

Abstract
Background
Infantile-onset glycogen storage disease type II (GSD-II; Pompe disease; MIM 232300) causes death early in childhood from cardiorespiratory failure in absence of effective treatment, whereas late-onset Pompe disease causes a progressive skeletal myopathy. The limitations of enzyme replacement therapy could potentially be addressed with adeno-associated virus (AAV) vector-mediated gene therapy.
Methods
AAV vectors containing tissue-specific regulatory cassettes, either liver-specific or muscle-specific, were administered to 12 and 17 month old Pompe disease mice to evaluate the efficacy of gene therapy in advanced Pompe disease. Biochemical correction was evaluated through GAA activity and glycogen content analyses of the heart and skeletal muscle. Western blotting, urinary biomarker, and Rotarod performance were evaluated following vector administration.
Results
The AAV vector containing the liver-specific regulatory cassette secreted high-level hGAA into the blood and corrected glycogen storage in the heart and diaphragm. The biochemical correction of the heart and diaphragm was associated with efficacy, as reflected by increased Rotarod performance; however, the clearance of glycogen from skeletal muscles was relatively impaired, in comparison with younger Pompe disease mice. An alternative vector containing a muscle-specific regulatory cassette transduced skeletal muscle with high efficiency, but also failed to achieve complete clearance of accumulated glycogen. Decreased transduction of the heart and liver in older mice, especially in females, was implicated as a cause for reduced efficacy in advanced Pompe disease.
Conclusion
The impaired efficacy of AAV vector-mediated gene therapy in old Pompe disease mice emphasized the need for early treatment to achieve full efficacy.
Keywords: Glycogen storage disease type II, adeno-associated virus, acid alpha-glucosidase, acid maltase, Pompe disease
Introduction
Infantile-onset glycogen storage disease type II (glycogen storage disease type II; MIM 232300) causes death early in childhood from cardiorespiratory failure related to an underlying hypertrophic cardiomyopathy, if enzyme replacement therapy (ERT) is delayed or the patient fails to respond sustainably to ERT [1,2]. The deficiency of acid alpha-glucosidase (GAA; acid maltase; EC 3.2.1.20) in Pompe disease affects the heart and skeletal muscle primarily, and infants with Pompe disease develop profound weakness and hypotonia. Late-onset forms of Pompe disease feature progressive weakness without significant cardomyopathy, and patients with juvenile-onset Pompe disease typically become ventilator-dependent due to respiratory and or cardiac muscle involvement. Late-onset Pompe disease (MIM 232300) typically presents with proximal leg weakness that progresses to leg, arm, and accessory respiratory muscle weakness [1,3]. Premature death occurs from cardiorespiratory failure in these patients. The pathophysiology of Pompe disease stems from acid α-glucosidase (GAA) deficiency and glycogen accumulation in striated muscles; however, the heart is spared in late-onset Pompe disease [1]. The diagnosis of late-onset Pompe disease is complicated by its close resemblance to the muscular dystrophies, although muscle histology reveals unique glycogen vacuolization in Pompe disease [3].
GAA normally functions as an acid hydrolase that metabolizes lysosomal glycogen, and GAA deficiency causes lysosomal glycogen accumulation in virtually all tissues [4–6]. The availability of ERT with recombinant human (rh) GAA has prolonged survival and ameliorated the cardiomyopathy of infantile Pompe disease [7]; however, it remains to be determined if ERT will be efficacious in late-onset Pompe disease. Documented limitations of ERT in Pompe disease include the requirement for frequent intravenous infusions of high levels of GAA to achieve efficacy, and the possibility of humoral immunity [8]. The rhGAA doses were markedly higher than doses required for ERT in other lysosomal storage disorders, reflecting the high threshold for correction of GAA deficiency in the skeletal muscle of Pompe disease patients [7–9].
We have evaluated the efficacy of sustained hGAA production with adeno-associated virus (AAV) type 2 vector containing liver and muscle-specific regulatory cassettes, both pseudotyped with AAV type 8 (AAV2/8), in older Pompe disease mice. Partial correction was demonstrated with surrogate markers, including the reduction of lysosomal-associated membrane protein 2 (LAMP-2) in the heart and skeletal muscle.
Materials and methods
Preparation of AAV 2/8 vector
Briefly, 293 cells were transfected with the vector plasmid [10,12, 13], the AAV packaging plasmid p5E18-VD 2/820 (courtesy of Dr. James M. Wilson, University of Pennsylvania, Philadelphia, PA), and pAdHelper (Stratagene, La Jolla, CA). Cell lysate was harvested 48 hours following infection and freeze-thawed 3 times, and isolated by sucrose cushion pelleting followed by 2 cesium chloride gradient centrifugation steps. AAV stocks were dialyzed against 3 changes of Hanks buffer, and aliquots were stored at −80°C. The number of vector DNA containing-particles was determined by DNase I digestion, DNA extraction, and Southern blot analysis. All viral vector stocks were handled according to Biohazard Safety Level 2 guidelines published by the NIH.
Analysis of AAV 2/8 vector in vivo
The AAV vector stocks were administered intravenously (via the retroorbital sinus) in 12 month-old GAA-KO mice [11]. The AAV2/8 vector encoding highly secreted hGAA driven by a liver-specific regulatory cassette (AAV-SPhGAApA [6]) was administered intravenously to 12 month-old GAA-KO mice (5×1011 vector particles/mouse; n=5) or saline (n=4). An alternative AAV2/8 vector containing a muscle-specific regulatory cassette [12] was administered to 12 month-old GAA-KO mice (AAV-MHCK7hGAApA; 7×1011 vector particles/mouse; n=5). Two AAV2/8 vectors were administered simultaneously to 17 month-old GAA-KO mice (n=4), both AAV-LSPhGAApA (5×1011 vector particles/mouse; see [13]) and (AAV-MHCK7hGAApA; 7×1011 vector particles/mouse). At the indicated time points post-injection, plasma or tissue samples were obtained and processed as described below. All animal procedures were done in accordance with Duke University Institutional Animal Care and Use Committee-approved guidelines.
Rotarod testing was performed as described [14]. Urinary Glc4 concentrations were determined relative to creatinine by stable isotope-dilution electrospray tandem mass spectrometry as previously described.[15] GAA activity and glycogen content were analyzed as described [16]. Vector genome quanification was performed as described [12]. A P value of <0.05 indicated a significant difference between the observed values for each group of GAA-KO mice following AAV vector administration and the control group of PBS-injected GAA-KO mice.
Western blotting analysis of striated muscle homogenates was performed as described [14] using the hGAA monoclonal antibody (courtesy of Genzyme Corp., Framingham, MA), LAMP-2 rabbit polyclonal antibody (Abcam, Cambridge, MA), and GAPDH monoclonal antibody (Abcam, Cambridge, MA). The ELISA was performed as described [17]. All samples yielded absorbance values that were within the linear range of the assay at this dilution.
Results
Efficacy of an AAV vector in older Pompe disease mice
Twelve month-old GAA-KO mice were administered either the AAV vector encoding highly secreted hGAA driven by a liver-specific regulatory cassette (AAV-SPhGAApA [6]; 5×1011 vector particles/mouse; n=5) or saline (PBS; n=4), and monitored for 24 weeks to evaluate the efficacy of vector treatment in old Pompe disease mice. Multiple endpoints were evaluated over the 6 month duration of the study, including changes in Rotarod time, weight and the urinary biomarker, Glc4.
Efficacy was demonstrated during 24 weeks of observation following AAV2/8 vector administration by observing changes in the parameters evaluated. The absolute change in Rotarod time, the time mice were able to run on a rotating rod, reflected the increase in endurance for each mouse. Endurance increased for vector-treated mice during each of four intervals from 2 to 24 weeks following vector administration, in contrast to sham-treated controls that exhibited a slight decrease in Rotarod time (Fig. 1A: p<0.05 for each interval). Despite the increase in Rotarod times following vector administration, the mean time was much lower than that for wildtype mice at 15 months of age (85 +/− 18 sec versus 181 +/− 35 sec). Individual weights varied at the start of the study, and the mean weight for the two groups was similar; however, weight gain was greater for AAV vector-injected GAA-KO mice during each interval, in comparison with PBS-injected GAA-KO mice (Fig. 1B: p<0.05 for each interval).
Fig. 1. Endpoint analysis following AAV vector administration.

Twelve-month old male GAA-KO mice were injected intravenously with AAV-SPhGAApA (n=5, except for the last interval n=4) or PBS (n=3). A single AAV vector-treated mouse died in the sixth month of the study of an undetermined cause, at 18 months old when GAA-KO mice are reaching the end of their lifespan [35]. Mean +/− s.d shown, and significant differences indicated (*) based upon P < 0.05 calculated with an unpaired T-test with Welch's correction (A) Rotarod testing. The change in Rotarod time was calculated for each mouse during the indicated time intervals. (B) Weight. The change is weight was calculated for each mouse during the indicated time intervals. Each line represents an individual mouse. (C) Urinary biomarker, [Glc4], was analyzed for each mouse (n=4 in each group).
Urinary Glc4 has been developed as a noninvasive biomarker that correlated with lower muscle glycogen content in Pompe disease patients [18]. Surprisingly, urinary Glc4 decreased in PBS-injected old Pompe disease mice between 2 and 22 weeks (Fig. 1C). Importantly, Glc4 was significantly decreased in AAV vector-treated mice, in comparison with age-matched, PBS-treated mice at both 2 and 22 weeks following vector administration (Fig. 1C; p<0.05 at each time). Glc4 remained elevated in the urine of AAV-treated GAA-KO mice at 22 weeks, in comparison with 15 month-old wildtype mice (12.9 +/− 7.6 versus 4.5 +/− 0.7 mmol/mole creatinine). Therefore, the reduction in urinary Glc4 levels following AAV vector administration, in comparison with age-matched control Pompe mice, was another indication of partial efficacy from gene therapy
Biochemical correction of striated muscle
Therapy in lysosomal storage disorders, such as Pompe disease, has been advanced by the correction of an enzyme deficiency through receptor-mediated uptake of the introduced therapeutic enzyme from the blood, a strategy that underlies both ERT and gene therapy for Pompe disease [8,19]. The quantification of GAA activity in plasma and tissues demonstrated the sustained production of GAA with the AAV vector in older GAA-KO mice (Fig. 2). Plasma GAA activity was significantly elevated over the background level by the AAV vector, although a slight decline was observed between 2 and 22 weeks post-injection (Fig. 2A). The slight loss of plasma GAA activity was not explained by anti-GAA antibody formation, because IgG was undetectable as analyzed by ELISA at 22 weeks (not shown).
Fig. 2. Biochemical correction with chimeric hGAA in GAA-KO mice.
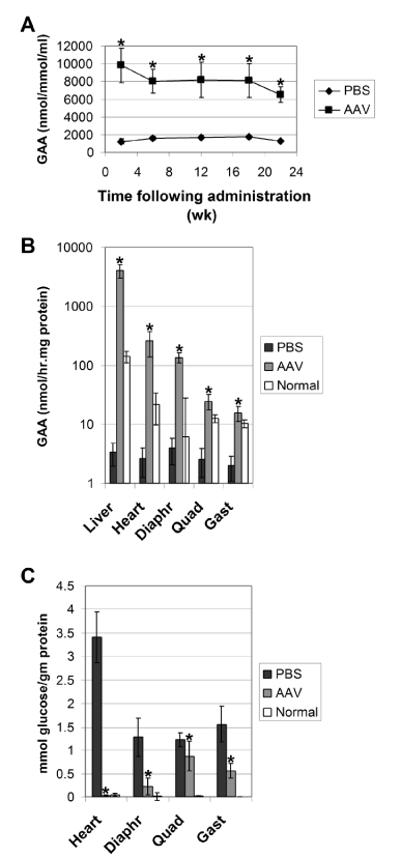
Mean +/−s.d. is shown. Control mice were age-matched, PBS-injected GAA-KO mice (n=4). Significant differences indicated (*), based upon P < 0.05 calculated with a two-tailed homoscedastic Student's t-test. (A) Plasma GAA was assayed for GAA-KO mice following administration of the AAV vector (n=5, except for the last time point n=4). (B) Tissue GAA was assayed for GAA-KO mice 24 weeks following administration of the AAV vector (n=4). (C) Glycogen content in tissues for mice in (B).
GAA activity was significantly elevated in the liver and striated muscle at 24 weeks following vector administration (Fig. 2B). Glycogen content was significantly reduced in striated muscle by the elevation of GAA activity accompanying AAV vector administration. The glycogen content was reduced to nearly normal levels in the heart and diaphragm, and significantly reduced in the gastrocnemius; however, the quadriceps retained higher glycogen accumulations (Fig. 2C).
The heart and diaphragm were cross-corrected very effectively by levels 12 to 22-fold above normal, respectively (Table 1). The AAV vector-transduced liver exhibited the highest GAA activity, where activity exceeded the level assayed in normal mice by 28-fold. As reported, skeletal muscle was resistant to cross-correction, including the quadriceps and gastrocnemius muscles (Table 1) [20,21]. The glycogen content was reduced proportionally more for the gastrocnemius than for the quadriceps at 18 months of age, in comparison with PBS-treated controls; moreover, the greater clearance of glycogen in the gastrocnemius occurred despite equivalent levels of GAA activity in both muscle groups following AAV vector administration (Table 1). The biochemical correction of muscles other than the quadriceps was approximately equivalent regardless of whether the vector was administered at 3 months or 12 months of age (Table 1).
Table 1.
Correction of glycogen storage in GAA-KO mice.1
| Vector | Age at vector administration | Quadriceps | Gastrocnemius | Diaphragm | Heart |
|---|---|---|---|---|---|
| AAV-SPhGAApA | 12 months | 33%* | 67% | 84% | 99% |
| 3 months 2 | 75% | 79% | 91% | 100% | |
| AAV-MHCK7hGAApA | 12 months | 40%* | 28% | 48%* | 95% |
| 3 months 3 | 82% | 36% | 82% | 98% |
Reduction compared to sham-treated GAA-KO mice. Significant difference in older, versus younger, GAA-KO mice indicated
p<0.05.
Previously reported GAA-KO mice following administration of an equivalent number of AAV2/8 vector particles at 3 month of age [6].
Previously reported GAA-KO mice following administration of an equivalent number of AAV2/8 vector particles at 3 month of age [12].
Western blot analysis of striated muscle revealed that lysosomal protein, LAMP-2 was markedly elevated in heart for PBS-injected GAA-KO mice, either 9 or 18 months of age. The ~110 kD signal for LAMP-2 was reduced to approximately the normal level observed for wildtype mice following AAV2/8 vector administration (Fig. 3; lanes 3–4 and 7–8, versus lane 9). Thus, LAMP-2 could be considered a surrogate marker for the reversal of accumulated lysosomal glycogen in the striated muscle in Pompe disease.
Fig. 3. Western blot detection of hGAA in tissues.
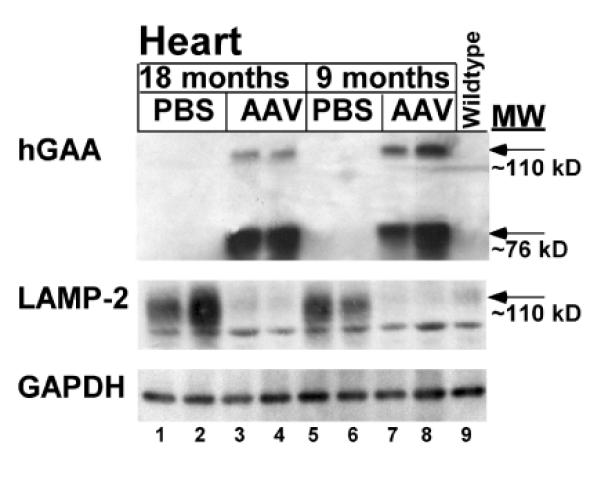
Tissues analyzed 24 weeks following administration of the AAV vector to 12 month-old (18 months) or 3 month-old (9 months) GAA-KO mice. Three different proteins were detected, hGAA, lysosomal associated membrane protein 2 (LAMP-2), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). GAPDH served as a control to indicate equal loading of each lane. Heart from GAA-KO mice, following administration of the AAV2/8 vector (AAV) or mock treatment (PBS). Each lane represents an individual mouse. Controls were a PBS-injected, age-matched GAA-KO mice, and a C57BL/6 mouse (wildtype).
Transduction of skeletal muscles with a muscle-specific transgene
An alternative AAV2/8 vector containing a muscle-specific regulatory cassette [12] was administered to 12 month-old GAA-KO mice (AAV-MHCK7hGAApA; 7×1011 vector particles/mouse; n=5). The follow-up period was abbreviated due to the demise of two vector-treated mice several weeks following vector administration; therefore, the degree of biochemical correction was evaluated in the remaining three mice at 16.5 months of age. Evidence of functional improvement was demonstrated by the Rotarod performance of the vector-treated mice, in comparison with age-matched PBS-injected GAA-KO mice at 16.5 months of age (117 +/− 14 sec versus 74 +/− 25 sec; p<0.05). The GAA activity was significantly increased in the liver, heart, and skeletal muscle of vector-treated mice, in comparison with sham-treated controls (Fig. 4A). However, the glycogen content was decreased significantly only in the heart and quadriceps of the vector-treated mice, in comparison with sham-treated GAA-KO mice, not in the gastrocnemius and diaphragm (Fig. 4B). Anti-GAA antibodies were detected within 6 weeks of AAV-MHCK7hGAA administration (not shown), which suggested that secretion and uptake of hGAA was unlikely to accomplish the correction of untransduced myofibers [12]. In comparison with young, 3 month old GAA-KO mice, the 12 month old mice had significantly less clearance of glycogen storage in the quadriceps and diaphragm (Table 1). Thus, advanced Pompe disease was associated with the resistance of critical muscle groups to correction by intramuscular hGAA production.
Fig. 4. Tissue analysis following AAV vector administration.
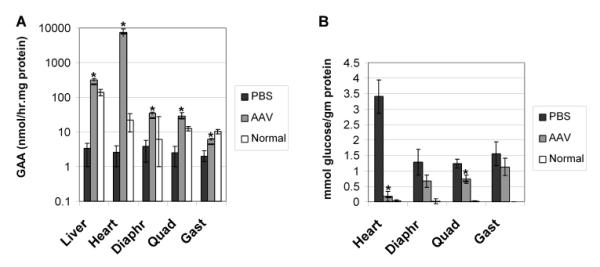
Twelve-month old male GAA-KO mice were injected intravenously with AAV-MHCK7hGAApA (n=5), or PBS (n=3). Two AAV vector-treated mouse died in the second month of the study of an undetermined cause, at an age when GAA-KO mice are reaching the end of their lifespan [35]. Mean +/− s.d shown, and significant differences indicated (*) based upon P < 0.05 calculated with an unpaired T-test with Welch's correction. (A) GAA activity in the indicated tissues. (B) Glycogen content for the mice shown in (A).
Transduction with AAV2/8 vectors was decreased in old GAA-KO mice
In an attempt to maximize biochemical correction in old GAA-KO mice, a group of 17 month-old GAA-KO mice were treated with two AAV2/8 vectors, AAV-LSPhGAApA (5×1011 vector particles/mouse) and AAV-MHCK7hGAApA (7×1011 vector particles/mouse). Somewhat surprisingly, GAA expression was increased only in the heart and liver, not in the diaphragm or skeletal muscles of these very old GAA-KO mice (Fig. 5A). The dosage administered for each of the two vectors had been sufficient to reduce the glycogen content of heart >90% and to partially correct glycogen storage in skeletal muscle of young GAA-KO mice [12,13]. Mice were evaluated early, at 6 weeks following vector administration, to allow valid comparison with age-matched PBS-injected GAA-KO mice and to avoid loss of mice due to age-related mortality. Previous studies revealed high-level hGAA expression with AAV vectors within two to three weeks following AAV 2/8 or 2/6 vector administration, indicating that vector transduction was effective by 6 weeks post-administration [13,22]. However, glycogen content was not reduced in any of the muscles examined from these very old GAA-KO mice following dual vector administration, in comparision with PBS-injected 18 month-old GAA-KO mice (Fig. 5B).
Fig. 5. Tissue analysis following dual vector administration to 17 month-old GAA-KO mice.

Seventeen-month old female GAA-KO mice were injected intravenously with AAV-LSPhGAApA and AAV-MHCK7hGAApA (n=4). (A) GAA activity in the indicated tissues. (B) Glycogen content for the mice shown in (A). (C) Vector genome quantification by Realtime PCR. Detection of the hGAA cDNA in GAA-KO mice following AAV-SPhGAApA administration at 3 months old (Young, n=4) or 12 months old (Old, n=4 for liver, n=2 for heart), or AAV-LSPhGAApA and AAV-MHCK7hGAApA co-administration at 17 months old (Very Old, n=4). Mean +/− s.d shown, and significant differences indicated by (*) indicating p < 0.05, (**) indicating p<0.01, and (***) indicating p<0.001 as calculated with an unpaired T-test.
An explanation for the lack of biochemical correction in the mice treated at 17 months was revealed by vector genome quantification (Fig. 5C). Vector genomes were quantified by Realtime PCR to detect the hGAA cDNA in AAV-treated mice, which revealed transduction with either of the AAV2/8 vectors administered. The transduction of liver was significantly lower following treatment of 12 month-old mice with AAV-SPhGAApA (Old; p<0.05) or 17 month-old mice with both AAV-LSPhGAApA and AAV-MHCK7hGAApA (Very Old; p<0.001), in comparison with three month-old (Young) GAA-KO mice following administration of AAV-SPhGAApA (Fig. 5C). The transduction of the heart was also reduced in 17 month-old mice with both AAV-LSPhGAApA and AAV-MHCK7hGAApA (Very Old; p<0.001), in comparison with three month-old (Young) GAA-KO mice following administration of only AAV-SPhGAApA (Fig. 5C). The 17 month-old mice were female, as no male very old GAA-KO mice were available, and AAV2/8 vectors previously were demonstrated to transduce liver less efficiently in female Pompe disease mice [17]. Nonetheless, the level of transduction was also significantly reduced in the liver of 12 month-old male mice (Old, p<0.05), in comparison with young male mice, following administration of AAV-SPhGAApA (Fig. 5C); therefore, decreased transduction in the liver due to advanced age demonstrated one factor underlying the low efficacy of AAV vector-mediated gene therapy in old mice.
Discussion
Introduced hGAA had sustained efficacy in young Pompe disease mice, when expressed either from a liver-specific transgene or an LSP-containing AAV vector [10,13,23]. In older Pompe disease mice the skeletal muscles were more resistant to the correction of accumulated glycogen by either ERT or transgene-expressed hGAA, which failed to correct the type II fibers of skeletal muscle [23,24]. Sustained hGAA expression with an AAV vector has now demonstrated the partial correction of all striated muscles examined in older Pompe disease mice, with the notable exception of the quadriceps. The quadriceps were resistant to correction in a short-term study with a second generation Ad vector, which raises the concern that very weak proximal leg muscles in late-onset Pompe disease patients will not respond to gene therapy with AAV or Ad vectors [3,20,21,25]. The current study evaluated an AAV vector encoding a chimeric, highly secreted hGAA created by substitution of the human α-1-antitrypsin leader sequence for that of native hGAA in 12 month-old GAA-KO mice [10]. This modified hGAA achieved higher plasma levels of hGAA, lower liver hGAA activity, and high efficacy in young GAA-KO mice in an earlier study. Administration of the above-mentioned AAV vector has now confirmed the increased resistance of proximal leg muscles to cross-correction with introduced hGAA, although the responsiveness of heart and diaphragm to sustained, liver-restricted hGAA production led to functional recovery through improved endurance and weight gain. A second vector, containing a muscle-specific regulatory cassette to drive high-level hGAA expression, was less efficacious than the first, reducing in the diaphragm and gastrocnemius much less efficiently than in younger Pompe disease mice. One significant age-related factor was identified in the current study, namely the progressive decrease in transduction of target tissues with the AAV2/8 vectors in older mice.
Preclinical studies in tolerant Pompe disease mice have illuminated the high dosage requirements for ERT in this disorder. Tolerant GAA-KO mice demonstrated resistance to the correction of glycogen storage in skeletal muscle, as compared to the heart and diaphragm [23]. Specifically, skeletal muscle containing type I myofibers responded to ERT, in contrast to type II myofibers that were resistant to correction [23,25]. Type II myofibers exhibited abnormal autophagy in Pompe disease mice, resulting in the accumulation of glycogen-filled endosomes as well as lysosomes [26,27].
Gene therapy in old Pompe disease mice has been attempted through transient production of hGAA with a second-generation adenovirus vector, confirming the relative resistance of skeletal muscle to correction [21]. When the glycogen content of striated muscle was analyzed at 17 days following vector administration, the heart retained significant residual stored glycogen in comparison to the current study. The above-mentioned adenovirus vector provoked immune responses that attenuated the efficacy of introduced hGAA at later time-points [28]. By contrast, AAV2/8 vectors containing a liver-specific regulatory cassette evaded the immune responses against hGAA with accompanying long-term efficacy in young Pompe disease mice [10,13]. Thus, the reduced efficacy of AAV vector-mediated gene therapy in old GAA-KO mice does not stem from immune responses against hGAA per se.
The AAV2/8 vector containing a muscle-specific regulatory cassette to drive hGAA expression previously reduced glycogen accumulations in young GAA-KO mice by >95% in heart and >75% in the diaphragm and quadriceps [12]. This vector has now been administered to 12 month-old GAA-KO mice, and glycogen content was reduced by only ~50%. Not only impaired autophagy, but also reduced receptor mediated uptake of hGAA, lower transgene expression, or decreased transduction could underlie the low efficacy of AAV vectors in old GAA-KO mice. Receptor-mediated uptake of hGAA by skeletal muscle could affect the efficacy of muscle-targeted gene therapy, as it does for liver-targeted gene therapy, and decreased expression of the cation-independent mannose-6-phosphate receptor in skeletal muscle could complicate gene therapy in advanced Pompe disease [29–31].
Decreased AAV vector-mediated gene therapy was identified in the current study through vector genome quantification, which revealed age-related decreases in the transduction of the liver with AAV2/8 vectors. Inclusion of 17 month-old female GAA-KO mice revealed a complete absence of efficacy in this group, the oldest GAA-KO mice studied to date. Ten-month old GAA-KO mice responded to an AAV2/8 vector with biochemical correction of lysosomal storage of glycogen, although no functional improvement was demonstrated in that earlier study [32]. We found that liver GAA transduction was decreased two-fold in young female GAA-KO mice, in comparison with male GAA-KO mice, which does not seem to explain the complete lack of efficacy observed in these very old female mice [14]. The liver is relatively spared in Pompe disease, and thus factors other than lysosomal glycogen accumulation must be considered as a cause for reduced transduction in old mice [1,3]. The receptor binding and uptake of AAV8 has not been fully delineated, and therefore studies to determine any age-related declines in AAV2/8 transduction are not yet feasible. The level of transduction in the liver is a critical factor in determining the efficacy of AAV vectors containing liver-specific regulatory cassettes, which implies that AAV vectors should be further evaluated in old mice given that such vectors might be considered for clinical trials in adult patients with other lysosomal storage disorders or hemophilia [33,34].
The current liver-targeted approach has the significant advantage of long-term, high-level hGAA expression that could explain improved biochemical correction of the heart and skeletal muscles in old GAA-KO mice, in comparison to previous reports of adenovirus vector-mediated gene therapy, liver-specific transgene expression, or ERT [21,23,27]. These experiments confirmed that delayed treatment of Pompe disease caused irreversible glycogen accumulation in the quadriceps despite, the correction of GAA deficiency at physiological levels, although the accumulation of endolysosomes in other muscles was still possible in older Pompe disease mice. Taken together, these data confirm that early treatment would be preferable in Pompe disease, either through gene therapy or ERT. In summary, the reduction of glycogen and lysosomal accumulations demonstrated here, especially prominent in the heart, confirmed that sustained expression of hGAA could partially reverse signature biochemical defects even in advanced Pompe disease; however, glycogen storage in very old femaile Pompe disease mice was completely resistant to correction.
Acknowledgements
This work was supported by NIH Grant R01 HL081122-01A1 from the National Heart, Lung, and Blood Institute. DDK was supported by the Muscular Dystrophy Association and Genzyme Corporation. B.S. was supported by a Development Grant from the Muscular Dystrophy Association. GAA-KO mice were provided courtesy of Dr. Nina Raben at the National Institutes of Health (Bethesda, MD). The AAV8 packaging plasmid, p5E18-VD 2/8, was provided courtesy of Dr. James M. Wilson at the University of Pennsylvania (Philadelphia, PA).
References
- 1.Hirschhorn R, Reuser AJJ. Glycogen Storage Disease Type II: Acid α-Glucosidase (Acid Maltase) Deficiency. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Basis for Inherited Disease. 8th ed. McGraw-Hill; New York: 2001. pp. 3389–419. [Google Scholar]
- 2.Kishnani PS, Hwu WL, Mandel H, Nicolino M, Yong F, Corzo D. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr. 2006;148:671–6. doi: 10.1016/j.jpeds.2005.11.033. [DOI] [PubMed] [Google Scholar]
- 3.Engel AG, Gomez MR, Seybold ME, Lambert EH. The spectrum and diagnosis of acid maltase deficiency. Neurology. 1973;23:95–106. doi: 10.1212/wnl.23.1.95. [DOI] [PubMed] [Google Scholar]
- 4.Martiniuk F, Mehler M, Tzall S, Meredith G, Hirschhorn R. Sequence of the cDNA and 5'-flanking region for human acid alpha-glucosidase, detection of an intron in the 5' untranslated leader sequence, definition of 18-bp polymorphisms, and differences with previous cDNA and amino acid sequences. DNA Cell Biol. 1990;9:85–94. doi: 10.1089/dna.1990.9.85. [DOI] [PubMed] [Google Scholar]
- 5.Hoefsloot LH, Hoogeveenwesterveld M, Reuser AJJ, Oostra BA. Characterization of the Human Lysosomal Alpha-Glucosidase Gene. Biochem J. 1990;272:493–7. doi: 10.1042/bj2720493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ponce E, Witte DP, Hirschhorn R, Huie ML, Grabowski GA. Murine acid alpha-glucosidase: cell-specific mRNA differential expression during development and maturation. Am J Pathol. 1999;154:1089–96. doi: 10.1016/s0002-9440(10)65361-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kishnani PS, Corzo D, Nicolino M, et al. Recombinant human acid {alpha}-glucosidase: Major clinical benefits in infantile-onset Pompe disease. Neurology. 2007;68:99–109. doi: 10.1212/01.wnl.0000251268.41188.04. [DOI] [PubMed] [Google Scholar]
- 8.Amalfitano A, Bengur AR, Morse RP, et al. Recombinant human acid alpha-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genet Med. 2001;3:132–8. [PubMed] [Google Scholar]
- 9.Desnick RJ. Enzyme replacement and enhancement therapies for lysosomal diseases. J Inher Met Dis. 2004;27:385–410. doi: 10.1023/B:BOLI.0000031101.12838.c6. [DOI] [PubMed] [Google Scholar]
- 10.Sun B, Zhang H, Benjamin DK, Jr., et al. Enhanced Efficacy of an AAV Vector Encoding Chimeric, Highly Secreted Acid alpha-Glucosidase in Glycogen Storage Disease Type II. Mol Ther. 2006;14:822–30. doi: 10.1016/j.ymthe.2006.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raben N, Nagaraju K, Lee E, et al. Targeted disruption of the acid alpha-glucosidase gene in mice causes an illness with critical features of both infantile and adult human glycogen storage disease type II. J Biol Chem. 1998;273:19086–92. doi: 10.1074/jbc.273.30.19086. [DOI] [PubMed] [Google Scholar]
- 12.Sun B, Young SP, Li P, et al. Correction of multiple striated muscles in murine Pompe disease through adeno-associated virus-mediated gene therapy. Mol Ther. 2008;16:1366–71. doi: 10.1038/mt.2008.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Franco LM, Sun B, Yang X, et al. Evasion of immune responses to introduced human acid alpha-glucosidase by liver-restricted expression in glycogen storage disease type II. Mol Ther. 2005;12:876–84. doi: 10.1016/j.ymthe.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 14.Sun B, Zhang H, Franco LM, et al. Efficacy of an adeno-associated virus 8-pseudotyped vector in glycogen storage disease type II. Mol Ther. 2005;11:57–65. doi: 10.1016/j.ymthe.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 15.Young SP, Stevens RD, An Y, Chen YT, Millington DS. Analysis of a glucose tetrasaccharide elevated in Pompe disease by stable isotope dilution-electrospray ionization tandem mass spectrometry. Anal Biochem. 2003;316:175–80. doi: 10.1016/s0003-2697(03)00056-3. [DOI] [PubMed] [Google Scholar]
- 16.Amalfitano A, McVie-Wylie AJ, Hu H, et al. Systemic correction of the muscle disorder glycogen storage disease type II after hepatic targeting of a modified adenovirus vector encoding human acid-alpha-glucosidase. Proc Natl Acad Sci U S A. 1999;96:8861–6. doi: 10.1073/pnas.96.16.8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun B, Chen YT, Bird A, et al. Packaging of an AAV vector encoding human acid alpha-glucosidase for gene therapy in glycogen storage disease type II with a modified hybrid adenovirus-AAV vector. Mol Ther. 2003;7:467–77. doi: 10.1016/s1525-0016(03)00022-4. [DOI] [PubMed] [Google Scholar]
- 18.An Y, Young SP, Kishnani PS, et al. Glucose tetrasaccharide as a biomarker for monitoring the therapeutic response to enzyme replacement therapy for Pompe disease. Mol Genet Metab. 2005;85:247–54. doi: 10.1016/j.ymgme.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 19.Cresawn KO, Fraites TJ, Wasserfall C, et al. Impact of humoral immune response on distribution and efficacy of recombinant adeno-associated virus-derived acid alpha-glucosidase in a model of glycogen storage disease type II. Hum Gene Ther. 2005;16:68–80. doi: 10.1089/hum.2005.16.68. [DOI] [PubMed] [Google Scholar]
- 20.Raben N, Lu N, Nagaraju K, et al. Conditional tissue-specific expression of the acid alpha-glucosidase (GAA) gene in the GAA knockout mice: implications for therapy. Hum Mol Genet. 2001;10:2039–47. doi: 10.1093/hmg/10.19.2039. [DOI] [PubMed] [Google Scholar]
- 21.Xu F, Ding EY, Migone F, et al. Glycogen storage in multiple muscles of old GSD-II mice can be rapidly cleared after a single intravenous injection with a modified adenoviral vector expressing hGAA. J Gene Med. 2005;7:171–8. doi: 10.1002/jgm.660. [DOI] [PubMed] [Google Scholar]
- 22.Sun B, Zhang H, Franco LM, et al. Correction of glycogen storage disease type II by an adeno-associated virus vector containing a muscle-specific promoter. Mol Ther. 2005;11:889–98. doi: 10.1016/j.ymthe.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 23.Raben N, Jatkar T, Lee A, et al. Glycogen stored in skeletal but not in cardiac muscle in acid alpha-glucosidase mutant (Pompe) mice is highly resistant to transgene-encoded human enzyme. Mol Ther. 2002;6:601–8. [PubMed] [Google Scholar]
- 24.Raben N, Fukuda T, Gilbert AL, et al. Replacing acid alpha-glucosidase in Pompe disease: Recombinant and transgenic enzymes are equipotent, but neither completely clears glycogen from type II muscle fibers. Mol Ther. 2005;11:48–56. doi: 10.1016/j.ymthe.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 25.Radojevic V, Humm AM, Rosler KM, Lauterburg T, Burgunder JM. Abnormal trafficking of sarcolemmal proteins in alpha-glucosidase deficiency. Acta Neuropathol (Berl) 2003;105:373–80. doi: 10.1007/s00401-002-0656-z. [DOI] [PubMed] [Google Scholar]
- 26.Fukuda T, Ewan L, Bauer M, et al. Dysfunction of endocytic and autophagic pathways in a lysosomal storage disease. Ann Neurol. 2006;59:700–8. doi: 10.1002/ana.20807. [DOI] [PubMed] [Google Scholar]
- 27.Fukuda T, Ahearn M, Roberts A, et al. Autophagy and mistargeting of therapeutic enzyme in skeletal muscle in pompe disease. Mol Ther. 2006;14:831–9. doi: 10.1016/j.ymthe.2006.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ding EY, Hodges BL, Hu H, et al. Long-term efficacy after [E1-, polymerase-] adenovirus-mediated transfer of human acid-alpha-glucosidase gene into glycogen storage disease type II knockout mice. Hum Gene Ther. 2001;12:955–65. doi: 10.1089/104303401750195917. [DOI] [PubMed] [Google Scholar]
- 29.Raben N, Danon M, Gilbert AL, et al. Enzyme replacement therapy in the mouse model of Pompe disease. Molecular Genetics and Metabolism. 2003;80:159–69. doi: 10.1016/j.ymgme.2003.08.022. [DOI] [PubMed] [Google Scholar]
- 30.Winkel LPF, Van den Hout JMP, Kamphoven JHJ, et al. Enzyme replacement therapy in late-onset Pompe's disease: A three-year follow-up. AnnNeurol. 2004;55:495–502. doi: 10.1002/ana.20019. [DOI] [PubMed] [Google Scholar]
- 31.Zhu YX, Li XM, Kyazike J, et al. Conjugation of mannose 6-phosphate-containing oligosaccharides to acid alpha-glucosidase improves the clearance of glycogen in Pompe mice. J Biol Chem. 2004;279:50336–41. doi: 10.1074/jbc.M409676200. [DOI] [PubMed] [Google Scholar]
- 32.Ziegler RJ, Bercury SD, Fidler J, et al. Ability of adeno-associated virus serotype 8-mediated hepatic expression of acid alpha-glucosidase to correct the biochemical and motor function deficits of presymptomatic and symptomatic Pompe mice. Hum Gene Ther. 2008;19:609–21. doi: 10.1089/hum.2008.010. [DOI] [PubMed] [Google Scholar]
- 33.Ziegler RJ, Cherry M, Barbon CM, et al. Correction of the Biochemical and Functional Deficits in Fabry Mice Following AAV8-mediated Hepatic Expression of alpha-galactosidase A. Mol Ther. 2007;15:492–500. doi: 10.1038/sj.mt.6300066. [DOI] [PubMed] [Google Scholar]
- 34.Wang L, Nichols TC, Read MS, Bellinger DA, Verma IM. Sustained expression of therapeutic level of factor IX in hemophilia B dogs by AAV-mediated gene therapy in liver. Mol Ther. 2000;1:154–8. doi: 10.1006/mthe.2000.0031. [DOI] [PubMed] [Google Scholar]
- 35.Raben N, Nagaraju K, Lee E, Plotz P. Modulation of disease severity in mice with targeted disruption of the acid alpha-glucosidase gene. Neuromuscul Disord. 2000;10:283–91. doi: 10.1016/s0960-8966(99)00117-0. [DOI] [PubMed] [Google Scholar]