

Abstract
Rituximab efficacy in cancer therapy depends in part on induction of complement-dependent cytotoxicity (CDC). Human CD59 (hCD59) is a key complement regulatory protein that restricts the formation of the membrane attack complex, thereby inhibiting induction of CDC. hCD59 is highly expressed in B-cell non-Hodgkin's lymphoma (NHL) and up-regulation of hCD59 is an important determinant of the sensitivity of NHL cells to rituximab treatment. Here we report that the potent hCD59 inhibitor rILYd4 enhances CDC in vitro and in vivo, thereby sensitizing rituximab-resistant lymphoma cells and primary chronic lymphocytic leukemia cells (CLL) to rituximab treatment. By defining PK/PD profiles of rILYd4 in mice, we showed that by itself rILYd4 does not adversely mediate in vivo hemolysis of hCD59-expressing erythrocytes. Increasing expression levels of the complement regulators CD59 and CD55 in rituximab-resistant cells occurs due to selection of pre-existing clones, rather than de novo induction of these proteins. Moreover, lymphoma cells overexpressing CD59 were directly responsible for the resistance to rituximab-mediated CDC therapy. Our results rationalize the use of rILYd4 as a therapeutic adjuvant for rituximab treatment of rituximab-resistant lymphoma and CLL. Further, they suggest that preemptive elimination of CD59 overexpressing subpopulations along with rituximab treatment may be a useful approach to ablate or conquer rituximab resistance.
Keywords: Intermedilysin, CD59, complement, rituximab, Lymphoma and chronic lymphocytic leukemia
Introduction
In the last 10 years, the chimeric antibody of rituximab, which specifically targets CD20 on the B lymphocyte membrane, has led to significant progress in the treatment of B-cell non-Hodgkin’s lymphoma (NHL)(1). However, a subset of NHL patients do not respond to rituximab, despite expressing CD20(2), and many patients who initially respond develop resistance to further treatment over time(3). The mechanisms suspected to mediate rituximab’s therapeutic effect include 1) complement-dependent cytotoxicity (CDC)(4–7); 2) antibody-dependent cellular cytotoxicity (ADCC)(2, 8–10) involving phagocytosis(11) and/or Fc:FcR dependent mechanisms(12); and 3) apoptosis(1, 9, 12).
The role of the CDC on rituximab-mediated lymphoma therapy has been extensively investigated in vivo and in vitro(9). Complement depletion by cobra venom factor or C1q-deficiency significantly reduces the antitumor activity of rituximab in mouse models(6, 13–15). Consistently, complement consumption has been observed in vitro and in vivo after rituximab administration(5, 16), and addition of fresh frozen plasma as a source of complement is able to increase the therapeutic response to rituximab in refractory-CLL patients(17, 18). The importance of CDC in B-cell lymphoma response to rituximab was further confirmed by the finding that antibodies that abrogate the function of membrane complement regulatory proteins (mCRPs) such as CD46, CD55 and CD59 enhance the therapeutic effect of rituximab in animal models of the disease(2, 4, 19–24).
The complement system is the principal part of the innate immune system and plays an important role in host defense. To prevent the potentially harmful effect of complement activation on normal cells, some mCRPs including CD46, CD55 and CD59 have evolved to restrict complement activation at different stages of the complement cascades(9, 25). CD59, a glycosylphosphatidylinositol (GPI)-anchored mCRP, restricts formation of the membrane attack complex (MAC) by preventing C9 polymerization through binding to C8 and C9(26). CD55, another GPI-anchored mCRP, inactivates the C3 and C5 convertases by accelerating the decay of those proteases(27–29), while CD46, a non GPI-anchored membrane protein, acts as a cofactor for inactivation of cell-bound C4b and C3b by serum factor I(30). Not only do these mCRPs protect normal cells from bystander complement attack, but they also confer protection to cancer cells by limiting complement activation by a therapeutic antibody such as rituximab. Numerous findings indicate that CD59 is the most effective mCRP protecting B cell lymphomas from rituximab-mediated CDC(2, 4, 21, 31). Dalle et al. have recently found that CD59 but neither CD46 nor CD55 is over-expressed in an in vivo model of rituximab resistant (RR) follicular lymphoma (FL)-derived tumor cells isolated from a patient(32). Moreover, in a clinical study of chronic lymphocytic leukemia (CLL), Bannerji et al found a significant increase in hCD59 expression in patients who failed to clear CLL cells from peripheral blood after initiation of rituximab treatment (33). Taken together, these results suggest that the over-expression of mCRPs, and especially CD59, contributes to the resistance of lymphoma and CLL cells to rituximab therapy(34, 35). For these reasons, the development of a molecule capable of abrogating CD59 function in cancer cells is likely to fulfill an unmet clinical need.
Recently, we have generated a specific high affinity inhibitor of hCD59 denoted as rILYd4(36). rILYd4 is the recombinant 114 amino acid peptide representing domain four (D4) of intermedilysin (ILY), a cytolytic toxin secreted by Streptococcus intermedius. We have demonstrated that rILYd4 binds to the hCD59 functional site and thereby abrogates hCD59 function(36). Here, we further demonstrate that rILYd4 sensitizes RR B-cell NHL and primary CLL cells to rituximab treatment in vitro, ex vivo, and in vivo. The results indicate that rILYd4 may provide a novel therapeutic approach as an adjuvant to rituximab for treatment of lymphoma.
Materials and Methods
Additional information is available in Supplementary Materials and Methods.
Mice
Animal studies were approved by the Harvard Medical School Institutional Animal Care and Use Committee.
Balb/C nude mice used for the determination of rILYd4 in vivo efficacy were purchased from Charles River Laboratory (Wilmington, MA). Mice specifically expressing hCD59 as a transgene in the erythrocytes of mCd59a and mCd59b double knockout mice (hCD59RBC+/−/mCd59ab−/−) were used for PK/PD (pharmcokinetic/pharmacodynamic) and toxicity studies. hCD59RBC+/−/mCd59ab−/− were generated by crossing the mCd59a and mCd59b knockout mouse (mCd59ab−/−)(37) with a hCD59 transgenic mouse (ThCD59RBC), in which hCD59 was specifically expressed under the control of the hemoglobin promoter, at a level comparable to that seen in human erythrocytes(38).
B-lymphoma and primary CLL cells and reagents
Human Burkitt’s B-cell lymphoma Ramos, Daudi and Raji cells were purchased from ATCC (Manassas, VA), and cultured in the RPMI-1640 medium supplemented with 10% heat-inactivated fetal bovine serum, 100 U/ml penicillin and 100 μg/ml streptomycin. These cell lines were authenticated by the supplier, obtained within 6 months of their use, and passaged less than 50 times. We did not re-authenticate the cell lines.
The CLL patients had been previously enrolled on Dana-Farber Harvard Cancer Center (DFHCC) protocol 99–224. All participants signed informed consent prior to sample collection. The blood from CLL patients was then separated on a Ficoll gradient and peripheral blood mononuclear cells (PBMCs) were frozen. The frozen PBMCs from six patients participating in this study (supplementary Table 1) were cultured in IMDM medium (Invitrogen) supplemented with 10% human AB serum (GemCell, Gemini Co., West Sacramento, CA), 50 μg/ml transferrin (Roche Applied Science, Indianapolis, IN), 5 μg/ml human insulin (Roche Applied Science, Indianapolis, IN), 100 U/ml penicillin and 100 μg/ml streptomycin. Intact ILY and rILYd4 tagged with HisX6 at its N-terminals were purified as described in (36, 38) .
Generation of RR and hCD59 negative Ramos cell lines
We used a previously reported procedure (31) to develop Ramos cells resistant to CDC at different concentrations of rituximab. The cells generated were denoted as RR0.2, RR0.8, RR3.2, RR12.8 and RR51.2 because they could survive complement attack induced by rituximab (Biogen Idec, Cambridge, MA) at concentrations of 0.2, 0.8, 3.2, 12.8 and 51.2 μg/ml, respectively, all in the presence of 10% normal human serum (NHS) (Valley Biomedical, Winchester, VA). We followed the same procedures to generate rituximab-resistant NHL Daudi and Raji cells.
The hCD59-expressing subpopulations in all of the above Ramos cell lines were removed with repeated intact ILY treatment (5 μg/ml, on 3–5 occasions), and the surviving Ramos cell subpopulations were regarded as hCD59 negative cells, which was further confirmed by FACS analysis.
Isolation of CD59-expressing population by cell sorting
Parental or original Ramos cells (OR) were stained with anti-hCD59 antibody and FITC-conjugated secondary antibody. Cells staining positive for CD59 (0.38% of the total population) were collected in the first round by cell sorting (CS) and then grown in culture medium. When the cells reached 5 × 106 cells/ml density, a second round of CS was performed to collect cells positive for CD59 (5% of the total population) for further experiments. Cells sorted through two rounds of selection of the CD59 positive population are termed CS cells.
Rituximab-mediated CDC on Ramos cells and primary CLL cells
Cell viability was determined by either Trypan blue or Alamar blue assay(4). 5 × 104 Ramos cells per well were seeded on 96-well plates. Rituximab (10 μg/ml) and 10% NHS were added to the wells (total volume 200 μl/well) in the absence and presence of different concentrations of rILYd4, and plates were incubated at 37°C. For the Trypan blue assay, complement activation was stopped after an one hour incubation on ice followed by staining with 0.04% Trypan blue. The dead cells identified by the blue staining were counted in a blinded fashion and cytolysis expressed as the fraction of dead cells out of the total number of cells. For the Alamar blue assay, cells were treated for 4 hours and then 30 μl Alamar blue and 70 μl culture medium were added to each well and incubated overnight. Cytolysis was assessed by reading the plates in an F-2000 fluorescence spectrophotometer (Hitachi) (excitation: 560 nm; emission: 590 nm). The medium alone (without cells), NHS alone, and heat-inactivated human serum (IHS) alone were used as control for background in the relevant calculation. The positive control, considered 100% cytolysis, was Triton X-100 (0.1% in DPBS) treated wells and negative control were cells without any treatment. Percent lysis in each well was calculated as: (fluorescence in negative control well – fluorescence in test well)/fluorescence in negative control well X 100.
The primary CLL cells spontaneously die after culturing for several days. Thus, we had to perform the experiments individually. Primary CLL cells were seeded on 96-well plates at 2 × 106 cells/ml. Different concentrations of rILYd4, 20 μg/ml rituximab, and 20% NHS (total 200 μl/well) were added to the corresponding wells and incubated at 37°C. After 3 hours incubation, plates were placed on ice to stop complement activation followed by staining with 0.04% Trypan blue. Blue stained dead cells were counted by two independent investigators and cytolysis was estimated from the fraction of dead cells as above.
Anti-cancer efficacy of rILYd4 in vivo
To assure the resistance of the RR51.2 cell line, we treated RR51.2 cells with 51.2 μg/ml rituximab and 10% NHS for 1 hour at 37°C before xenografting the cells into the nude mice. 1 × 107 cells suspended in 100 μl 50% matrigel (BD Biosciences, Rockville, MD) diluted in serum-free culture medium were implanted subcutaneously on the left flank of each Balb/C nude mouse. An early development (the mice had no-measurable tumor) or established tumor model (the mice had an average 0.15 g tumor) were used to evaluate the in vivo efficacy of rILYd4. After grafting, treatment was performed at day 6 in the development model and at day 18 in the established tumor model. Rituximab (2 mg/kg) without or with rILYd4 (2 mg/kg) was injected intraperitoneally (i.p.) on 3 occasions 4 days apart (Q4D). Tumors size was measured with calipers and tumor mass was calculated by the formula: (width)2 × length/2 (39, 40). Tumor free rate in the established tumor model expressed as the fraction of the number of the mice without the tumor at 35 days after the treatment out of the total number of mice with the tumor before the treatment.
Data analysis
The differences between means of paired samples on primary CLL cells was evaluated by Wilcoxon’s signed rank test. In the other experiments, the comparison between two or three groups was examined with a nonparametric Mann-Whitney test. A P value <0.05 was considered significant.
Results
rILYd4 sensitizes RR cells to a rituximab-mediated CDC effect in vitro
We generated five RR Ramos cell lines and demonstrated that the increase in CD59 expression levels correlated with the degree of resistance to rituximab-mediated CDC (Figure 1A, 1B). In contrast, the level of CD20 expression was not altered by the selection of rituximab resistance (Supplementary Figure 1). These results confirmed functionally the generation of RR cells.
Figure 1. Expression of CD59 on OR and RR cells with or without ILY pretreatment and their response to rituximab-mediated CDC in vitro.

(A) Physical detection of CD59 expression on OR and RR cells by FACS. Cells were stained with anti-hCD59 (black lines) or isotype-matched Ab (solid gray lines). (B) Functional confirmation of RR cell lines in CDC treatment (n=4). (C) Physical absence of hCD59 expression on ILY-pretreated OR and RR cells. (D) The hCD59 negative cells enriched by ILY pretreatment (5 μg/ml for 5 times) are very sensitive to rituximab-mediated CDC. Results are presented as mean ± s.e.m of four independent Alamar blue assays.
Intact ILY binds exclusively to hCD59 and rapidly lysis hCD59-expressing cells(38, 41). Repetitive exposure of OR and RR Ramos cells to ILY was used to enrich for the subpopulation of CD59 negative cells. Elimination of CD59-expressing cells was verified by FACS analysis (Figure 1C). All these hCD59 negative cells were very sensitive to rituximab-mediated CDC as shown by the complete lysis obtained even with the lowest dose of rituximab (0.2 μg/ml) (Figure 1D). Taken together, these in vitro results indicate that the CD59-expressing subpopulation in Ramos cells may be responsible for the resistance to rituximab-mediated CDC. This interpretation provides a strong rationale for the utilization of rILYd4 as an adjuvant in rituximab therapy: rILY4 would sensitize RR cells by abrogating hCD59 function and thereby enhancing rituximab-mediated CDC.
The effectiveness of this approach was tested using RR51.2 cells, which express the highest level of CD59 and are resistant to CDC effect mediated by rituximab at the highest concentration (51.2 μg/ml). The results showed that rILYd4 increased rituximab-mediated CDC in a dose-dependent manner (ED50 of rILYd4 = 0.614μg/ml or 33nmol/L) (Figure 2A and Supplementary Figure 2A). Treatment of the same cells with escalating concentrations of rILYd4 in the absence of a complement source (either no addition of NHS or addition of inactivated HS) did not induce cell lysis (Supplementary Figure 2B). This result rules out a direct toxic effect of rILYd4 and confirms the complement-dependent nature of the rituximab-mediated cytolysis (Supplementary Figure 2B).
Figure 2. rILYd4 is an effective adjuvant for rituximab against RR cells or CLL cells.
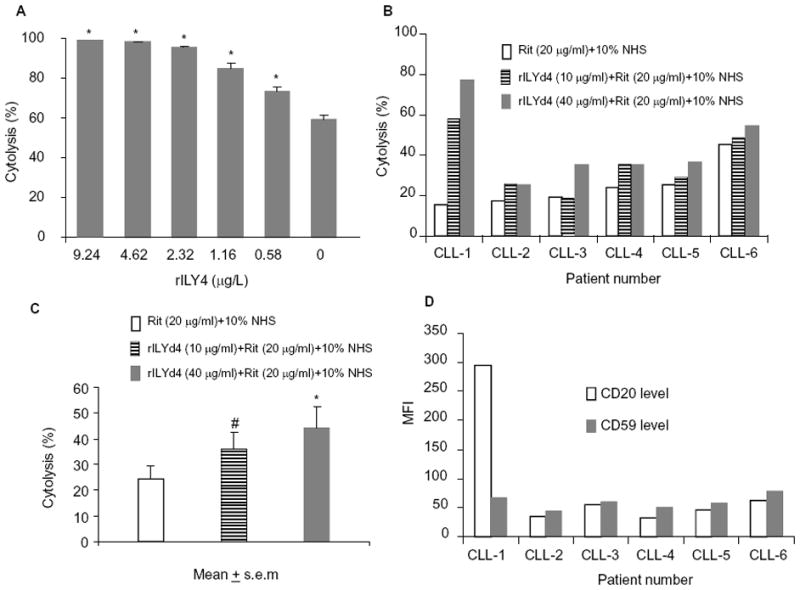
(A) rILYd4 sensitizes RR51.2 cells to rituximab (Rit) (10 μg/ml)-mediated CDC effects in vitro as measured by Alamar blue assay. *: P<0.05 vs no rILYd4 treatment. The data represents mean ± s.e.m from four experiments. (B) rILYd4 sensitizes CLL cells to rituximab-mediated CDC effects ex vivo and (C) in a dose dependent manner. (D) Expression levels of CD20 and CD59 were detected by FACS analysis. #: P<0.05 vs no rILYd4 treatment. *: P<0.05 vs no rILYd4 treatment cells. Results are presented as mean ± s.e.m from the six patients CLL-1-CLL-6.
rILYd4 sensitizes primary CLL cells to rituximab-mediated CDC ex vivo
The clinical relevance of the above findings was demonstrated by the capacity of rILYd4 to sensitize primary CLL cells to rituximab-mediated CDC effects ex vivo. The selection of primary CLL cells instead of B-cell NHL was dictated by their greater accessibility. Treatment of CLL primary cells from 6 different patients (Supplementary Table 1) with escalating doses of rILYd4 resulted in a dose-dependent increase in the rituximab-mediated CDC effect in all the samples (Figure 2B and 2C). Determination of the levels of CD20 and CD59 on the surface of the CLL cells from each patient by FACS analysis showed that the cells from subject CLL-1 that express CD20 at the highest level are also the most sensitive to the induction of rituximab-mediated CDC by rILYd4 (Figure 2D and Supplementary Figure 3). This observation suggests that CD20 level may be associated with the sensitivity to rILYd4 treatment. Taken together, these results indicate that rILYd4 sensitizes RR NHL and CLL cells to rituximab-mediated CDC in vitro and ex vivo, respectively.
Potential mechanism of up-regulation of CD59 in RR Ramos cells
Here, we document that the levels of GPI-anchored proteins such as mCRPs CD59 (Figure 1) and CD55, as well as non-mCRP CD48, but not non GPI-anchored proteins such as CD20 and mCRP CD46 gradually increased on the surface of these RR Ramos cells (Supplementary Figure 1). These results are consistent with the findings as reported previously by Takei et al(31). More interestingly, after removing the CD59-expressing subpopulation with ILY pretreatment, the expression of the other GPI-anchored proteins CD55 and CD48 disappeared simultaneously in the residual CD59 negative subpopulation, while the expression of non-GPI anchored proteins such as CD20 and CD46 remained unchanged (Figure 1C and Supplementary figure 4). To investigate the potential mechanism of the up-regulation of CD59 in RR Ramos cells, we used a CS (cell sorting) method to enrich and characterize the CD59-expressing population from OR cell. The enriched-CD59 positive cell population expressed CD20, CD46, CD59, CD55 and CD48 (Figure 3A) at a similar level and had the same sensitivity to rituximab-mediated CDC as the RR51.2 cells did (Figure 1A, 2B, 3C and Supplementary Figure 1). In addition, rILYd4 also sensitized CD59-enriched CS cells to rituximab-mediated CDC in a dose-dependent manner (Figure 3D). Furthermore, the CD59 negative subpopulation obtained from ILY-treated CS cells (Figure 3B) was sensitive to rituximab-mediated CDC (Figure 3E). Taken together, these results indicate that increased detection of GPI-anchored proteins in CD59-positive Ramos cells by multiple challenges with rituximab and complement may result from serial enrichment of the CD59-positive cells but not from induction of CD59 expression. Therefore, the elimination of the CD59 expressing subpopulation by combination treatment with rituximab and CD59 inhibitor such as rILYd4 may be a powerful approach to conquer rituximab resistance.
Figure 3. Expression of CD20, CD46, CD59, CD55 and CD48 on CD59-enriched CS cells with and without ILY pretreatment and their response to CDC effect induced by rituximab in vitro.

FACS analysis on CS cells without ILY pretreatment (A) and with ILY pretreatment (B). Cells were stained with anti-CD20, CD46, CD59, CD55, and CD48 Ab (black lines) or isotype-matched Ab (solid gray lines). CS cells are resistant to CDC effect as measured by Alamar blue assay, which is similar to RR51.2 cells (C). rILY4 sensitizes CD59-expressing cells to the CDC effect of rituximab as measured by Alamar blue assay (D). The hCD59 negative cells enriched from CS cells by the ILY pretreatment are very sensitive to rituximab-mediated CDC (E). Results are mean ± s.e.m of four different experiments.
rILYd4 sensitizes RR51.2 cells to rituximab-mediated CDC effect in vivo
Next, we used both developing and established orthotopic xenograft models to investigate whether rILYd4 sensitizes RR Ramos cells to rituximab treatment in vivo. We implanted RR51.2 cells into nude mice following published protocols(39, 40). In the developing xenograft model, we observed that combination treatment with rituximab and the adjuvant rILYd4 dramatically slowed down tumor growth as compared with rituximab alone (Figure 4A). Importantly, treatment with only rILYd4 did not affect tumor growth significantly (Figure 4A). A similar observation was also made in the established xenograft model where combination treatment with rituximab and the adjuvant rILYd4 resulted in significant reduction in tumor size as compared with treatment with rituximab alone (Figure 4B). Further, the tumor-free rates in the established xenograft model after treatment with vehicle, rILYd4 alone, rituximab alone and combination of rituximab with rILYd4 were 0%, 0%, 8.3% and 50%, respectively, suggesting treatment with rILYd4 as adjuvant can lead to elimination of tumor in 50% of the animals (Figure 4C).
Figure 4. rILYd4 is an effective adjuvant against RR cells in vivo.
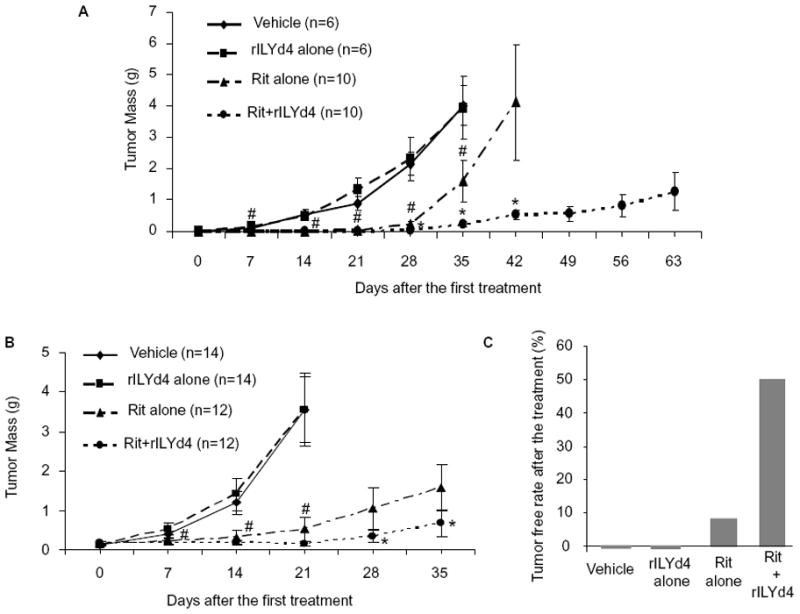
(A) In a developing xenograft mouse model. (B) In an established xenograft mouse model. (C) Tumor free rate in an established xenograft mouse model after treatment. #: P<0.05 vs vehicle treatment. *: P<0.05 vs rituximab alone treatment. The data represents as mean ± s.e.m.
PK/PD profiles of rILYd4
The circulating half life and volume of distribution of rILYd4 were determined after tail vein (i.v.) injection of rILYd4 into mCd59ab−/−/ThCD59RBC+/− mice. The serum level of rILYd4 showed a biphasic decay curve, with a fast initial distribution followed by a slower elimination phase. PK parameters are shown in Figure 5A. The half life (t1/2) of rILYd4 in vivo was 2.71 hr with a distribution volume of 383 ml/kg. The distribution of rILYd4 indicated by the Vss (distribution volume) is likely due to rILYd4’s binding to hCD59 on the erythrocytes of compound mice mCd59ab−/−/ThCD59RBC+/−.
Figure 5. PK and PD profiles of rILYd4.
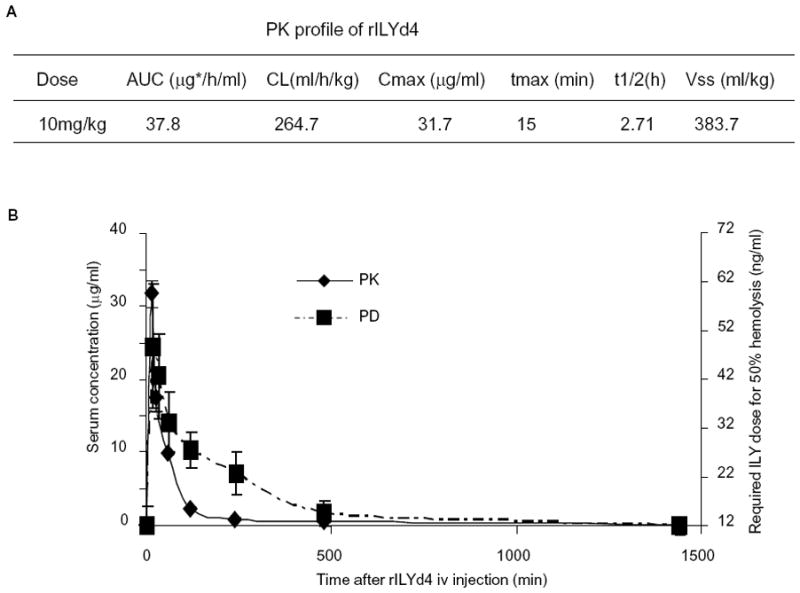
(A) PK profile of rILYd4. AUC: Area under the curve, CL: clearance, Cmax: maximum concentration of a compound in blood, Tmax: time at which Cmax is reached, t1/2: time required for the concentration of a compound in blood or plasma to decrease by 50%, and Vss: distribution volume. (B) Correlation between the PK and PD profile of rILYd4. The data represents as mean ± s.e.m.
PD study was carried out to evaluate the functionality of rILYd4. Theoretically, the rILYd4 bound to hCD59 should inhibit the hemolysis induced by intact ILY(36). Erythrocytes collected at different time points following treatment with 10 mg/kg rILYd4 showed a clearly time-dependent decline in protection against ILY-induced hemolysis as measured by the concentration of ILY required for 50% hemolysis (Figure 5B). Concomitant decay of rILYd4 function, as demonstrated by the PD study, largely overlapped with the decline in rILYd4 exposure in serum as determined in the PK study. 24 hours after one 10 mg/kg i.v. dose of rILYd4, we could not detect any additional protection by rILYd4 against the hemolytic activity of ILY (Figure 5B).
rILYd4 alone did not mediate hemolytic anemia in mCd59ab−/−/ThCD59RBC+/− mice
We have previously reported(36) and also confirmed in this study that rILYd4 alone has no direct lytic effect on hCD59-expressing cells in vitro (Supplementary Figure 2B). To further assess its toxicity profile in vivo, we utilized mCd59ab−/−/ThCD59RBC+/−. In an acute toxicity study, we found that there were no significant differences in the levels of hemoglobin content or free plasma hemoglobin (data not shown) between mCd59ab−/−/ThCD59RBC+/− mice treated with rILYd4 or vehicle (Figure 6A). In the sub-chronic toxicity study with administration of rILYd4 for one month, rILYd4 did not result in any significant difference in hemoglobin levels, reticulocyte counts, and body weight or pathological changes in tissues (data not shown) as compared to vehicle alone (Figure 6B-D). These results suggest that transient inhibition of hCD59 function by rILYd4 does not induce hemolysis or any negative side effects, and that rILYd4 is neither acutely nor sub-chronically toxic.
Figure 6. rILYd4 does not mediate acute or sub-chronic toxicity in mCd59ab−/− /ThCD59RBC+/− mice.
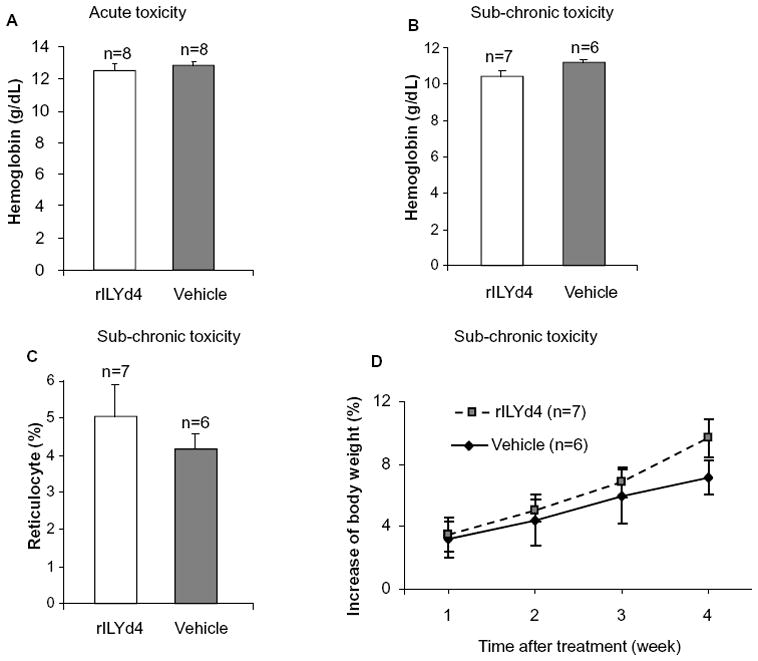
(A) Hemoglobin content in acute toxicity study. (B–D) Hemoglobin content (B), reticulocyte count (C) and body weight (D) in sub-chronic toxicity study. Results are presented as mean ± s.e.m.
Discussion
Here, we demonstrate that the administration of rILYd4 abrogates hCD59 function in RR cells and restores the sensitivity of these resistant Ramos and primary CLL cells to rituximab-mediated CDC effects in vitro, ex vivo and in vivo. We also document that the hCD59 over-expressing population of Ramos cells is responsible for the resistance to rituximab-mediated CDC in vitro. These results highlight the critical role of CD59 in the development of rituximab resistance, and indicate that rILYd4 may provide a new approach to enhance the therapeutic efficacy of rituximab by abrogating hCD59 activity. These interpretations are also supported by our recent findings that rILYd4 was able to sensitize the lymphoma cell line RL-7 or the multiple myeloma cell line ARH-7 to rituximab-mediated CDC(42).
The role of CDC in rituximab therapy was challenged previously by the findings that the expressions of mCRPs does not predict clinical outcome after rituximab treatment in follicular NHL(43). Recently, Weiner’s group demonstrate that the C3b component of complement could inhibit NK cell activation and ADCC effects during rituximab treatment(10), and thus C3 depletion improves rituximab antitumor activity(20). Also, several researches have shown that both CDC and ADCC play major roles in the antitumor activity of rituximab(2, 8–10). Furthermore, CDC-resistant cells are sensitive to ADCC and vice versa(9, 14, 44). Differences in the relative importance of CDC and ADCC following rituximab treatment may result from the different types of tumors used, expression levels of CD20 and mCRP, tumor-inoculating methods, and tumor growth period(9, 11). Although the investigation of the relative roles of CDC and ADCC is beyond the scope of our studies, it will be very helpful for the future of drug design and therefore warrants further investigation.
Since CD59 is universally expressed in human cells with a relative high levels in erythrocytes, potential side effects such as hemolysis are the potential hurdles for the development of clinically-useful hCD59 inhibitors. Monoclonal antibodies directed against hCD59 are useful tools for the study of CD59 function in vitro(4, 19, 31). In spite of that, it is impossible to apply them to cancer therapy, since they bind not only cancer cells, but also many other cells that express hCD59, potentially inducing ADCC, or CDC or apoptosis in normal cells. To address this problem, Ziller et al. identified mini-antibodies that specifically blocked hCD59 and hCD55 function(23). They reported that these mini-antibodies facilitated rituximab-mediated lymphoma treatment in vivo; however, although alone they failed to activate complement, they killed about 25% of cancer cells through ADCC(24). Therefore, to limit this unwanted side effect on normal cells and improve specific targeting of cancer cells, they administered biotin-labeled rituximab in combination with avidin-labeled MB55–MB59 to target the mini-anti CD59 antibodies to the cancer cells(24). In contrast with this approach, rILYd4 restores sensitivity of the RR lymphoma cells to rituximab treatment in vitro and in vivo. Moreover, rILYd4 itself does not trigger lysis(36) or ADCC effect in cells that are non targeted by the antibody (data not shown) ex vivo and in vivo. Therefore, rILYd4 may be a safe and effective adjuvant for rituximab therapy.
The higher expression of CD59 in cancer cells such as lymphoma and rituximab pretreated lymphoma cells has been long recognized but its mechanism remains unclear(32, 33). Our results indicate that RR Ramos cells over-expressing GPI-anchored proteins including CD59, CD55 and CD48, may result from the positive selection of pre-existing cells that highly express GPI-anchored proteins, rather than from the induction of GPI-anchored proteins. We therefore postulate that tumor heterogeneity may be responsible for the increase in the population of highly expressing CD59 lymphoma cells, a contributor to rituximab resistance. In support of this hypothesis, we demonstrate that two other NHL cell lines (Raji and Daudi) resistant to rituximab-mediated CDC also express a higher level of CD59 than their parental cell lines (Supplementary Figure 5). Together, these results indicate that the ablation of those pre-existing resistant tumor cells by early treatment with combination of rILYd4 and rituximab may effectively limit the expansion of RR lymphoma cells through the abrogation of this subpopulation of cells expressing high levels of hCD59. Further, rILYd4 may also sensitize tumor cells that have acquired resistance to rituximab after multiple therapies to the anti-cancer activity of rituximab.
We also designed and executed a PK/PD study to evaluate drug exposure and −/−other kinetic parameters such as t1/2, Cmax and volume of distribution in mCd59ab /ThCD59RBC+/− mice. The avid binding of rILY4 to hCD59 most likely explains its larger volume of distribution (Vss=383 ml/kg). For the PD study, we took advantage of the fact that the pre-formed rILYd4-hCD59 interaction on erythrocytes was able to compete with binding of the full length ILY to hCD59 and exert protection against ILY-mediated hemolysis(36). The comparable PK and PD profiles suggest that rILYd4 bound to erythrocytes remains functional throughout the in vivo exposure. These data provide insight and guidance for the further engineering of rILYd4 to better suit biologic therapy.
The toxicity profile is critical for identifying a dosing window to get good efficacy with tolerable side effects. Here, we demonstrate that i.p. injection of 3-fold the effective dose (6 mg/kg, Q4D X 8 times) of rILYd4 did not induce erythrocyte lysis in hCD59 transgenic mice in a mCd59 deficient background. No other notable pathological changes have been observed either. Overall, at the doses tested, rILYd4 did not show any sign of unwanted side effects. It remains to be seen whether other toxic effects emerge upon reaching the maximum tolerated dose in mice. Immunogenicity is another critical aspect for the effective development of a protein drug. Previous findings indicate that rILYd4 has low immunogenicity(45, 46). However, a low- or even non-immunogenic form of rILYd4 or ILYd4-derived peptides will be essential for clinical application, and requires further investigation and development.
Supplementary Material
Acknowledgments
We are grateful to Ms. Annie Qin for helpful editorial assistance.
Grant support
NIHRO1AI061174 (X.B.) and NIHR21CA141324 (X.B.), Harvard Technology Development Accelerator Fund (X.B), and Natural Science Foundation of China (30972952) (Z.P.), China National key Technology R&D Program (2008BAI60B03) (Z.P.), and China Scholarship Council (2009628090) (X.G.). Dr Jennifer Brown is supported by NIH K23, as well as by an ASH Scholar Award and the Leukemia and Lymphoma Society Scholar in Clinical Research Award.
Footnotes
Disclosure of Conflicts of Interest
The authors declare that they have no conflict of interests.
References
- 1.Glennie MJ, French RR, Cragg MS, Taylor RP. Mechanisms of killing by anti-CD20 monoclonal antibodies. Molecular immunology. 2007;44:3823–37. doi: 10.1016/j.molimm.2007.06.151. [DOI] [PubMed] [Google Scholar]
- 2.Fishelson Z, Donin N, Zell S, Schultz S, Kirschfink M. Obstacles to cancer immunotherapy: expression of membrane complement regulatory proteins (mCRPs) in tumors. Molecular immunology. 2003;40:109–23. doi: 10.1016/s0161-5890(03)00112-3. [DOI] [PubMed] [Google Scholar]
- 3.Bonavida B. Rituximab-induced inhibition of antiapoptotic cell survival pathways: implications in chemo/immunoresistance, rituximab unresponsiveness, prognostic and novel therapeutic interventions. Oncogene. 2007;26:3629–36. doi: 10.1038/sj.onc.1210365. [DOI] [PubMed] [Google Scholar]
- 4.Golay J, Lazzari M, Facchinetti V, et al. CD20 levels determine the in vitro susceptibility to rituximab and complement of B-cell chronic lymphocytic leukemia: further regulation by CD55 and CD59. Blood. 2001;98:3383–9. doi: 10.1182/blood.v98.12.3383. [DOI] [PubMed] [Google Scholar]
- 5.Kennedy AD, Beum PV, Solga MD, et al. Rituximab infusion promotes rapid complement depletion and acute CD20 loss in chronic lymphocytic leukemia. J Immunol. 2004;172:3280–8. doi: 10.4049/jimmunol.172.5.3280. [DOI] [PubMed] [Google Scholar]
- 6.Golay J, Cittera E, Di Gaetano N, et al. The role of complement in the therapeutic activity of rituximab in a murine B lymphoma model homing in lymph nodes. Haematologica. 2006;91:176–83. [PubMed] [Google Scholar]
- 7.Beum PV, Lindorfer MA, Beurskens F, et al. Complement activation on B lymphocytes opsonized with rituximab or ofatumumab produces substantial changes in membrane structure preceding cell lysis. J Immunol. 2008;181:822–32. doi: 10.4049/jimmunol.181.1.822. [DOI] [PubMed] [Google Scholar]
- 8.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat Med. 2000;6:443–6. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 9.Zhou X, Hu W, Qin X. The role of complement in the mechanism of action of rituximab for B-cell lymphoma: implications for therapy. Oncologist. 2008;13:954–66. doi: 10.1634/theoncologist.2008-0089. [DOI] [PubMed] [Google Scholar]
- 10.Wang SY, Racila E, Taylor RP, Weiner GJ. NK-cell activation and antibody-dependent cellular cytotoxicity induced by rituximab-coated target cells is inhibited by the C3b component of complement. Blood. 2008;111:1456–63. doi: 10.1182/blood-2007-02-074716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Introna M, Golay J. Complement in antibody therapy: friend or foe? Blood. 2009;114:5247–8. doi: 10.1182/blood-2009-10-249532. [DOI] [PubMed] [Google Scholar]
- 12.Lim SH, Beers SA, French RR, Johnson PW, Glennie MJ, Cragg MS. Anti-CD20 monoclonal antibodies: historical and future perspectives. Haematologica. 2010;95:135–43. doi: 10.3324/haematol.2008.001628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cragg MS, Glennie MJ. Antibody specificity controls in vivo effector mechanisms of anti-CD20 reagents. Blood. 2004;103:2738–43. doi: 10.1182/blood-2003-06-2031. [DOI] [PubMed] [Google Scholar]
- 14.Cittera E, Leidi M, Buracchi C, et al. The CCL3 family of chemokines and innate immunity cooperate in vivo in the eradication of an established lymphoma xenograft by rituximab. J Immunol. 2007;178:6616–23. doi: 10.4049/jimmunol.178.10.6616. [DOI] [PubMed] [Google Scholar]
- 15.Di Gaetano N, Cittera E, Nota R, et al. Complement activation determines the therapeutic activity of rituximab in vivo. J Immunol. 2003;171:1581–7. doi: 10.4049/jimmunol.171.3.1581. [DOI] [PubMed] [Google Scholar]
- 16.Winkler U, Jensen M, Manzke O, Schulz H, Diehl V, Engert A. Cytokine-release syndrome in patients with B-cell chronic lymphocytic leukemia and high lymphocyte counts after treatment with an anti-CD20 monoclonal antibody (rituximab, IDEC-C2B8) Blood. 1999;94:2217–24. [PubMed] [Google Scholar]
- 17.Klepfish A, Gilles L, Ioannis K, Eliezer R, Ami S. Enhancing the action of rituximab in chronic lymphocytic leukemia by adding fresh frozen plasma: complement/rituximab interactions & clinical results in refractory CLL. Annals of the New York Academy of Sciences. 2009;1173:865–73. doi: 10.1111/j.1749-6632.2009.04803.x. [DOI] [PubMed] [Google Scholar]
- 18.Xu W, Miao KR, Zhu DX, et al. Enhancing the action of rituximab by adding fresh frozen plasma for the treatment of fludarabine refractory chronic lymphocytic leukemia. International journal of cancer. 2010 doi: 10.1002/ijc.25560. [DOI] [PubMed] [Google Scholar]
- 19.Golay J, Zaffaroni L, Vaccari T, et al. Biologic response of B lymphoma cells to anti-CD20 monoclonal antibody rituximab in vitro: CD55 and CD59 regulate complement-mediated cell lysis. Blood. 2000;95:3900–8. [PubMed] [Google Scholar]
- 20.Wang H, Liu Y, Li ZY, Fan X, Hemminki A, Lieber A. A recombinant adenovirus type 35 fiber knob protein sensitizes lymphoma cells to rituximab therapy. Blood. 2010;115:592–600. doi: 10.1182/blood-2009-05-222463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harjunpaa A, Junnikkala S, Meri S. Rituximab (anti-CD20) therapy of B-cell lymphomas: direct complement killing is superior to cellular effector mechanisms. Scandinavian journal of immunology. 2000;51:634–41. doi: 10.1046/j.1365-3083.2000.00745.x. [DOI] [PubMed] [Google Scholar]
- 22.Bellosillo B, Villamor N, Lopez-Guillermo A, et al. Complement-mediated cell death induced by rituximab in B-cell lymphoproliferative disorders is mediated in vitro by a caspase-independent mechanism involving the generation of reactive oxygen species. Blood. 2001;98:2771–7. doi: 10.1182/blood.v98.9.2771. [DOI] [PubMed] [Google Scholar]
- 23.Ziller F, Macor P, Bulla R, Sblattero D, Marzari R, Tedesco F. Controlling complement resistance in cancer by using human monoclonal antibodies that neutralize complement-regulatory proteins CD55 and CD59. European journal of immunology. 2005;35:2175–83. doi: 10.1002/eji.200425920. [DOI] [PubMed] [Google Scholar]
- 24.Macor P, Tripodo C, Zorzet S, et al. In vivo targeting of human neutralizing antibodies against CD55 and CD59 to lymphoma cells increases the antitumor activity of rituximab. Cancer Res. 2007;67:10556–63. doi: 10.1158/0008-5472.CAN-07-1811. [DOI] [PubMed] [Google Scholar]
- 25.Fisicaro N, Aminian A, Hinchliffe SJ, et al. The pig analogue of CD59 protects transgenic mouse hearts from injury by human complement. Transplantation. 2000;70:963–8. doi: 10.1097/00007890-200009270-00014. [DOI] [PubMed] [Google Scholar]
- 26.Sugita Y, Tobe T, Oda E, et al. Molecular cloning and characterization of MACIF, an inhibitor of membrane channel formation of complement. J Biochem (Tokyo) 1989;106:555–7. doi: 10.1093/oxfordjournals.jbchem.a122893. [DOI] [PubMed] [Google Scholar]
- 27.Medof ME, Lublin DM, Holers VM, et al. Cloning and characterization of cDNAs encoding the complete sequence of decay-accelerating factor of human complement. Proceedings of the National Academy of Sciences of the United States of America. 1987;84:2007–11. doi: 10.1073/pnas.84.7.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davitz MA, Low MG, Nussenzweig V. Release of decay-accelerating factor (DAF) from the cell membrane by phosphatidylinositol-specific phospholipase C (PIPLC) J Exp Med. 1986;163:1150–61. doi: 10.1084/jem.163.5.1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nicholson-Weller A, Spicer DB, Austen KF. Deficiency of the complement regulatory protein, “decay-accelerating factor,” on membranes of granulocytes, monocytes, and platelets in paroxysmal nocturnal hemoglobinuria. The New England journal of medicine. 1985;312:1091–7. doi: 10.1056/NEJM198504253121704. [DOI] [PubMed] [Google Scholar]
- 30.Brodbeck WG, Mold C, Atkinson JP, Medof ME. Cooperation between decay-accelerating factor and membrane cofactor protein in protecting cells from autologous complement attack. J Immunol. 2000;165:3999–4006. doi: 10.4049/jimmunol.165.7.3999. [DOI] [PubMed] [Google Scholar]
- 31.Takei K, Yamazaki T, Sawada U, Ishizuka H, Aizawa S. Analysis of changes in CD20, CD55, and CD59 expression on established rituximab-resistant B-lymphoma cell lines. Leuk Res. 2006;30:625–31. doi: 10.1016/j.leukres.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 32.Dalle S, Dupire S, Brunet-Manquat S, Reslan L, Plesa A, Dumontet C. In vivo model of follicular lymphoma resistant to rituximab. Clin Cancer Res. 2009;15:851–7. doi: 10.1158/1078-0432.CCR-08-1685. [DOI] [PubMed] [Google Scholar]
- 33.Bannerji R, Kitada S, Flinn IW, et al. Apoptotic-regulatory and complement-protecting protein expression in chronic lymphocytic leukemia: relationship to in vivo rituximab resistance. J Clin Oncol. 2003;21:1466–71. doi: 10.1200/JCO.2003.06.012. [DOI] [PubMed] [Google Scholar]
- 34.Coral S, Fonsatti E, Sigalotti L, et al. Overexpression of protectin (CD59) down-modulates the susceptibility of human melanoma cells to homologous complement. Journal of cellular physiology. 2000;185:317–23. doi: 10.1002/1097-4652(200012)185:3<317::AID-JCP1>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 35.Fonsatti E, Altomonte M, Coral S, et al. Emerging role of protectin (CD59) in humoral immunotherapy of solid malignancies. Clin Ter. 2000;151:187–93. [PubMed] [Google Scholar]
- 36.Hu W, Yu Q, Hu N, et al. A High-Affinity Inhibitor of Human CD59 Enhances Complement-Mediated Virolysis of HIV-1: Implications for Treatment of HIV-1/AIDS. J Immunol. 2010;184:359–68. doi: 10.4049/jimmunol.0902278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qin X, Hu W, Song W, et al. Generation and phenotyping of mCd59a and mCd59b double-knockout mice. American journal of hematology. 2009;84:65–70. doi: 10.1002/ajh.21319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu W, Ferris SP, Tweten RK, et al. Rapid conditional targeted ablation of cells expressing human CD59 in transgenic mice by intermedilysin. Nat Med. 2008;14:98–103. doi: 10.1038/nm1674. [DOI] [PubMed] [Google Scholar]
- 39.DiJoseph JF, Dougher MM, Kalyandrug LB, et al. Antitumor efficacy of a combination of CMC-544 (inotuzumab ozogamicin), a CD22-targeted cytotoxic immunoconjugate of calicheamicin, and rituximab against non-Hodgkin's B-cell lymphoma. Clin Cancer Res. 2006;12:242–9. doi: 10.1158/1078-0432.CCR-05-1905. [DOI] [PubMed] [Google Scholar]
- 40.Dijoseph JF, Dougher MM, Armellino DC, et al. CD20-specific antibody-targeted chemotherapy of non-Hodgkin's B-cell lymphoma using calicheamicin-conjugated rituximab. Cancer Immunol Immunother. 2007;56:1107–17. doi: 10.1007/s00262-006-0260-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Giddings KS, Zhao J, Sims PJ, Tweten RK. Human CD59 is a receptor for the cholesterol-dependent cytolysin intermedilysin. Nat Struct Mol Biol. 2004;11:1173–8. doi: 10.1038/nsmb862. [DOI] [PubMed] [Google Scholar]
- 42.You Tao, Hu Weiguo, Ge Xiaowen, Shen Jinnan, Qin Xuebin. Application of a novel inhibitor of human CD59 for the enhancement of complement-dependent cytolysis on cancer cells. Cellular & molecular immunology. 2010 doi: 10.1038/cmi.2010.35. (In press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weng WK, Levy R. Expression of complement inhibitors CD46, CD55, and CD59 on tumor cells does not predict clinical outcome after rituximab treatment in follicular non-Hodgkin lymphoma. Blood. 2001;98:1352–7. doi: 10.1182/blood.v98.5.1352. [DOI] [PubMed] [Google Scholar]
- 44.van Meerten T, van Rijn RS, Hol S, Hagenbeek A, Ebeling SB. Complement-induced cell death by rituximab depends on CD20 expression level and acts complementary to antibody-dependent cellular cytotoxicity. Clin Cancer Res. 2006;12:4027–35. doi: 10.1158/1078-0432.CCR-06-0066. [DOI] [PubMed] [Google Scholar]
- 45.Ohkura K, Nagamune H, Kourai H. Structural analysis of human specific cytolysin intermedilysin aiming application to cancer immunotherapy. Anticancer research. 2004;24:3343–53. [PubMed] [Google Scholar]
- 46.Nagamune H, Ohkura K, Umezu K, Shouji H, Kourai H. A cell membrane modification technique using domain 4 of intermedilysin for immunotherapy against cancer. Anticancer research. 2004;24:3367–72. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.