

Abstract
Epigenetic information is an important mediator of the relationship between genotype and phenotype in eukaryotic organisms. One of the most important and widely conserved forms of epigenetic information is the methylation of genes. However, the function of intragenic DNA methylation remains poorly understood. The goal of this study was to gain greater understanding of the nature of intragenic methylation by determining its role in the multilayered epigenetic landscape of insects. We first investigated the evolutionary lability of DNA methylation by examining whether methylation patterns were conserved in the fire ant and honey bee. We found that DNA methylation was targeted to largely overlapping sets of orthologs in both species. Next, we compared intragenic DNA methylation levels in the fire ant and honey bee to comprehensive epigenetic and gene-regulatory data from Drosophila melanogaster orthologs. We observed striking evidence of a conserved association between DNA methylation in fire ants and honey bees, and several active histone modifications, constitutive gene expression, and “broad” promoter architecture in D. melanogaster. Overall, our study illustrates that DNA methylation is a single component of a conserved, integrated, multilayered epigenetic and regulatory landscape in insect genomes.
Keywords: DNA methylome, epigenetics, eusocial, gene regulation, Solenopsis invicta
Introduction
A wide array of animals and plants display intragenic DNA methylation. Intragenic methylation is the methylation of exons and introns (Zemach et al. 2010) and is targeted to active genes (Zemach et al. 2010; Jjingo et al. 2012), in contrast to promoter methylation, which is linked to gene repression (Bird and Wolffe 1999; Pai et al. 2011; Zeng et al. 2012). Intragenic methylation has been hypothesized to affect a wide array of processes including the regulation of messenger RNA splicing (Shukla et al. 2011), the initiation of transcription within genes (Zilberman et al. 2007; Maunakea et al. 2010), and transcriptional elongation (Lorincz et al. 2004; Zilberman et al. 2007).
Given the important regulatory roles suggested for intragenic DNA methylation, it is somewhat perplexing that DNA methylation has been lost entirely in some eukaryotic lineages, including several clades of insects (Zemach et al. 2010; Glastad et al. 2011). Insight into the loss of DNA methylation may be found in the presence of functional overlap with histone modifications (Cedar and Bergman 2009; Nanty et al. 2011; Suganuma and Workman 2011). The regulatory roles of histone modifications include the mediation of binding affinities of protein complexes, such as those related to transcriptional and splicing machinery (Kolasinska-Zwierz et al. 2009; Luco et al. 2010, 2011; Negre et al. 2011). Thus, similar to DNA methylation, histone modifications convey important epigenetic information, which affects gene regulation and organismal development. Indeed, one of the most exciting developments in the field of epigenetics was the realization that patterns of DNA methylation interact with histone modifications and nucleosome positioning in a multifaceted epigenetic landscape (Cedar and Bergman 2009; Chodavarapu et al. 2010).
This study is focused on understanding the patterning and regulatory significance of intragenic DNA methylation in insect genomes. We first examine the extent to which DNA methylation targeting is conserved between species. This provides insight into the evolutionary lability of DNA methylation. Second, we determine whether conserved evolutionary associations exist between DNA methylation patterning and histone modifications. The associations between these epigenetic marks have remained largely unexplored in insects because the primary model for understanding insect histone modifications, Drosophila melanogaster, lacks DNA methylation (Zemach et al. 2010). To address this issue, we compare DNA methylation data from the fire ant Solenopsis invicta and the honey bee Apis mellifera to extensive histone modification data from D. melanogaster. Finally, we explore associations between gene expression, DNA methylation, and histone modifications, and how these associations relate to other regulatory features in insect genomes. Overall, our investigation provides important insight into how different layers of epigenetic information are linked to gene regulation.
Materials and Methods
DNA Methylomes
We generated DNA methylation data from the fire ant S. invicta to compare with existing data from the honey bee A. mellifera (detailed materials and methods provided in Supplementary Material online). We used whole bodies of four different adult fire ant morphs (queens, workers, haploid males, and diploid males) to generate single-base resolution DNA methylation maps (DNA methylomes). From these data, we generated a species-level S. invicta DNA methylome (supplementary table S1, Supplementary Material online). For comparative purposes, we mapped a species-level DNA methylome of A. mellifera using adult queen and worker brain DNA methylomes (Lyko et al. 2010) and a whole body worker DNA methylome (Zemach et al. 2010) (supplementary table S2, Supplementary Material online).
Bisulfite conversion and Illumina paired end sequencing of S. invicta genomic DNA were performed by Beijing Genomics Institute (Zhenzhen, China). Reads from S. invicta and A. mellifera (Lyko et al. 2010; Zemach et al. 2010) were aligned to reference genomes (Honeybee Genome Sequencing Consortium 2006; Wurm et al. 2011) using Bismark (Krueger and Andrews 2011). Fractional methylation values were calculated for each CpG site as mCG/CGall, where mCG is the number of reads with a methylated cytosine at a CpG site (according to nonconversion) and CGall is the total number of reads mapped to the site. Fractional methylation values were then averaged across each annotated element. Solenopsis invicta DNA methylation data are available from Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/; GSE39959).
Histone Modification Data
Drosophila melanogaster preprocessed Chromatin ImmunoPrecipitation using genomic tiling arrays (ChIP-chip) data files identifying regions of significant enrichment for each histone modification were obtained from modEncode (Celniker et al. 2009) (supplementary table S3, Supplementary Material online). We used ChIP-chip data from the S2-DRSC late-stage embryonic cell line. We determined the proportion of an element overlapped by significant regions of enrichment (overlap), the mean enrichment score for these regions, and a composite metric, which is the product of score and overlap metrics. The composite metric was used in all analyses unless otherwise indicated.
Gene Expression
Solenopsis invicta gene expression data were obtained from a prior cDNA microarray study (Ometto et al. 2011). For each gene, we assessed the coefficient of variation (standard deviation/mean; CV) of expression values as the mean of CV values calculated separately for whole body S. invicta adult and pupal workers, queens, and haploid males (median of 5 biological replicates per morph) (Ometto et al. 2011).
Drosophila melanogaster gene expression data were obtained from FlyAtlas (Chintapalli et al. 2007). We estimated the overall CV in FlyAtlas expression values as the mean of CV values calculated separately for each of 10 tissues (based on 4 biological replicates per tissue). The “tissue specificity index” of each gene was calculated using data from 10 tissues.
Orthology
We used OrthoDB (Waterhouse et al. 2011) 12-insect orthology data to assign orthologous genes between S. invicta, A. mellifera, and D. melanogaster.
Gene Ontology
Solenopsis invicta and A. mellifera Gene Ontology (GO) was assigned, and GO term enrichment determined, using Blast2GO (Conesa et al. 2005).
Results
DNA Methylation Targets Are Conserved in the Fire Ant S. invicta and Honey Bee A. mellifera
We found that DNA methylation was targeted primarily to exons in the fire ant and the honey bee (supplementary figs. S1–S3, Supplementary Material online) (Lyko et al. 2010; Bonasio et al. 2012). DNA methylation was lower in introns than exons, but the discrepancy between exon and intron methylation level was much larger in the honey bee (supplementary fig. S2, Supplementary Material online) (Lyko et al. 2010). We also observed a pronounced preference for DNA methylation targeting to 5′-intragenic regions in both species (supplementary figs. S2 and S4, Supplementary Material online) (Zemach et al. 2010; Bonasio et al. 2012).
The fire ant displayed substantially lower levels of intragenic DNA methylation than the honey bee (supplementary fig. S1, Supplementary Material online). There were also minor, but statistically significant, differences in coding sequence DNA methylation levels among morphs within both species (all comparisons between morphs within the fire ant and within the honeybee differed significantly, P < 0.01, Wilcoxon signed-rank tests, n = 5,203 orthologous genes) (Lyko et al. 2010; Bonasio et al. 2012).
Comparative analyses using empirical DNA methylation data revealed that the localization of DNA methylation was highly similar in the fire ant and the honey bee (Spearman’s rho between species-level DNA methylation levels of orthologous coding sequences = 0.717, P < 2.2 × 10−16, n = 5,203; supplementary table S4 and figs. S2–S4, Supplementary Material online). DNA methylation was targeted to fewer genes in fire ants than in honey bees (table 1). However, 93% of orthologous genes methylated in the fire ant were also targeted by DNA methylation in the honey bee (table 1). Thus, the genes targeted by DNA methylation in the fire ant were largely a subset of those targeted in the honey bee (table 1). Furthermore, as in the honey bee and other invertebrates (Hunt et al. 2010; Sarda et al. 2012), genes targeted by DNA methylation in the fire ant exhibited enrichment of functional annotations linked to cellular “housekeeping,” including an 8-fold enrichment of “translation” annotations (supplementary table S5, Supplementary Material online).
Table 1.
DNA Methylation Largely Targets the Same Genes in the Fire Ant and the Honey Bee
| Species | Methylated Genes | Unmethylated Genes | Uniquely Methylated in This Species (%) |
|---|---|---|---|
| Fire ant | 2,581 | 2,622 | 174 (6.7) |
| Honey bee | 3,030 | 2,173 | 623 (20.6) |
DNA Methylation Is Targeted to Genes Marked by Active Histone Modifications in Insects
The genomic localizations of many histone modifications are highly conserved among taxa (Bernstein et al. 2005; Feng and Jacobsen 2011; Woo and Li 2012), suggesting that insight into histone modification targeting can be gleaned from distantly related organisms (Nanty et al. 2011). Moreover, the presence of many histone modifications, previously identified in diverse eukaryotes, was recently confirmed in the honey bee (Dickman et al. 2013). Accordingly, we compared patterns of DNA methylation in the fire ant and honey bee to histone modifications in D. melanogaster (Celniker et al. 2009). The goal of this analysis was to determine whether intragenic DNA methylation was associated with particular histone modifications in insect genomes (Nanty et al. 2011).
We observed striking positive correlations between DNA methylation levels, in the fire ant and honey bee, and several histone modifications associated with active transcriptional states in D. melanogaster (fig. 1 and table 2) (Filion et al. 2010; Kharchenko et al. 2011; Nanty et al. 2011). The correlation coefficients describing these associations varied by exon position according to the spatial localization of each histone modification (fig. 1 and supplementary fig. S5, Supplementary Material online). In particular, active histone modifications that were localized to 5′-gene regions in D. melanogaster (e.g., histone H3 lysine 4 di- and trimethylation [H3K4me2/me3]) (Kharchenko et al. 2011) were most strongly correlated with DNA methylation in 5′-gene regions following the translation start site in the fire ant (fig. 1). By comparison, active histone modifications associated with transcriptional elongation (e.g., H3K79me2 and H3K36me3) (Kharchenko et al. 2011) exhibited greater homogeneity in correlations with DNA methylation across exon positions (fig. 1). In addition, the modifications H3K27me2/me3, which are associated with domains of polycomb-mediated repression and the regulation of tissue-specific gene expression (Bell et al. 2010; Kharchenko et al. 2011; Lafos et al. 2011), were negatively correlated with DNA methylation in the fire ant and honey bee (fig. 1). Overall, these correlations, which were largely unchanged when controlling for sequence length (supplementary table S6, Supplementary Material online), suggest remarkable concordance between the patterning of DNA methylation, when present, and histone modifications across evolutionary time (Nanty et al. 2011).
Fig. 1.—
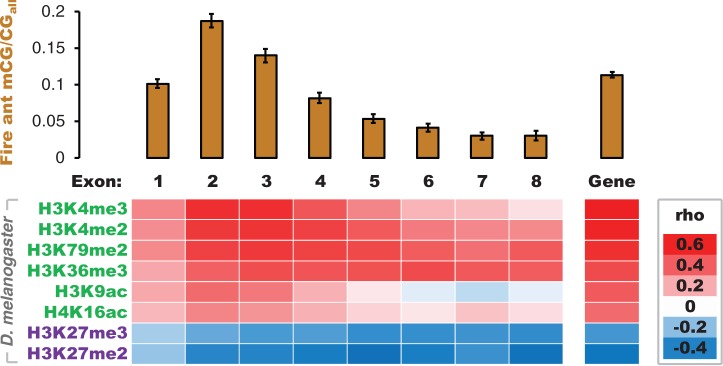
DNA methylation is targeted to genes marked by active histone modifications. Bars indicate mean DNA methylation level (mCG/CGall) in the fire ant with 95% confidence intervals. The heatmap conveys the strength and direction of Spearman’s rank correlation coefficients (rho) between DNA methylation levels in the fire ant Solenopsis invicta and enrichment of histone modifications targeted to orthologs in the fly Drosophila melanogaster. Histone modifications in green are associated with active transcription and those in purple are associated with repression of transcription (Kharchenko et al. 2011). Exon position is indicated from 5′ to 3′ relative to the translation start site in each species and “gene” indicates the gene body (introns included but excluding untranslated regions).
Table 2.
Spearman’s Rank Correlations (ρ) between Coding Sequence DNA Methylation Level in the Fire Ant or Honey Bee and Histone Modifications Associated with Orthologous DNA Sequences in Drosophila melanogaster
| Drosophila melanogaster Histone Modification (X) | Active/Repressivea | Genomic Associationa | Fire Ant Methylation (ρ X)b | Honey Bee Methylation (ρ X)b |
|---|---|---|---|---|
| H3K4me3 | Active | Transcription start site-proximal regions | 0.618**** | 0.529**** |
| H3K4me2 | Active | Transcription start site-proximal regions | 0.572**** | 0.505**** |
| H3K79me2 | Active | Transcription start site-proximal regions, transcriptional elongation of exonic regions, and intronic regions | 0.527**** | 0.484**** |
| H3K9ac | Active | Transcription start site-proximal regions | 0.436**** | 0.354**** |
| H3K36me3 | Active | Transcriptional elongation of exonic regions | 0.397**** | 0.392**** |
| H4K16ac | Active | Chromosome X genes and male dosage compensation | 0.362**** | 0.333**** |
| H3K79me1 | Active | Transcriptional elongation of exonic regions and intronic regions | 0.301**** | 0.357**** |
| H3K4me1 | Active | Intronic regions | 0.159**** | 0.198**** |
| H3K18ac | Active | Intronic regions | 0.007NS | 0.014NS |
| H3K9me2 | Repressive | Pericentromeric heterochromatin and heterochromatin-like domains | −0.032* | −0.010NS |
| H3K9me3 | Repressive | Pericentromeric heterochromatin and heterochromatin-like domains | −0.068**** | −0.025NS |
| H3K9me1 | −0.151**** | −0.120**** | ||
| H3K36me1 | Active | Intronic regions | −0.157**** | −0.139**** |
| H3K27me3 | Repressive | Polycomb-mediated repression | −0.316**** | −0.297**** |
| H3K27me2 | −0.432**** | −0.398**** |
bn = 5,619 orthologs for the honeybee and n = 4,674 orthologs for the fire ant.
****P < 0.0001.
*P < 0.05.
NSP ≥ 0.05.
DNA Methylation and Active Histone Modifications Are Associated with Constitutive Gene Expression
We investigated the relationship between intragenic DNA methylation and gene expression in the fire ant (Ometto et al. 2011). We observed a generally positive but bell-shaped relationship between levels of DNA methylation and overall gene expression levels (fig. 2A), as observed in previous studies in diverse eukaryotes (Zemach et al. 2010; Bonasio et al. 2012; Jjingo et al. 2012). We further observed a striking negative correlation between variability in gene expression, as measured by the CV among individuals, and DNA methylation (fig. 2B).
Fig. 2.—

Both DNA methylation and active histone modifications are similarly associated with transcriptional regulation. Mean fire ant coding sequence DNA methylation level (mCG/CGall) exhibits (A) a generally positive but bell-shaped relationship with gene expression level, and (B) a negative relationship with the CV in gene expression (CV expression). Error bars indicate 95% confidence intervals (n = 3,201 for panels A and B). (C) Gene expression measures in Drosophila melanogaster are strongly negatively (CV expression and tissue specificity) or positively (expression level) correlated with enrichment of active histone modifications in D. melanogaster and DNA methylation in Solenopsis invicta and Apis mellifera (n = 3,892 orthologous genes). All correlations are highly significant (P < 2.2 × 10−16).
We next investigated the transcriptional correlates of active histone modifications in D. melanogaster. Specifically, we examined whether particular D. melanogaster histone modifications were associated with D. melanogaster gene expression measures (Chintapalli et al. 2007) including expression level, tissue specificity, and variability of gene expression among individuals as measured by CV. We found that active histone modifications were positively correlated with overall gene expression level and negative correlated with tissue specificity of expression in D. melanogaster (fig. 2C), as observed previously (Filion et al. 2010; Kharchenko et al. 2011). We also observed negative correlations between active histone modifications and variability of gene expression among D. melanogaster individuals.
We next investigated whether DNA methylation levels in fire ant and honey bee genes were correlated to gene expression measures for D. melanogaster orthologs. Remarkably, we found that measures of DNA methylation in both the fire ant and honey bee were highly associated with D. melanogaster gene expression metrics. Specifically, DNA methylation in the fire ant and honeybee was negatively associated with CV and tissue specificity but positively associated with expression level, in D. melanogaster (fig. 2C). These results support the concordance between DNA methylation and histone modifications, not only spatially (fig. 1) but also functionally (fig. 2C).
Promoter Architecture and Intragenic Epigenetic Modifications
Recent studies in mammals and flies revealed that core promoters of genes fall into distinct classes defined by their RNA polymerase II (Pol II) initiation profiles (Lenhard et al. 2012). “Broad” core promoters exhibit multiple RNA Pol II initiation sites within the promoter and are associated with genes exhibiting cellular housekeeping functions and constitutive expression (Engström et al. 2007; Lenhard et al. 2012). In contrast, “sharp” core promoters exhibit a more focused RNA Pol II initiation site and are associated with tissue-specific expression (Engström et al. 2007; Lenhard et al. 2012).
To explore potential associations between core promoter type and intragenic DNA methylation, we compared coding sequence DNA methylation levels in the fire ant and honey bee to promoter classifications of D. melanogaster orthologs (Hoskins et al. 2011). We found that fire ant and honey bee orthologs of D. melanogaster genes with broad promoters showed considerably more intragenic methylation than orthologs classified as having sharp promoters (fig. 3 and supplementary fig. S6, Supplementary Material online).
Fig. 3.—

DNA methylation patterns differ based on promoter architectural classification. (A) Mean DNA methylation level in fire ant coding sequences differ according to sharp (n = 530) and broad (n = 2,919) core promoter architecture classifications of Drosophila melanogaster orthologs (Mann–Whitney U test, P < 2.2 × 10−16). (B) Density graph comparison of fire ant DNA methylation distributions for each promoter class-based gene category, illustrating high relative representation of methylated genes associated with broad core promoters.
Discussion
Intragenic DNA Methylation and Active Histone Modifications
The targeting of DNA methylation appears to be highly conserved across invertebrate taxa that possess functional DNA methylation systems (table 1) (Sarda et al. 2012). This targeting also appears to be strongly associated with the presence of several active histone modifications in insects (fig. 1 and table 2). Moreover, DNA methylation is preferentially targeted to 5′-regions within insect genes (fig. 1) (Zemach et al. 2010; Bonasio et al. 2012). H3K4me2/me3, which is associated with 5′-gene regions in D. melanogaster (Kharchenko et al. 2011), exhibits the strongest correlations with DNA methylation in our analyses (fig. 1).
The associations between DNA methylation and H3K4me2/me3 are particularly remarkable because H3K4 methylation is negatively correlated with intragenic DNA methylation in mammals (supplementary fig. S7, Supplementary Material online) (Ooi et al. 2007; Meissner et al. 2008; Maunakea et al. 2010). One possible explanation for this difference is functional divergence of DNA methyltransferase enzymes between insects and mammals (Ooi et al. 2007), facilitating differential preference to methylated versus unmethylated H3K4 in the two taxa. Regardless, in contrast to mammals, intragenic DNA methylation appears to be consistently positively associated with a wide array of active histone modifications in insect genomes (fig. 1, supplementary fig. S7, Supplementary Material online; see supplementary discussion, Supplementary Material online).
Intragenic DNA Methylation and Gene Regulation
Intragenic DNA methylation is targeted to genes with low variability of expression among fire ant individuals (fig. 2B). DNA methylation is also associated with genes displaying ubiquitous expression among tissues in the honey bee (Foret et al. 2009) and morphs in ants (Bonasio et al. 2012), as well as with orthologs that exhibit ubiquitous expression among D. melanogaster tissues (fig. 2C). These results, coupled with the broad core promoter architecture of orthologs of methylated genes (fig. 3), provide strong evidence that intragenic DNA methylation is preferentially targeted to constitutively expressed genes in insects.
DNA methylation is known to influence transcriptional regulation in a number of ways. For example, DNA methylation may directly alter transcription factor binding (Bird 2002; Shukla et al. 2011; Wang et al. 2012). In addition, DNA methylation may increase the compaction and rigidity of nucleosome-DNA complexes (Choy et al. 2010; Lee JY and Lee T-H 2011), thereby indirectly altering DNA accessibility to transcription factors (Li and Widom 2004; Bintu et al. 2012). In this manner, intragenic DNA methylation may shield cryptic binding sites within gene bodies, thus preventing spurious transcription initiation in constitutively expressed genes (Zilberman et al. 2007; Maunakea et al. 2010). Intragenic DNA methylation is also preferentially targeted to exons (supplementary figs. S2 and S3, Supplementary Material online), where RNA Pol II is most prevalent (Yin et al. 2011) and nucleosomes are preferentially positioned in diverse eukaryotes (Schwartz et al. 2009). This underscores the possibility that intragenic DNA methylation acts to enhance or modify transcript integrity and nucleosome positioning in insects (Chodavarapu et al. 2010).
The relationships between DNA methylation, histone modifications, nucleosome positioning, and histone variants (Coleman-Derr and Zilberman 2012) suggest that DNA methylation is complementary, and potentially redundant, to other forms of epigenetic information in eukaryotes. We speculate that the multilayered nature of the eukaryotic epigenome may help to explain how the loss of DNA methylation has been tolerated in some insect lineages (Zemach et al. 2010; Glastad et al. 2011).
Histone Modifications, DNA Methylation Patterning, and Gene Expression Profiles Define Constitutive and Variable Transcription States in Insect Genomes
The association between the patterning of DNA methylation and active histone modifications (fig. 1), along with their similar regulatory relationships (figs. 2 and 3), suggests that genes can generally be partitioned into two highly conserved epigenomic states (fig. 4). These states can be described as “constitutive” and “variable” (fig. 4). Constitutive and variable states differ in transcriptional activity (Filion et al. 2010; Kharchenko et al. 2011; Hunt, Ometto, et al. 2013), promoter architecture (Hoskins et al. 2011; Lenhard et al. 2012), epigenetic modifications (Foret et al. 2009; Filion et al. 2010; Kharchenko et al. 2011), physical domains of the genome (Sexton et al. 2012), gene function (Filion et al. 2010; Sarda et al. 2012), and selective constraint (Sarda et al. 2012; Hunt, Ometto, et al. 2013). The fact that we were able to infer constitutive and variable states from analyses of DNA methylation data from Hymenoptera (S. invicta and A. mellifera) and diverse data sets from Diptera (D. melanogaster), two insect orders that diverged approximately 350 Ma (Wiegmann et al. 2009), suggests that such regulatory properties may be highly conserved over evolutionary time (Engström et al. 2007; Hunt, Glastad, et al. forthcoming).
Fig. 4.—
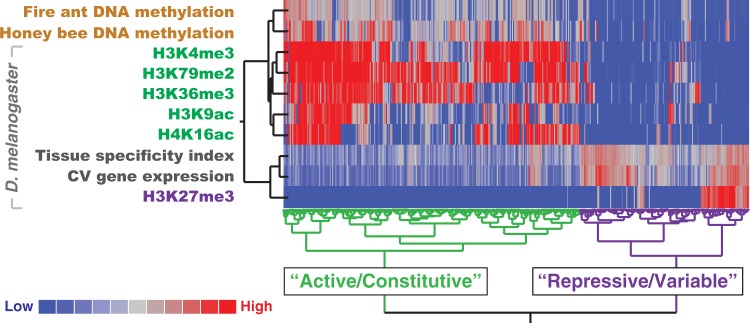
Histone modifications, DNA methylation patterning, and gene expression profiles define constitutively and variably expressed genes in insects. A heatmap of coding sequence DNA methylation levels, histone modification enrichment, and gene expression measures produced by the Ward hierarchical clustering method with dendrograms proportional to distance. Two clusters emerge (n = 3,892 orthologous genes).
Conclusions
Intragenic DNA methylation exhibits limited variation among insect taxa with functional DNA methylation systems and is strongly associated with multiple histone modifications targeted to orthologous D. melanogaster loci. Many regulatory properties of DNA methylation are potentially driven, at least in part, by the association of DNA methylation with conserved epigenetic and regulatory genomic domains, illustrating that DNA methylation is a single component of a conserved, complex, multilayered epigenome. Overall, our results support the view that the regulatory roles of intragenic DNA methylation cannot be fully understood without considering the tight integration of DNA methylation with histone modifications, nucleosome positioning, and RNA Pol II kinetics.
Supplementary Material
Supplementary materials and methods, discussion, figures S1–S7, and tables S1–S6 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org).
Acknowledgments
This work was supported by the US National Science Foundation (grant numbers DEB-1011349, DEB-0640690, IOS-0821130, and MCB-0950896) and the Georgia Tech-Elizabeth Smithgall Watts endowment. The authors thank Lino Ometto for gene expression data and Jia Zeng for code used to parse SAM pileup files.
Literature Cited
- Bell O, et al. Accessibility of the Drosophila genome discriminates PcG repression, H4K16 acetylation and replication timing. Nat Struct Mol Biol. 2010;17:894–900. doi: 10.1038/nsmb.1825. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120:169–181. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Bintu L, et al. Nucleosomal elements that control the topography of the barrier to transcription. Cell. 2012;151:738–749. doi: 10.1016/j.cell.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- Bird AP, Wolffe AP. Methylation-induced repression—belts, braces, and chromatin. Cell. 1999;99:451–454. doi: 10.1016/s0092-8674(00)81532-9. [DOI] [PubMed] [Google Scholar]
- Bonasio R, et al. Genome-wide and caste-specific DNA methylomes of the ants Camponotus floridanus and Harpegnathos saltator. Curr Biol. 2012;22:1755–1764. doi: 10.1016/j.cub.2012.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet. 2009;10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- Celniker SE, et al. Unlocking the secrets of the genome. Nature. 2009;459:927–930. doi: 10.1038/459927a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chintapalli VR, Wang J, Dow JAT. Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat Genet. 2007;39:715–720. doi: 10.1038/ng2049. [DOI] [PubMed] [Google Scholar]
- Chodavarapu RK, et al. Relationship between nucleosome positioning and DNA methylation. Nature. 2010;466:388–392. doi: 10.1038/nature09147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy JS, et al. DNA methylation increases nucleosome compaction and rigidity. J Am Chem Soc. 2010;132:1782–1783. doi: 10.1021/ja910264z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman-Derr D, Zilberman D. DNA methylation, H2A.Z, and the regulation of constitutive expression. Cold Spring Harbor Symp Quant Biol. 2012 doi: 10.1101/sqb.2012.77.014944. Advance access published December 18, 2012, doi:10.1101/sqb.2012.77.014944. [DOI] [PubMed] [Google Scholar]
- Conesa A, et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- Dickman MJ, Kucharski R, Maleszka R, Hurd PJ. Extensive histone post-translational modification in honey bees. Insect Biochem Mol Biol. 2013;43:125–137. doi: 10.1016/j.ibmb.2012.11.003. [DOI] [PubMed] [Google Scholar]
- Engström PG, Ho Sui SJ, Drivenes Ø, Becker TS, Lenhard B. Genomic regulatory blocks underlie extensive microsynteny conservation in insects. Genome Res. 2007;17:1898–1908. doi: 10.1101/gr.6669607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Jacobsen SE. Epigenetic modifications in plants: an evolutionary perspective. Curr Opin Plant Biol. 2011;14:179–186. doi: 10.1016/j.pbi.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filion GJ, et al. Systematic protein location mapping reveals five principal chromatin types in Drosophila cells. Cell. 2010;143:212–224. doi: 10.1016/j.cell.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foret S, Kucharski R, Pittelkow Y, Lockett G, Maleszka R. Epigenetic regulation of the honey bee transcriptome: unravelling the nature of methylated genes. BMC Genomics. 2009;10:472. doi: 10.1186/1471-2164-10-472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glastad KM, Hunt BG, Yi SV, Goodisman MAD. DNA methylation in insects: on the brink of the epigenomic era. Insect Mol Biol. 2011;20:553–565. doi: 10.1111/j.1365-2583.2011.01092.x. [DOI] [PubMed] [Google Scholar]
- Honeybee Genome Sequencing Consortium. Insights into social insects from the genome of the honeybee Apis mellifera. Nature. 2006;443:931–949. doi: 10.1038/nature05260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins RA, et al. Genome-wide analysis of promoter architecture in Drosophila melanogaster. Genome Res. 2011;21:182–192. doi: 10.1101/gr.112466.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt BG, Brisson JA, Yi SV, Goodisman MAD. Functional conservation of DNA methylation in the pea aphid and the honeybee. Genome Biol Evol. 2010;2:719–728. doi: 10.1093/gbe/evq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt BG, Ometto L, Keller L, Goodisman MAD. Evolution at two levels in fire ants: the relationship between patterns of gene expression and protein sequence evolution. Mol Biol Evol. 2013;30:263–271. doi: 10.1093/molbev/mss234. [DOI] [PubMed] [Google Scholar]
- Hunt BG, Glastad KM, Yi SV, Goodisman MAD. The function of intragenic DNA methylation: insights from insect epigenomes. Integr Comp Biol. Forthcoming doi: 10.1093/icb/ict003. [DOI] [PubMed] [Google Scholar]
- Jjingo D, Conley AB, Yi SV, Lunyak VV, Jordan IK. On the presence and role of human gene-body DNA methylation. Oncotarget. 2012;3:462–474. doi: 10.18632/oncotarget.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko PV, et al. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature. 2011;471:480–485. doi: 10.1038/nature09725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolasinska-Zwierz P, et al. Differential chromatin marking of introns and expressed exons by H3K36me3. Nat Genet. 2009;41:376–381. doi: 10.1038/ng.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–1572. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafos M, et al. Dynamic regulation of H3K27 trimethylation during Arabidopsis differentiation. PLoS Genet. 2011;7:e1002040. doi: 10.1371/journal.pgen.1002040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Lee T-H. Effects of DNA methylation on the structure of nucleosomes. J Am Chem Soc. 2011;134:173–175. doi: 10.1021/ja210273w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenhard B, Sandelin A, Carninci P. Metazoan promoters: emerging characteristics and insights into transcriptional regulation. Nat Rev Genet. 2012;13:233–245. doi: 10.1038/nrg3163. [DOI] [PubMed] [Google Scholar]
- Li G, Widom J. Nucleosomes facilitate their own invasion. Nat Struct Mol Biol. 2004;11:763–769. doi: 10.1038/nsmb801. [DOI] [PubMed] [Google Scholar]
- Lorincz MC, Dickerson DR, Schmitt M, Groudine M. Intragenic DNA methylation alters chromatin structure and elongation efficiency in mammalian cells. Nat Struct Mol Biol. 2004;11:1068–1075. doi: 10.1038/nsmb840. [DOI] [PubMed] [Google Scholar]
- Luco RF, Allo M, Schor IE, Kornblihtt AR, Misteli T. Epigenetics in alternative pre-mRNA splicing. Cell. 2011;144:16–26. doi: 10.1016/j.cell.2010.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luco RF, et al. Regulation of alternative splicing by histone modifications. Science. 2010;327:996–1000. doi: 10.1126/science.1184208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyko F, et al. The honey bee epigenomes: differential methylation of brain DNA in queens and workers. PLoS Biol. 2010;8:e1000506. doi: 10.1371/journal.pbio.1000506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maunakea AK, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466:253–257. doi: 10.1038/nature09165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner A, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanty L, et al. Comparative methylomics reveals gene-body H3K36me3 in Drosophila predicts DNA methylation and CpG landscapes in other invertebrates. Genome Res. 2011;21:1841–1850. doi: 10.1101/gr.121640.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negre N, et al. A cis-regulatory map of the Drosophila genome. Nature. 2011;471:527–531. doi: 10.1038/nature09990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ometto L, Shoemaker D, Ross KG, Keller L. Evolution of gene expression in fire ants: the effects of developmental stage, caste, and species. Mol Biol Evol. 2011;28:1381–1392. doi: 10.1093/molbev/msq322. [DOI] [PubMed] [Google Scholar]
- Ooi SKT, et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–717. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai AA, Bell JT, Marioni JC, Pritchard JK, Gilad Y. A genome-wide study of DNA methylation patterns and gene expression levels in multiple human and chimpanzee tissues. PLoS Genet. 2011;7:e1001316. doi: 10.1371/journal.pgen.1001316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarda S, Zeng J, Hunt BG, Yi SV. The evolution of invertebrate gene body methylation. Mol Biol Evol. 2012;29:1907–1916. doi: 10.1093/molbev/mss062. [DOI] [PubMed] [Google Scholar]
- Schwartz S, Meshorer E, Ast G. Chromatin organization marks exon-intron structure. Nat Struct Mol Biol. 2009;16:990–995. doi: 10.1038/nsmb.1659. [DOI] [PubMed] [Google Scholar]
- Sexton T, et al. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell. 2012;148:458–472. doi: 10.1016/j.cell.2012.01.010. [DOI] [PubMed] [Google Scholar]
- Shukla S, et al. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature. 2011;479:74–79. doi: 10.1038/nature10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suganuma T, Workman JL. Signals and combinatorial functions of histone modifications. Annu Rev Biochem. 2011;80:473–499. doi: 10.1146/annurev-biochem-061809-175347. [DOI] [PubMed] [Google Scholar]
- Wang H, et al. Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res. 2012;22:1680–1688. doi: 10.1101/gr.136101.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse RM, Zdobnov EM, Tegenfeldt F, Li J, Kriventseva EV. OrthoDB: the hierarchical catalog of eukaryotic orthologs in 2011. Nucleic Acids Res. 2011;39:D283–D288. doi: 10.1093/nar/gkq930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegmann B, et al. Single-copy nuclear genes resolve the phylogeny of the holometabolous insects. BMC Biol. 2009;7:34. doi: 10.1186/1741-7007-7-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo YH, Li W-H. Evolutionary conservation of histone modifications in mammals. Mol Biol Evol. 2012;29:1757–1767. doi: 10.1093/molbev/mss022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurm Y, et al. The genome of the fire ant Solenopsis invicta. Proc Natl Acad Sci U S A. 2011;108:5679–5684. doi: 10.1073/pnas.1009690108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Sweeney S, Raha D, Snyder M, Lin H. A high-resolution whole-genome map of key chromatin modifications in the adult Drosophila melanogaster. PLoS Genet. 2011;7:e1002380. doi: 10.1371/journal.pgen.1002380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemach A, McDaniel IE, Silva P, Zilberman D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science. 2010;328:916–919. doi: 10.1126/science.1186366. [DOI] [PubMed] [Google Scholar]
- Zeng J, et al. Divergent whole-genome methylation maps of human and chimpanzee brains reveal epigenetic basis of human regulatory evolution. Am J Hum Genet. 2012;91:455–465. doi: 10.1016/j.ajhg.2012.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberman D, Gehring M, Tran RK, Ballinger T, Henikoff S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat Genet. 2007;39:61–69. doi: 10.1038/ng1929. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.