause M, Svitkina TM, Borisy GG. Critical role of Ena/VASP proteins for filopodia formation in neurons and in function downstream of netrin-1. Neuron. 2004;42:37–49. doi: 10.1016/s0896-6273(04)00108-4. [DOI] [PubMed] [Google Scholar]
- Leyssen M, Ayaz D, Hebert SS, Reeve S, De Strooper B, Hassan BA. Amyloid precursor protein promotes post-developmental neurite arborization in the Drosophila brain. EMBO J. 2005;24:2944–2955. doi: 10.1038/sj.emboj.7600757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majocha RE, Agrawal S, Tang JY, Humke EW, Morotta CA. Modulation of the PC12 cell response to nerve growth factor by antisense oligonucleotide to amyloid precursor protein. Cell Mol Neurobiol. 1994;14:425–437. doi: 10.1007/BF02088829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merdes G, Soba P, Loewer A, Bilic MV, Beyreuther K. Interference of human and Drosophila APP and APP-like proteins with PNS development in Drosophila. EMBO J. 2004;23:4082–4095. doi: 10.1038/sj.emboj.7600413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milward EA, Papadopoulos R, Fuller SJ, Moir RD, Small D, Beyreuther K. The amyloid protein precursor of Alzheimer’s disease is a mediator of the effects of nerve growth factor on neurite outgrowth. Neuron. 1992;9:129–137. doi: 10.1016/0896-6273(92)90228-6. [DOI] [PubMed] [Google Scholar]
- Moya KL, Benowitz LI, Schneider GE, Allinquant B. The amyloid precursor protein is developmentally regulated and correlated with synaptogenesis. Dev Biol. 1994;161:597–603. doi: 10.1006/dbio.1994.1055. [DOI] [PubMed] [Google Scholar]
- Perez RG, Zheng H, Van der Ploeg LH, Koo EH. The beta-amyloid precursor protein of Alzheimer’s disease enhances neuron viability and modulates neuronal polarity. J Neurosci. 1997;17:9407–9414. doi: 10.1523/JNEUROSCI.17-24-09407.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabo SL, Lanier LM, Ikin AF, Khorkova O, Sahasrabudhe S, Greengard P, Buxbaum JD. Regulation of beta-amyloid secretion by FE65, an amyloid protein precursor-binding protein. J Biol Chem. 1999;274:7952–7957. doi: 10.1074/jbc.274.12.7952. [DOI] [PubMed] [Google Scholar]
- Sabo SL, Ikin AF, Buxbaum JD, Greengard P. The Alzheimer amyloid precursor protein (APP) and FE65, an APP-binding protein, regulate cell movement. J Cell Biol. 2001;153:1403–1414. doi: 10.1083/jcb.153.7.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabo SL, Ikin AF, Buxbaum JD. The amyloid precursor protein and its regulatory protein, FE65, in growth cones and synapses in vitro and in vivo. J Neurosci. 2003;23:5407–5415. doi: 10.1523/JNEUROSCI.23-13-05407.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small DH, Nurcombe V, Ree G, Clarris H, Moi R, Beyreuther K, Masters CL. A heparin-binding domain in the amyloid protein precursor of Alzheimer’s disease is involved in the regulation of neurite outgrowth. J Neurosci. 1994;14:2117–2127. doi: 10.1523/JNEUROSCI.14-04-02117.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szebenyi G, Callaway JL, Dent EW. Interstitial branches develop from active regions of the axon demarcated by the primary growth cone during pausing behaviors. J Neurosci. 1998;18:7930–7940. doi: 10.1523/JNEUROSCI.18-19-07930.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]