

Abstract
Background:
Brain arteriovenous malformations (AVMs) produce circulatory and functional disturbances in adjacent as well as in remote areas of the brain, but their physiological effect on the cerebrospinal fluid (CSF) pressure is not well known.
Methods:
The hypothesis of an intrinsic disease mechanism leading to increased CSF pressure in all patients with brain AVM is outlined, based on a theory of hemodynamic control of intracranial pressure that asserts that CSF pressure is a fraction of the systemic arterial pressure as predicted by a two-resistor series circuit hydraulic model. The resistors are the arteriolar resistance (that is regulated by vasomotor tonus), and the venous resistance (which is mechanically passive as a Starling resistor). This theory is discussed and compared with the knowledge accumulated by now on intravasal pressures and CSF pressure measured in patients with brain AVM.
Results:
The theory provides a basis for understanding the occurrence of pseudotumor cerebri syndrome in patients with nonhemorrhagic brain AVMs, for the occurrence of local mass effect and brain edema bordering unruptured AVMs, and for the development of hydrocephalus in patients with unruptured AVMs. The theory also contributes to a better appreciation of the pathophysiology of dural arteriovenous fistulas, of vein of Galen aneurismal malformation, and of autoregulation-related disorders in AVM patients.
Conclusions:
The hydraulic hypothesis provides a comprehensive frame to understand brain AVM hemodynamics and its effect on the CSF dynamics.
Keywords: Brain arteriovenous malformation, cerebral autoregulation, cerebrospinal fluid pressure, dural arteriovenous fistula, intracranial hypertension, Starling resistor
INTRODUCTION
Brain arteriovenous malformations (AVMs) are physiologically active lesions due to the artery-to-vein shunt flow. Circulatory and functional disturbances may be produced in adjacent as well as in remote areas of the brain. Intracranial pressure is well-known to be increased in patients with brain AVM in connection with acute bleeding episodes and in case of associated obstructive hydrocephalus (intracranial pressure is by classic definition the cerebrospinal fluid pressure [Pcsf], and these terms are used interchangeably in this text). Otherwise intracranial pressure has commonly not been a major concern in the management of AVM patients, especially in the adult patient group. However, increased Pcsf and papilledema clinically resembling pseudotumor cerebri has been reported in patients with unruptured brain AVMs, and symptom resolution is known to occur after AVM removal.[4,8,65,66] Increased cerebral blood volume (CBV) resulting from venous hypertension, and increased cerebrospinal fluid (CSF) volume due to impaired absorption, were suggested as probable major factors in the development of intracranial hypertension in these patients, but the fact that symptoms of increased Pcsf would occur in some cases and not in others with similar angiographic findings was considered an enigma.[3] Instead, it seems reasonable to presume that these sporadic reports of intracranial hypertension do not represent exceptional cases but indicate the existence of an intrinsic disease mechanism leading to increased Pcsf in all AVM patients. The same disease mechanisms could be involved in the occurrence of mass effect and brain edema in patients with unruptured brain AVMs.
The purpose of this article is to present a theory for the pathophysiology of development of increased Pcsf in patients with brain AVMs applying a basic hydraulic hypothesis relating cerebral intravasal and CSF pressures.
THEORY OF HEMODYNAMIC CONTROL OF INTRACRANIAL PRESSURE
In the early literature,[11,15] it was recognized that most instantaneous, physiological variations of Pcsf must be a reflection of the pulsatile alterations in pressure of the intracranial blood compartment (other immediate Pcsf changes were recognized to be produced by changes in posture), but that in normal conditions there is no constant relationship of steady state Pcsf and the systemic arterial pressure. In 1973 Löfgren[35,36] proposed a basic hydraulic hypothesis to relate the cerebral blood circulation and the Pcsf [Figure 1]. The premises, deliberately oversimplified, were: (a) the Monro–Kellie theory (i.e., the cranial compartment is incompressible, the total volume of its contents – brain tissue, blood, CSF, and any expansive process when such is the case – is constant, and any increase in volume of one of the cranial constituents has to be compensated by a decrease in volume of another); (b) the electrical circuits analogy of the law of Poiseuille for flow of an ideal fluid in a cylindrical pipe (i.e., the pressure drop is the volumetric flow times the resistance to fluid movement); and (c) the outlet venous pressure is zero. Given these premises, we have:
Figure 1.
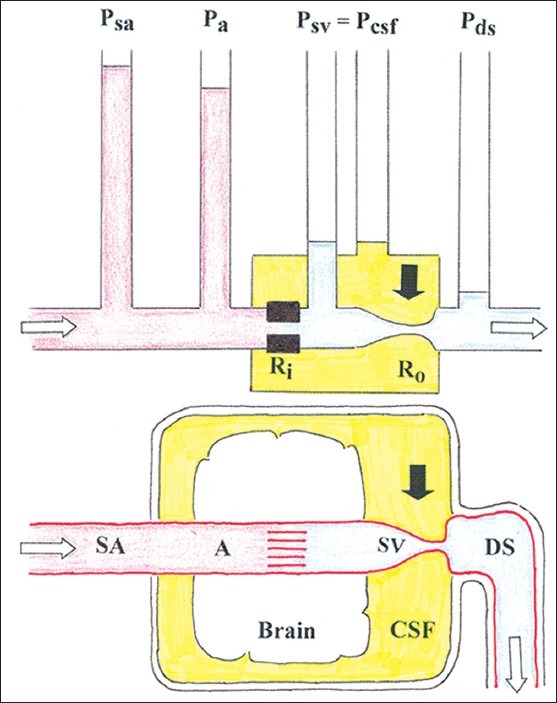
Basic hydraulic model of the relation between arterial pressure and cerebrospinal fluid pressure. A = cerebral artery; DS = dural venous sinus; CSF = cerebrospinal fluid; Pa = cortical artery blood pressure; Pcsf = cerebrospinal fluid pressure; Pds = dural sinus pressure; Psa= systemic arterial blood pressure Psv= subarachnoid venous pressure; Ra = inflow (arteriolar) resistance; Rv = outflow (venous) resistance; SA = systemic (precerebral) artery; SV = subarachnoid vein; a black arrow indicates the site of venous collapse; the white arrows indicate the direction of blood flow
Pa = F (Ra + Rv)
and
Psv = R v. F
where F is the cerebral blood flow (CBF), and the other definitions are provided in Figure 1. Ra is regulated by vasomotor tonus and, in conditions of functioning cerebral autoregulation, it adapts continuously to keep constant blood flow rate. At the venous outflow a state of zero transmural pressure in the subarachnoid veins related to the CSF space is assumed, thus
Psv = Pcsf.
Eliminating F and solving for Pcsf gives:
Pa (Ra + Rv)-1 = Pcsf. Rv-1
Pcsf = Pa. Rv (Ra + Rv)-1
And finally:
Pcsf = Pa [1 + Ra/Rv]-1
The latter equation describes a linear relationship between Pa and Pcsf with a slope varying with the ratio of the cerebrovascular resistances Ra (which is regulated by vasomotor tonus and cerebral autoregulation) and Rv (which is passive). In other words, intracranial pressure is at any moment a fraction of the arterial blood pressure quantitatively determined by the coordinated action of the inflow and outflow cerebrovascular resistances. The pressure transmission from Pa to Pcsf increases when Ra does not adapt (i.e., in case of maximum dilation or maximum constriction of the afferent arterioles), and theoretically P a = Pcsf when Ra is zero.[17,35,36,55]
The venous resistance Rv has the properties of a Starling resistor.[26] The subarachnoid veins are valveless, collapsible vessels that are distally kept open by their dural attachments as they enter the dural sinuses. The terminal vein segment just proximal to the sinus entrance has special pressure-flow properties. The pressure in the compartment bordering these veins (i.e., Pcsf) is in equilibrium with the inward-acting pressure (Psv) over a wide range of pressures. At their end segment the subarachnoid veins can be either open, or partially collapsed, or almost completely collapsed. When partially collapsed, the veins widen when more fluid is entering than is leaving, and they close when more fluid is leaving than entering. Under normal baseline conditions Pcsf is considerably higher than the outflow pressure (i.e., Pds), and F is independent of the latter. Thus, under normal conditions Psv = Pcsf > Pds, the driving force across the system (Pa – Pcsf) is less than the total pressure drop (Pa – Pds), and F is largely independent of the downstream pressure in the dural sinuses Pds, a phenomenon that has been referred to as the vascular waterfall.[14,37,50] The occurrence of a pressure drop or vascular waterfall in the intracranial venous drainage was demonstrated in animal experiments in the baboon[14] and dog.[37] Due to the vascular waterfall, increase in Pds does not influence cerebral venous outflow as long as Psv >Pds. The magnitude of the waterfall (the pressure difference Psv – Pds) probably works as a buffer in the immediate regulation of the intracranial pressure, and provides an explanation on why the brain is not drained of venous blood each time a person sits up.[37]
The concept of a hydraulic model of the cerebrovascular bed consisting of a Starling resistor coupled in series with an upstream resistance has been studied by Chopp, et al.[9] in a physical model involving fluid flow in a Starling resistor (a collapsible tube encased in a rigid fluid-filled chamber) in series with an upstream adjustable clamp (a resistor corresponding to Ra in the nomenclature of the present text), and performing volume-pressure tests by injecting fluid into the rigid chamber. The volume-pressure curves obtained in this model were in agreement with volume-pressure curves of the CSF obtained in animal experiments, including data obtained in experimental work by the same group and from the literature, supporting the hypothesis that the exponential pressure response of Pcsf to a transient increase in CSF volume observed in in vivo results from compression of the cerebral vessels, presumably the veins. Some aspects of the hydraulic hypothesis were investigated in experiments on animals: Impaired cerebral autoregulation, induced in dogs by pharmacological manipulations of Pa or by hypovolemic hypotension, was associated with a rise in Pcsf,[52] and a close dependence of Pcsf on Pa was seen to emerge after a global ischemia-induced impairment of the cerebral autoregulation in rabbits and in pigs.[17,55] In clinical practice, the hydraulic model provides a theoretical background for the increase in intracranial pressure provoked by an increase in SAP that can be observed in patients with acute, severe brain injury and sustained intracranial hypertension, and endorses the use of induced arterial hypotension and precapillary vasoconstriction to reduce the hydrostatic capillary pressure for reduction of brain edema as part of the management of this category of patients.[17]
Before discussing the relevance of the hydraulic hypothesis to AVM physiopathology, the role of a related physiological phenomenon has to be emphasized: The limits put by venous tissue distensibility on the condition of zero transmural pressure between the bridging subarachnoid veins and CSF expressed as Psv = Pcsf. A short description of the volume-pressure relationship in veins can be elucidating.[2,6,67] In an isolated vein with fluid infusion, when the intraluminal pressure rises from zero, the intravasal volume increases by changing the vessel's cross-sectional shape from a more or less collapsed flat form to an elliptic form and then to a circular shape, which corresponds to the largest volume per unit of vessel length. The venous volume-pressure curve is sigmoid in shape with three defined segments. Beginning at the semi-collapsed zero-pressure condition up to the point where the luminal volume increase brings the venous cross-section to a completely circular shape the venous volume-pressure relationship is approximately linear (i.e., the vessel wall resistance to increasing luminal pressure is zero). Further venous pressure increase will lead to vessel wall distension and further venous dilation, but now intraluminal pressure increases more than vessel volume and the volume-pressure relationship becomes curvilinear, turning away from the volume axis. In this phase, the vein becomes less distensible the more it is stretched. When the maximum venous wall distension is reached the volume-pressure curve becomes parallel to the pressure axis, and further intravenous pressure increase will no longer increase venous volume. Let us now place this isolated vein preparation in a fluid-filled, closed compartment, such as the intracranial cavity: The extravasal fluid pressure will fluctuate parallel with venous pressure as long as the venous volume-pressure relationship is linear (a situation that is analog to the condition when Psv = Pcsf), and further increase in intravasal pressure will have a decreasing effect and finally no effect on the extravasal pressure.
BRAIN ARTERIOVENOUS MALFORMATIONS
Brain AVMs are congenital lesions that morphologically consist of three components: The afferent or feeding arteries, the nidus, and the draining veins.[19,38,40,43,49,70] Compared with ordinary arteries, the feeding arteries are usually dilated, tortuous, and elongated. Instead of a capillary network connecting arterial and venous circulations, in an AVM inflowing arteries supply a nidus consisting of a coiled tangle of primitive, malformed arterioles (50-200 micra in diameter), that branch off the feeding arteries and shunt directly into malformed venous loops (0.5-2 mm in diameter), which in their turn run into the main draining vein (s). The draining vein is as a rule particularly dilated, sometimes aneurismal, and reversal of blood flow direction may occur in tributaries of the draining vein (s).[49] Microscopically, AVM vessel structure may vary from fairly well differentiated arteries and veins to unmistakably abnormal vessels that resemble neither artery nor vein, and present degenerative changes such as hyalinized walls, fibrosis, atheromatosis, and thrombosis, as well as necrotic areas after minor hemorrhages. Lack of a high-resistance arteriolar bed and presence of a low-resistance nidus generates high blood flow rate and hypotension in the feeding artery combined to intranidal and draining vein hypertension. Brain AVMs are known to increase in size on serial angiography, which has been more extensively documented in small ones.[27,61] AVM growth occurs due to enlargement of its arterial and venous components, recruitment of new arterial feeders (including dural feeders), and growth of the nidus. AVM feeder arteries are known to dilate with time. Studies on normal cerebral arteries[56] and on arteries feeding cerebral AVMs[57] seem to support the hypothesis that each segment of the cerebral arteries adapts the luminal diameter locally in response to sustained changes of blood flow so that wall shear stress (the force exerted along the endothelium by the viscous drag of flowing blood) on the long term is kept within optimum limits. Those studies[56,57] were done using mathematical calculations on measurements of cerebral arterial diameter in digital subtraction angiography, and measurements of cerebral artery blood flow velocity using transcranial Doppler sonography. Wall shear stress can be estimated from velocity measurements from phase-contrast magnetic resonance angiography, but only recently the technical limitations of this technique were overcome, making possible to test the hypothesis of wall shear stress remodelling in cerebral AVM feeders.[7] In a study of whole-brain velocity-encoded angiograms obtained with phase-contrast magnetic resonance angiography,[7] AVM patients who were asymptomatic or had mild symptoms did not present any significant difference in wall shear stress in feeding vessels compared with normal contralateral vessels, whereas patients presenting with severe symptoms (hemorrhage, severe headache, intractable seizures, or focal neurologic deficits) had significantly higher wall shear stress in AVM feeding vessels compared with contralateral vessels. After removal of the AVM the feeder artery walls are known to remodel again, this time slowly reducing their caliber.[45,70] Blood flow is laminar in the feeding artery[57] but has been suggested to go over to turbulence inside the nidus and in the proximal draining vein.[43] Some conversion of the kinetic energy of high flow velocities and laminar flow in the feeder arteries into potential energy with further increase in lateral pressure probably occurs within and just beyond the nidus.[43] Intraoperative Doppler measurements showed that laminar blood flow in the draining vein is disturbed just distal to the nidus, becoming more distally pulsatile with velocities and waveforms as in arteries.[19,46] Arterialized Doppler waveforms were recorded also in the dural sinuses in patients with brain AVM.[44] The draining vein is subjected to intensive pressure- and flow-related strains that may lead to structural fatigue, focal dilation, and varix formation.
THE HYDRAULIC HYPOTHESIS AND BRAIN ARTERIOVENOUS MALFORMATIONS
The following is a theory for the pathophysiology of occurrence of increased Pcsf in patients with unruptured brain AVMs applying the basic hydraulic hypothesis [Figure 2]. In a brain AVM the vascular resistance in the arterial feeders (inflow resistance, or Ra) is very low, and the increased blood pressure in the high-flow draining vein is transmitted to the subarachnoid veins and to the CSF, thus increasing Psv and Pcsf. Given the principle of communicating vessels, the locally increased pressure transmission Psv to Pcsf at the draining vein is transmitted to the whole CSF space, which can induce general subarachnoid vein compression (and thus generally increased Rv) far away from the vein draining the arteriovenous shunt. For each patient with AVM the magnitude of pressure transmission Psv to Pcsf and global increase in Rv will depend on the pattern of the AVM venous drainage (number of veins and presence of anastomoses with veins draining normal brain) and whether maximum draining vein distensibility is reached of not. Globally increased Pcsf decreases cerebral perfusion pressure, and whether this leads to decreased CBF will depend by and large on the functional status of cerebral autoregulation. At the point when maximum AVM draining vein distension is achieved the pressure transmission Psv to Pcsf is limited, but upstream venous pressure increase (to the AVM nidus and connected venous territories) is not limited, and brain edema can evolve. Shunt flow to the dural sinus increases Pds. When Pds is increased the pressure gradient across the arachnoid villi decreases, leading to a relative hinder of CSF absorption that can result in increase in CSF volume and further increase in Pcsf. On the long run, any effect of Pds on Pcsf probably depends on the magnitude of the shunt flow and pressure transmission, and on whether the vascular waterfall at the entry of the subarachnoid veins into the dural sinus is present or is abolished, either intermittently or permanently. In the extreme situation when Pds equals Pcsf and a steady state is achieved, Pds and Pcsf will oscillate together over time, and general cerebral venous engorgement and CSF outflow obstruction will develop that may result in hydrocephalus development when other CFS outflow mechanisms (e.g., CSF bulk flow to the spinal canal) become exhausted.
Figure 2.
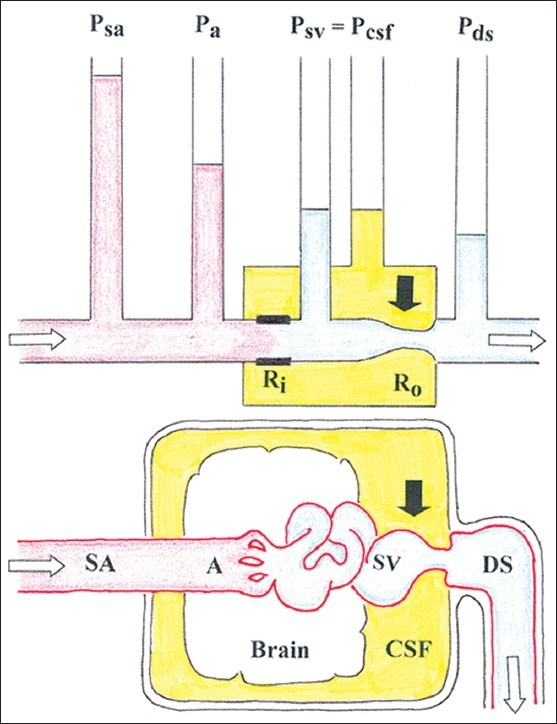
Basic hydraulic model of the relation between arterial pressure and cerebrospinal fluid pressure in the presence of a brain AVM. The system shown in this figure is to be coupled in parallel to a number of systems as in Figure 1. Pa represents the pressure in the terminal AVM feeder artery; empirically we know that Pa < Psa in most AVM cases. See the other abbreviations in Figure Figure 1
In the following sections AVM hemodynamics is reviewed and the physiological and clinical relevance of the basic hydraulic model are discussed.
CEREBRAL ARTERIAL AND VENOUS PRESSURES IN BRAIN AVMS
There are several studies reporting pressure measurements in brain AVM vessels during open surgery for AVM excision. The measurements were done by direct puncture of the feeding arteries,[3,8,20,33,41,46] and of the draining veins.[3,8,33,42,46] While in normal conditions cortical artery pressure was found to be in average 92% of the mean SAP,[34] these studies showed that in most cases the pressure in the AVM-feeding arteries measured just adjacent to the AVM nidus is substantially lower than SAP, and that AVM-draining vein pressure, measured just distal to the AVM nidus, is higher than normal cortical vein pressure. After removal of the AVM or clamping of the feeding artery, the pressure in the proximal stump of the feeding artery increased to approximately the level of the SAP, and the pressure in the draining vein decreased drastically. Pooling detailed data charts published in some of these studies[3,8,20,33,41,42,46,59] and not considering methodological discrepancies (the zero pressure level may have been calibrated using somewhat different reference sites), gives the following results: Before AVM excision the average feeding artery pressure was 49.3 mmHg (SD = 16.9; N = 75), corresponding to 64.1% (SD = 22.8; N = 75) of SAP, and the average draining vein pressure was 17.1 mmHg (SD = 6.1; N = 54). Draining vein pressure was in average 12.5 mmHg (SD = 6.6; N = 46) higher that the central venous pressure. After AVM excision the pressure in the artery stump was in average 72.2 mmHg (SD = 11; N = 19), or 84% (SD = 14.2; N = 18) of the SAP. Measurements of intravascular pressure done by microcatheterization of the feeders in transarterial embolization showed similar findings concerning feeder artery pressures, which in three representative studies[13,21,58] was in average 50%, 55%, and 68%, respectively, of the mean SAP before AVM embolization. A tendency for lower terminal feeder artery pressures to occur in longer feeding arteries was observed in one study,[46] but it was not confirmed in another study.[20] Feeding artery pressures were found to be significantly higher in AVMs with previous hemorrhage than in clinically unruptured AVMs.[24,41,59] A tendency to higher feeder artery pressures occurred in AVM feeders with brain-nourishing branches distal to the nidus[58] as well as in small AVMs (with nidus up to 3 cm in diameter).[59] The draining vein pressure was significantly higher in AVMs with previous hemorrhage than in unruptured AVMs in one study,[41] but this finding was not corroborated in a subsequent study.[24] A positive linear correlation between feeder artery pressure and draining vein pressure, without any apparent influence of venous stenosis on angiography, was reported;[71] this finding cannot be corroborated in the pool of data described earlier. In patients with unruptured AVMs presenting with mass effect, draining vein pressure before surgical removal of the AVM in 10 patients (22 mmHg in average) was significantly higher than in six patients with unruptured AVMs without mass effect (12 mmHg).[42] Summarizing, in terms of the hydraulic hypothesis these studies described an intravascular pressure drop from the circle of Willis to the terminal AVM feeding artery that is consistent with low Ra, and an increase in AVM draining vein pressure that according to elementary flow dynamics is in equilibrium with Pcsf.
CSF AND SUPERIOR SAGITTAL SINUS PRESSURES IN BRAIN AVMS
Löfgren's group started a study on Pcsf in patients with brain AVM two decades ago. A preliminary communication was published in abstract form,[68] but the study was never completed. The following is a summary of the available data of this unique study together with some complementary information (J Löfgren, personal communication, 2011). The patient population was a series of 50 patients with supratentorial cerebral AVMs undergoing endovascular treatment. One of the patients had papilledema. Pcsf measured by means of lumbar puncture in all patients before treatment was in average 15.1 mmHg (SD = 4.4; range 8-30 mmHg), in one-third of the cases Pcsf was higher than 15 mmHg, and after complete or partial AVM embolization Pcsf had decreased to 9.3 mmHg (SD = 1.5). Pcsf had a positive linear correlation (R = 0.64; P = 0.0001) with the volume of the AVM nidus estimated from angiographic images. A definitively raised Pcsf was the case in all patients with an AVM nidus measuring more than 4 cm in maximum diameter. Pcsf values were related to the various size groups used in the staging system of Spetzler and Martin,[60] that is, AVMs with angiographic diameters 0-3, 3-6, and larger than 6 cm. Normal or nearly normal Pcsf was observed in patients with AVMs less than 3 cm, and at a size larger than 6 cm Pcsf was higher than 15 mmHg. The interpretation is possible that even an AVM of smallest size has a definite effect on the fluid pressure and that this effect increases linearly with the size, if initially almost imperceptibly, with about 1 mmHg/ml AVM volume. In a subset of 14 patients the CSF dynamics was evaluated with the constant rate infusion method; the CSF outflow resistance was in average 8.5 mmHg/min/ml (SD = 3.9), that was mostly within the normal range, and did not correlate thoroughly to Pcsf. In another subset of 20 patients the pressure in the superior sagittal sinus (Psss), with zero at the level of the jugular vein, was measured by means of transfemoral venous catheterization both before and after complete or partial AVM embolization. In these 20 patients Psss was in average 9 mmHg (SD = 3.24; range 4-15 mmHg) before embolization, and 4.8 mmHg (SD = 1.88; range 2-9 mmHg) after embolization; the corresponding Pcsf measured via lumbar puncture in this patient subset was 14.8 mmHg (SD = 4.5; range 8-24 mmHg) before embolization, and 11.05 mmHg (SD = 3.86; range 5-19 mmHg) after embolization; Pcsf related linearly with Psss before embolization (R = 0.79; P = 0.0001). Summarizing, the main observations were that brain AVMs, even in the absence of hemorrhage or obstructive hydrocephalus, cause a disturbance of the CSF circulation in the direction of increased Pcsf. In this study, the increased level of lumbar Pcsf was in direct proportion to the volume of the AVM, and there was a proportional elevation of the dural sinus pressure that can be secondary to increased blood flow. The increase of Psss probably resulted of the increased shunt flow, and the venous waterfall was diminished but not erased, as shown by a positive gradient Pcsf – Psss and average CSF outflow resistance within normal values in the patients where the appropriate measurements were done. The increase of Pcsf can be explained by the basic hydraulic hypothesis, and the overall results are not in discordance with this hypothesis.
MASS EFFECT AND BRAIN EDEMA BORDERING UNRUPTURED BRAIN AVMS
Imaging studies of clinically unruptured brain AVMs show evidence of mass effect (i.e., compression, distortion, and displacement of normal anatomic structures by the AVM nidus, its arterial feeders and efferent veins) in 44-55% of patients[28,42] and brain edema in 3.3-3.9% of patients.[25,29,42] This is an intriguing observation, since in craniotomy for nonhemorrhagic AVMs the dura mater is usually loose, and the brain pulsates normally. It is likely that in this situation brain swelling is suppressed by properly controlled ventilation and some degree of hypotension during general anesthesia (see below) that decrease the shunt-flow through the AVM, which is blood pressure-dependent, in this way also decreasing the pressure transmission from Pa to Pcsf (according to the hydraulic hypothesis). Abnormal venous outflow resulting in venous hypertension has been suggested to be the cause of brain edema.[25] In the context of a pressure transmission mechanism as described in the hydraulic hypothesis the development of parenchymal brain edema is dependent on the magnitude of venous hypertension resulting from the arteriovenous shunt, and on the presence of venous outflow obstruction. Since the terminal venous orifices at the entrance in the dural sinuses are not known to dilate in the presence of AVM, venous outflow obstruction can be in part determined by normal venous anatomy in the presence of venous hypertension. In an arteriovenous shunt the draining vein is the driving source of transmural pressure input to the CSF space, and the association of cerebral venous hypertension with venous outflow obstruction can both increase the pressure transmission to CSF and, by further increasing the tension on the venous wall, accelerate the development of venous pathology (degenerative mural changes, varicosities and dilated venous sacs, and thrombosis). In the presence of venous outflow obstruction the pressure transmission from Psv to Pcsf is limited to the point when maximum vein dilation is achieved, but pressure redistribution upstream to the AVM nidus and to adjacent venous territories through anastomoses is not limited, leading to the development of perinidal and perivenous vasogenic brain edema.
DURAL ARTERIOVENOUS FISTULAS
Brain edema, symptomatic increased intracranial pressure, and papilledema are common presentations in patients with unruptured dural arteriovenous fistulas (DAVF), both in those DAVFs with direct cortical vein drainage and in those draining into a venous sinus with retrograde drainage into subarachnoid veins.[5,10,54] Application of the basic hydraulic model seems to be straightforward in DAVFs with direct cortical venous drainage with reference to the pressure transmission at the draining vein, but the situation is more dramatic since the draining vein of a DAVF often exhibits more profound pathological changes than the draining vein of a brain AVM. The reason is that the etiology of DAVF is usually thrombosis of a dural sinus or a cerebral vein, and the aggressive development of brain edema in DAVF compared with brain AVM is probably intensified by local postthrombotic venous stenosis and congestion. DAVFs did not arouse the same interest as brain AVMs for studies of intravasal pressure. Meticulous measurements of intraventricular Pcsf measured via an external ventriculostomy, and Pds measured via a catheter placed into the surgically exposed superior sagittal sinus, were reported in an adult patient with severe intracranial hypertension due to a DAVF.[30] In that case, the DAVF had feeders from the right external carotid artery drained to the ipsilateral sinus transversus (that presented with irregular lumen and stenosis in the images provided in the case report) with insufficient antegrade venous drainage and reflux to the contralateral sinus transversus and to cortical veins, that is, it was a DAVF that could be classified[10] as type IIa + b. The mean Pcsf was 30 mmHg, and Pds was clearly above normal values but constantly 2-3 mmHg lower than Pcsf. Both Pcsf and Pds normalized after embolization through, and ligature of the external carotid artery feeding the DAVF. The authors concluded that increased Pds was the cause of intracranial hypertension,[30] but it is more probable that in this case the cause of increased Pcsf was blood pressure transmission as proposed by the hydraulic model aggravated by blood flow stagnation in cortical veins, since the vascular waterfall was not erased even if lowered, and the patient did not present with hydrocephalus.
A single-case report illustrated particularly well the role of venous hypertension in arteriovenous shunt-related brain edema:[23] A patient presenting with headache and progressive lower extremity weakness was found to present edema and contrast-agent enhancement of the medulla on magnetic resonance imaging (MRI), and a small, nonruptured cerebellar AVM with drainage to the anterior spinal vein on angiography. This case's clinical presentation and initial imaging findings (MRI) are typically associated to spinal DAVFs or to intracranial DAVFs with spinal perimedullary drainage (classified as type V[10] ), but the pathophysiological mechanism of brain edema (localized venous hypertension) can be the same in DAVF and in AVM.
HYDROCEPHALUS IN PATIENTS WITH NONRUPTURED AVMS
The occurrence of hydrocephalus in patients with unruptured brain AVMs is uncommon. When present in these patients, hydrocephalus usually results of obstruction of the CSF space, typically obstruction of the interventricular foramen or of the Sylvian aqueduct, either by an enlarged draining vein or by the AVM nidus itself.[16] Appreciable pressure increase in the dural sinus is expected for sure in patients with brain AVM because of direct shunting of arterial blood into the sinus lumen. That was the case in patients reported with symptomatic intracranial hypertension associated with high-flow, unruptured brain AVMs,[3,8,30,65,66] but these patients did not develop hydrocephalus. In contrast, noncommunicating hydrocephalus is common in the pediatric AVM population with huge arteriovenous shunts. Hydrocephalus occurs in 38% of infants (aged from 1 month to 2 years) with high-flow brain AVMs.[53] Rare cases of hydrocephalus associated with nonruptured AVMs in adult patients with no evidence of obstruction of the CSF space were recently reported: One patient with AVM in the cerebellum presenting with large varicosities in the draining veins,[16] and one patient with a choroidal-type AVM.[12] In these cases development of hydrocephalus was attributed to pressure increase in the deep cerebral venous system.[12,16] Unfortunately neither Pds nor other relevant pressure measurements were done in these patients, but those reports provide imaging evidence of engorged periventricular and transmedullary veins.[12,16]
Considering the basic hydraulic hypothesis, it is appealing to hypothesize that the development of nonobstructive hydrocephalus in patients with unruptured brain AVM can be related to the absence of a vascular waterfall (i.e., a positive pressure gradient Pcsf–Pds) at the venous entry into the dural sinuses. It is likely that CSF resorption will not be appreciably disturbed and communicating hydrocephalus will not develop in AVM patients as long as the vascular waterfall is preserved. Since locally increasing Pds to the Pcsf level will result in increased CSF absorption elsewhere, it is reasonable to suppose that the vascular waterfall has to be eliminated globally for hydrocephalus development. Attempts to produce experimental hydrocephalus by occluding large venous conduits have usually failed because of the development of venous collaterals and enlargement of alternative routes of CSF drainage.[48] This concept seems to apply consistently to well-established concepts of CSF secretion, bulk flow and absorption (hydrocephalus would in this case develop due to increased resistance to CSF absorption in the arachnoid villi, which is pressure-dependent) as well as to recent hypotheses of CSF formation and absorption at the cerebral capillaries (hydrocephalus development due to CSF absorption disturbance because of increased hydrostatic pressure in the cerebral venules and capillaries). In most adult patients with nonruptured AVM, the pressure input into the dural sinus is presumably not large enough to abolish the vascular waterfall enduringly, and these patients usually do not develop communicating hydrocephalus. The situation is different in the pediatric AVM population in which Pds can be high enough, and steadily so, to erase the vascular waterfall and to stop CSF absorption in the arachnoid villi; in addition, increased venous pressure per se would contribute to development of macrocrania in neonates and infants, when the skull sutures are still not closed. A similar situation occurs in infants with obstruction of the superior vena cava in infants that develop nonobstructive hydrocephalus that is attributed to global increase in the cranial venous sinus pressure.[18,22] This is probably also the case in the few documented cases of communicating hydrocephalus in adults with nonruptured AVM, as indicated by the presence of engorged deep cerebral veins in these patients.
VEIN OF GALEN ANEURISMAL MALFORMATION
Hydrocephalus occurs in 46.8% of patients with vein of Galen aneurismal malformation (VGAM), and increased venous sinus pressure has been considered of primary importance in the development of hydrocephalus in these patients.[73] Huge dural sinus pressures, well over Pcsf, were measured in two patients with VGAM and hydrocephalus.[39] In this study, intraventricular pressure (Pcsf) and superior sagittal sinus pressure (Pds) were measured before and after transvenous embolization of the VGAMs. Preembolization Pds was very high in both patients (450 mmH2O and 500 mmH2O, respectively) and about five times higher than Pcsf (which was 90 mmH2 O and 110 mmH2O). Embolization led to a remarkable fall of Pds in both patients (down to 20 mmH2O and zero, respectively), and to slightly decreased Pcsf (80 mmH2O and 100 mmH2O). From the point of view of the basic hydraulic hypothesis a VGAM represent a focus of extremely elevated blood flow and venous pressure that faraway exceeds the limit of pressure transmission from Psv to Pcsf due to maximum vein distension. A factor that could limit the pressure transmission in VGAMs is the atypical character of its venous drainage. A VGAMs draining vein is not an otherwise normal brain vein that also drains an arteriovenous shunt, but it is more accurately an anomalous vein (the median prosencephalic vein, that is the embryological forerunner of the vein of Galen) that does not collect normal brain veins, and that often drains into a persistent falcine sinus.[73] Other venous anomalies that can influence the distribution of the shunt-related venous hypertension are absence of the straight sinus in most cases of VGAM, persistence of other embryonic sinuses such as the occipital and marginal sinuses, and an ill-defined anatomical configuration of the remainder of the venous system.[31,73] Moreover, in young patients with distensible skull high draining venous pressure alone is probably a major determinant of the development of macrocephaly.
AUTOREGULATION-RELATED PHENOMENA
Cerebral autoregulation is the intrinsic property of the brain to maintain constant blood flow despite changes in arterial perfusion pressure by modulation of brain artery vascular tone, especially in small arterioles. In healthy individuals, cerebral autoregulation mechanisms are effective when the SAP is kept within approximate lower and upper limits of 60 and 150 mmHg, respectively, but these limits are not at all inflexible; they vary with the level of arterial Pco2, they are higher in patients with chronic hypertension, and autoregulation is abolished in acute conditions with associated brain tissue lactacidosis.[2,32,63] The upper limit of auroregulation is particularly variable, since SAP levels well above this theoretical threshold occur in daily life without generating hypertensive encephalopathy.[2] There is evidence suggesting that autoregulation in the AVM feeding artery territories undergoes adaptive changes due to chronic hypoperfusion, with the lower limit shifted to a lower SAP level in patients with brain AVM studied under neuroleptic counscious sedation.[72] In humans, under neuroleptanalgesia cerebral autoregulation persists to the mean SAP level of 40 mmHg, below which it disappears.[51] The existence of a shift of the autoregulation with tolerance of lower arterial pressure levels under general anesthesia or in chronic cerebral hypoperfusion makes sense, since the average AVM feeding artery pressure was 49.3 mmHg (SD = 16.9) in measurements pooled from the literature (presented earlier), and thus under conditions of ordinary autoregulation limits many of those patients would present with overt vasomotor paralysis in the AVM-related vascular territories.
The concept cerebrovascular steal refers to the regional hypoperfusion of the brain parenchyma around an AVM. The clinical correlate of steal is the finding of patients with unruptured AVM presenting with progressive or variable neurological deficits. The steal results from two factors, namely the pressure difference between arteries feeding the AVM and arteries feeding the brain that brings on redistribution of blood flow from the brain parenchyma sharing the same feeding arteries as the AVM to the arteriovenous shunt, and the local reduction of the cerebral perfusion pressure (i.e., inflow pressure minus outflow pressure) because of the increase in the venous pressure combined to decrease of the arterial pressure.[43,70] Considering basic principles of hemodynamics there should be no question that the vascular steal occurs,[70] and actually it can be visualized on angiography of high-flow AVMs, when a rapid filling of the feeding arteries (often also of collateral arteries communicating with the feeders) and early filling of the draining veins is associated to poor filling of the normal cerebral arteries bordering the AVM in the midarterial phase. Within its intrinsic limits, cerebral autoregulation mechanisms make possible to compensate for the cerebrovascular steal by promoting dilation of the arteriolar resistance vessels in response to the regional decrease in perfusion pressure close to the AVM. Regional CBF can be quantified with good spatial resolution around brain AVMs in studies using either the 133Xe inhalation method or the stable xenon computed tomography (Xe-CT). In a pioneer study using both these methods,[47] the local CBF in 15 patients with supratentorial AVMs (nine were large AVMs, with nidus measuring 6-14 cm) was compared with measurements in nine normal volunteers, and the following results were found: Compared with the normal group, the local CBF values for gray matter and for white matter in AVM patients were both significantly reduced in the brain parenchyma adjacent to the AVM, and in the contralateral brain hemisphere local CBF was significantly reduced in the gray matter but not in the white matter; the hemispheres containing an AVM presented with greater reduction in gray matter local CBF than the contralateral hemisphere; in five patients the measurements were repeated after successful AVM excision, and all local CBF reductions in gray and white matter improved to normal values.
A remarkable blood pressure-flow relation to a hypotensive challenge in AVM patients was observed in a study using the 133Xe method by Taneda, et al.[64] In this study, regional CBF (gray matter flow only) was measured in eight patients with AVMs at least 4 cm in size, first at normotension and later during pharmacologically induced decrease in systemic arterial blood pressure. In six patients the hemispheric CBF paradoxically increased on both brain hemispheres in response to hypotension (the CBF increase being larger in the AVM hemisphere than in the opposite hemisphere), in one patient with a hemorrhagic lesion the hemispheric CBF increased in the AVM hemisphere and decreased on the opposite side, and in one patient (also with a hemorrhagic lesion) the hemispheric CBF decreased on both sides but the reduction was milder on the AVM side; moreover, an increase of more than 50% of CBF in response to hypotension was recorded in at least a single detector in all cases (even in the case with average CBF reduction). The CBF measurements were repeated in five patients after excision of the AVMs, and then no increase was observed in response to hypotension. In explaining this phenomenon Taneda, et al.[64] emphasized the role of the venous side of the shunt system: Since the arterial pressure in an AVM is directly mediated to the venous side, decreasing the arterial pressure decreases the venous back-pressure and improves CBF even in the brain hemisphere contralateral to the AVM. This paradoxical blood pressure-flow relation can be simply understood in the context of the hydraulic hypothesis: Reducing the venous back-pressure at the AVM draining vein also reduces Pcsf globally, and thus the cerebral perfusion pressure in the underperfused brain parenchyma can increase despite a decrease of SAP.
These considerations on cerebral autoregulation and paradoxical blood pressure-flow responses in patients with brain AVMs can be further scrutinized considering the use of profound systemic arterial hypotension during surgery for AVM excision. Pertuiset et al.[51] reported on their experience and on the theoretical and experimental background of setting the level of mean SAP between 40 and 50 mmHg in AVM surgery. In a group of patients with large, superficial AVMs regional CBV was monitored using an isotope technique during blood pressure manipulations. It was shown that when SAP decreases to <50 mmHg the CBV decreases in the AVM and increases in the normal brain surrounding the AVM as a result of normal autoregulation, and when the mean SAP rises again to baseline levels CBV in the AVM becomes quite high, and decreases significantly everywhere else. The brain surrounding an AVM had a lesser CBV response compared to the response in a group of patients with saccular aneurysms.
The term normal perfusion pressure breakthrough (NPPB) refers to the onset of acute or sub-acute brain swelling, either with or without hemorrhage, following resection or embolization of large, high-flow brain AVMs despite complete obliteration of the AVM nidus.[62,69] The pathophysiology of NPPB is supposed to be the restoration of normal perfusion pressure in the arteries feeding the brain tissue surrounding an AVM after its obliteration. In the presence of the AVM that parenchyma is chronically hypoperfused and its arteries’ autoregulatory capacity is impaired after being chronically operating at its lower limit (i.e., maximally dilated), which renders the parenchyma vulnerable to the restoration of normal perfusion pressure when failure of the inflow arteries to constrict leads to foci of disruption in the capillary bed, resulting in brain edema, hemorrhage, or both.[62] In theory, this concept is in full agreement with the hydraulic hypothesis, the segmental lack of vessel tonus (low, nonreactive Ra) explaining the development of hyperemia and brain edema. However, NPPB occurs rarely in practice and should not be used to explain away faulty surgical technique.[69] Pressure autoregulation and CO2 reactivity in cerebral arteries remote from the AVM is intact in most patients after AVM resection.[19,71] Probably most hyperemic complications of AVM therapy can be explained by obstruction of the venous system of the brain adjacent to the AVM, either resulting from the therapeutic procedure or from postoperative thrombosis.[1] The occurrence of NPPB in a patient with unruptured brain AVM and intracranial hypertension was well-documented in at least one publication. Barrow[4] reported a patient with subjective bruit, visual loss and papilledema, and a high-flow AVM draining into the superior sagittal sinus, and an opening Pcsf of 500 mmH2O (i.e., 35 mmHg) on lumbar puncture that seemingly developed white matter edema and temporary neurological deficits about 36 hours after removal of the AVM. Briefly, NPPB is a rare event that has a sound theoretical basis that is in agreement with the hydraulic hypothesis. It is appealing to speculate whether the pretreatment level of Pcsf could be a predictive factor for posttreatment NPPB.
CONCLUDING REMARKS
The indication for treatment in patients with brain AVMs has been focused mainly on the mortality and permanent neurological morbidity associated to AVM bleeding and rebleeding, and to a minor extent to the occurrence of epilepsy, headache, and progressive neurological deficits due to perfusion steal phenomenon. The basic hydraulic hypothesis points to another underlying effect of brain AVM on CSF dynamics whose significance for symptom development, nonhemorrhagic and hemorrhagic as well, has not been examined properly. A chronic increase in Pcsf, especially in the cases where pressure level approaches the breakpoint of the CSF pressure-volume curve (when the system passes from a high-compliant to a low-compliant state) can have an aggravating effect on several well-known pathophysiological mechanisms related to brain AVM. A most obvious implication is that the possibility of a habitually increased Pcsf must be considered in measurements and monitoring of the intracranial pressure in AVM patients, when indicated, for proper interpretation of the pressure values. The baseline Pcsf level can influence the severity of spontaneous brain AVM bleeding. The intracranial pressure rise in such events may start at an abnormally high level and with a compromised volume reserve that would make the pressure effect greater than otherwise would have been the case. The basic hydraulic hypothesis provides a comprehensive frame to understand the knowledge accumulated by now on Pcsf, pressure measurements in AVM vessels, and hydrocephalus development (or not) in patients with brain AVM. There is no reason not to retain it as a tentative hypothesis for future mathematical models of brain AVM dynamics as well as for prospective data collection and investigation of its relevance for the natural history and management strategies in patients with AVM or other arteriovenous shunts in the brain.
ACKNOWLEDGMENT
The author would like to express his sincere gratitude to Professor emeritus Jan Löfgren for stimulating criticism, encouragement, and support.
Footnotes
Available FREE in open access from: http://www.surgicalneurologyint.com/text.asp?2013/4/1/42/109657
REFERENCES
- 1.Al-Rodhan NR, Sundt TM, Jr, Piepgras DG, Nichols DA, Rüfenacht D, Stevens LN. Occlusive hyperemia: A theory for the hemodynamic complications following resection of intracerebral arteriovenous malformations. J Neurosurg. 1993;78:167–75. doi: 10.3171/jns.1993.78.2.0167. [DOI] [PubMed] [Google Scholar]
- 2.Auer LM. The pathogenesis of hypertensive encephalopathy: Experimental data and their clinical relevance with special reference to neurosurgical patients. Acta Neurochir (Wien) 1978;(Suppl 27):1–111. [PubMed] [Google Scholar]
- 3.Barnett GH, Little JR, Ebrahim ZY, Jones SC, Friel HT. Cerebral circulation during arteriovenous malformation operation. Neurosurgery. 1987;20:836–42. doi: 10.1227/00006123-198706000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Barrow DL. Unruptured cerebral arteriovenous malformations presenting with intracranial hypertension. Neurosurgery. 1988;23:484–90. doi: 10.1227/00006123-198810000-00014. [DOI] [PubMed] [Google Scholar]
- 5.Borden JA, Wu JK, Shucart WA. A proposed classification for spinal and cranial dural arteriovenous fistulous malformations and implications for treatment. J Neurosurg. 1995;82:166–79. doi: 10.3171/jns.1995.82.2.0166. [DOI] [PubMed] [Google Scholar]
- 6.Burton AC. Relation of structure to function of the tissues of the wall of blood vessels. Physiol Rev. 1954;34:619–42. doi: 10.1152/physrev.1954.34.4.619. [DOI] [PubMed] [Google Scholar]
- 7.Chang W, Loecher MW, Wu Y, Niemann DB, Ciske B, Aagaard-Kienitz B, et al. Hemodynamic changes in patients with arteriovenous malformations assessed using high-resolution 3D radial phase-contrast magnetic resonance angiography. AJNR Am J Neuroradiol. 2012;33:1565–72. doi: 10.3174/ajnr.A3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chimowitz MI, Little JR, Awad I, Sila CA, Kosmorsky G, Furlan AJ. Intracranial hypertension associated with unruptured cerebral arteriovenous malformations. Ann Neurol. 1990;27:474–9. doi: 10.1002/ana.410270504. [DOI] [PubMed] [Google Scholar]
- 9.Chopp M, Portnoy HD, Branch C. Hydraulic model of the cerebrovascular bed: An aid to understanding the volume-pressure curve test. Neurosurgery. 1983;13:5–11. doi: 10.1227/00006123-198307000-00002. [DOI] [PubMed] [Google Scholar]
- 10.Cognard C, Gobin YP, Pierot L, Bailly AL, Houdart E, Casasco A, et al. Cerebral dural arteriovenous fistulas: Clinical and angiographic correlation with a revised classification of venous drainage. Radiology. 1995;194:671–80. doi: 10.1148/radiology.194.3.7862961. [DOI] [PubMed] [Google Scholar]
- 11.Davson H. Physiology of the Cerebrospinal Fluid. London: Churchill; 1967. [Google Scholar]
- 12.Ebinu JO, Matouk CC, Wallace MC, Terbrugge KG, Krings T. Hydrocephalus secondary to hydrodynamic disequilibrium in an adult patient with a choroidal-type arteriovenous malformation. Interv Neuroradiol. 2011;17:212–6. doi: 10.1177/159101991101700212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fogarty-Mack P, Pile-Spellman J, Hacein-Bey L, Osipov A, DeMeritt J, Jackson EC, et al. The effect of arteriovenous malformations on the distribution of intracerebral arterial pressures. AJNR Am J Neuroradiol. 1996;17:1443–9. [PMC free article] [PubMed] [Google Scholar]
- 14.Fry DL, Thomas LJ, Greenfield JC. Flow in collapsible tubes. In: Patel DJ, Vaishnav RN, editors. Basic Hemodynamics and its Role in Disease Processes. Baltimore: University Park Press; 1980. pp. 407–24. [Google Scholar]
- 15.Gardner WJ. Cerebrospinal Fluid: Dynamics. In: Glasser O, editor. Medical Physics. Vol. 1. Chicago: The Year Book Publishers; 1944. pp. 148–52. [Google Scholar]
- 16.Geibprasert S, Pereira V, Krings T, Jiarajongmun P, Lasjaunias P. Hydrocephalus in unruptured brain arteriovenous malformations: Pathomechanical considerations, therapeutic implications, and clinical course. J Neurosurg. 2009;110:500–7. doi: 10.3171/2008.7.JNS0815. [DOI] [PubMed] [Google Scholar]
- 17.Gustafsson O, Rossitti S. Intracranial pressure is a fraction of arterial blood pressure. Eur J Neurol. 1995;2:31–7. doi: 10.1111/j.1468-1331.1995.tb00090.x. [DOI] [PubMed] [Google Scholar]
- 18.Haar FL, Miller CA. Hydrocephalus resulting from superior vena cava thrombosis in an infant. J Neurosurg. 1975;42:597–601. doi: 10.3171/jns.1975.42.5.0597. [DOI] [PubMed] [Google Scholar]
- 19.Hassler W. Hemodynamic aspects of cerebral angiomas. Acta Neurochir Suppl (Wien) 1986;37:1–136. [PubMed] [Google Scholar]
- 20.Hassler W, Steinmetz H. Cerebral hemodynamics in angioma patients: An intraoperative study. J Neurosurg. 1987;67:822–31. doi: 10.3171/jns.1987.67.6.0822. [DOI] [PubMed] [Google Scholar]
- 21.Henkes H, Gottwald TF, Brew S, Kaemmerer F, Miloslavski E, Kuehne D. Pressure measurements in arterial feeders of brain arteriovenous malformations before and after endovascular embolization. Neuroradiology. 2004;46:673–7. doi: 10.1007/s00234-004-1229-8. [DOI] [PubMed] [Google Scholar]
- 22.Hooper R. Hydrocephalus and obstruction of the superior vena cava in infancy. Clinical study of the relationship between cerebrospinal fluid pressure and venous pressure. Pediatrics. 1961;28:792–9. [PubMed] [Google Scholar]
- 23.Jayaraman MV, McTaggart RA, Sachs GM, Doberstein CE. Cerebellar pial arteriovenous malformations presenting with medullary venous hypertension: Imaging and endovascular treatment. J Neurointervent Surg. 2012;2:38–40. doi: 10.1136/jnis.2009.000471. [DOI] [PubMed] [Google Scholar]
- 24.Kader A, Young WL, Pile-Spellman J, Mast H, Sciacca RR, Mohr JP, et al. The Columbia University AVM Study Project. The influence of hemodynamic and anatomic factors on hemorrhage from cerebral arteriovenous malformations. Neurosurgery. 1994;34:801–8. doi: 10.1227/00006123-199405000-00003. [DOI] [PubMed] [Google Scholar]
- 25.Kim BS, Sarma D, Lee SK, TerBrugge KG. Brain edema associated with unruptured brain arteriovenous malformations. Neuroradiology. 2009;51:327–35. doi: 10.1007/s00234-009-0500-4. [DOI] [PubMed] [Google Scholar]
- 26.Knowlton FP, Starling EH. The influence of variations in temperature and blood-pressure on the performance of the isolated mammalian heart. J Physiol. 1912;44:206–10. doi: 10.1113/jphysiol.1912.sp001511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krayenbühl HA. Angiographic contribution to the problem of enlargement of cerebral arteriovenous malformations. Acta Neurochir (Wien) 1977;36:215–42. doi: 10.1007/BF01405393. [DOI] [PubMed] [Google Scholar]
- 28.Kumar AJ, Viñuella F, Fox AJ, Rosenbaum AE. Unruptured intracranial arteriovenous malformations do cause mass effect. AJNR Am J Neuroradiol. 1985;6:29–32. [PMC free article] [PubMed] [Google Scholar]
- 29.Kurita H, Shin M, Ueki K, Kawamoto S, Kirino T. Congestive brain oedema associated with a pial arteriovenous malformation with impaired venous drainage. Acta Neurochir (Wien) 2001;143:339–42. doi: 10.1007/s007010170087. [DOI] [PubMed] [Google Scholar]
- 30.Lamas E, Lobata RD, Esparza J, Escudero L. Dural posterior fossa AVM producing raised sagittal sinus pressure. J Neurosurg. 1977;46:804–10. doi: 10.3171/jns.1977.46.6.0804. [DOI] [PubMed] [Google Scholar]
- 31.Lasjaunias PL, Chng SM, Sachet M, Alvarez H, Rodesch G, Garcia-Monaco R. The management of vein of Galen aneurismal malformations. Neurosurgery. 2006;59:S184–94. doi: 10.1227/01.NEU.0000237445.39514.16. [DOI] [PubMed] [Google Scholar]
- 32.Lassen NA. Control of cerebral circulation in health and disease. Circ Res. 1974;34:749–60. doi: 10.1161/01.res.34.6.749. [DOI] [PubMed] [Google Scholar]
- 33.Leblanc R, Little JR. Hemodynamics of arteriovenous malformations. Clin Neurosurg. 1990;36:299–317. [PubMed] [Google Scholar]
- 34.Little JR, Tomsak RL, Ebrahim ZY, Furlan AJ. Retinal artery pressure and cerebral artery perfusion pressure in cerebrovascular occlusive disease. Neurosurgery. 1986;18:716–20. doi: 10.1227/00006123-198606000-00006. [DOI] [PubMed] [Google Scholar]
- 35.Löfgren J. Pressure-volume relationships of the cerebrospinal fluid system: An experimental analysis in dogs. PhD Thesis. Göteborg: Göteborgs Universitet; 1973. [Google Scholar]
- 36.Löfgren J. Effects of variation in arterial pressure and carbon dioxide tension on the cerebrospinal fluid pressure-volume relationships. Acta Neurol Scand. 1973;49:586–98. doi: 10.1111/j.1600-0404.1973.tb01332.x. [DOI] [PubMed] [Google Scholar]
- 37.Luce JM, Huseby JS, Kirk W, Butler J. A Starling resistor regulates cerebral venous outflow in dogs. J Appl Physiol. 1982;53:1496–503. doi: 10.1152/jappl.1982.53.6.1496. [DOI] [PubMed] [Google Scholar]
- 38.McCormick WF. The pathology of vascular (“arteriovenous”) malformations. J Neurosurg. 1966;24:807–16. doi: 10.3171/jns.1966.24.4.0807. [DOI] [PubMed] [Google Scholar]
- 39.Mickle JP, Quisling RG. The transtorcular embolization of vein of Galen aneurysms. J Neurosurg. 1986;64:731–5. doi: 10.3171/jns.1986.64.5.0731. [DOI] [PubMed] [Google Scholar]
- 40.Mingrino S. Supratentorial arteriovenous malformations of the brain. Adv Tech Stand Neurosurg. 1978;5:93–123. [Google Scholar]
- 41.Miyasaka Y, Yada K, Kurata A, Tokiwa K, Irikura K, Tanaka R, et al. Correlation between intravascular pressure and risk of hemorrhage due to arteriovenous malformations. Surg Neurol. 1993;39:370–3. doi: 10.1016/0090-3019(93)90202-c. [DOI] [PubMed] [Google Scholar]
- 42.Miyasaka Y, Kurata A, Tanaka R, Nagai S, Yamada M, Irikura K, et al. Mass effect caused by clinically unruptured cerebral arteriovenous malformations. Neurosurgery. 1997;41:1060–4. doi: 10.1097/00006123-199711000-00008. [DOI] [PubMed] [Google Scholar]
- 43.Morgan M, Winder M. Haemodynamics of arteriovenous malformations of the brain and consequences of resection: A review. J Clin Neurosci. 2001;8:216–24. doi: 10.1054/jocn.2000.0795. [DOI] [PubMed] [Google Scholar]
- 44.Murayama Y, Usami S, Hata Y, Ganaha F, Hasegawa Y, Terao T, et al. Transvenous hemodynamic assessment of arteriovenous malformations and fistulas: Preliminary clinical experience in Doppler guidewire monitoring of embolotherapy. Stroke. 1996;27:1358–64. doi: 10.1161/01.str.27.8.1358. [DOI] [PubMed] [Google Scholar]
- 45.Norlén G. Proceedings of the 1st International Congress of Neurosurgery. Brussels: Les Editions “Acta Medica Belgica”; 1957. The cerebral circulation in supratentorial angiomas as studied by angiography before and after removal; pp. 217–22. [Google Scholar]
- 46.Nornes H, Grip A. Hemodynamic aspects of cerebral arteriovenous malformations. J Neurosurg. 1980;53:456–64. doi: 10.3171/jns.1980.53.4.0456. [DOI] [PubMed] [Google Scholar]
- 47.Okabe T, Meyer JS, Okayasu H, Harper R, Rose J, Grossman RG, et al. Xenon-enhanced CT CBF measurements in cerebral AVMs before and after excision. Contribution to pathogenesis and treatment. J Neurosurg. 1983;59:21–31. doi: 10.3171/jns.1983.59.1.0021. [DOI] [PubMed] [Google Scholar]
- 48.Owler BK, Parker G, Halmagyi GM, Johnston IH, Besser M, Pickard JD, et al. Cranial venous outflow obstruction and pseudotumor cerebri syndrome. Avd Tech Stand Neurosurg. 2005;30:107–74. doi: 10.1007/3-211-27208-9_4. [DOI] [PubMed] [Google Scholar]
- 49.Pellettieri L, Carlsson CA, Grevsten S, Norlén G, Uhlemann C. Surgical versus conservative treatment of intracranial arteriovenous malformations: A study in surgical decision making. Acta Neurochir (Wien) 1980;(Suppl 29):1–86. [PubMed] [Google Scholar]
- 50.Permutt S, Riley RL. Hemodynamics of collapsible vessels with tone: The vascular waterfall. J Appl Physiol. 1963;18:924–32. doi: 10.1152/jappl.1963.18.5.924. [DOI] [PubMed] [Google Scholar]
- 51.Pertuiset B, Ancri D, Lienhart A. Profound arterial hypotension (MAP<50 mmHg) induced with neuroleptanalgesia and sodium nitroprusside (series of 531 cases). Reference to vascular autoregulation mechanism and surgery of vascular malformations of the brain. Adv Tech Stand Neurosurg. 1981;8:75–122. [Google Scholar]
- 52.Portnoy HD, Chopp M, Branch C. Hydraulic model of myogenic autoregulation and the cerebrovascular bed: The effects of altering systemic arterial pressure. Neurosurgery. 1983;13:482–98. doi: 10.1227/00006123-198311000-00002. [DOI] [PubMed] [Google Scholar]
- 53.Rodesch G, Malherbe V, Alvarez H, Zerah M, Devictor D, Lasjaunias P. Nongalenic cerebral arteriovenous malformations in neonates and infants. Review of 26 consecutive cases (1982-1992) Child's Nerv Syst. 1995;11:231–41. doi: 10.1007/BF00277659. [DOI] [PubMed] [Google Scholar]
- 54.Rossitti S. Transarterial embolization of intracranial dural arteriovenous fistulas with direct cortical venous drainage with ethylene vinyl alcohol copolymer (Onyx) Klin Neuroradiol. 2009;19:122–8. doi: 10.1007/s00062-009-8031-2. [DOI] [PubMed] [Google Scholar]
- 55.Rossitti S, Gustafsson O. The basic relation between the systemic arterial pressure and the intracranial pressure: A study on Löfgren's hydraulic model of the cerebral circulation. Biomech Sem. 1994;8:75–86. [Google Scholar]
- 56.Rossitti S, Löfgren J. Vascular dimensions of the cerebral arteries follow the principle of minimum work. Stroke. 1993;24:371–7. doi: 10.1161/01.str.24.3.371. [DOI] [PubMed] [Google Scholar]
- 57.Rossitti S, Svendsen P. Shear stress in cerebral arteries supplying arteriovenous malformations. Acta Neurochir (Wien) 1995;137:138–45. doi: 10.1007/BF02187185. [DOI] [PubMed] [Google Scholar]
- 58.Sorimachi T, Takeuchi S, Koike T, Minakawa T, Abe H, Tanaka R. Blood pressure monitoring in feeding arteries of cerebral arteriovenous malformations during embolization: A preventive role in hemodynamic complications. Neurosurgery. 1995;37:1041–8. doi: 10.1227/00006123-199512000-00002. [DOI] [PubMed] [Google Scholar]
- 59.Spetzler RF, Hargraves RW, McCormick PW, Zabramski JM, Flom RA, Zimmermann RS. Relationship of perfusion pressure and size to risk of hemorrhage from arteriovenous malformations. J Neurosurg. 1992;76:918–23. doi: 10.3171/jns.1992.76.6.0918. [DOI] [PubMed] [Google Scholar]
- 60.Spetzler RF, Martin NA. A proposed grading system for arteriovenous malformations. J Neurosurg. 1986;65:476–83. doi: 10.3171/jns.1986.65.4.0476. [DOI] [PubMed] [Google Scholar]
- 61.Spetzler RF, Wilson CB. Enlargement of an arteriovenous malformation documented bt angiography. Case report. J Neurosurg. 1975;43:767–9. doi: 10.3171/jns.1975.43.6.0767. [DOI] [PubMed] [Google Scholar]
- 62.Spetzler RF, Wilson CB, Weinstein P, Mehdorn M, Towsend J, Telles D. Normal perfusion pressure breakthrough theory. Clin Neurosurg. 1978;25:651–72. doi: 10.1093/neurosurgery/25.cn_suppl_1.651. [DOI] [PubMed] [Google Scholar]
- 63.Strandgaard S, Olesen J, Skinoj E, Lassen NA. Autoregulation of brain circulation in severe arterial hypertension. BMJ. 1973;1:507–10. doi: 10.1136/bmj.1.5852.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Taneda M, Hayakawa T. The paradoxical blood pressure-flow relationship in the brain with an arteriovenous malformation. Surg Neurol. 1993;40:390–4. doi: 10.1016/0090-3019(93)90218-p. [DOI] [PubMed] [Google Scholar]
- 65.Vassilouthis J. Cerebral arteriovenous malformation with intracranial hypertension. Surg Neurol. 1979;11:402–4. [PubMed] [Google Scholar]
- 66.Vorstman EB, Niemann DB, Molyneux AJ, Pike MG. Benign intracranial hypertension associated with arteriovenous malformation. Dev Med Child Neurol. 2001;44:133–5. doi: 10.1017/s0012162201001803. [DOI] [PubMed] [Google Scholar]
- 67.Weale FE. An Introduction to Surgical Haemodynamics. London: Loyd-Luke; 1966. [Google Scholar]
- 68.Widlund M, Löfgren J, Svendsen P. Mechanism of increased cerebrospinal fluid pressure in cerebral arteriovenous malformations. Upsala J Med Sci. 1993;(Suppl 52):54. [Google Scholar]
- 69.Wilson CB, Hieshima G. Occlusive hyperemia: A new way to think about an old problem. J Neurosurg. 1993;78:165–6. doi: 10.3171/jns.1993.78.2.0165. [DOI] [PubMed] [Google Scholar]
- 70.Yasargil MG. Microneurosurgery IIIA: AVM of the Brain, History, Embryology, Pathological Considerations, Hemodynamics, Diagnostic Studies, Microsurgical Anatomy. Stuttgart, New York: Georg Thieme Verlag; 1987. [Google Scholar]
- 71.Young WL, Kader A, Pile-Spellman J, Ornstein E, Stein BM The Columbia University AVM Study Project. Arteriovenous malformation draining vein physiology and determinants of transnidal pressure gradients. Neurosurgery. 1994;35:389–96. doi: 10.1227/00006123-199409000-00005. [DOI] [PubMed] [Google Scholar]
- 72.Young WL, Pille-Spellman J, Ptohovnik I, Kader A, Stein BM The Columbia University AVM Study Project. Evidence for adaptive autoregulatory displacement in hypotensive cortical territories adjacent to arteriovenous malformations. Neurosurgery. 1994;34:601–11. doi: 10.1227/00006123-199404000-00006. [DOI] [PubMed] [Google Scholar]
- 73.Zerah M, Garcia-Monaco R, Rodesch G, Terbrugge K, Tardieu M, de Victor D, et al. Hydrodynamics in vein of Galen malformations. Child's Nerv Syst. 1992;8:111–7. doi: 10.1007/BF00298261. [DOI] [PubMed] [Google Scholar]