

Abstract
We describe herein the assembly of hEPO(79–166), a key glycopeptide segment en route to erythropoietin, in minimally protected form. Key to the success of this synthetic endeavor was the application of our two-step cysteine-free native chemical ligation strategy, by which we achieved formal ligation at alanine and proline residues through the use of an N-terminal amino acid surrogate presenting a readily removable thiol functionality.
Introduction
Our laboratory has long been engaged in the development and application of powerful new technologies for the chemical synthesis of “biologic” level structures, such as peptides and glycopeptides.[i] From a target perspective, we have defined, as an orienting goal, the de novo total synthesis of the glycoprotein, erythropoietin (EPO), in homogeneous form. It was clear from the outset that reaching this target would prove to be a formidable exercise, and that meaningful progress toward this goal would be predicated on the development of a range of enabling methods in oligosaccharide and polypeptide synthesis. Indeed, our ongoing pursuit of synthetic, homogeneous EPO has inspired the development of a range of broadly useful ligation and aspartylation[ii] reactions.[i]
A major advance in the capacity to synthesize homogeneous polypeptides arose from the seminal discovery of native chemical ligation (NCL) by Kent and colleagues.[iii] As outlined in Figure 1a, the C-terminus of one fragment (Peptide 1) possesses a thioester, while the coupling partner (Peptide 2) incorporates an N-terminal cysteine residue. Intermolecular trans-thioesterification generates a unimolecular intermediate that is well-positioned to undergo intramolecular S→N acyl transfer, generating the coupled peptide fragment. The elegant and powerful Kent NCL method has been widely used for the merger of peptides bearing suitably placed cysteine residues. Early on, we recognized that the requirement that a cysteine residue be situated at the site of peptide ligation would represent a significant obstacle in the synthesis of EPO, whose four cysteines are not located at logical disconnection points. To address this challenge, we and others[iv] conceived of a two-step cysteine-free NCL strategy, which involves incorporation, on the N-terminus of one peptide fragment, of an amino acid surrogate possessing a temporary activating thiol functionality (Figure 1b). Following ligation, the thiol group is removed to reveal the target peptide sequence, bearing the native amino acid at the site of ligation. Key to the application of this strategy to complex systems was our discovery of a mild, free radical-based desulfurization protocol, which is highly tolerant of a diverse array of structural motifs commonly found in peptide and glycopeptide substrates.[v] Through recourse to this approach, we and others have thus far established the means to achieve ligation at alanine,[v] valine,[vi] threonine,[vii] leucine,[viii] lysine,ix and proline[x] residues. As will be shown (vide infra), our two-step cysteine-free NCL strategy greatly facilitated the assembly of the target hEPO(79–166) glycopeptide segment. As compared to the strategy used for the preparation of synthetic erythropoiesis protein (SEP), the strategy employed herein has the advantage of producing hEPO without changing its natural amino acid sequence.xi
Figure 1.
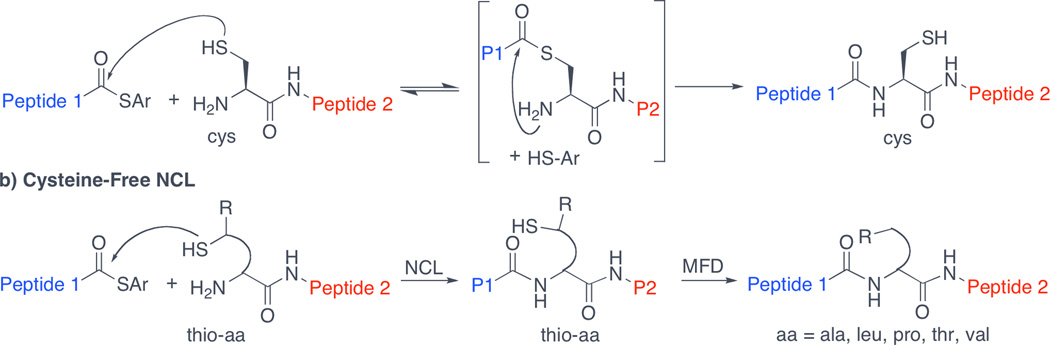
NCL and cysteine-free NCL.
Erythropoietin (EPO), encoded as a 166-residue conserved polypeptide chain and found in nature as a 165 residue mature protein bearing four sites of glycosylation, was selected as our orienting synthetic target on the basis of both its challenging structure and its compelling therapeutic profile. As the primary regulator of erythropoiesis, EPO elevates or maintains red-blood cell levels through a feedback mechanism involving the EPO receptor (EPOR) and the carbohydrate domains covalently attached to EPO.[xii] Previous studies have suggested that the carbohydrates can have significant regulatory effects on the biological properties of hEPO, including its biosynthesis, secretion, conformation, and stability.xiii However, efforts to rigorously elucidate the structure-function relationships of individual hEPO glycoforms have thus far been limited by the fact that natural and recombinant EPO are currently obtained as heterogeneous mixtures of glycoforms. Access to homogeneous hEPO glycoforms with defined glycan structures is thus considered key to evaluating the impact of glycosylation on biological properties.[xiv] Depending upon the size and glycosylation pattern of the target protein, homogeneous glycoproteins may theoretically be accessed by either synthetic or enzymatic means.[xv] Despite the attendent challenges, the synthetic approach offers much greater flexibility with respect to structural modification of the glycan.
Our original synthetic approach toward EPO envisioned assembling three glycopeptide fragments – hEPO(78–166),[xvi] hEPO(29–77),[xvii] and hEPO(1–28)[xviii] – which together span the entire expanse of erythropoietin. Under this plan, the individual glycopeptide fragments would be merged through recourse to sequential Blake-Aimoto coupling[xix] and cysteine-based NCL. Toward this end, we were able to prepare the constituent glycopeptide domains. However, all attempts to accomplish the requisite Blake-Aimoto condensation of partially protected hEPO(29–77) and hEPO(78–166) have been unsuccessful, resulting in extensive aggregation and decomposition of the glycopeptide substrates.[xx] Disappointingly, the attempted condensation reaction failed even in organic solvents such as dimethyl sulfoxide (DMSO), dimethylformamide (DMF), and trifluoroethanol (TFE).
In light of these findings, we devised a modified strategy toward EPO that would employ the two-step formal alanine version of the Cys-free NCL method recently developed in our laboratory. As shown in Scheme 1, the synthetic target, a hEPO glycoform bearing N-linked chitobiose glycans, would be assembled from three glycopeptide fragments: hEPO(79–166) (1), hEPO(29–78) (2), and hEPO(1–28) (3). Thus, the point of ligation between fragments 1 and 2 has been shifted by one residue so that the N-terminal amino acid at the ligation site is an alanine residue in the native sequence. Under our plan, a cysteine residue, located at the N-terminus of glycopeptide 1 (in red), would serve as a surrogate for alanine, to facilitate NCL of 1 and 2. It was our hope that the aggregation problems encountered in previous attempts at traditional fragment coupling would be circumvented through recourse to unprotected peptide segments under the solublizing standard NCL conditions, which use buffered solution containing high concentrations of guanidine hydrochloride, conditions designed to minimize aggregation problems.[xxi] Following ligation, the cysteine residue will be converted to the native alanine residue through exposure to our metal-free dethiylation (MFD) conditions.[vi] Following merger of fragments 1 and 2, standard cysteine-based NCL would serve to join fragment 3 [hEPO(1–28)], thus completing the synthesis of a full length polypeptide chain corresponding to [Arg166]EPO with single glycoform.
Scheme 1.

Retrosynthetic analysis of hEPO. Acm, acetamidomethyl, is a thiol protecting group for cysteine. R can be alkyl or aryl groups. Z is thiazolidine-4-(R)-carboxylic acid, a protected cysteine residue.
We describe herein a straightforward synthesis of the most complex of these fragments, glycopeptide 1, which is composed of an 88-amino acid chain bearing one N-linked and one O-linked glycan.
Experimental Section
Materials
All solvents were reagent grade or HPLC grade (Fisher). The following Fmoc and Boc amino acids and dipeptides from Sigma-Aldrich, EMD Biosciences, and Chem-Impex International were used as building blocks for solid phase peptide synthesis: Fmoc-Ala-OH, Boc-Ala-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Asp(OtBu)-OH, Fmoc-Cys(Acm)-OH, Fmoc-Cys(StBu)-OH, Fmoc-Thz-OH, Boc-Thz-OH, Fmoc-Gln(Trt)-OH, Boc-Gln(Trt)-OH, Fmoc-Glu(OtBu)-OH, Fmoc-Gly-OH, Fmoc-His(Trt)-OH, Fmoc-Ile-OH, Fmoc-Leu-OH, Fmoc-Lys(Alloc)-OH, Fmoc-Lys(Boc)-OH, Fmoc-Phe-OH, Fmoc-Pro-OH, Fmoc-Ser(tBu)-OH, Fmoc-Thr(tBu)-OH, Fmoc-Trp(Boc)-OH, Fmoc-Val-OH, Fmoc-Asp(tBu)-Ser(ΨMe, MePro)-OH, Fmoc-Ile-Ser(ΨMe, MePro)-OH, Fmoc-Leu-Thr(ΨMe, MePro)-OH, Fmoc-Tyr(tBu)-Ser(ΨMe, MePro)-OH, Fmoc-Tyr(tBu)-Thr(ΨMe, MePro)-OH, Fmoc-Val-Ser(ΨMe, MePro)-OH. The fully protected glycophorinxxii building block was prepared according to the published procedure.xxiii
Analysis and Purification Methods
All separations involved a mobile phase of 0.05% TFA (v/v) in water (solvent A)/0.04% TFA in acetonitrile (solvent B). LCMS analyses were performed using a Waters 2695 Separations Module and a Waters 996 Photodiode Array Detector equipped with Varian Microsorb C18 and C4 columns at a flow rate of 0.2 mL/min. UPLC-MS analyses were performed using a Waters AcquityTM Ultra Preformance LC system equipped with Acquity UPLC® BEH C18 and C4 columns at a flow rate of 0.3 mL/min. Preparative separations were performed using a Ranin HPLC solvent delivery system equipped with a Rainin UV-1 detector and Varian Microsorb C18 and C4 columns at a flow rate of 16.0 mL/min.
General Procedure for Solid Phase Peptide Synthesis
Automated peptide synthesis was performed on an Applied Biosystems Pioneer continuous flow peptide synthesizer. Peptides were synthesized using the standard automated Fmoc protocol (HATU, DIEA, DMF). The deblocking solution was a mixture of DMF/piperidine/DBU (100/5/5). The NovaSyn® TGT resins from EMD Biosciences were employed for the synthesis. Upon completion of the automated synthesis on a 0.05 mmol scale, the peptide resin was washed into a peptide cleavage vessel with DCM. The resin cleavage was performed with TFA/H2O/TIS (95:2.5:2.5) solution or DCM/AcOH/TFE (8:1:1) for 45 min. The liquid was blown off with nitrogen. The oily residue was extracted with diethyl ether and centrifuged to give a white pellet. After the ether was decanted, the solid was lyophilized or purified for further use.
A Procedure for the Preparation of Peptide p-cyanophenyl Esters
1.0 equiv of fully protected peptides and 6.0 equiv of p-cyanophenol were mixed with 0.28 equiv of DMAP (26 mM in DCM), 3.0 equiv of DCC (29 mM in DCM) was then added to the mixture at room temperature. After stirring for 2 hr, the solvent was removed by nitrogen stream and the oily residue was treated as above with ether. The pellet was dissolved in TFA/H2O/TIS (95:2.5:2.5) and stirred at room temperature for 45 min. The liquid was blown off and the residue was treated with ether. The pellet was dissolved in MeCN/H2O (1:1) for further analysis and purification.
General Procedure for the Preparation of Peptide Esters
The fully protected peptidyl acid (1.1 equiv) and the amino acid ester hydrochloride (1.0 equiv) were dissolved in CHCl3/TFE (3:1) and cooled to −10 °C. HOOBt (1.1 equiv) and EDCI (1.1 equiv) were then added. The reaction mixture was stirred at room temperature for 2.5 h. The solvent was gently blown off by a nitrogen stream and the residue was washed with H2O/AcOH (95:5). After centrifugation, the pellet was dissolved in TFA/H2O/TIS (95:2.5:2.5) and stirred at room temperature for 45 min. The solvent was removed and the residue was treated as above with ether. The pellet was dissolved in MeCN/H2O (1:1) for further analysis and purification.
General Procedure for Direct Aminolysis
Synthetic peptide ester (1.0 equiv), peptide (1.0–2.0 equiv), nBu3P (1.5–2.0 equiv) and HOOBt (33 equiv) were carefully dissolved in DMSO. The mixture was stirred at room temperature for 10 min, after which time, 22 quiv of DIEA was added in one portion. The reaction progress was monitored by LCMS or UPLC-MS. After completion of the reaction, it was quenched by the addition of EtOAc/AcOH (95:5, 20 times the reaction volume). The precipitate was collected by centrifugation, dissolved in MeCN/H2O (1:1), and purified by HPLC.
Native Chemical Ligation and Metal-Free Dethiylation
N-terminal peptide ester (1.5 equiv) and C-terminal peptide (1.0 equiv) were dissolved in ligation buffer (6 M Gdn·HCl, 100 mM Na2HPO4, 50 mM TCEP, pH 7.5). The resulting solution was stirred at room temperature. When there was no increase in product formation, the reaction was quenched with MeCN/H2O/AcOH (47.5:47.5:5) and purified by HPLC.
To a solution of the purified ligation product in degassed CH3CN/H2O (v/v = 1:1, 0.2 ml) was added 0.2 ml of 0.5 M bond-breaker® TCEP solution (Pierce), 0.02 ml of 2-methyl-2-propanethiol and 0.2 ml of radical initiator VA-044 (0.1 M in H2O). The reaction mixture was stirred at 37 °C and monitored by LCMS or UPLC MS. Upon completion, the reaction was quenched by the addition of MeCN/H2O/AcOH (47.5:47.5:5). The desired product was isolated by HPLC.
Results and Discussion
Our original strategy envisioned an iterative direct aminolysis approach toward 1, with points of ligation at the Pro87–Trp88, Ala125–Ser126, and Ala127–Ala128 sites (Scheme 2).[xvi,xixc] Thus, glycopeptide 1 would be prepared under modified Blake-Aimoto conditions16 from four fragments: two long polypeptides – hEPO(128–166) (6) and hEPO(88–125) (8) – and two shorter glycopeptides – hEPO(126–127) (7) and hEPO(79–87) (9). In designing this route, we hoped to minimize loss of the precious glycan domains by appending the oligosaccharides to short peptide fragments. Moreover, the direct aminolysis–based fragment condensation reaction is resistant to epimerization at the proline and alanine sites, thus allowing for the production of enantiomerically pure glycopeptides.[xix]
Scheme 2.

Direct aminolysis based strategy for the synthesis of glycopeptide 1. Fmoc (fluorenyl-9-methoxycarbonyl) and Alloc (allyloxycarbonyl) are the protecting groups for the amine functionality. The inset is ESI-MS for glycopeptide 10 (Chemical Formula: C509H819N131O171S3, calc. Exact Mass: 11598.86, [M+6H]6+: 1934.14, [M+7H]7+: 1657.98, [M+8H]8+: 1450.85, [M+9H]9+: 1289.76, [M+10H]10+: 1160.89, [M+11H]11+: 1055.44, [M+12H]12+: 967.57; Observed: [M+6H]6+: 1935.55, [M+7H]7+: 1659.33, [M+8H]8+: 1452.21, [M+9H]9+: 1290.96, [M+10H]10+: 1161.96, [M+11H]11+: 1056.45, [M+12H]12+: 968.69). Reagents and conditions: (a) HATU, DIEA, DMSO, 39%; (b) 1. nBu3P, HOOBt, DIEA, DMSO; 2. piperidine, DMSO, 58% for the first step, 35% for the second step; (c) 1. HOOBt, DIEA, DMSO; 2. piperidine, DMSO, 70%.
Peptide 6 was obtained by solid phase peptide synthesis (SPPS), using commercially available building blocks and Fmoc chemistry. Following HPLC purification, the partially protected peptide was used directly in the condensation reaction without further modification (23%, isolated yield). Peptide 8 was prepared from the fully protected peptidyl acid using the method developed by Sakakibara et al.[xxiv] Briefly, following cleavage from the highly acid labile trityl ester resin, the protected hEPO(88–124) fragment was coupled with L-alanine-2-(ethyldithiophenyl) ester hydrochloride to give rise to fully protected peptidyl phenyl ester, which, upon one-pot side chain deprotection, afforded the desired product 8 in 12% yield.
The next subgoal was that of synthesizing the two glycopeptides, 7 and 9. As described previously, we had much earlier demonstrated the ability to produce O-linked glycopeptidyl esters, even in the presence of multiple hydroxyl groups and secondary and tertiary carboxyl groups.[xvi] We applied this technique to the synthesis of 7. As expected, substrates 4 and 5 rapidly underwent coupling to afford glycopeptide 7 in two forms, with the major one bearing a 6-membered lactone ring between the hydroxyl group at the C-4 of galactose and the carboxyl group of sialic acid. The structures of the two products were confirmed through analysis of their fragmentation patterns in electrospray ionization-mass spectrometry (ESI-MS). As precedented, the lactone can be hydrolyzed under mild conditions to generate the structure of natural glycophorin.[xxv] In our experiments, we found that formation of the lactone ring served to beneficially prevent the cleavage of the labile α-2,3-glycosidic bond during both the condensation reaction and HPLC purification. The ratio of the lactone product generated can be enhanced by increasing the amount of HATU used in the coupling reaction. The preparation of the second glycopeptide fragment, 9, was straightforward and high yielding. Under Lansbury aspartylation conditions, the disaccharide glycosylamine could be efficiently attached to the corresponding peptide p-cyanophenyl ester, affording compound 9 in 64% yield.
With the four component glycopeptides segments in hand, the stage was now set for the assembly of the glycopeptide 10 by aminolysis of the activated esters. Coupling of 7 with acceptor 6 was first investigated. The reaction was carried out as previously described, with the exception that nBu3P was used as the reducing agent in place of tris(2-carboxyethyl)phosphine (TCEP). This modification served to eliminate competing side reactions caused by the carboxyl groups of TCEP. In addition, nBu3P has better solubility in DMSO, making it easier to achieve the high concentration of reactants required for the bimolecular coupling reaction.[xvi] Indeed, under the optimized conditions, the condensation reaction between 6 and 7 proceeded smoothly and cleanly to give the desired glycopeptide product, as shown. Deprotection of the N-terminal Fmoc group was effected by treatment with piperidine. Next, condensation with 8 and subsequent Fmoc removal provided partially protected acceptor hEPO(88–166). Finally, formation of glycopeptide 10 was accomplished through aminolysis of the p-cyanophenyl ester 9 with this acceptor, followed by unmasking of the N-terminal amine functional group.[xvii] As expected, in the absence of phosphines, the disulfide bond was stable under the experimental conditions.
Having accomplished the synthesis of glycopeptide 10, we next attempted to condense this fragment to a lysine-protected hEPO(29–78) glycopeptide. The segment condensation reaction was carefully studied under a variety of conditions. Unfortunately, all of our attempts to effect the union of these two fragments were unsuccessful. At best, only a trace amount of presumed coupled product could be observed by examination of crude reaction mixtures using LCMS. It appears that the failure of this reaction can be attributed to the poor handling properties of the lysine-protected glycopeptide substrates.
This disappointing finding was seen to arise from protection of the amino groups of lysine residues, leading to destabilization of the synthetic glycopeptide fragments.[xx] We thus sought to deprotect the Alloc groups of 10. However, because of complications apparently caused by the presence of the thiol group and the O-linked glycan, the attempted palladium-mediated deprotection reaction was also unsuccessful.
While the possibilities for removing the Alloc groups by non-palladium based reactions have not yet been exhausted, we began to investigate an alternative NCL–based route toward glycopeptide 1. Because NCL is highly selective for the N-terminal amino groups, even in the presence of unprotected lysine residues, this option would allow us to circumvent the difficulties arising from introduction of lysine protecting groups.[xxvi] As described above, our laboratory has developed a series of two-step cysteine-free NCL methods which enable formal ligation at non-cysteine residues through emplacement of a thiol-containing surrogate on the N-terminal peptide fragment. We now sought to apply these methods to the synthesis of fragment 1 en route to hEPO. As shown in Scheme 3, our modified route to 1 would require the synthesis of two glycopeptide substrates, 12 and 14, and the peptide segments 11 and peptide thioester 13. Peptide 11 and glycopeptide 12 each present an N-terminal cysteine, which, following ligation, must be converted to the native alanine residue through metal-free dethiylation. Similarly, the N-terminus of fragment 13 incorporates a thio-proline amino acid surrogate. As we recently reported, provided it is of the correct stereochemical configuration, the resident thiol functionality effectively promotes ligation, and is readily removed to reveal the natural proline residue.[x]
Scheme 3.

Native chemical ligation strategy for the synthesis of glycopeptide 1. Reagents and conditions: (a) 1. 6 M Gn•HCl, 100 mM NaH2PO4, 50 mM TCEP, pH 7.5, 67%; 2. piperidine, DMSO, 61%; 3. 0.2M MeONH2, 60%; (b) 6 M Gn•HCl, 100 mM NaH2PO4, 50 mM TCEP, pH 7.5, 23%; (c) 6 M Gn•HCl, 100 mM NaH2PO4, 50 mM TCEP, 200 mM MPAA, pH 7.8, 40%; (d) TCEP, VA-044, tBuSH.
Fragments, 11, 13, and 14, were prepared in an analogous manner to those described above. In the modified syntheses, Boc-Cys(StBu)-OH, (2S,4R)-Boc-Mpt(Trt)-OH, and Boc-Thz-OH were used as building blocks for the N-terminal residues, in place of the Fmoc amino acids. The synthesis of glycopeptide 12, which contains an unprotected glycophorin moiety, is shown in Scheme 4. In order to avoid manipulation of the protecting groups in the late stages of the synthesis, we elected to prepare glycopeptide 12 from the fully unprotected glycosylamino acid 18 (Scheme 4). In this route, the key step is the selective formation of the amide bond in 19. In the event, coupling between activated ester, Fmoc-Thz-OSu, and 18 proceeded very efficiently under slightly modified Schotten-Baumann conditions[xxvii] to provide di-glycopeptide 19 in 36% yield. Under the previously described conditions, 4 was coupled to 19 to provide the desired product 12 in 21% yield.
Scheme 4.
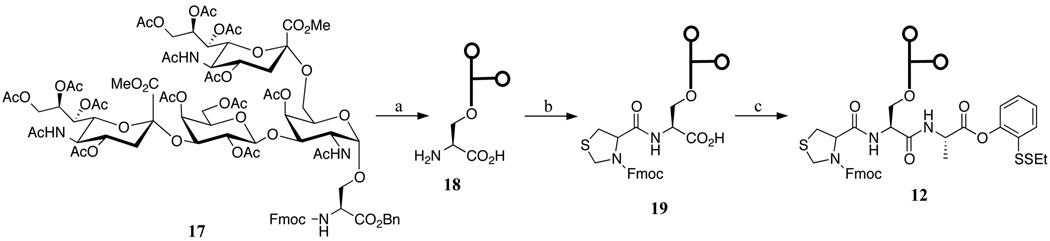
Chemical synthesis of glycopeptide 12. Reagents and conditions: (a) 1. 1.0 M NaOH, MeOH; 2. Fmoc-Thz-OSu, Na2CO3, DMF, DME, H2O, 36% for 2 steps; (b) 4, HATU, DIEA, DMSO, 21%.
With the requisite fragments in hand, we next investigated the synthesis of 15. In the event, peptide 11 was coupled with 12, followed by Fmoc deprotection and thiazolidine ring-opening, to provide the ligation acceptor corresponding to hEPO(125–166). As a key step in the synthesis, the proline-mediated ligation of acceptor 13 and donor 14 was carefully studied. This type of merger, though with much simpler substrates, had recently been demonstrated.[9] We were pleased to find that, under our standard conditions, the union of these two substrates could be achieved in a reasonable isolated yield (23%). With this ligation donor, the coupling to acceptor hEPO(125–166) proceeded smoothly in the presence of 4-mercapto-phenylacetic acid (MPAA), to deliver the desired product 15.
The next challenge would be to effect the removal of the three erstwhile thiol groups located at positions 87, 125, and 128. We had not previously had the opportunity to evaluate the efficiency of our metal-free desulfurization reaction in a complex glycopeptide of this size. In the event, we were pleased to observe that, upon exposure to our standard aqueous, metal-free dethiylation (MFD) conditions,xxviii complete desulfurization was achieved in 2h, yielding the target glycopeptide 16. Although we have not yet attempted to open the thiazolidine ring of 16, based on our previous experience, we expect that treatment of 16 with excess methoxyamine at pH 4 should afford the desired product 1.[xxix]
Conclusions
In summary, we have prepared complex glycopeptide 1 en route to homogeneous EPO (Figure 3). Through the course of our synthesis, we demonstrated the power and versatility of our recently developed two-step cysteine-free NCL strategy for the assembly of large peptide and glycopeptide fragments. Current efforts are focused on the preparation of fragments 2 and 3 and the ligation of these fragments to generate synthetic EPO.
Figure 3.

Chemical synthesis of hEPO. The residues in yellow color are the disconnection sites.
Figure 2.

ESI-MS for glycopeptides 16. Chemical formula: C486H791N131O161S2, calc. Mass: 11102.72, [M+6H]6+: 1851.45, [M+7H]7+: 1587.10, [M+8H]8+: 1388.84, [M+9H]9+: 1234.64, [M+10H]10+: 1111.27, [M+11H]11+: 1010.34, [M+12H]12+: 926.23; Observed mass: [M+6H]6+: 1852.31, [M+7H]7+: 1588.08, [M+8H]8+: 1389.84, [M+9H]9+: 1235.09, [M+10H]10+: 1111.95, [M+11H]11+: 1010.94, [M+12H]12+: 927.18
Acknowledgments
Financial support was provided by the National Institutes of Health (CA28824 to SJD). We thank Ms. Rebecca Lambert for valuable discussions. We also thank Drs. George Sukenick, Sylvi Rusli, and Ms. Hui Fang of SKI’s NMR core facility for mass spectral and NMR assistance and Ms. Laura Wilson for assistance with the preparation of the manuscript.
References
- i.a) Danishefsky SJ, Bilodeau MT. Angew. Chem. Int. Ed. 1996;35:1380–1419. [Google Scholar]; b) Wilson RM, Danishefsky SJ. Pure Appl. Chem. 2007;79:2189–2216. [Google Scholar]; c) Kan C, Danishefsky SJ. Tetrahedron. 2009;65:9047–9065. doi: 10.1016/j.tet.2009.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ii.Aspartylation is a reaction that involves the coupling of a glycan with an aspartate residue of a peptide under certain coupling conditions to afford an asparagine-linked (N-linked) glycan.
- iii.Dawson PE, Muir TW, Clark-Lewis I, Kent SBH. Science. 1994;266:776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- iv.a) Yan LZ, Dawson PE. J. Am. Chem. Soc. 2001;123:526–533. doi: 10.1021/ja003265m. [DOI] [PubMed] [Google Scholar]; b) Crich D, Banerjee A. J. Am. Chem. Soc. 2007;129:10064–10065. doi: 10.1021/ja072804l. [DOI] [PubMed] [Google Scholar]; c) Okamoto R, Kajihara Y. Angew. Chem. Int. Ed. 2008;47:5402–5406. doi: 10.1002/anie.200801097. [DOI] [PubMed] [Google Scholar]
- v.Wan Q, Danishefsky SJ. Angew. Chem. Int. Ed. 2007;46:9248–9252. doi: 10.1002/anie.200704195. [DOI] [PubMed] [Google Scholar]
- vi.a) Haase C, Rohde H, Seitz O. Angew. Chem. Int. Ed. 2008;47:6807–6810. doi: 10.1002/anie.200801590. [DOI] [PubMed] [Google Scholar]; b) Chen J, Wan Q, Yuan Y, Zhu JL, Danishefsky SJ. Angew. Chem. Int. Ed. 2008;47:8521–8524. doi: 10.1002/anie.200803523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vii.Chen J, Wang P, Zhu J, Wan Q, Danishefsky SJ. Tetrahedron. 2010;66:2277–2283. doi: 10.1016/j.tet.2010.01.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- viii.a) Harpaz Z, Siman P, Kumar KS, Brick A. ChemBioChem. 2010;11:1232–1235. doi: 10.1002/cbic.201000168. [DOI] [PubMed] [Google Scholar]; b) Tan Z, Shang S, Danishefsky SJ. Angew. Chem. Int. Ed. 2010;49:9500–9503. doi: 10.1002/anie.201005513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ix.a) Yang R, Pasunooti KK, Li F, Liu XW, Liu CF. J. Am. Chem. Soc. 2009;131:13592–13593. doi: 10.1021/ja905491p. [DOI] [PubMed] [Google Scholar]; b) Kumar KSA, Haj-Yahya M, Olschewski D, Lashuel HA, Brik A. Angew. Chem. Int. Ed. 2009;48:8090–8094. doi: 10.1002/anie.200902936. [DOI] [PubMed] [Google Scholar]
- x.Shang S, Tan Z, Dong S, Danishefsky SJ. J. Am. Chem. Soc. 2011;133:10784–10786. doi: 10.1021/ja204277b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xi.Kochendoerfer GG, Chen SY, Mao F, Cressman S, Traviglia S, Shao H, Hunter CL, Low DW, Cagle EN, Carnevali M, Gueriguian V, Keogh PJ, Porter H, Stratton SM, Wiedeke MC, Wilken J, Tang J, Levy JJ, Miranda LP, Crnogorac MM, Kalbag S, Botti P, Schindler-Horvat J, Savatski L, Adamson JW, Kung A, Kent SB, Bradburne JA. Science. 2003;299:884–887. doi: 10.1126/science.1079085. [DOI] [PubMed] [Google Scholar]
- xii.a) Egrie JC, Browne JK. Nephrol. Dial. Transplant. 2001;16(Suppl 3):3–13. [PubMed] [Google Scholar]; b) Toyoda T, Arakawa T, Yamaguchi H. J. Biochem. 2002;131:511–515. doi: 10.1093/oxfordjournals.jbchem.a003128. [DOI] [PubMed] [Google Scholar]; c) Jelkmann W. Intern. Med. 2004;43:649–659. doi: 10.2169/internalmedicine.43.649. [DOI] [PubMed] [Google Scholar]
- xiii.Dube S, Fisher JW, Powell JS. J. Biol. Chem. 1988;263:17516–17521. [PubMed] [Google Scholar]
- xiv.a) Pratt MR, Bertozzi CR. Chem. Soc. Rev. 2005;34:58–68. doi: 10.1039/b400593g. [DOI] [PubMed] [Google Scholar]; b) Rich JR, Withers SG. Nat. Chem. Biol. 2009;5:206–215. doi: 10.1038/nchembio.148. [DOI] [PubMed] [Google Scholar]; c) Gamblin DP, Scanlan EM, Davis BG. Chem. Rev. 2009;109:131–163. doi: 10.1021/cr078291i. [DOI] [PubMed] [Google Scholar]
- xv.a) Bernardes GJ, Castagner B, Seeberger PH. ACS Chem. Biol. 2009;4:703–713. doi: 10.1021/cb900014n. [DOI] [PubMed] [Google Scholar]; b) Kiessling LL, Splain RA. Annu. Rev. Biochem. 2010;79:619–653. doi: 10.1146/annurev.biochem.77.070606.100917. [DOI] [PubMed] [Google Scholar]
- xvi.Tan Z, Shang S, Halkina T, Yuan Y, Danishefsky SJ. J. Am. Chem. Soc. 2009;131:5424–5431. doi: 10.1021/ja808704m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xvii.Yuan Y, Chen J, Wan Q, Tan Z, Chen G, Kan C, Danishefsky, SJ. J. Am. Chem. Soc. 2009;131:5432–5437. doi: 10.1021/ja808705v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xviii.Kan C, Trzupek JD, Wu B, Wan Q, Chen G, Tan Z, Yuan Y, Danishefsky, SJ. J. Am. Chem. Soc. 2009;131:5438–5443. doi: 10.1021/ja808707w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xix.a) Blake J, Li CH. Proc. Natl. Acad. Sci. USA. 1981;78:4055–4058. doi: 10.1073/pnas.78.7.4055. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Aimoto S. Curr. Org. Chem. 2001;5:45–87. [Google Scholar]; c) Chen G, Wan Q, Tan Z, Kan C, Hua Z, Ranganathan K, Danishefsky SJ. Angew. Chem. Int. Ed. 2007;46:7383–7387. doi: 10.1002/anie.200702865. [DOI] [PubMed] [Google Scholar]
- xx.Tan Z, Shang S, Danishefsky SJ. Proc. Natl. Acad. Sci. USA. 2011;108:4297–4302. doi: 10.1073/pnas.1100195108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxi.Lyutova EM, Kasakov AS, Gurvits BY, Y. B. Biotechnol. Prog. 2007;23:1411–1416. doi: 10.1021/bp070209h. [DOI] [PubMed] [Google Scholar]
- xxii.Thomas DB, Winzler RJ. J. Biol. Chem. 1969;244:5943–5946. [PubMed] [Google Scholar]
- xxiii.Schwarz JB, Kuduk SD, Chen X-T, Sames D, Glunz PW, Danishefsky SJ. J. Am. Chem. Soc. 1999;121:2662–2673. [Google Scholar]
- xxiv.Sakakibara S. Biopolymers. 1995;37:17–28. doi: 10.1002/bip.360370105. [DOI] [PubMed] [Google Scholar]
- xxv.Ando S, Nakahara Y, Ito Y, Ogawa T, Nakahara Y. Carbohydr. Res. 2000;329:773–780. doi: 10.1016/s0008-6215(00)00235-4. [DOI] [PubMed] [Google Scholar]
- xxvi.Kent SB. Chem. Soc. Rev. 2009;38:338–351. doi: 10.1039/b700141j. [DOI] [PubMed] [Google Scholar]
- xxvii.a) Schotten C. Ber. Dtsch. Chem. Ges. 1884;17:2544–2547. [Google Scholar]; b) Baumann E. Ber. Dtsch. Chem. Ges. 1886;19:3218–3222. [Google Scholar]
- xxviii.Shang S, Tan Z, Danishefsky SJ. Proc. Natl. Acad. Sci. USA. 2011;108:5986–5989. doi: 10.1073/pnas.1103118108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxix.Bang D, Kent SB. Angew. Chem. Int. Ed. 2004;43:2534–2538. doi: 10.1002/anie.200353540. [DOI] [PubMed] [Google Scholar]