

Abstract
Modulating bile acid synthesis has long been considered a good strategy by which to improve cholesterol homeostasis in humans. The farnesoid X receptor (FXR), the key regulator of bile acid synthesis, was, therefore, identified as an interesting target for drug discovery. We compared the effect of four, structurally unrelated, synthetic FXR agonists in two fat-fed rodent species and observed that the three most potent and selective agonists decreased plasma cholesterol in LDL receptor-deficient (Ldlr −/−) mice, but none did so in hamsters. Detailed investigation revealed increases in the expression of small heterodimer partner (Shp) in their livers and of intestinal fibroblast growth factor 15 or 19 (Fgf15/19) in mice only. Cyp7a1 expression and fecal bile acid (BA) excretion were strongly reduced in mice and hamsters by all four FXR agonists, whereas bile acid pool sizes were reduced in both species by all but the X-Ceptor compound in hamsters. In Ldlr −/− mice, the predominant bile acid changed from cholate to the more hydrophilic β-muricholate due to a strong repression of Cyp8b1 and increase in Cyp3a11 expression. However, FXR agonists caused only minor changes in the expression of Cyp8b1 and in bile acid profiles in hamsters. In summary, FXR agonist-induced decreases in bile acid pool size and lipophilicity and in cholesterol absorption and synthesis could explain the decreased plasma cholesterol in Ldlr −/− mice. In hamsters, FXR agonists reduced bile acid pool size to a smaller extent with minor changes in bile acid profile and reductions in sterol absorption, and consequently, plasma cholesterol was unchanged.
Keywords: cholic acid, chenodeoxycholic acid, muricholic acid, LDL receptor-deficient
Elevated LDL cholesterol (LDL-C) in the blood is a known risk factor for cardiovascular disease (CVD), the predominant cause of mortality in the Western world (1). Bile acids (BA) are the major metabolites of cholesterol in mammals. They are produced in the liver and secreted into the small intestine where, because of their amphipathic properties, they facilitate the absorption of dietary and biliary lipids, including cholesterol (2). Indeed, secretion of biliary BA and cholesterol into the intestine is the major route by which cholesterol is excreted from the body. As some of the cholesterol for biliary BA and the cholesterol supply derives from LDL-C in the blood, increases in BA and cholesterol excretion are associated with decreases in LDL-C and the risk of CVD (3). Alternatively, when BA secretion is reduced, this may also lead to a lowering of LDL-C. In this situation, a negative cholesterol balance may be achieved because BAs are required for the absorption of dietary and biliary cholesterol from the intestine. Targeting BA metabolism and enterohepatic circulation has, therefore, long been considered an attractive mechanism for treating dyslipidemia (4).
Cholesterol is converted into the primary BA cholic acid (CA) and chenodeoxycholic acid (CDCA) in the liver by a series of enzyme-catalyzed reactions that are summarized in Fig. 1 and have been well reviewed previously (2, 5). The initial, common, rate-limiting step in the classical synthetic pathway is the conversion of cholesterol into 7α-hydroxycholesterol by the cytochrome P450 enzyme cholesterol 7α-hydroxylase (Cyp7a1) (5, 6). The ratio of the two primary BAs is then determined by the activity of sterol 12α-hydroxylase (Cyp8b1), which is essential for the synthesis of CA (7). When Cyp8b1 expression is reduced or absent, CDCA and its metabolites become the dominant or only fraction in the BA pool. In mice, but much less so in hamsters or humans, CDCA is converted into muricholic acid (MCA) through the activity of other cytochrome P450 enzymes, the sterol-6β-hydroxylases (potentially Cyp3a11 and Cyp2b10) (8). The number and orientation of the hydroxyl groups on the BA determine their lipophilicity, which is in the order CDCA > CA > MCA (9). Consequently, it is the combined activities of Cyp7a1, Cyp8b1, Cyp3a11, and Cyp2b10 that determine the quantities, proportions, and properties of the BA that is synthesized in these species (Fig. 1).
Fig. 1.

Main steps in the BA synthesis pathway in mice and hamsters, including the relevant enzymes. The initial, rate-limiting step in BA synthesis is controlled by CYP7A1, while CYP8B1 is responsible for the conversion of a common precursor of CA and CDCA into CA and, hence, the CA/CDCA ratio. Finally, enzymes, including CYP3A11 and CYP2B10, are involved in the conversion of CDCA into α-, β-, and ω-MCA in mice but not in hamsters.
Both the size and composition of the BA pool have been shown to influence the ability of BA to facilitate dietary lipid absorption. In Cyp7a1 −/− mice, for example, BA pool size was reduced by about 80% and cholesterol absorption by more than 97% (10). Wang et al. showed that the more hydrophobic BAs are better at emulsifying dietary lipids in the gut prior to absorption (9). Modifying the synthesis and composition of the BA in mice and hamsters and, potentially, in humans may, therefore, have beneficial effects on cholesterol homeostasis.
The farnesoid X receptor (FXR, NR1H4) is a nuclear hormone receptor that is activated by BA in proportion with its lipophilicity (11–13) and regulates the expression of the key genes involved in BA synthesis in the liver, namely Cyp7a1 (11, 14, 15), Cyp8b1 (7), and Cyp3a11 (16). BAs have also been shown to change the expression of the pregnane X receptor (PXR)-regulated genes Cyp3a11 (17) and Cyp2b10 (8, 17). However, FXR is the main regulator of BA synthesis and profile under normal (i.e., noncholestatic) conditions (14, 16, 18–20). Repression/feedback regulation of BA synthesis by FXR agonists is mediated by increased expression of small heterodimer partner (Shp, NR0B2) in the liver and by increased expression and secretion of fibroblast growth factor 15 (Fgf15), the mouse homolog of FGF19/Fgf19 in humans and hamsters, in the small intestine. SHP acts as a corepressor of liver receptor homolog 1 (LRH-1/NR5A2)- or hepatocyte nuclear factor 4α (HNF4α/NR2A1)-induced transcription of Cyp7a1 and Cyp8b1 (14, 18, 21–23). On the other hand, FGF15/19 activates its receptor FGFR4 located on the plasma membrane of hepatocytes, which leads to the inhibition of Cyp7a1 and Cyp8b1 expression either via SHP by substantially increasing the stability of SHP protein or by SHP-independent means (19, 20, 24–27). To better define the role of FXR, we determined the effect of three potent and selective, synthetic FXR agonists (RO5186026, the X-Ceptor compound, and GW4064) and one less potent and less selective BA derivative (6-ECDCA, INT-747) on BA and cholesterol metabolism in mice and hamsters. This report provides, for the first time, a comprehensive picture of the FXR-mediated differential regulation of BA synthesis in LDL receptor-deficient (Ldlr −/−) mice and hamsters and the consequences of such modulations on dietary cholesterol absorption and plasma cholesterol concentrations.
METHODS
FXR agonists used in animal studies
RO5186026, Exelixis X-Ceptor, 6-ECDCA, and GW4064 were synthesized by Roche (Table 1) (28).
TABLE 1.
Potencies of FXR agonists in human and mouse SP-binding assays and potencies and relative efficacies in human and mouse transactivation assays
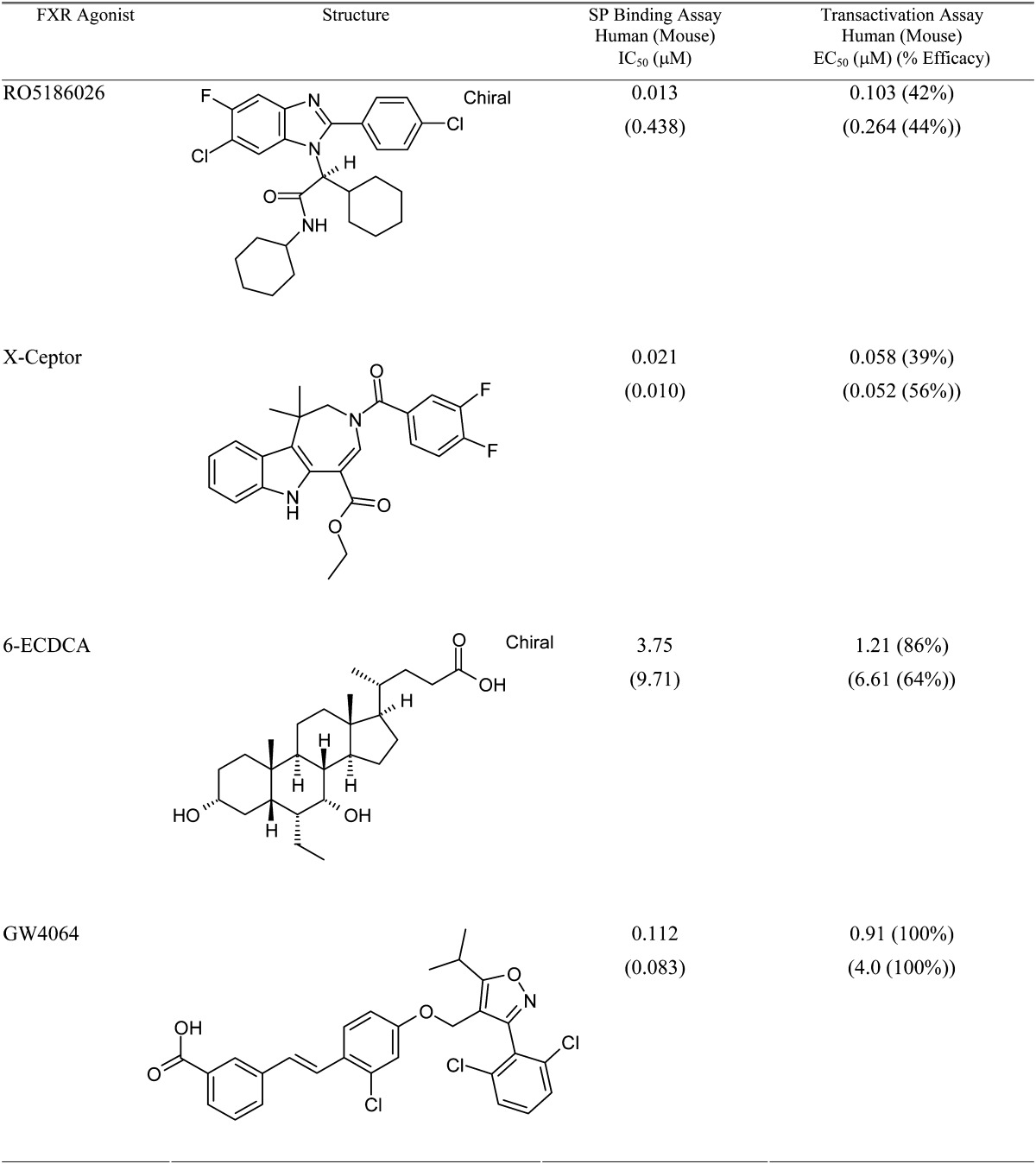
Potencies of the FXR agonists and efficacies relative to that achieved by GW4064 were measured as described in Methods. The values are means of at least two separate measurements.
Scintillation proximity-binding assay
Binding of test substances to human or murine FXR ligand-binding domains (LBD) was measured in a radioligand-displacement scintillation proximity (SP)-binding assay exactly as described previously (28).
Gene transactivation assay in BHK21 cells
The pFA-FXR-LBD plasmid constructs used express the DNA binding domain of yeast Gal4 transcription factor fused, in-frame, N-terminally to the LBDs of either human or mouse FXR. Baby hamster kidney cells (BHK21 ATCC CCL10) were grown in DMEM medium containing 10% FBS at 37° in a 95% O2:5% CO2 atmosphere. Cells were seeded in 6-well plates at a density of 105 cells/well and then batch-transfected with the pFA-FXR-LBD expression plasmids plus reporter plasmids expressing luciferase under the control of five copies of the yeast GAL4 promoter-response elements (pFR-Luc). Transfection was performed using FuGENE 6 (Roche Molecular Biochemicals) according to the suggested protocol. Six hours after transfection, the cells were harvested by trypsinization and seeded in 96-well plates (104 cells/well). After 24 h, medium was removed and replaced with 100 μl of phenol red-free medium containing the test substances or control ligands (final DMSO concentration: 0.1%). After another 24 h incubation, 50 μl of the supernatant was discarded, and 50 μl of Luciferase Constant-Light Reagent (Roche Molecular Biochemicals) was added to lyse the cells and initiate the luciferase reaction. Luminescence, as a measure of luciferase activity, was detected using a Packard TopCount. Potencies for half-maximal activation of receptor transcriptional activity (EC50 values) and percentage efficacies relative to the efficacy of GW4064 (100%) were calculated using using a customised XLfit template.
Animal studies
Main LDLR −/− mouse and hamster studies.
Male C57BL/6J Ldlr −/− mice (B6.129S7- Ldlrtm1Her/J, 9–10 weeks old) from the Jackson Laboratory (Bar Harbor, ME) were fed a high-fat diet (Provimi Kliba AG #2157, Kaiseraugst, Switzerland), containing 18% fat (soybean oil/pork fat 1.375:1 + 0.02% cholesterol), from 2 weeks prior to treatment and throughout the study. Mice were assigned to groups (n = 6/group) based on plasma HDL-cholesterol (HDL-C), LDL-C, triglycerides (TG), and body weight. They were dosed for 3 days by oral gavage with vehicle alone (7.5% gelatin) to acclimatize them to the dosing regimen. They were then treated with compounds or vehicle alone once daily by gavage for 5 days. Feces were collected during the final 3 days of the study for BA determination. At the end of the treatment period, mice were fasted for 4 h, then anesthetized 2 h after the final dose, and then bled retro-orbitally to measure plasma lipid parameters and plasma neutral sterols. They were then euthanized while under anesthesia. Gallbladders with surrounding liver tissue and intestinal contents were collected and placed in 10 ml ethanol awaiting BA analysis. Remaining liver tissue was frozen in liquid nitrogen and stored at −80°C for mRNA expression and cholesterol and TG determination. Small intestines were flushed with ice-cold 0.9% NaCl and divided into five parts of equal length. Parts 1, 3, and 5, corresponding to duodenum, jejunum, and ileum, respectively (29), were frozen in liquid nitrogen and stored at −80°C for mRNA extraction.
Male hamsters (8–9 weeks old from Charles River Laboratories, Sulzfeld, Germany) were housed individually and fed a high-fat diet as pellets (Provimi Kliba #2453, Kaiseraugst, Switzerland), containing 14.5% coconut oil and 0.05% cholesterol, for 17 days prior to treatment and throughout the study. Hamsters were then assigned to treatment groups (n = 8/group in the vehicle-treated group and n = 6/group for other groups) according to their plasma HDL-C, LDL-C, and TG levels and body weights and dosed for 3 days by oral gavage with vehicle alone (7.5% gelatin) to acclimatize them to the dosing regimen. They were then treated with compounds or vehicle alone once daily by gavage for 11 days. Feces were collected during the final 3 days of the study for BA determination. Terminal blood samples were collected, animals were euthanized, and gallbladder plus liver tissue and small intestinal content, duodenum, jejunum, and ileum were collected and frozen as described for mice.
Single- and multiple-dose subsidiary transcription studies.
Male Ldlr −/− mice and hamsters (both 8–9 weeks old) were fed the diets, assigned to treatments (n = 6–8/group) and predosed with vehicle by gavage for 3 days to acclimatize them to the dosing regimen. In the first study, animals were given a single dose of RO5186026 (30 mg/kg) or vehicle, and then blood was collected and plasma was frozen 4 h or 8 h following a fasting period of 4 h. The animals were euthanized, and liver, duodenum, jejunum, and ileum were collected and frozen for mRNA extraction as described for the previous study. Hepatic Shp, Cyp7A1, Cyp8B1, Cyp3A11 (mice only), Hmgcr (mice only) and Insig-2 (mice only) as well as duodenal, jejunal, and ileal Ibabp and Fgf15/19 expression were measured as described below. In a second study, after the vehicle predosing period, the Ldlr −/− mice and hamsters were dosed once daily for either 5 or 8 days, respectively, with either vehicle or RO5186026 (30 mg/kg/day). The animals were euthanized 2 h postdosing, following a fasting period of 4 h, and duodenum, jejunum, and ileum were collected and frozen for mRNA extraction as described for the main study. Ibabp and Fgf15/19 expression were measured in the duodenum, jejunum, and ileum as described below.
Intestinal cholesterol absorption using the fecal dual isotope method.
Male Ldlr −/− mice and hamsters were fed the diets and then assigned to treatments (n = 7 mice/group and 8 hamsters/group) as described above. They were then treated for 5 and 7 days, respectively, with either RO5186026 (30 mg/kg/day) or ezetimibe (3 mg/kg/day), a known inhibitor of intestinal cholesterol absorption (30–33). On the third day (mice) or fifth day (hamsters) of treatment, animals were administered a mixture containing 1 μCi of [14C]cholesterol (Perkin-Elmer) and 2 μCi of [3H]Sitostanol (American Radiolabeled Chemicals) in 100 μl medium chain-length TG (MCT) oil. Feces were collected during the last 2 days of the experiment, then dried and ground to a fine powder. Radioactivity in the feces was measured using a β-counter. Cholesterol absorption was calculated as follows: % Absorption = (([14C]Cholesterol / [3H]Sitostanol in the dosing mixture) – ([14C]Cholesterol / [3H]Sitostanol in the feces) / ([14C]Cholesterol / [3H]Sitostanol in the dosing mixture)) × 100.
In vivo study protocols were approved by the Veterinarian Central Office of Kanton Basel-Stadt, Switzerland, to F. Hoffmann-La Roche, AG, Basel, Switzerland.
Total and lipoprotein cholesterol and TG determination
Plasma total cholesterol and TG were measured using standard enzymatic assays on a Hitachi 912 analyzer (Roche Diagnostics AG). For the lipoprotein cholesterol profiles, either equal volumes of mouse plasma were pooled from all the animals in each group prior to analysis or plasma from each hamster was analyzed separately, and the mean profile was calculated. Plasma lipoproteins were separated into fractions by size-exclusion chromatography (Superose-6 gel FPLC, AKTA system, Pharmacia), and their cholesterol content was quantified using a fluorometric assay (34). Lipoprotein distribution was calculated assuming a Gaussian distribution for each peak, using a nonlinear, least-squares curve fitting procedure to calculate area under the curve.
BA pool size and composition
BA pool sizes were determined in the gallbladder plus surrounding liver tissue (L+GB) and small intestine (SI) contents. Tissues were cut into small pieces and homogenized in ethanol. After 5 min of boiling, samples were cooled to room temperature and filtered through a Whatman filter paper. Total BA concentrations were determined enzymatically using aliquots of the ethanolic extracts (Total BA Assay, Diazyme Labs, Ref. DZ042A-K, and a Hitachi 912 analyzer). CA, CDCA, deoxycholic acid (DCA), ursodeoxycholic acid (UDCA), lithocholic acid (LCA), α-MCA, β-MCA, ω-MCA, and hyodeoxycholic acid (HDCA) were determined by GC-MS in the liver plus gallbladder extracts. After addition of norcholic acid as an internal standard, samples were heated for 3 h at 120°C in 1.25 M NaOH. The BAs were then extracted using C18 silica cartridges (Strata C18-E, 500 mg/6 ml) and eluted using methanol. The eluate was dried and the BA was methylated by redissolving in methanol (400 µl), adding acetyl chloride (10 µl) and heating (70°C, 2 h). The samples were then dried and silylated using 250 µl pyridine/bis-(trimethylsilyl)trifluoroacetamide (BSTFA, 30:70, v:v) containing 1% trimethylchlorosilane (TMCS) at 70°C for 45 min. The reaction mixture (1 µl) was then injected into an Agilent 5973i GC MSD using a narrow bore DB-1MS column.
Plasma neutral sterols
Plasma lathosterol, desmosterol, campesterol, and β-sitosterol were determined using GC-MS. After addition of epicholesterol as an internal standard, plasma samples (20 μl) were saponified in 0.35 M ethanolic KOH, and the unesterified neutral sterols were extracted into cyclohexane and dried. After silylation in pyridine/BSTFA, samples were analyzed on an Agilent 5973i GC MSD using a narrow bore DB-1MS. Results were expressed as a ratio with plasma cholesterol levels quantified enzymatically using a standard assay on a Hitachi 912 auto analyzer (Roche Diagnostics AG).
Liver cholesterol and TG determination
Liver lipids were determined according to the method described by Carr et al. (35) with some minor modifications. Liver samples (∼200 mg) were extracted with 4 ml of chloroform:methanol (2:1), and the phases were separated by the addition of 0.05% H2SO4 (0.8 ml). A 0.5 ml aliquot of the chloroform phase was collected and dried under nitrogen, and the residue dissolved in 0.5 ml of 1% Triton X-100 in chloroform. The chloroform was removed under nitrogen, and the residue dissolved in 1 ml of water. Cholesterol and TG were measured as described above.
Fecal BA determination
Dried feces (50 mg) were added to 2.2 ml ethanolic NaOH (0.08M) and heated (95°C) for 2 h. After cooling, neutral sterols were extracted three times with 5 ml hexane. After acidification of the hydrolysate using 2.5 ml of 0.16M HCI, BAs were extracted five times with 5 ml of ethyl acetate. The dried extract was solubilized in 1.25% Triton X-100 in 20% methanol. BAs were quantified using the enzymatic assay described above.
Gene expression
Frozen tissues (30 mg) were homogenized in 1 ml Trizol (Invitrogen) in a Fastprep machine (Qbiogene). After a chloroform extraction, RNA was extracted from lysates using PureLink Micro-to-Midi kit (Invitrogen) following the instructions given by the manufacturer. mRNA was reverse transcribed using Transcriptor cDNA first strand synthesis kit (Roche Applied Science, Rotkreutz, Switzerland). qPCR reactions were performed in a LC480 (Roche Applied Science). In the case of mice, specific primers and probes were ordered from Applied Biosystems (Life Technology), and reactions were prepared with LC480 Probes Master (Roche Applied Science) following procedures supplied by the manufacturer. Order numbers for probes and primers were mm99999915_g1 (mouse Gapdh), mm00442278_m1 (mouse Shp), mm00501637_s1 (mouse Cyp8b1), mm00731567_m1 (mouse Cyp3a11), mm00433278_m1 [mouse ileal bile acid-binding protein (Ibabp)], mm00434316_m1 (mouse Fgf15), mm01282501_m1 [mouse hydroxymethylglutaryl-CoA reductase (Hmgcr)], and mm01308255_m1 [mouse insulin-induced gene 2 (Insig-2)].
Hamster primers were derived from published sequences and ordered from Microsynth AG (Balgach, Switzerland). qPCR reactions were performed using LC480 SYBR Green I Master (Roche Applied Science). Sequences were Gapdh forward 5′-AGGTTGTCTCCTGCGACTTCA-3′, Gapdh reverse 5′-GCATCAAAGGTGGAAGAGTGG-3′ Shp forward 5′-AGGGAGGCCTTGGATGTC-3′, Shp reverse 5′-AGAAGGACGGCAGGTTCC-3′ Cyp8b1 forward 5′-GATGGCACCCGGAAAGTGGA-3′, Cyp8b1 reverse 5′-TAGTGGTGGATCTTCTTGCC-3′ Cyp7a1 forward 5′-GATCAAGTCTTTCCGGCGTT-3′, Cyp7a1 reverse 5′-CTGTTCCCGGGCCTTATGT-3′ Ibabp forward 5′-TCATCACAGAGGTCCAGCAG-3′, Ibabp reverse 5′-CACATTCTTTGCCAATGGTG-3′ and Fgf19 forward 5′-ACTTTATTTATGCCCCAGATCA-3′, Fgf19 reverse 5′-TTTTGATACAAACCAACTCCTTTTT-3′.
Statistics
Statistically significant differences between control and treated groups were assessed by either a Wilcoxon/Kruskal-Wallis test or by one-way ANOVA (ANOVA) followed by Dunnett's test for posthoc comparisons using JMP6 software (SAS Institute Inc.). Significance was defined as P < 0.05.
RESULTS
In vitro characterization of FXR agonists
RO5186026, X-Ceptor, 6-ECDCA, and GW4064 are structurally unrelated FXR agonists (28, 36–38). Their potencies and relative efficacies for human and mouse FXR, measured using scintillation proximity binding and gene transactivation assays, are shown in Table 1. RO5186026, X-Ceptor, and GW4064 have strong affinities for human FXR (IC50 = 0.013 μM, 0.021 μM, and 0.12 μM, respectively), whereas 6-ECDCA, a BA derivative, has a lower affinity (IC50 = 3.75 μM). Although X-Ceptor and GW4064 showed similarly strong affinities for the mouse FXR receptor (0.010 μM and 0.083 μM, respectively), the affinities of 6-ECDCA and especially RO5186026 for the mouse receptor were lower (EC50s = 9.7 μM and 0.44 μM, respectively).
RO5186026, X-Ceptor and GW4064 were also more potent than 6-ECDCA in the human and mouse transactivation assays, although the difference between GW4064 and 6-ECDCA was small. As expected for a cell-based assay versus a protein-binding assay, the potencies of the three most potent FXR agonists in the transactivation assay were generally lower (2.8- to 19.7-fold) than those in the SP-binding assay, except for RO5186026 in the mouse transactivation assay, which was 1.7-fold more potent than in the SP-binding assay. 6-ECDCA was also slightly more potent in the transactivation assay than in the binding assay. Percentage efficacies relative to the efficacy of GW4064 were lower for all of the other compounds (39% to 86%).
Specificity of the four compounds for FXR was determined using functional reporter-gene transcription assays for PPARα, PPARδ, PPARγ, LXRα, LXRβ, and RXRα. All four compounds were unable to activate the expression of these reporter genes at 40 μM (data not shown).
As we and others have reported previously (28, 38), 6-ECDCA is an agonist of human GPBAR1 (EC50: 1 μM), whereas the other FXR agonists are not. Therefore, RO5186026, X-Ceptor, and GW4064 are potent, selective FXR agonists, and 6-ECDCA is less selective and less potent.
Effects of FXR agonist treatment on food intake, body weight, liver weight, and liver lipids
Male Ldlr −/− mice and male hamsters were treated daily for 5 and 11 days, respectively, with either RO5186026 (30 mg/kg), X-Ceptor (30 mg/kg), 6-ECDCA (30 mg/kg), or GW4064 (100 mg/kg).
None of the FXR agonists affected cumulative food intake in Ldlr −/− mice. However, body weight gain increased slightly following treatment with RO5186026, X-Ceptor, and 6-ECDCA compared with a small reduction in the vehicle-treated group (0.55–0.60 g versus −0.27 g in control group, all P < 0.05). In hamsters, RO5186026 increased body weight gain slightly (13.1 g versus 7.0 g in the control group, P < 0.01), whereas 6-ECDCA reduced cumulative food intake by 13% (P < 0.05) and reduced body weight gain (1.7 g versus 7.0 g in the control group, P < 0.05). Liver weights were unchanged in Ldlr −/− mice but were increased in RO5186026-, X-Ceptor-, and GW4064-treated hamsters (23%, P < 0.01; 13%, P < 0.05; and 14%, P < 0.01, respectively; supplementary table I).
In Ldlr −/− mice, liver cholesterol content was reduced by RO5186026 (−21%, P < 0.05) and X-Ceptor (−19%, P < 0.05); and liver TG was reduced by RO5186026 (−37%, P < 0.01), X-Ceptor (−41%, P < 0.01), 6-ECDCA (−39%, P < 0.01), and GW4064 (−29%, P < 0.01). In hamsters, liver cholesterol and TG were unaffected by any of the treatments (supplementary table I).
Potent FXR agonists decrease plasma cholesterol in Ldlr −/− mice but not in hamsters
RO5186026, X-Ceptor, and GW4064 were shown to significantly decrease total plasma cholesterol levels by 47% (P < 0.01), 31% (P < 0.01), and 14% (P < 0.05), respectively, in Ldlr −/− mice, with no change observed in 6-ECDCA-treated mice or in FXR agonist-treated hamsters (Fig. 2A). The decreases observed in mice treated with potent FXR agonists were mainly due to decreases in LDL-C as shown by the lipoprotein cholesterol profiles (Fig. 2B). VLDL-cholesterol (VLDL-C) and HDL-cholesterol (HDL-C) were also slightly decreased in animals treated with RO5186026 and X-Ceptor, the most potent antihypercholesterolemic compounds. No changes in lipoprotein cholesterol profiles were observed in FXR agonist-treated hamsters, except with 6-ECDCA, which decreased HDL-C and increased VLDL-C (Fig. 2B). As described above, the latter effects were associated with reduced food consumption and body weight gain.
Fig. 2.

Effect of FXR agonists on plasma cholesterol in Ldlr −/− mice and hamsters. Male Ldlr −/− mice and male Syrian hamsters were dosed daily by gavage with vehicle for 3 days prior to the study to acclimatize them to the dosing regimen. Thereafter, the animals were treated for 5 and 11 days, respectively, with vehicle alone, RO5186026, X-Ceptor, 6-ECDCA (all at 30 mg/kg/day), or GW4064 (100 mg/kg/day). The animals were bled and plasma was prepared. (A) Total plasma cholesterol was measured enzymatically. Values are means ± SE (n = 6 mice/group and n = 6–8 hamsters/group). (B) Plasma lipoprotein profiles were prepared (see Methods). Vehicle profiles are in black and compound profiles are colored. Significant differences between the vehicle- and compound-treated groups (*P < 0.05; **P < 0.01) were determined using the Wilcoxon/Kruskal-Wallis tests.
FXR agonists decrease BA pool size and excretion and modify BA distribution in Ldlr −/− mice and hamsters
We investigated whether the plasma cholesterol-lowering effect of FXR agonists in Ldlr −/− mice was associated with changes in the BA pool size. RO5186026, X-Ceptor, 6-ECDCA, and GW4064 strongly decreased BA pool size in Ldlr −/− mouse L+GB (−85, −69, −55, and −66%, respectively, all P < 0.05) and SI (−57, −53, −70, and −49%, respectively, all P < 0.05; Fig. 3A). In hamsters, RO5186026, 6-ECDCA, and GW4064 decreased BA pool size significantly in both the L+GB (−53, −74, and −39%, respectively, P < 0.05) and SI (−51, −80, and −38%, P < 0.05; Fig. 3B). Only X-Ceptor did not significantly modulate the BA pool size in hamsters. The ratio of BA in the L+GB versus the SI was reduced in mice treated with RO5186026, X-Ceptor, and GW4064, indicating a redistribution of BA into the SI (supplementary Fig. I). No differences in these ratios were seen in FXR agonist-treated hamsters. BA excretion in feces collected during the last 3 days of treatment was also reduced by all FXR agonists in both species. RO5186026, X-Ceptor, 6-ECDCA, and GW4064 strongly decreased BA excretion in the feces of Ldlr −/− mice (−44, −64, −50, and −40%, respectively, P < 0.01) and hamsters (−63, −46, −82, and −54%, respectively, P < 0.01 for all; supplementary Fig. II).
Fig. 3.

Effect of FXR agonists on BA pool size in Ldl −/− mice and hamsters. Male Ldlr −/− mice and male Syrian hamsters were dosed daily by gavage with vehicle for 3 days prior to the study to acclimatize them to the dosing regimen. Thereafter, the mice and hamsters were treated for 5 and 11 days, respectively, with vehicle alone, RO5186026, X-Ceptor, 6-ECDCA (all at 30 mg/kg/day), or GW4064 (100 mg/kg/day). Total BA was measured in the L+GB and SI of Ldlr −/− mice (A) and hamsters (B) as described in Methods. Values are means ± SE (n = 6/group). Significant differences between the vehicle- and compound-treated groups (*P < 0.05) were determined by ANOVA followed by Dunnett's test.
Together these data indicate that all of the FXR agonists strongly reduced BA pool size and excretion in Ldlr −/− mice and hamsters, except for the lack of effect of X-Ceptor on BA pool size in hamsters.
BA profile is modulated by FXR agonists in Ldlr −/− mice but not in hamsters
In Ldlr −/− mice, the BA pool is mainly composed of CA (∼70%) and α-, β-, plus ω-MCA (∼25% in total) (Fig. 4A). We showed that all four FXR agonists dramatically reduced the proportion of CA from ∼70% in vehicle-treated mice to 17, 26, 9, and 34%, respectively, in RO5186026-, X-Ceptor-, 6-ECDCA-, and GW4064-treated mice. α-, β-, and ω-MCA all increased upon treatment with FXR agonists until their joint contribution to the pool reached 73, 65, 83, and 61%, in RO5186026-, X-Ceptor-, 6-ECDCA-, and GW4064-treated mice, respectively.
Fig. 4.

Effect of FXR agonists on BA profiles in Ldlr −/− mice and hamsters. Male Ldlr −/− mice and male Syrian hamsters were dosed daily by gavage with vehicle for 3 days prior to the study to acclimatize them to the dosing regimen. Thereafter, the mice and hamsters were treated for 5 and 11 days, respectively, with vehicle alone, RO5186026, X-Ceptor, 6-ECDCA (all at 30 mg/kg/day), or GW4064 (100 mg/kg/day).The composition of the BA pool in the L+GB was determined using GC-MS in Ldlr −/− mice (A) and hamsters (B). The data are expressed as percentage of total BA.
In hamsters, the BA pool is mainly composed of the two primary BAs, CA (64%) and CDCA (19%) (Fig. 4B). Treatment with X-Ceptor did not modulate the composition of the BA pool significantly. However, the proportion of CDCA in the BA pool increased to 32, 63, and 27%, respectively, in hamsters treated with RO5186026, 6-ECDCA, and GW4064 (Fig. 4B).
These data demonstrate that FXR agonists modulate the composition of the BA pool in Ldlr −/− mice, with β-MCA becoming the predominant BA instead of CA. Interestingly, only minor or no changes in BA profile occurred in hamsters treated with the three most potent FXR agonists.
FXR agonists reduced cholesterol synthesis and absorption in Ldlr −/− mice, whereas in hamsters, most FXR agonists decreased cholesterol synthesis without affecting cholesterol absorption
We measured the effect of FXR agonists on plasma levels of noncholesterol neutral sterols, namely, lathosterol and desmosterol, as biomarkers of cholesterol synthesis and the plant sterols campesterol and β-sitosterol as biomarkers of intestinal cholesterol absorption (39). Fig. 5A shows plasma neutral sterol:cholesterol ratios in Ldlr −/− mice. Desmosterol was only modulated by 6-ECDCA (−39%), but lathosterol was reduced in mice treated with RO5186026 (−26%), X-Ceptor (−33%), 6-ECDCA (−39%), and GW4064 (−35%). In a subsidiary study, Ldlr −/− mice were given a single dose of RO5186026 (30 mg/kg) or vehicle, and changes in the hepatic expression of hydroxymethylglutaryl-CoA reductase (Hmgcr) and Insig-2 were measured 4 and 8 h postdosing to monitor any initial changes in gene expression (Fig. 6). Hmgcr is the rate-limiting enzyme in the cholesterol synthesis pathway, and Insig-2 is known to block the proteolytic processing of sterol regulatory element-binding proteins (SREBP) in the Golgi and to enhance the degradation of Hmgcr protein, thereby blocking cholesterol synthesis at both translational and posttranslational levels (40, 41). As reported by Hubbert et al., who treated mice with GW4064 daily for 5 days, Hmgcr expression was unaffected by FXR agonist treatment, but Insig-2 expression was increased (2.1- and 4.8-fold at 4 and 8 h posttreatment, respectively; Fig. 6) (41). These data suggest that FXR agonists decrease cholesterol synthesis and, consequently, plasma and hepatic cholesterol levels in Ldlr −/− mice partially by inducing Insig-2 expression and decreasing HMG-CoA reductase protein.
Fig. 5.

Effect of FXR agonists on plasma markers for cholesterol synthesis and dietary sterol absorption in Ldlr −/− mice and hamsters. Male Ldlr −/− mice and male Syrian hamsters were dosed daily by gavage with vehicle for 3 days prior to the study to acclimatize them to the dosing regimen. Thereafter, the mice and hamsters were treated for 5 and 11 days, respectively, with vehicle alone, RO5186026, X-Ceptor, 6-ECDCA (all at 30 mg/kg/day), or GW4064 (100 mg/kg/day). Plasma markers for cholesterol synthesis (lathosterol and desmosterol) and cholesterol absorption (campesterol and β-sitosterol) were measured by GC-MS and normalized to plasma cholesterol measured enzymatically in Ldlr −/− mice (A) and hamsters (B). Values are means ± SE (n = 6/group). (C) Cholesterol absorption was also measured using the dual isotope method (see Methods). Ldlr −/− mice and hamsters were treated for 5 and 7 days, respectively, with either vehicle, RO5186026 (30 mg/kg/day), or ezetimibe (3 mg/kg/day). Values are means ± SE (n = 7 mice/group; n = 8 hamsters/group). Significant differences between the groups were determined by ANOVA followed by a Dunnett's test (*P < 0.05; **P < 0.01, ***P < 0.001).
Fig. 6.

Effects of FXR agonists on Hmgcr, Insig-2, Shp, Cyp7a1, Cyp8b1, and Cyp3a11 gene expression in Ldlr −/− mouse livers. Male Ldlr −/− mice were dosed daily by gavage with vehicle for 3 days prior to the study to acclimatize them to the dosing regimen. Thereafter, the animals were given either a single dose of RO5186026 (30 mg/kg) or vehicle. Four and 8 h posttreatment, groups of mice were euthanized, and livers were removed and frozen. Gene expression was measured using quantitative PCR, and the data were normalized using the expression of Gapdh. Values are means ± SE (n = 6/group). Significant differences between the vehicle and compound-treated groups were determined by a Wilcoxon/ Kruskal-Wallis test (**P < 0.01).
All four FXR agonists also dramatically reduced plasma campesterol and β-sitosterol in Ldlr −/− mice, suggesting that treatment with FXR agonists reduced the intestinal absorption of cholesterol.
In hamsters, RO5186026 reduced cholesterol synthesis as demonstrated by the lower plasma lathosterol (−31%) and desmosterol (−27%), but it increased β-sitosterol (+63%) (Fig. 5B). The X-Ceptor compound slightly decreased desmosterol (−14%), whereas plasma biomarkers for intestinal cholesterol absorption were unchanged (Fig. 5B). 6-ECDCA showed a unique profile because markers for synthesis and absorption of sterols were both reduced (desmosterol, −22%; campesterol, −45%; and β-sitosterol, −32%; Fig. 5B). Treatment of hamsters with GW4064 caused no change in biomarkers for cholesterol synthesis but increased plasma β-sitosterol by 38%.
We also measured the absorption of radiolabeled cholesterol in Ldlr −/− mice and hamsters treated with either RO5186026 or ezetimibe, a well-known inhibitor of intestinal cholesterol absorption (33). As expected, ezetimibe reduced cholesterol absorption by 74% in Ldlr −/− mice (P < 0.01) and by 94% (P < 0.001) in hamsters (Fig. 5C) (42, 43). Although RO5186026 decreased cholesterol absorption by 56% in mice (P < 0.01), only a marginal decrease in cholesterol absorption (15%, P < 0.05) was observed in hamsters.
These data indicate that FXR agonists decrease cholesterol absorption and synthesis in Ldlr −/− mice. However, in hamsters, the picture is not so clear. Whereas there was a general trend toward reduced de novo cholesterol synthesis with all but GW4064, the effect of FXR agonists on intestinal cholesterol absorption differed. RO5186026 and GW4064 increased β-sitosterol, whereas X-Ceptor had no effect, and 6-ECDCA reduced both campesterol and β-sitosterol. Contrary to the interpretation of the β-sitosterol data, cholesterol absorption was slightly reduced by RO5186026 in hamsters when measured using radiotracers.
FXR agonists regulate BA pool size and composition at the transcriptional level in Ldlr −/− mice
In a separate study, Ldlr −/− mice were treated once with RO5186026 (30 mg/kg), and changes in the expression of the main genes responsible for BA synthesis and profile in the liver were measured 4 and 8 h posttreatment to monitor any initial changes in gene expression (Fig. 6). As expected with an FXR agonist, RO5186026 strongly activated Shp expression by 8-fold at both 4 and 8 h posttreatment. The expression of Cyp7a1 and Cyp8b1 was strongly decreased by 93 and 66% at 4 h posttreatment and by 97 and 95% at 8 h posttreatment, respectively. Interestingly, the expression of Cyp3a11 was increased by 2.2-fold 8 h posttreatment in RO5186026-treated mice.
Together these data indicate that FXR agonists reduce BA pool size by repressing the expression of Cyp7a1 and change BA composition by modulating Cyp8b1 and Cyp3a11 expression, effects that are sustained for prolonged periods.
FXR agonists regulate BA pool size but not BA profile at the transcriptional level in hamsters
As well as analyzing changes in the expression of genes responsible for BA synthesis and profile in the main study (i.e., 2 h postdosing on day 11 of dosing; Fig. 7A), a separate group of hamsters was treated with a single dose of RO5186026, and changes in gene expression were analyzed 4 and 8 h posttreatment to monitor any transient changes in gene expression (Fig. 7B). RO5186026 treatment increased hepatic Shp expression by 3.3-fold at 2 h posttreatment on day 11 and by 2.3- and 1.5-fold at 4 and 8 h posttreatment after a single dose. X-Ceptor, 6-ECDCA, and GW4064 all increased Shp expression by 1.5-, 1.8-, and 2.5-fold, respectively, at 2 h posttreatment on day 11.
Fig. 7.

Effects of FXR agonists on Shp, Cyp7a1, and Cyp8b1 gene expression in hamster livers. Male hamsters were dosed daily by gavage with vehicle for 3 days prior to the study to acclimatize them to the dosing regimen. Thereafter, the animals were treated with vehicle alone, RO5186026, X-Ceptor, or 6-ECDCA (all at 30 mg/kg/day), or GW4064 (100 mg/kg/day) for 11 days (n = 6–8/group) (A) or once with vehicle alone or RO5186026 (30 mg/kg, n = 6–8/group) (B). Groups of hamsters were then euthanized, and livers were collected 2 h posttreatment on day 11 or 4 or 8 h after the single dose. Gene expression was measured by quantitative PCR and normalized to Gapdh. Values are means ± SE. Significant differences between the vehicle and compound-treated groups were determined by Wilcoxon/ Kruskal-Wallis test (*P < 0.05, **P < 0.01).
RO5186026 strongly inhibited Cyp7a1 expression by 92% at 2 h posttreatment on day 11 and by 98 and 91% at 4 and 8 h after a single dose. All the other FXR agonists also reduced Cyp7a1 expression by similar amounts 2 h posttreatment on day 11. Interestingly, RO5186026 decreased the expression of Cyp8b1 by 46 and 52% at 4 and 8 h after a single dose, but it did not affect its expression at 2 h posttreatment on day 11. Of the other FXR agonists, only 6-ECDCA decreased Cyp8b1 expression by 33% at 2 h posttreatment on day 11. Hence, all of the compounds increased the expression of Shp at 2 h posttreatment on day 11, and all were able to suppress Cyp7a1 expression at the same time point, indicating that Shp is important in the repression of Cyp7a1 expression in hamsters.
Together these data indicate that FXR agonists regulate BA pool size at the transcriptional level in hamsters through the repression of Cyp7a1 expression. We also showed that Cyp8b1 expression was only slightly or not decreased by the FXR agonists 2 h after 11 daily doses, and in hamsters treated with a single dose of RO5186026, it was decreased less (by 46 and 52%) compared with the stronger (66 and 95%) inhibition observed in Ldlr −/− mice at 4 and 8 h postdosing, respectively.
Fgf15/19 is strongly activated in Ldlr −/−mouse small intestine upon treatment with FXR agonists but is unaffected in hamsters
Apart from affecting gene expression in the liver, FXR agonists have been shown to regulate gene expression in the intestine, especially in the ileum, where the majority of the BA are reabsorbed. We measured the expression of Ibabp and Fgf15/19, known direct target genes for FXR, in the duodenum, jejunum, and ileum of RO5186026-treated Ldlr −/− mice and hamsters. Gene expression was measured 4 and 8 h after a single dose (Fig. 8) or 2 h postdosing at the end of 5- or 8-day dosing studies in mice or hamsters, respectively (supplementary Fig. III). As reported previously, Ibabp was expressed at much higher levels in the ileum than in the duodenum and jejunum (Fig. 8A and supplementary Fig. III) (29). In both single- and multiple-dose studies, Ibabp expression was upregulated by RO5186026 in mice and hamsters, with the biggest effects seen 8 h posttreatment in the single-dose study. The expression of Ibabp was greatly upregulated in the duodenum and jejunum of both species while in the ileum upregulation was only seen in Ldlr −/− mice at 8 h posttreatment in the single dose study. As with Ibabp expression, Fgf15/19 was poorly expressed in the duodenum and jejunum, but it was reasonably well expressed in the ileum (Fig. 8B and supplementary Fig. III). Four and 8 h after a single dose of RO5186026, expression of Fgf15 was strongly upregulated in all three segments of the SI in Ldlr −/− mice. Levels of expression declined slightly between 4 and 8 h posttreatment. In the multiple-dose study (i.e., sampled 2 h postdosing), Fgf15 expression was upregulated in the jejunum and ileum of Ldlr −/− mice. On the other hand, Fgf19 expression was unchanged in the SI of RO5186026-treated hamsters in both studies, with the exception of small (∼2.5-fold) but statistically significant increases in the jejunum at 4 and 8 h after a single dose (Fig. 8B).
Fig. 8.

Effects of FXR agonists on the expression of Ibabp and Fgf15/19 in Ldlr −/− mouse and hamster SI. Male Ldlr −/− mice and Syrian hamsters were dosed daily by gavage with vehicle for 3 days prior to the study to acclimatize them to the dosing regimen. The animals were then given either a single dose of RO5186026 (30 mg/kg) or vehicle. Four and 8 h posttreatment, groups of animals were euthanized, and the SI was removed, dissected, and frozen as described in Methods. Ibabp (A) and Fgf15/19 (B) gene expression was measured in the duodenum, jejunum, and ileum using quantitative PCR, and the data were normalized using the expression of Gapdh. Values are means ± SE (n = 6/group). Significant differences between the groups were determined by Wilcoxon/Kruskal-Wallis test (*P < 0.05, **P < 0.01).
These data show that RO5186026, a potent and selective FXR agonist, can strongly increase the expression of Fgf15/19 in Ldlr −/− mice but not in hamsters. They also show that Ibabp and Fgf15 expression can be increased to physiologically relevant levels by small, non-BA FXR agonists in the more proximal segments of the SI in mice.
Hepatic Fgf15/19 expression in Ldlr −/− mice and hamsters
As described above, the expression of Fgf15/19 in the SI is thought to regulate bile acid metabolism in the liver. However, ever since the discovery of this regulatory function of Fgf15/19, there has been interest in its possible autocrine function in the liver (27). FGF15/19 is not normally expressed in the liver, but its expression has been shown to be upregulated by FXR agonists in human hepatocytes and in the livers of patients with extrahepatic cholestasis (27, 44–46). We measured the expression of Fgf15/19 in the livers of RO5186026-treated Ldlr −/− mice and hamsters in the timecourse study and found very low levels of expression that were unaffected by treatment (data not shown).
DISCUSSION
Complex feedback and buffering mechanisms have evolved so that suitable BA pool sizes, profiles, and bound/free ratios are maintained in the enterohepatic system. Under normal (i.e., noncholestatic) conditions, most of the feedback is believed to be initiated by endogenous BA acting as agonists to FXR, which upregulates the expression of target genes such as Shp and Fgf15/19. As summarized in Fig. 9, these in turn decrease BA synthesis and so maintain pool size by downregulating Cyp7a1 and control BA profile by downregulating Cyp8b1. The expression of Cyp3a11, which also modifies BA profile, is reported to be upregulated by FXR agonists in mice (16). We compared the effect of four synthetic FXR agonists on BA pool sizes, profiles, and excretion and on cholesterol synthesis, plasma lipoprotein profiles, and intestinal absorption in Ldlr −/− mice and Syrian hamsters fed high-fat diets.
Fig. 9.

Schematic diagram showing the regulation of BA pool size and composition and, consequently, dietary cholesterol absorption and plasma cholesterol concentrations by FXR agonists in Ldlr −/− mice and hamsters. Solid and dashed arrows represent strong and weak effects, respectively. −, negative; +, positive; X, no effect.
Characterization of FXR agonists
RO5186026, X-Ceptor, 6-ECDCA, and GW4064 are structurally unrelated FXR agonists which, as we partially reported previously (28), have different potencies, efficacies, and selectivities for FXR (Table 1). We showed that RO5186026, X-Ceptor, and GW4064 bind with high affinity and selectively to human and mouse FXR. On the other hand, 6-ECDCA, a modified BA, was more than 20-fold less potent and was less selective, being a 4-fold more potent GPBAR1 than FXR agonist. Activity in human and mouse cell-based transactivation assays confirmed the relative order of potencies of the compounds, although the activity of GW4064 was disproportionately lower and that of 6-ECDCA slightly higher than they were in the SP-binding assay.
As there is excellent sequence homology between human, mouse, and hamster FXR ligand-binding domains (supplementary Fig. IV) and the binding and transactivation data of individual compounds were similar in human and mouse assays, we would expect these compounds to bind to and transactivate hamster FXR with similar potencies and efficacies. From the results presented, there should be no doubt that all of the compounds are reasonably potent FXR agonists in hamsters because they all increased Shp and decreased Cyp7a1 expression and fecal BA, and RO5186026 increased Ibabp expression in hamsters. Also, three of the four compounds decreased BA pool size in hamsters.
These compounds are likely to have different pharmacokinetic properties that will influence their pharmacodynamics in vivo. BA and their analogs, such as 6-ECDCA, once conjugated with either taurine or glycine in the liver, will be secreted in bile and aid lipid absorption in the SI before being absorbed in the ileum where they will modify gene expression and then be returned to the liver. On the other hand, non-BA FXR agonists, such as RO5186026, X-Ceptor, and GW4064, are likely to be absorbed and influence gene expression in the proximal SI. This is corroborated by the big increases in duodenal and jejunal expression of Ibabp in both species and of Fgf15 in Ldlr −/− mice treated with RO5186026. Also, BAs will undergo enterohepatic circulation and, consequently, would be expected to have longer half-lives and more prolonged FXR agonist effects than non-BA FXR agonists.
Effects of FXR agonist treatment on food intake, body weight, liver weight, and liver lipids
We selected doses of the FXR agonists that had been shown to be effective at reducing plasma cholesterol in Ldlr −/− mice for use in both species. RO5186026, X-Ceptor, and 6-ECDCA were dosed at 30 mg/kg/day, and GW4064 at 100 mg/kg/day. Dosing periods were chosen to be sufficiently long for plasma lipids to reequilibrate.
None of the compounds, except 6-ECDCA, caused any adverse effects on food intake or body weight gain in either Ldlr −/− mice or hamsters. Hamsters treated with 6-ECDCA had reduced food intake and body weight gain that were associated with decreased HDL-C and increased VLDL-C. These adverse effects were most likely caused by the accumulation of 6-ECDCA in the enterohepatic system. Although not a consistent finding in different patient populations, 6-ECDCA increased pruritus in primary biliary cirrhosis (PBC) patients, an effect that may also have occurred in hamsters (47).
Interestingly, liver weights were increased by 13–23% in RO5186026-, X-Ceptor-, and GW4064-treated hamsters but were unaffected in Ldlr−/− mice. Liver weights have also been reported to be increased in hamsters fed BA (48). These increases in liver weight may be associated with reduced BA synthesis because, as we reported previously, treatment with nonabsorbable BA sequestrants, which increase BA synthesis, reduced liver weight by about 15% in hamsters (49).
As demonstrated previously with WAY-362450, a close analog of X-Ceptor, liver cholesterol was reduced by RO5186026 and X-Ceptor (about −20%) in Ldlr −/− mice, probably by a combination of decreased cholesterol synthesis and FXR-mediated upregulation of biliary export via ABCG5/ABCG8 (41, 50–53). Liver TG was decreased by all the FXR agonists (−29 to −40%) in Ldlr −/− mice, possibly by reducing Srebp1c expression (54). In contrast, liver cholesterol and TG were unchanged by FXR agonist treatment in hamsters. The latter is consistent with the lack of effect of CDCA on liver TG previously reported in fructose-fed hamsters (55). However, in contrast to the present results, CDCA was reported to inhibit the small diet-induced increase in hepatic cholesterol in the same model.
Potent FXR agonists decreased plasma cholesterol in Ldlr −/− mice but not in hamsters
The three most potent FXR agonists, RO5186026, X-Ceptor, and GW4064, reduced plasma cholesterol by 47, 31, and 14%, respectively, in Ldlr −/− mice, but none did so in hamsters. This is in accord with what others have shown with FXR agonists, including WAY-362450, in Ldlr −/− mice (52).
The lack of effect of 6-ECDCA on plasma cholesterol in Ldlr −/− mice was unexpected because it reduced cholesterol synthesis, BA pool size, and sterol absorption to a similar extent as the other, more potent FXR agonists. Also, lower doses of 6-ECDCA have been shown to reduce hepatic and/or plasma cholesterol in apoE −/− and Western diet-fed DBA/2J mice (56, 57). It is possible then that the adverse effects associated with the higher dose of 6-ECDCA may have counteracted its potential for reducing plasma cholesterol.
The high-fat/cholesterol-fed hamster model we used may be relatively unresponsive to the antihyperlipidemic effects of FXR agonists because WAY-362450 given orally or 0.1% CDCA in the diet partially or completely prevented diet-induced increases in plasma cholesterol and TG in fructose-fed hamsters (52, 55). The increased sensitivity of fructose-fed hamsters to FXR agonists might be related to the 80% decrease in expression of Cyp7a1 induced by diet alone and greater de novo lipogenesis and hepatic secretion of TG-rich lipoproteins (55). Hamsters seem to be relatively insensitive to FXR agonist treatment not just compared with Ldlr −/− mice but also compared with rhesus monkeys. Lundquist et al. reported a 63% reduction in LDL-C in rhesus monkeys treated with 14cc, another analog of the X-Ceptor compound (58).
FXR agonists decreased BA pool size and excretion in Ldlr −/− mice and hamsters
It is well recognized that inadequate intraluminal concentrations of BA in the SI and/or change to a more hydrophilic profile can reduce intestinal cholesterol absorption (7, 9, 10). In Cyp7a1 −/− mice, for example, BA pool size was reduced by about 80% and cholesterol absorption by more than 97% (10). In FXR agonist-treated Ldlr −/− mice and hamsters, intestinal BA pool sizes were reduced by 49–70% and 38–80%, respectively, by all except X-Ceptor in hamsters, which, strangely, did not modify the BA pool size. All of the FXR agonists also reduced fecal BA excretion in both species, mostly by similar proportions to the reductions in BA pool sizes. The exception was with X-Ceptor in hamsters that reduced fecal BA excretion by 46% with no change in pool size (Fig. 3 and supplementary Fig. II), indicating possible increased intestinal reclamation of BA to conserve them.
Some of the bigger decreases in BA pool size seen in this study might, therefore, be expected to reduce cholesterol absorption, at least in mice, independently of any effects caused by changes in BA profile.
BA profile was modulated by FXR agonists in Ldlr −/− mice but not in hamsters
All of the FXR agonists radically changed BA profiles in Ldlr −/− mice from mainly CA (about 70%) to mainly α-, β-, plus ω-MCA (61–83%). The effects of FXR activation in Ldlr −/− mice were similar in some respects to those seen in sterol 12α-hydroxylase knockout (Cyp8b1 −/−) mice (7). The change in the BA profile from mainly CA to mainly MCA was the same and can be attributed to the decreased expression of Cyp8b1 in FXR agonist-treated mice (Fig. 6). However, unlike in FXR agonist-treated mice, Cyp8b1 deficiency led to increased BA pool size and fecal excretion because of the diversion of BA synthesis from CA, a weak FXR agonist, to MCA, which does not activate FXR. With reduced FXR agonist activity present in the enterohepatic circulation, a derepression of BA synthesis occurred, as indicated by the upregulation of Cyp7a1 and hepatic cholesterol synthesis. In FXR agonist-treated mice, Cyp7a1 and cholesterol synthesis were both inhibited (Figs. 5 and 6), which led to a reduction in BA pool size and fecal BA excretion (Fig. 3 and supplementary Fig. II) and even a reduction in hepatic cholesterol (supplementary Table I). Despite the increased BA pool size in Cyp8b1 −/− mice, cholesterol absorption was still reduced because of the increased hydrophilicity of the BA pool. As highlighted by Li-Hawkins et al., this suggests that BA profiles can be more important than pool size in determining the solubilization and absorption of cholesterol in the intestine (7).
In contrast to the increase in Cyp3a11 expression seen with FXR agonist treatment, Cyp8b1 deficiency did not affect Cyp3a11 mRNA (7). This may be because of direct upregulation of Cyp3a11 by the potent FXR agonists (16) and could account for the smaller proportion of CDCA seen in these mice (∼4%) compared with Cyp8b1 knockout mice (∼15%).
In mice treated with FXR agonists, the BA profile is made more hydrophilic and pool size is reduced, which is potentially an even better combination for reducing cholesterol absorption. Indeed, cholesterol absorption measured using radiotracers was reduced by 56% by RO5186026 compared with ∼41% in male Cyp8b1 −/− mice.
It is also possible that, in FXR agonist-treated Ldlr −/− mice, the combination of high biliary MCA and upregulated transporters (BSEP, MDR2, and ABCG5/ABCG8) may help transport more biliary cholesterol and phospholipid into the intestine where less of it is subsequently reabsorbed. This could contribute to the decreases in hepatic cholesterol and TG (i.e., TG used in phospholipid synthesis) in FXR agonist-treated mice.
In hamsters, the normal CA + CDCA-dominated profiles (60% + 20%) were only slightly affected by FXR agonists, except for 6-ECDCA, which increased the proportion of CDCA to 63%, a probable consequence of the reduced Cyp8b1 expression seen only in this group (Figs. 4B and 7).
In RO5186026-, X-Ceptor-, and GW4064-treated hamsters, cholesterol synthesis was reduced by all but GW4064. Also, cholesterol absorption, as measured using radiotracers, was slightly reduced by RO5186026. Intriguingly, β-sitosterol, a plasma marker of cholesterol absorption, was increased 63 and 38% in RO5186026- and GW4064-treated hamsters, respectively, and there was also a trend for campesterol to be increased in the same animals (Fig. 5B). To explain the contradiction between the radiotracer and β-sitosterol results, we hypothesize that, in line with the radiotracer results, β-sitosterol absorption is slightly reduced but that the effect of this on plasma phytosterol levels is more than counterbalanced by a reduction in excretion. β-sitosterol from plasma is excreted in bile as a mixture of unmodified sterol and, depending on the species, highly hydrophilic, mainly trihydroxy, 21 carbon (C21) BA (59, 60). For example, in rats, β-sitosterol is converted into C21 BA via a pathway that does not involve 7α-hydroxylation (60). If the synthesis of these C21 BA from β-sitosterol occurs in hamsters and is downregulated by FXR agonists, then it may have led to its accumulation in plasma. Alternatively, the reduction in biliary secretion of normal BA in FXR agonist-treated hamsters may have led to a reduction in phytosterol secretion into the bile and so have reduced its excretion. The phytosterol results with 6-ECDCA lend support to this hypothesis, as both campersterol and β-sitosterol were reduced in 6-ECDCA-treated hamsters, a model in which 6-ECDCA itself would be expected to contribute to biliary bile acid flow.
The lack of effect of FXR agonists on plasma cholesterol in hamsters may be because their BA pool sizes were not sufficiently reduced (RO5186026, −51%; GW4064, −38%), especially when considering that their BA profiles are more lipophilic than those in mice. These results would suggest that higher doses of the FXR agonists or different dosing regimens might be required to reduce plasma cholesterol in this hamster model.
Biological rationale for FXR agonist-induced changes in hepatic gene expression compared with those in humans
Although both species responded in slightly different ways, they compensated for the perceived cholestatic conditions associated with FXR agonist treatment so as to reduce the anticipated adverse effects, that is, by decreasing BA pool size and, in mice, by reducing the hydrophobicity of the BA. The conversion of the potent FXR agonist CDCA into its inactive product MCA in mice potentially serves at least three functions. First, BA synthesis continues at a higher rate than if CDCA were the end product; consequently, MCA is available to transport biliary sterols into the gut and less of the toxic secondary BA LCA can be formed from CDCA. Indeed, synthesis of MCA may have additional advantages as cholesterol gallstones, a potential cause of cholestasis, have been shown to be dissolved even more rapidly using MCA than by using UDCA (61).
In hamsters all of the FXR agonists reduced Cyp7a1 expression and, therefore, BA synthesis, and all but X-Ceptor reduced BA pool size. The lack of effect of X-Ceptor on pool size was possibly because of transient effects on Shp/Cyp7a1 expression and/or by increasing the intestinal reclamation of BA. Consistent with the slight or zero reduction in Cyp8b1 expression, none of the more potent agonists substantially modified BA profiles. Interestingly, in bile duct-ligated hamsters, Cyp7a1 and Cyp8b1 mRNA, protein, and activities were reported to be unchanged (62), whereas in CDCA-fed hamsters, Cyp7a1 and Cyp8b1 activities were shown to be slightly downregulated (63, 64). The lack of effect of the nonBA FXR agonists on Cyp8b1 expression in hamsters is potentially beneficial as it avoids the accumulation of CDCA and its toxic microbial metabolite LCA.
The observed changes in Cyp7a1, Cyp8b1, and Cyp3a11 expression were, therefore, consistent with FXR agonist-treated mice and hamsters countering the perceived hazards of cholestasis using the metabolic pathways available to them.
Patients with extrahepatic cholestasis or gallstones and normal rhesus monkeys respond in the same way as hamsters to increases in BA/FXR agonist activity with 70–95% reductions in hepatic Cyp7a1 mRNA and no change or even an increase in Cyp8b1 mRNA (46, 58, 65). The one exception in vivo was in patients treated with CDCA in whom microsomal CYP8B1 activity was inhibited by 50% (66). In agreement with previous in vivo and in vitro studies in humans, the present work suggests that the SHP-dependent mechanism for regulating CYP8B1 by FXR agonists in hamsters is considerably less effective than the regulation of the same gene in mice and rats (67). Various mechanisms have been proposed for the reduced effect of BA on the expression CYP8B1 in humans compared with rats (22, 67, 68). If these are also proven to apply in hamsters, then the hamster may be an even better model for human BA metabolism than it was previously considered.
Intestinal but not hepatic expression of Fgf15/19 was greatly increased by FXR agonists in Ldlr −/− mice, but neither was affected in hamsters
Inhibition of Cyp7a1 and Cyp8b1 expression by FXR agonists has been shown to be mediated by intestinal expression and secretion of Fgf15/19 as well as hepatic expression of Shp, but the exact contribution of each, and any differences between species, are unclear. Upon FXR activation, FGF15/19 expression is upregulated, and it is released into the circulation where it activates its receptor FGFR4 in the liver. This leads to the activation of the mitogen-activated protein kinase (MAPK) signaling pathway that suppresses Cyp7a1 and Cyp8b1 expression (19, 20, 69). Kim et al. and Kong et al. have shown in mice that the FXR/Fgf15/19 pathway is critical for suppressing both Cyp7a1 and Cyp8b1, whereas the FXR/Shp pathway suppresses mainly Cyp8b1 expression (70, 71). This was probably the case in FXR agonist-treated Ldlr −/− mice, in which both hepatic Shp and intestinal Fgf15 expression were increased and both Cyp7a1 and Cyp8b1 expression reduced. However, in hamsters, Shp expression was upregulated while Fgf19 expression was almost completely unchanged, and Cyp7a1 was reduced while Cyp8b1 was relatively unchanged. This indicates a different role for Shp in regulating these CYPs in hamsters versus mice. Whereas Shp plays a crucial role in suppressing Cyp8b1 but less so Cyp7a1 in mice, it probably suppresses Cyp7a1 but less so Cyp8b1 in hamsters, although we cannot exclude that other mechanisms might be involved. One possible explanation for the lack of effect of RO5186026 on Fgf15/19 expression is that the ileum is the major site of Fgf15/19 expression in hamsters, and as evidenced by the lack of effect on Ibabp expression in the ileum (Fig. 8A and supplementary Fig. III), it may not have been sufficiently exposed to RO5186026, possibly because all of the compound that was going to be absorbed was absorbed in the more proximal segments of the SI. Consequently, FXR agonists that have a sustained effect in the ileum as well as the liver in hamsters may have greater effects on BA pool size than those that primarily target the liver. Alternatively, it is possible that FXR agonists do not stimulate the expression of Fgf19 mRNA in hamster SI.
In summary, we report a thorough analysis of the differential regulation of BA and cholesterol metabolism by four structurally dissimilar FXR agonists in high-fat/cholesterol-fed Ldlr −/− mice and Syrian hamsters. Our findings are summarized in Fig. 9. In both species, FXR agonists decreased the BA pool size by increasing Shp and reducing Cyp7a1 expression. In addition, but only in Ldlr −/− mice, FXR agonists increased Fgf15 and Cyp3a11 expression and substantially repressed Cyp8b1 expression. They, therefore, generated a more hydrophilic BA pool composed mainly of MCA. The absorption of dietary cholesterol and plasma cholesterol levels were, consequently, reduced in Ldlr −/− mice. Unexpectedly, increased expression of Fgf15 was not only seen in the ileum but also in the more proximal segments of the SI. In hamsters, increases in Shp expression and big reductions in Cyp7a1 expression and BA pool sizes were accompanied, interestingly, by no change in Fgf19 expression and only minor changes in Cyp8b1 expression and the hydrophobicity of the BA profiles and dietary cholesterol absorption and plasma cholesterol levels that were unchanged.
Supplementary Material
Acknowledgments
The authors thank Corinne Handschin, Marie-Thérèse Traendlin, Heinz Meyer, and Sabine Weiss for their excellent technical assistance with the in vivo experiments and analytics; Martin Ebeling for his help with aligning the FXR sequences; and Matthew Wright and Michael Pech for their strong support.
Footnotes
Abbreviations:
- BA
- bile acid
- CA
- cholic acid
- CDCA
- chenodeoxycholic acid
- CVD
- cardiovascular disease
- CYP7A1/Cyp7a1
- cholesterol-7α-hydroxylase
- CYP8B1/Cyp8b1
- sterol-12α-hydroxylase
- DCA
- deoxycholic acid
- FGF15/19
- fibroblast growth factor 15 or 19
- FXR
- farnesoid X receptor
- HDCA
- hyodeoxycholic acid
- HDL-C
- HDL cholesterol
- L+GB
- liver plus gallbladder
- LCA
- lithocholic acid
- LDL-C
- LDL cholesterol
- Ldlr−/−
- LDL receptor-deficient
- MCA
- muricholic acid
- Shp
- small heterodimer partner
- SI
- small intestine
- SP
- scintillation proximity
- TG
- triglyceride
- UDCA
- ursodeoxycholic acid
- VLDL-C
- VLDL cholesterol
The online version of this article (available at http://www.jlr.org) contains supplementary data in the form of one table and four figures.
REFERENCES
- 1.Roger V. L., Go A. S., Lloyd-Jones D. M., Benjamin E. J., Berry J. D., Borden W. B., Bravata D. M., Dai S., Ford E. S., Fox C. S., et al. 2012. Executive summary: heart disease and stroke statistics—2012 update. Circulation. 125: 188–197 [DOI] [PubMed] [Google Scholar]
- 2.Lefebvre P., Cariou B., Lien F., Kuipers F., Staels B. 2009. Role of bile acids and bile acid receptors in metabolic regulation. Physiol. Rev. 89: 147–191 [DOI] [PubMed] [Google Scholar]
- 3.Shanes J. G. 2012. A review of the rationale for additional therapeutic interventions to attain lower LDL-C when statin therapy is not enough. Curr. Atheroscler. Rep. 14: 33–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Insull W. 2006. Clinical utility of bile acid sequestrants in the treatment of dyslipidemia: a scientific review. South. Med. J. 99: 257–273 [DOI] [PubMed] [Google Scholar]
- 5.Russell D. W., Setchell K. D. R. 1992. Bile acid biosynthesis. Biochemistry. 31: 4737–4749 [DOI] [PubMed] [Google Scholar]
- 6.Vlahcevic Z. R., Heuman D. M., Hylemon P. B. 1991. Regulation of bile acid synthesis. Hepatology. 13: 590–600 [PubMed] [Google Scholar]
- 7.Li-Hawkins J., Gafvels M., Olin M., Lund E. G., Andersson U., Schuster G., Bjorkhem I., Russell D. W., Eggertsen G. 2002. Cholic acid mediates negative feedback regulation of bile acid synthesis in mice. J. Clin. Invest. 110: 1191–1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zollner G., Wagner M., Moustafa T., Fickert P., Silbert D., Gumhold J., Fuchsbichler A., Halilbasic E., Denk H., Marschall H-U., et al. 2006. Coordinated induction of bile acid detoxification and alternative elimination in mice: role of FXR-regulated organic solute transporter-α/β in the adaptive response to bile acids. Am. J. Physiol. Gastrointest. Liver Physiol. 290: G923–G932 [DOI] [PubMed] [Google Scholar]
- 9.Wang D. Q., Tazuma S., Cohen D. E., Carey M. C. 2003. Feeding natural hydrophilic bile acids inhibits intestinal cholesterol absorption: studies in the gallstone-susceptible mouse. Am. J. Physiol. Gastrointest. Liver Physiol. 285: G494–G502 [DOI] [PubMed] [Google Scholar]
- 10.Schwarz M., Russell D. W., Dietschy J. M., Turley S. D. 1998. Marked reduction in bile acid synthesis in cholesterol 7α-hydroxylase-deficient mice does not lead to diminished tissue cholesterol turnover or to hypercholesterolemia. J. Lipid Res. 39: 1833–1843 [PubMed] [Google Scholar]
- 11.Makishima M., Okamoto A. Y., Repa J. J., Tu H., Learned R. M., Luk A., Hull M. V., Lustig K. D., Mangelsdorf D. J., Shan B. 1999. Identification of a nuclear receptor for bile acids. Science. 284: 1362–1365 [DOI] [PubMed] [Google Scholar]
- 12.Parks D. J., Blanchard S. G., Bledsoe R. K., Chandra G., Consler T. G., Kliewer S. A., Stimmel J. B., Willson T. M., Zavacki A. M., Moore D. D., et al. 1999. Bile acids: natural ligands for an orphan nuclear receptor. Science. 284: 1365–1368 [DOI] [PubMed] [Google Scholar]
- 13.Wang H., Chen J., Hollister K., Sowers L. C., Forman B. M. 1999. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol. Cell. 3: 543–553 [DOI] [PubMed] [Google Scholar]
- 14.Lu T. T., Makishima M., Repa J. J., Schoonjans K., Kerr T. A., Auwerx J., Mangelsdorf D. J. 2000. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol. Cell. 6: 507–515 [DOI] [PubMed] [Google Scholar]
- 15.Repa J. J., Turley S. D., Lobaccaro J. A., Medina J., Li L., Lustig K., Shan B., Heyman R. A., Dietschy J. M., Mangelsdorf D. J. 2000. Regulation of absorption and ABC1-mediated efflux of cholesterol by RXR heterodimers. Science. 289: 1524–1529 [DOI] [PubMed] [Google Scholar]
- 16.Gnerre C., Blättler S., Kaufmann M. R., Looser R., Meyer U. A. 2004. Regulation of CYP3A4 by the bile acid receptor FXR: evidence for functional binding sites in the CYP3A4 gene. Pharmacogenetics. 14: 635–645 [DOI] [PubMed] [Google Scholar]
- 17.Schuetz E. G., Strom S., Yasuda K., Lecureur V., Assem M., Brimer C., Lamba J., Kim R. B., Ramachandran V., Komoroski B. J., et al. 2001. Disrupted bile acid homeostasis reveals an unexpected interaction among nuclear hormone receptors, transporters, and cytochrome P450. J. Biol. Chem. 276: 39411–39418 [DOI] [PubMed] [Google Scholar]
- 18.Goodwin B., Jones S. A., Price R. R., Watson M. A., McKee D. D., Moore L. B., Galardi C., Wilson J. G., Lewis M. C., Roth M. E., et al. 2000. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol. Cell. 6: 517–526 [DOI] [PubMed] [Google Scholar]
- 19.Inagaki T., Choi M., Moschetta A., Peng L., Cummins C. L., McDonald J. G., Luo G., Jones S. A., Goodwin B., Richardson J. A., et al. 2005. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2: 217–225 [DOI] [PubMed] [Google Scholar]
- 20.Song K. H., Li T., Owsley E., Strom S., Chiang J. Y. 2009. Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibit cholesterol 7α-hydroxylase gene expression. Hepatology. 49: 297–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.del Castillo-Olivares A., Gil G. 2001. Suppression of sterol 12α-hydroxylase transcription by the short heterodimer partner: insights into the repression mechanism. Nucleic Acids Res. 29: 4035–4042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang M., Chiang J. Y. L. 2001. Transcriptional regulation of the human sterol 12α-hydroxylase gene (CYP8B1). J. Biol. Chem. 276: 41690–41699 [DOI] [PubMed] [Google Scholar]
- 23.Nitta M., Ku S., Brown C., Okamoto A. Y., Shan B. 1999. CPF: An orphan nuclear receptor that regulates liver-specific expression of the human cholesterol 7α-hydroxylase gene. Proc. Natl. Acad. Sci. USA. 96: 6660–6665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shin D. J., Osborne T. F. 2009. FGF15/FGFR4 integrates growth factor signaling with hepatic bile acid metabolism and insulin action. J. Biol. Chem. 284: 11110–11120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jung D., Inagaki T., Gerard R. D., Dawson P. A., Kliewer S. A., Mangelsdorf D. J., Moschetta A. 2007. FXR agonists and FGF15 reduce fecal bile acid excretion in a mouse model of bile acid malabsorption. J. Lipid Res. 48: 2693–2700 [DOI] [PubMed] [Google Scholar]
- 26.Miao J., Xiao Z., Kanamaluru D., Min G., Yau P. M., Veenstra T. D., Ellis E., Strom S., Suino-Powell K., Xu H. E., et al. 2009. Bile acid signaling pathways increase stability of Small Heterodimer Partner (SHP) by inhibiting ubiquitin-proteasomal degradation. Genes Dev. 23: 986–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holt J. A., Luo G., Billin A. N., Bisi J., McNeill Y. Y., Kozarsky K. F., Donahee M., Wang D. Y., Mansfield T. A., Kliewer S. A., et al. 2003. Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev. 17: 1581–1591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gardès C., Blum D., Bleicher K., Chaput E., Ebeling M., Hartman P., Handschin C., Richter H., Benson G. M. 2011. Studies in mice, hamsters, and rats demonstrate that repression of hepatic apoA-I expression by taurocholic acid in mice is not mediated by the farnesoid-X-receptor. J. Lipid Res. 52: 1188–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grober J., Zaghini I., Fujii H., Jones S. A., Kliewer S. A., Willson T. M., Ono T., Besnard P. 1999. Identification of a bile acid-responsive element in the human ileal bile acid-binding protein gene. Involvement of the farnesoid X receptor/9-cis-retinoic acid receptor heterodimer. J. Biol. Chem. 274: 29749–29754 [DOI] [PubMed] [Google Scholar]
- 30.Wang D. Q-H., Carey M. C. 2003. Measurement of intestinal cholesterol absorption by plasma and fecal dual-isotope ratio, mass balance, and lymph fistula methods in the mouse: an analysis of direct versus indirect methodologies. J. Lipid Res. 44: 1042–1059 [DOI] [PubMed] [Google Scholar]
- 31.Zilversmit D. B., Hughes L. B. 1974. Validation of a dual-isotope plasma ratio method for measurement of cholesterol absorption in rats. J. Lipid Res. 15: 465–473 [PubMed] [Google Scholar]
- 32.Sudhop T., Lutjohann D., Kodal A., Igel M., Tribble D. L., Shah S., Perevozskaya I., von Bergmann K. 2002. Inhibition of intestinal cholesterol absorption by ezetimibe in humans. Circulation. 106: 1943–1948 [DOI] [PubMed] [Google Scholar]
- 33.van Heek M., Compton D. S., Davis H. R. 2001. The cholesterol absorption inhibitor, ezetimibe, decreases diet-induced hypercholesterolemia in monkeys. Eur. J. Pharmacol. 415: 79–84 [DOI] [PubMed] [Google Scholar]
- 34.Heider J. G., Boyett R. L. 1978. The picomole determination of free and total cholesterol in cells in culture. J. Lipid Res. 19: 514–518 [PubMed] [Google Scholar]
- 35.Carr T. P., Andresen C. J., Rudel L. L. 1993. Enzymatic determination of triglyceride, free cholesterol, and total cholesterol in tissue lipid extracts. Clin. Biochem. 26: 39–42 [DOI] [PubMed] [Google Scholar]
- 36.Maloney P. R., Parks D. J., Haffner C. D., Fivush A. M., Chandra G., Plunket K. D., Creech K. L., Moore L. B., Wilson J. G., Lewis M. C., et al. 2000. Identification of a chemical tool for the orphan nuclear receptor FXR. J. Med. Chem. 43: 2971–2974 [DOI] [PubMed] [Google Scholar]
- 37.Richter H. G., Benson G. M., Blum D., Chaput E., Feng S., Gardes C., Grether U., Hartman P., Kuhn B., Martin R. E., et al. 2011. Discovery of novel and orally active FXR agonists for the potential treatment of dyslipidemia & diabetes. Bioorg. Med. Chem. Lett. 21: 191–194 [DOI] [PubMed] [Google Scholar]
- 38.Pellicciari R., Fiorucci S., Camaioni E., Clerici C., Costantino G., Maloney P. R., Morelli A., Parks D. J., Willson T. M. 2002. 6α-Ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J. Med. Chem. 45: 3569–3572 [DOI] [PubMed] [Google Scholar]
- 39.Miettinen T. A., Tilvis R. S., Kesaniemi Y. A. 1990. Serum plant sterols and cholesterol precursors reflect cholesterol absorption and synthesis in volunteers of a randomly selected male population. Am. J. Epidemiol. 131: 20–31 [DOI] [PubMed] [Google Scholar]
- 40.Goldstein J. L., DeBose-Boyd R. A., Brown M. S. 2006. Protein sensors for membrane sterols. Cell. 124: 35–46 [DOI] [PubMed] [Google Scholar]
- 41.Hubbert M. L., Zhang Y., Lee F. Y., Edwards P. A. 2007. Regulation of hepatic Insig-2 by the farnesoid X receptor. Mol. Endocrinol. 21: 1359–1369 [DOI] [PubMed] [Google Scholar]
- 42.Niesor E. J., Chaput E., Staempfli A., Blum D., Derks M., Kallend D. 2011. Effect of dalcetrapib, a CETP modulator, on non-cholesterol sterol markers of cholesterol homeostasis in healthy subjects. Atherosclerosis. 219: 761–767 [DOI] [PubMed] [Google Scholar]
- 43.Sudhop T., Reber M., Tribble D., Sapre A., Taggart W., Gibbons P., Musliner T., von Bergmann K., Lutjohann D. 2009. Changes in cholesterol absorption and cholesterol synthesis caused by ezetimibe and/or simvastatin in men. J. Lipid Res. 50: 2117–2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lundåsen T., Gälman C., Angelin B., Rudling M. 2006. Circulating intestinal fibroblast growth factor 19 has a pronounced diurnal variation and modulates hepatic bile acid synthesis in man. J. Intern. Med. 260: 530–536 [DOI] [PubMed] [Google Scholar]
- 45.Mitro N., Godio C., De Fabiani E., Scotti E., Galmozzi A., Gilardi F., Caruso D., Chacon A. B. V., Crestani M. 2007. Insights in the regulation of cholesterol 7α-hydroxylase gene reveal a target for modulating bile acid synthesis. Hepatology. 46: 885–897 [DOI] [PubMed] [Google Scholar]
- 46.Schaap F. G., van der Gaag N. A., Gouma D. J., Jansen P. L. M. 2009. High expression of the bile salt-homeostatic hormone fibroblast growth factor 19 in the liver of patients with extrahepatic cholestasis. Hepatology. 49: 1228–1235 [DOI] [PubMed] [Google Scholar]
- 47.Intercept Pharmaceuticals Study of INT-747 as monotherapy in patients with PBC. ClinicalTrails.gov Identifier: NCT00570765. Accessed September 20, 2012, at http://www.clinicaltrials.gov/ct2/show/results/NCT00570765?term=Study+of+INT-747+as+Monotherapy+in+Patients+With+PBC.&rank=1§=X4015#othr
- 48.Oda H., Kuroki S., Yamashita H., Nakayama F. 1990. Effects of bile acid feeding on hepatic deoxycholate 7α-hydroxylase activity in the hamster. Lipids. 25: 706–710 [DOI] [PubMed] [Google Scholar]
- 49.Benson G. M., Alston D. R., Bond B. C., Gee A. N., Glen A., Haynes C., Hickey D. M. B., Iqbal S., Jackson B., Jaxa-Chamiec A. A., et al. 1993. SK&F 97426-A a more potent bile acid sequestrant and hypocholesterolaemic agent than cholestyramine in the hamster. Atherosclerosis. 101: 51–60 [DOI] [PubMed] [Google Scholar]
- 50.Repa J. J., Berge K. E., Pomajzl C., Richardson J. A., Hobbs H., Mangelsdorf D. J. 2002. Regulation of ATP-binding cassette sterol transporters ABCG5 and ABCG8 by the liver X receptors α and β. J. Biol. Chem. 277: 18793–18800 [DOI] [PubMed] [Google Scholar]
- 51.Zhang Y., Yin L., Anderson J., Ma H., Gonzalez F. J., Willson T. M., Edwards P. A. 2010. Identification of novel pathways that control farnesoid X receptor-mediated hypocholesterolemia. J. Biol. Chem. 285: 3035–3043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Evans M. J., Mahaney P. E., Borges-Marcucci L., Lai K., Wang S., Krueger J. A., Gardell S. J., Huard C., Martinez R., Vlasuk G. P., et al. 2009. A synthetic farnesoid X receptor (FXR) agonist promotes cholesterol lowering in models of dyslipidemia. Am. J. Physiol. Gastrointest. Liver Physiol. 296: G543–G552 [DOI] [PubMed] [Google Scholar]
- 53.Yu L., Gupta S., Xu F., Liverman A. D., Moschetta A., Mangelsdorf D. J., Repa J. J., Hobbs H. H., Cohen J. C. 2005. Expression of ABCG5 and ABCG8 is required for regulation of biliary cholesterol secretion. J. Biol. Chem. 280: 8742–8747 [DOI] [PubMed] [Google Scholar]
- 54.Watanabe M., Houten S. M., Wang L., Moschetta A., Mangelsdorf D. J., Heyman R. A., Moore D. D., Auwerx J. 2004. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J. Clin. Invest. 113: 1408–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bilz S., Samuel V., Morino K., Savage D., Choi C. S., Shulman G. I. 2006. Activation of the farnesoid X receptor improves lipid metabolism in combined hyperlipidemic hamsters. Am. J. Physiol. Endocrinol. Metab. 290: E716–E722 [DOI] [PubMed] [Google Scholar]
- 56.Mencarelli A., Renga B., Distrutti E., Fiorucci S. 2009. Antiatherosclerotic effect of farnesoid X receptor. Am. J. Physiol. Heart Circ. Physiol. 296: H272–H281 [DOI] [PubMed] [Google Scholar]
- 57.Wang X. X., Jiang T., Shen Y., Adorini L., Pruzanski M., Gonzalez F. J., Scherzer P., Lewis L., Miyazaki-Anzai S., Levi M. 2009. The farnesoid X receptor modulates renal lipid metabolism and diet-induced renal inflammation, fibrosis, and proteinuria. Am. J. Physiol. Renal Physiol. 297: F1587–F1596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lundquist J. T., Harnish D. C., Kim C. Y., Mehlmann J. F., Unwalla R. J., Phipps K. M., Crawley M. L., Commons T., Green D. M., Xu W., et al. 2010. Improvement of physiochemical properties of the tetrahydroazepinoindole series of farnesoid X receptor (FXR) agonists: beneficial modulation of lipids in primates. J. Med. Chem. 53: 1774–1787 [DOI] [PubMed] [Google Scholar]
- 59.Boberg K. M., Einarsson K., Björkhem I. 1990. Apparent lack of conversion of sitosterol into C24-bile acids in humans. J. Lipid Res. 31: 1083–1088 [PubMed] [Google Scholar]
- 60.Boberg K. M., Lund E., Olund J., Björkhem I. 1990. Formation of C21 bile acids from plant sterols in the rat. J. Biol. Chem. 265: 7967–7975 [PubMed] [Google Scholar]
- 61.Wang D. Q-H., Tazuma S. 2002. Effect of β-muricholic acid on the prevention and dissolution of cholesterol gallstones in C57L/J mice. J. Lipid Res. 43: 1960–1968 [DOI] [PubMed] [Google Scholar]
- 62.Matsuzaki Y., Bouscarel B., Ikegami T., Honda A., Doy M., Ceryak S., Fukushima S., Yoshida S., Shoda J., Tanaka N. 2002. Selective inhibition of CYP27A1 and of chenodeoxycholic acid synthesis in cholestatic hamster liver. Biochim. Biophys. Acta. 1588: 139–148 [DOI] [PubMed] [Google Scholar]
- 63.Kuroki S., Hoshita T. 1983. Effect of bile acid feeding on hepatic steroid 12 α-hydroxylase activity in hamsters. Lipids. 18: 789–794 [DOI] [PubMed] [Google Scholar]
- 64.Goldstein L. I., Bonorris G. G., Coyne M. J., Schoenfield L. J. 1975. Persistent effects of chenodeoxycholic acid on biliary lipids in the hamster. J. Lab. Clin. Med. 85: 1032–1041 [PubMed] [Google Scholar]
- 65.Abrahamsson A., Gustafsson U., Ellis E., Nilsson L-M., Sahlin S., Björkhem I., Einarsson C. 2005. Feedback regulation of bile acid synthesis in human liver: importance of HNF-4α for regulation of CYP7A1. Biochem. Biophys. Res. Commun. 330: 395–399 [DOI] [PubMed] [Google Scholar]
- 66.Ahlberg J., Angelin B., Björkhem I., Einarsson K., Gustafsson J-Å., Rafter J. 1980. Effects of treatment with chenodeoxycholic acid on liver microsome metabolism of steroids in man. J. Lab. Clin. Med. 95: 188–194 [PubMed] [Google Scholar]
- 67.Ellis E., Axelson M., Abrahamsson A., Eggertsen G., Thörne A., Nowak G., Ericzon B-G., Björkhem I., Einarsson C. 2003. Feedback regulation of bile acid synthesis in primary human hepatocytes: evidence that CDCA is the strongest inhibitor. Hepatology. 38: 930–938 [DOI] [PubMed] [Google Scholar]
- 68.Sanyal S., Båvner A., Haroniti A., Nilsson L-M., Lundåsen T., Rehnmark S., Witt M. R., Einarsson C., Talianidis I., Gustafsson J-å., et al. 2007. Involvement of corepressor complex subunit GPS2 in transcriptional pathways governing human bile acid biosynthesis. Proc. Natl. Acad. Sci. USA. 104: 15665–15670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yu C., Wang F., Kan M., Jin C., Jones R. B., Weinstein M., Deng C-X., McKeehan W. L. 2000. Elevated cholesterol metabolism and bile acid synthesis in mice lacking membrane tyrosine kinase receptor FGFR4. J. Biol. Chem. 275: 15482–15489 [DOI] [PubMed] [Google Scholar]
- 70.Kim I., Ahn S-H., Inagaki T., Choi M., Ito S., Guo G. L., Kliewer S. A., Gonzalez F. J. 2007. Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J. Lipid Res. 48: 2664–2672 [DOI] [PubMed] [Google Scholar]
- 71.Kong B., Wang L., Chiang J. Y. L., Zhang Y., Klaassen C. D., Guo G. L. 2012. Mechanism of tissue-specific farnesoid X receptor in suppressing the expression of genes in bile-acid synthesis in mice. Hepatology. 56: 1034–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.