

Abstract
Introduction and Hypothesis:
Nuclear atypia with features of multi nuclei have been detected in human melanoma specimens. We found that the K type human endogenous retroviral element (HERV K) is expressed in such cells. Since cellular syncytia can form when cells are infected with retroviruses, we hypothesized that HERV K expressed in melanoma cells may contribute to the formation of multinuclear atypia cells in melanoma.
Experiments and Results:
We specifically inhibited HERV K expression using RNA interference (RNAi) and monoclonal antibodies and observed dramatic reduction of intercellular fusion of cultured melanoma cells. Importantly, we identified loss of heterozygosity (LOH)of D19S433 in a cell clone that survived and proliferated after cell fusion.
Conclusion:
Our results support the notion that proteins encoded by HERV K can mediate intercellular fusion of melanoma cells, which may generate multinuclear cells and drive the evolution of genetic changes that provide growth and survival advantages.
Keywords: Cell fusion, K-type human endogenous retroviral deoxyribonucleic acid, melanoma
INTRODUCTION
Melanoma is the most lethal skin malignancy, notorious for aggressive growth, and resistance to therapy. Patients with invasive and metastatic melanomas may die within 6-8 months after diagnosis. Recently, Yervoy (Ipilimumab), an inhibitor of cytotoxic T-lymphocyte antigen 4 (CTLA-4) and Zelboraf (vemurafenib), an inhibitor of mutant v-Raf murine sarcoma viral oncogene homologue B1 (BRAF), obtained Food and Drug Administration (FDA) approval to treat metastatic melanomas.[1,2] Although both drugs may prolong the lives of patients with advanced melanoma, therapeutic efficacy is limited. Both drugs typically lengthen life by only several months in patients that initially respond to the treatment. Further studies are necessary to overcome the aggressive growth of melanoma cells and their intrinsic and acquired resistance to cancer therapies.[1,2]
In melanomas, constitutive deregulation of BRAF-mitogen-activated protein kinase/extracellular signal regulated (ERK) kinase (MEK)-ERK and p16-cyclin dependent kinase 4 (CDK4)-retinoblastoma (RB) pathways occurs at high frequencies. We have reported that simultaneous inhibition of mutant BRAF and expression of wild-type inhibitor of CDK4A (INK4A, encoding p16) or combinatorial application of MEK and CDK4 inhibitors in melanoma cells significantly suppresses cell proliferation and enhances apoptosis.[3,4] We have also reported that the expression of K type human endogenous retrovirus (HERV-K) correlates with MEK-ERK activation and loss of p16 in melanoma specimens and can be suppressed by MEK and CDK4 inhibitors in cultured melanoma cells.[5] Our studies should help the design of novel combination of drug strategies to treat melanoma.[3–5]
BACKGROUND
HERVs are thought to be germline-integrated genetic remnants of exogenous retroviral infections and comprise approximately 8% of the human genome.[6,7] HERVs can be classified into over 20 families based on transfer RNA (tRNA) specificity of the primer binding site used to initiate reverse transcription; thus, HERV-K would use lysine and HERV-W tryptophan if they were replicating viruses.[8] Through millions of years of evolution, HERVs have become indispensible parts of the human genome. For example, syncytin-1, encoded by the envelope (ENV) gene of HERV-W, mediates intercellular fusion of trophoblast cells to form syncytiotrophoblast as well as preventing maternal immune attack against the developing embryo, thereby facilitating implantation of the embryo.[6,7] Similar to exogenous retroviruses such as human immunodeficiency virus (HIV) and human T cell leukemia virus (HTLV), a complete HERV sequence is composed of group-specific antigen (GAG), protease ( PRO), polymerase ( POL), and ENV genes flanked by two long terminal repeats (LTRs). Although most HERVs are degenerated with disruptive open reading frames, a few proviruses have retained intact genes, and the corresponding proteins can thus be expressed.[6–9]
HERVs have been implicated in the etiology of cancer, chronic inflammation, and other diseases,[7] and emerging data support a role of HERV-K in melanomagenesis. For example, HERV-K is activated in melanomas but not in melanocytes,[10,11] and inhibition of HERV-K by RNAi suppresses the in vivo growth of melanoma cells.[12–15] There are several potential mechanisms to explain the role of HERV-Ks in melanomagenesis. First, HERV-K ENV, which is homologous to syncytin, may have fusogenic activity to mediate melanoma-melanoma and melanoma-target cell intercellular fusions, and therefore can be the molecular link in the melanoma cell fusion theory of metastasis.[16,17] Second, HERV-K sequences may relocate in the genome by retrotransposition leading to mutagenesis.[14,18] Third, HERV-K proteins can be immunosuppressive and may facilitate tumor progression by providing a critical survival/escape mechanism for tumor invasion and metastasis.[12,14]
In this study, we show that HERV-K is expressed in human melanoma cells with features of nuclear atypia and has a role in mediating melanoma intercellular fusion in vitro. We also show that a proliferating cell clone which emerged from cell-cell fusion in cell culture gained loss of heterozygosity (LOH) of D19S433 locus.
MATERIALS AND METHODS
Patient specimens
A total of 72 formalin-fixed, paraffin-embedded pathological specimens from the archives at the University of Texas Medical Branch (UTMB) were available for the study. The samples included 34 melanomas and 38 specimens that contained nevi as described previously.[5] The study was approved by the UTMB institutional review board (IRB) for the protection of human subjects.
Immunohistochemistry
Immunohistochemical staining of HERV-K ENV was performed as described.[5] Basically, the staining was modified from the protocol of All-in-One Kit for immunohistochemical staining for tissues with antibodies (Invitrogen, Carlsbad, CA). Four-micron-thick sections of paraffin blocks were dewaxed with three changes of xylene and rehydrated through a graded series of alcohol concentrations into water. Sections were washed with phosphate buffered saline (PBS) Tween (PBST; Sigma, St Louis, MO) three times, each for 5 min, and then blocked with horse serum for 30 min at room temperature. For antigen retrieval, sections were heat-treated in a microwave oven for 20 min in 0.01 M citrate buffer (pH 6.0, 100 mM stock) and cooled for 20 min in a beaker. For immunostaining, the slides were incubated at 4°C overnight with HERV-K ENV antibody (Cat. # 1811-5, Austral Biologicals, San Ramon, CA) following the manufacturer's instructions. The slides were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies, followed by signal development, counterstaining, and mounting using All-in-One Kit for immunohistochemical staining for tissues with antibodies (Invitrogen, Carlsbad, CA). HERV-K ENV cytoplasmic and/or nuclear staining was dichotomized as negative (<30% of tumor cells staining positively) and positive (>30% of tumor cells staining positively) and scored by two pathologists (Z.L. and X.W.).
Cell culture
Human melanoma cell lines 624Mel and A375 were kindly provided by Dr. Stuart Aaronson (Mount Sinai School of Medicine, New York, NY). Cells were maintained in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 50 units/ml penicillin-streptomycin (Invitrogen, Carlsbad, CA) in a humidified incubator at 37°C with 5% CO2.
Plasmids, transient transfection, and generation of stable cell lines
The HERV-K shRNA retroviral vector pS-puro-H-Ki[13,15] and pS-puro-scrambled containing a non-targeting sequence as a control[19] were used for RNA interference. Both plasmids carry puromycin (puro) selection marker. These plasmids were generously provided by Dr. Corrado Spadafora (Italian National Institute of Health, Rome, Italy) and Dr. Christopher Counter (Duke University, Durham, NC), respectively. The pS-puro-H-Ki construct carried a 19-bp siRNA oligonucleotide specific for HERV-K GAG (5′-UCCCAGUAACGUUAGAACC-3′),[13] that is expressed in human melanoma cell lines A375 and 624Mel.[5,15] The pEYFP-neo-N3 plasmid (Clontech, Mountain View, CA) was used for double selection; it carried yellow fluorescent protein (YFP) with a neomycin (neo) marker for G418 resistance. Cells were seeded in tissue culture plates the day before transfection. Transient transfection was carried out using LipofectAMINE 2000 reagent according to the manufacturer's instructions (Invitrogen, Carlsbad, CA). Stable A375 and 624Mel cell lines expressing pS-puro-H-Ki, pS-puro-scrambled, or pEYFP-neo-N3 were generated after transfection and selection with media containing either 1 μg/ml puromycin or 700 μg/ml G418 as described.[20]
Ribonucleic acid extraction and reverse transcriptase polymerase chain reaction (PCR)
Total ribonucleic acid (RNA) was isolated from cultured cells using RNeasy Plus Mini kit (Qiagen Inc, Valencia, CA). To remove possible DNA contamination, extracted RNA was treated with 5 U/μg RNase free DNase (New England Biolabs, Ipswich, MA) for 1 h at 37°C; DNAse was inactivated by adding 10 mM EDTA and heating at 70°C for 10 min. 500 ng RNA was used as RT-polymerase chain reaction (RT-PCR) template. PCR primer sequences were HERV-K POL, 5′-CCACTGTAGAGCCTCCTAAACCC-3′ and 5′-GCTGGTATAGTAAAGGCAAATTTTTC-3′; HERV-K ENV, 5′-GTACCACTCCTCAGATGCAA-3′ and 5′-GTGACATCCCGCTTACCATG-3′; and GAPDH, 5′-TGGTATCGTGAAAGGACT-3′ and 5′-ATGCAAGTGAGCTTCCCG-TTC-3′ as described.[15] PCR amplification parameters were as described.[5,15] RT-PCR was performed using OneStep RT-PCR kit (Qiagen, Valencia, CA) on a MasterCycler Personal (Eppendorf, Hauppauge, NY). PCR products were analyzed by fractionation in 1.5% (w/v) agarose gel and visualized by GelRed DNA stain (Phenix Research Products, Candler, NC). Images were captured using a gel documentation system (Alpha Innotech, San Leandro, CA).
Immunoblotting assay
Western blots were performed as described.[3,5] Briefly, harvested cells were lysed in lysis Solution (Cell Signaling, Danvers, MA) supplemented with complete mini protease inhibitor Cocktail Tablets (Roche Diagnostics, Indianapolis, IN). Protein concentration of lysates was determined using Quick Start Bradford 1 × Dye Reagent (Bio-Rad, Hercules, CA). Lysates were separated in 10% SDS-polyacrylamide gel, electrophoretically transferred to Immobilon-P membrane (Millipore Corp, Billerica, MA), probed with primary antibodies, HERV-K ENV (Austral Biologicals, San Ramon, CA) and β-actin (Abcam, Cambridge, MA) followed by washes and hybridization with horseradish peroxidase (HRP)-conjugated secondary antibody (Jackson Immunoresearch, West Grove, PA). The membrane was incubated with Super Signal chemiluminescence substrate (Pierce, Rockford, IL) followed by exposure to blue sensitive X-ray film (Phenix Research, Candler, NC).
Cell fusion dependent colony formation assay
Colony formation assay was used to count “fused” clones under double selection as described.[20] Briefly, 106 of A375 or 624Mel cells stably expressing pEYFP-neo-N3 were mixed with equal numbers of the same cell types stably expressing either HERV-K shRNA pS-puro-H-Ki or control pS-puro-scrambled. The mixed cells were cultured to confluency in medium without puromycin or G418 selection. Cells were then trypsinized, plated in triplicate in 35 mm culture plates, and grown in medium that included both puromycin and G418 to select pS-puro-scrambled-pEYFP-neo-N3 and pS-puro-H-Ki-pEYFP-neo-N3 fused cells. Approximately 3 weeks later, the plates were rinsed with PBS and fixed in 10% methanol for 10 min, stained with a solution containing 0.5% crystal violet, 10% methanol, 10% acetic acid in distilled water for 30 min, and then rinsed three times with water to reveal stained colonies.
CellTracker live cell labeling assay
CellTracker probes for long-term tracing of living cells (Invitrogen, Carlsbad, CA), a series of fluorescent derivatives that could be retained in living cells through several generations were used to label live cells. Hoechst 33342 emitted blue fluorescence when bound to dsDNA, whereas Green boron-dipyrromethene (BODIPY) exhibited green fluorescence in the cytoplasm (Invitrogen, Carlsbad, CA). Live cell staining was performed according to manufacturer's instructions (Invitrogen, Carlsbad, CA). Cells were detached by trypsinization and washed in DMEM supplemented with 10% FBS and 50 units/ml penicillin-streptomycin (Invitrogen, Carlsbad, CA). Hoechst 33342 or Green BODIFY was added to yield a final concentration of 2 μg/ml Hoechst 33342 and 10 μM Green BODIFY. Staining was performed for 30 min at 37°C in the dark. Stained cells were washed in DMEM medium twice and then resuspended in DMEM medium at a density of 2 × 106 /ml. 107 Hoechst 33342-labeled A375 cells stably expressing pS-puro-scrambled or pS-puro-H-Ki were then each mixed with 107 Green BODIFY-labeled A375 pEYFP-neo-N3 expressing cells and seeded into 10 cm plates in medium without puromycin or G418 selection. After 12 h medium was refreshed and cells were cultured in medium containing 1 μg/ml puromycin and 700 μg/ml G418 for 48 h to select pS-puro-scrambled-pEYFP-neo-N3 and pS-puro-H-Ki-pEYFP-neo-N3 fused cells. Cells were subsequently trypsinized and examined under Nikon Eclipse TE 2000-U fluorescent microscope (Nikon, Melville, NY) using software Metamorph (Universal Imaging, Media, PA).
Antibody neutralization assay
104 each of A375 cells stably expressing pS-puro-scrambled and pEYFP-neo-N3 were added per well in 96-well plate. Different dilutions of two HERV-K ENV monoclonal antibodies (HERM-1811-5 and HERM-1821-5, obtained by immunization of mice with 79.5 kD full length and 42.8 kD C-terminal part of HERV-K ENV antigens, respectively; Austral Biologicals, San Ramon, CA) were added to culture medium. PBS was used as control. Mixed cells were cultured for 48 h without puro-G418 selection, followed by selection of pS-puro-scrambled-pEYFP-neo-N3 fused cells using 1 μg/ml puromycin and 700 μg/ml G418 for 48 h.
Microsatellite marker analysis
Microsatellite markers were analyzed using ABI AmpFISTR Identifiler PCR Amplification kit (Life Technologies, Carlsbad, CA) that simultaneously amplified 15 short tandem repeat (STR) loci in chromosomes 2, 3, 4, 5, 7, 8, 11, 12, 13, 16, 18, 19, and 21, plus the amelogenin gender-determining marker. One ng genomic DNA was used per PCR reaction. Multiplex PCR was performed at 95°C for 11 min, followed by 28 cycles at 94°C for 1 min, 59°C for 1 min, 72°C for 1 min, and 60°C for 60 min. PCR products were separated by capillary electrophoresis using ABI 3130 genetic analyzer, and data analyzed using GeneMapper ID software (Life Technologies, Carlsbad, CA).
Statistical analysis
Statistical analyses were performed using SPSS 13.0 software (SPSS Inc., Chicago, IL). Wilcoxon signed-rank test was used to detect differences in multinuclear cells and ENV immunostaining between melanomas and nevi.
RESULTS
Human endogenous retroviral element K envelop is expressed in melanoma cells of nuclear atypia
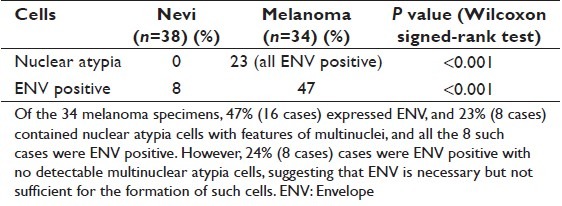
A total of 72 formalin-fixed, paraffin-embedded pathologic specimens were included in this study including 35 cutaneous melanomas (from 35 patients) and 38 benign nevi (from 38 patients). Nuclear atypia with features of multinuclear cells were detected in 23% (8 of 34) of melanoma but not in the nevi. Immunohistochemical analysis showed that human endogenous retroviral element K (HERV-K) envelop (ENV) was expressed in all the eight cases with multinuclear atypia cells [Figure 1, Table 1]. However, HERV-K ENV was also detected in eight melanoma cases without nuclear atypia for a total of 47% (16 of 34) in melanoma specimens; HERV-K ENV was also positive in nevus cells in 8% of the cases (3 of 38) without detectable multinuclear cells [Figure 1, Table 1]. These data suggest that ENV is linked to multinuclear atypia cells but not sufficient for its formation.
Figure 1.
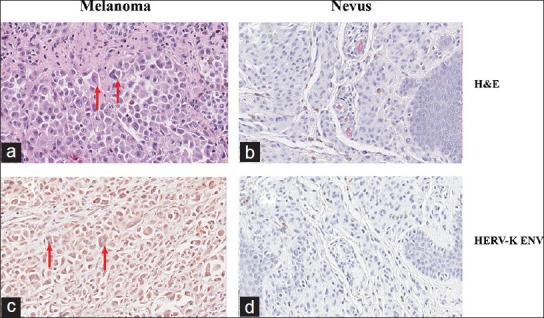
K type human endogenous retrovirus envelope (HERV-K ENV) is expressed in nuclear atypia melanoma cells. Formalin-fixed, paraffin embedded microscopic sections of melanoma (a, c) and nevus (b, d) were analyzed to identify multinuclear cells (a, b) and HERV-K ENV expression. (c, d) Shown is a representative staining pattern. Nuclear atypia with features of multinuclear cells were easily detected (arrows) in melanoma (a, c) but not in nevus (b, d) cells. In melanomas, such cells were positive for HERV-K by immunohistochemistry using a monoclonal HERV-K ENV antibody (Austral Biologicals, San Ramon, CA). Nevus cells did not express HERV-K ENV. (d) Magnification: × 400
Table 1.
Expression of human endogenous retroviral element-K envelope in nuclear atypia melanoma cells
Inhibition of intercellular fusion by blocking HERV-K
To examine a potential role of HERV-K in the formation of multinuclear cells through intercellular fusion, we first examined whether inhibiting HERV-K using RNAi would affect intercellular fusion in a colony formation assay. A375 and 624Mel human melanoma cells were transfected and selected by puromycin to stably express either control pS-puro-scrambled or HERV-K shRNA pS-puro-H-Ki as described previously.[13,15] Consistent with previous reports,[13,15] the expression of HERV-K RNA and protein was reduced in cells expressing pS-puro-H-Ki but not in control pS-puro-scrambled expressing cells [Figure 2a and b]. To examine cell fusion, we also generated A375 and 624Mel cell lines stably expressing pEYFP-neo-N3 (Clontech, Mountain View, CA) that carried yellow fluorescent protein (YFP) with a neomycin (G418) selection marker. 106 A375 or 624Mel cells stably expressing pEYFP-neo-N3 were mixed with equal numbers of the cells expressing either pS-puro-scrambled or pS-puro-H-Ki. Mixed cells were cultured to confluency in medium without puromycin or G418 selection. Cells were then trypsinized and passaged in culture medium containing both puromycin and G418 to select for pS-puro-scrambled-pEYFP-neo-N3 and pS-puro-H-Ki-pEYFP-neo-N3 fused cells. Inhibition of HERV-K almost completely abolished colony formation in both A375 and 624Mel cells [Figure 2c, control vs. shRNA expressing cells]. This result supports a role of HERV-K in mediating cell-cell fusion of melanoma cells.
Figure 2.
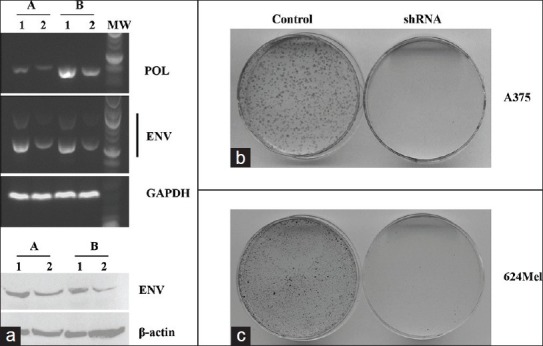
Inhibition of K type human endogenous retrovirus (HERV-K) expression blocks intercellular fusion-mediated colony formation. Human melanoma cell line A375 (a) and 624Mel (b) was transiently transfected and selected to stably express either control pS-puro-scrambled (1) or HERV-K shRNA pS-puro-H-Ki construct (2). (a). RT-polymerase chain reaction (RT-PCR) using total cellular RNA was performed using specific HERV-K POL and envelope (ENV) primers as described (15). Gel electrophoresis of RT-PCR products revealed that POL and ENV RNA levels were reduced in shRNA expressing cells of both A375 (a) and 624Mel (b) cells. GAPDH was used as internal control (b). Western blot analysis of HERV-K ENV protein. Total cellular protein was separated on SDS-PAGE, and blotted to Nilon membrane, and incubated with HERV-K ENV antibody. Actin was used as loading control. As expected, HERV-K POL and ENV transcripts (a) and ENV protein (b) levels were reduced in cells expressing shRNA. The knockdown levels of both RNA (a) and protein (b) were estimated as more than 50% (c). To examine cell fusion, 106 A375 or 624Mel cells stably expressing pEYFP-N3 were mixed with equal numbers of the same cells expressing either pS-puro-scrambled or pS-puro-H-Ki. Mixed cells were cultured to confluency in medium without puromycin or G418 selection. Cells were then trypsinized and passaged in culture medium that includes both puromycin and G418 to select pS-puro-scrambled-pEYFP-N3 and pS-puro-H-Ki-pEYFP-N3 fused cells. Colony formation assay was used to count “fused” clones under double selection
Here we used a colony formation assay to measure “fused” clones under double selection. Colony formation is a general feature of malignant transformation and is commonly used to measure malignant growth in vitro. It can reflect the activities of either cell cycle arrest or cell death when a reduction in colony number is observed. Colony formation assay is also used traditionally to measure contact inhibition and anchorage-independent growth. It is worth mentioning that the above cell fusion-dependent colony formation assay was designed so that only the puro-neo intercellular fused cells can survive puromycin-G418 double selection; unfused puro or neo as well as puro-puro and neo-neo fused cells will be killed by puromycin-G418 double selection. We also performed a routine colony formation assay without selecting for intercellular fused cells. In this experiment, pS-puro-scrambled and pS-puro-H-Ki cells were plated and cultured in medium supplemented with puromycin. Under such conditions, comparable numbers of colonies were observed in the control and HERV-K shRNA expressing cells (data not shown). This result is consistent with the published studies demonstrating that expression of pS-puro-H-Ki does not have significant impact on the in vitro proliferation and survival of melanoma cells.[13,15] We also found that similar numbers of colonies were produced when approximately 103-fold fewer cells were plated in the routine cell fusion-independent colony formation assay than those generated by pS-puro-scrambled-pEYFP-neo-N3 fusion, suggesting that the frequency of cell-cell fusion as measured by our colony formation assays is rare, approximately 1 in 1000.
Next, we used live cell labeling to observe cell-cell fusion. A375 cells stably expressing pS-puro-scrambled or pS-puro-H-Ki were labeled with Hoechst 33342, and A375 cells stably expressing pEYFP-N3 were labeled with Green BODIFY using CellTracker probes for long-term tracing of living cells reagents (Invitrogen, Carlsbad, CA). After dye labeling, 107 Hoechst 33342-labeled pS-puro-scrambled or pS-puro-H-Ki cells were each mixed with 107 Green BODIFY-labeled pEYFP-neo-N3 expressing cells. Mixed cells were cultured for 12 h without puromycin or G418 selection. Cells were then cultured in medium containing both puromycin and G418 for 48 h to select for puro-neo fused green-blue double stained cells. As shown in Figures 3a and b, green-blue double colored cells were present when green pEYFP-neo-N3 cells were mixed with blue pS-puro-scrambled [Figure 3a], but blocked with blue pS-puro-H-Ki cells [Figure 3b].
Figure 3(a, b).
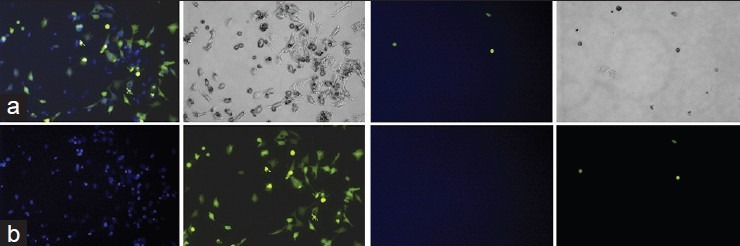
Live cell two color fusion assay and immune-neutralization of cell fusion using K type human endogenous retrovirus envelope monoclonal antibodies. Two color fusion assay (a and b). A375 cells stably expressing pS-puro-scrambled or pS-puro-H-Ki were labeled with blue Hoechst 33342, and A375 cells stably expressing pEYFP-N3 were labeled with green BODIFY using CellTracker probes for long-term tracing of living cells reagents according to manufacturer's instruction (Invitrogen, Carlsbad, CA). After dye labeling, 107 Hoechst 33342-labeled pS-puro-scrambled or pS-puro-H-Ki cells were each mixed with 107 green BODIFY-labeled pEYFP-N3 expressing cells. Mixed cells were cultured for 12 h without puromycin or G418 selection. Cells were then cultured in medium containing both puromycin and G418 for 48 h to select for puro-neo fused green (Green BODIFY) and blue (Hoechst 33342) double stained cells. Upper left, blue-green double colored cells were present when pEYFP-N3 expressing cells were mixed with pS-puro-scrambled (a), but blocked with pS-puro-H-Ki expressing cells (b). Lower left and right, blue and green stained cells, respectively. Photographed by fluorescence microscope. Upper right, photographed through phase contrast microscope
Like other enveloped viruses, HERV-K ENV protein may mediate cell-cell fusion.[21–23] HERV-K ENV is prominently expressed in A375 cells as detected using antibody that recognizes a 37 kD spliced transmembrane domain of HERV-K ENV.[5,10,11,24] To examine whether ENV plays a role in intercellular fusion, we performed an immunoneutralization assay using commercially available HERV-K ENV monoclonal antibodies HERM-1811-5 and HERM-1821-5 (Austral Biologicals, San Ramon, CA). We added PBS control or HERV-K ENV monoclonal antibodies to culture medium and examined the effect on HERV-K mediated cell-cell fusion using the two-color live cell labeling assay [Figures 3a and b]. As shown in Figure 3c, cell fusion was inhibited by both ENV antibodies and the efficiencies of neutralization were approximately 15-25% and 25-30% at 1:100 and 1:10 antibody dilutions, respectively. Detailed information about the two antibodies were not available to us, except that the antibodies were obtained by immunization of mice with 79.5 kD full length (HERM-1811-5) and 42.8 kD C-terminal part (HERM-1821-5) of HERV-K ENV antigens, respectively (Austral Biologicals, San Ramon, CA). Further studies are necessary to identify the neutralizing epitopes and additional factors that influence HERV-K ENV-mediated intercellular fusion.
Figure 3c.
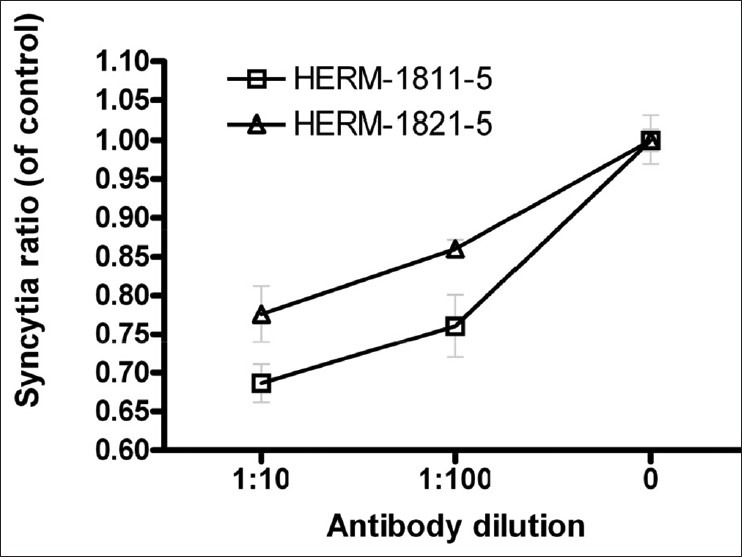
Antibody neutralization assay. 104 each of A375 cells stably expressing pS-puro-scrambled and pEYFP-N3-neo were added per well in 96 well plate. Different dilutions of two K type human endogenous retrovirus envelope monoclonal antibodies HERM-1811-5 and HERM-1821-5 were added to culture medium. PBS was used as control. Cells were cultured for 48 h, followed by selection of pS-puro-scrambled-pEYFP-N3 fused cells using puromycin and G418 double selection for 48 h. The numbers of blue-green double color cells (fused cells) as shown in 3a upper left were calculated and compared between antibody treated and control
D19S433 loss of heterozygosity in a proliferating post-fusion cell clone
Although cell fusion can lead to cell death and growth arrest due to massive aneuploidy and mitotic catastrophe,[25,26] cells that survive cell fusion may also gain growth advantage and functional novelty through genetic changes.[23,27,28] We performed DNA profiling of post-fusion proliferating cells to detect new genetic changes not present in the parental melanoma cells. We randomly picked 10 individual A375 cell colonies as the control colonies in Figure 2c; however, we plated fewer cells to allow the formation of well-separated individual colonies, including 2 pS-puro-scrambled, 2 pEYFP-neo-N3, and 6 pS-puro-scrambled-pEYFP-neo-N3 cell colonies. Each of the colonies was cultured in a 6-well plate for 2 weeks to obtain cells for DNA profiling. Genomic DNA was extracted and examined using AmpFISTR Identifiler PCR Amplification kit (Life Technologies, Carlsbad, CA). As shown in Figure 4a, the fragment height of one of the D19S433 alleles (allele a, in the red box) appeared lower in fused clones (clones 5-9) than the controls (clones 1-4), and clone number 10 showed LOH of D19S433. D19S433 is located at chromosomal 19q12-13.1. D19S433 has shown significant linkage to aggressive disease and adverse clinical outcome of prostate[29] and ovarian cancers,[30] supporting the notion that changes in this region may contribute to the malignant growth of tumor cells. Further studies are necessary to understand the exact nature and extent of the changes in chromosome 19, for example, whether it affects segmental or whole chromosome 19, and whether such changes occur in patients’ specimens during melanoma progression,
Figure 4.
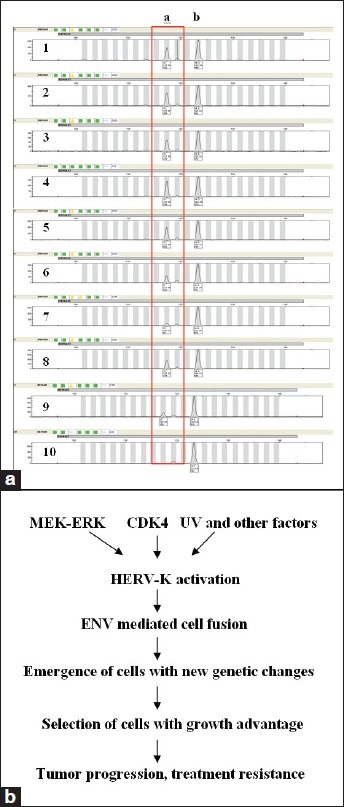
Detection of genetic changes in post-cell fusion cells and a proposed model of cell fusion in tumor progression (a). Ten single cell clones including 2 pS-puro-scrambled (clones 1-2), 2 pEYFP-N3-neo (clones 3-4), and 6 pS-puro-scrambled-pEYFP-N3-neo double selected clones (clones 5-10) were selected at random and amplified in cell culture. Genomic DNA was extracted from the cells. AmpFlSTR Identifiler PCR Amplification kit was used to examine microsatellite markers. Shown are fragment analysis of the D19S433 alleles. Note the fragment height of allele a (in the red box) was decreased in the fusion cells (6-9), and lost completely in clone #10 (b). A proposed model of K type human endogenous retrovirus in mediating intercellular fusion, evolution of genetic changes, and malignant progression
DISCUSSION
Nuclear atypia with features of multinuclear cells have been observed in a variety of tumors including melanoma,[31–33] and has been linked with poor disease prognosis of malignant melanomas.[32] Nuclear atypia may result from cell fusion, mitotic slippage, cytokinesis failure, and phagocytosis of apoptotic bodies.[27,28] Since cellular syncytia can form when cells are infected with retroviruses and all the melanoma multinuclear cells express HERV-K ENV, we hypothesized that HERV-K contributes to the formation of multinuclear atypia cells in melanoma. Our studies have demonstrated a potential link between HERV-K activation and nuclear atypia in melanoma and provide evidence supporting the fusogenic activity of HERV-K in melanoma cells in vitro. Of note, although we showed that stable inhibition of HERV-K ENV resulted in suppression of intercellular fusion, most melanoma cells that express HERV-K ENV do not become multinuclear atypia cells. This suggests that HERV-K ENV is necessary but not sufficient for intercellular fusion. Further studies are necessary to understand the molecular mechanisms of HERV-K ENV-mediated intercellular fusion including co-factors that modify HERV-K ENV-mediated intercellular fusion.
The identification of LOH of D19
S433 in a proliferating post-fusion clone supports the notion that cell fusion may trigger evolution and selection of genetic changes to provide survival and growth advantages. This is consistent with a speculation that HERV-K mediates cell fusion leading to enhanced survival/fitness of melanoma cells promoting malignant transformation, tumor progression, and therapy resistance. If proven true, we can even envision novel therapeutic strategies such as blocking HERV-K, for example, by immunoneutralization of ENV, to prevent and treat melanomas. However, we need to bear in mind that cell-cell fusion is fundamental to the development of multicellular organisms, such as fertilization, placentation, and development of skeletal muscle and bone.[34,35] Intercellular fusion may result in cell death due to “mitotic catastrophe” or growth arrest,[23,27,28] which has even been explored as a way to reduce tumor burden.[36]
Constitutive activation of the RAS-RAF-MEK pathway has been shown to drive chromosome abnormality and aneuploidy, which then contribute to malignant transformation and tumor progression.[37–39] We have reported the regulation of HERV-K by MEK-ERK and p16-CDK4 pathways in melanoma cells.[5] It is conceivable that the effect of RAS-RAF-MEK on chromosomal changes may be mediated, at least partly, through HERV-K ENV-mediated intercellular fusion. HERV-K can also be regulated by other factors in melanoma cells, for example, UV radiation, methylation of CpG site, and other transcription modulators.[40–43]
CONCLUSION
As summarized in Figure 4b, when activated, HERV-K may drive the evolution of cells with new genetic changes through HERV-K-mediated cell fusion, resulting in the emergence of cells that gain survival and growth advantage based on the cellular environment, which may contribute to tumor progression and treatment resistance. Further studies are warranted to further examine the molecular mechanisms and biological consequences of HERV-K activation in melanoma cells.
AUTHOR'S PROFILE
Dr. Gengming Huang, Senior Research Associate, Department of Pathology and Sealy Center for Cancer Cell Biology at University of Texas Medical Branch in Galveston, Texas.
Dr. Zhongwu Li, Key Laboratory of Carcinogenesis and Translational Research, Department of Pathology, Peking University School of Oncology, Beijing Cancer Hospital & Institute, Beijing, China
Dr. Xiaohua Wan, Senior Medical Technologist, Department of Laboratory Medicine, Beijing Tongren Hospital, Capital Medical University, Beijing, China.
Dr. Yue Wang, Director of Molecular Genetics at the Center for Medical Genetics in Houston, TX.
Dr. Jianli Dong, Associate Professor and Director Molecular Diagnostics in the Department of Pathology and Sealy Center for Cancer Biology at University of Texas Medical Branch in Galveston, Texas.
ACKNOWLEDGMENTS
We thank Drs. David Walker, Brad Thompson, and Benjamin Gelman for critical comments and helpful discussions. We also thank Dr. Stuart Aaronson for providing human melanoma cell lines. The pS-puro-H-Ki and pS-puro-scrambled plasmids were generously provided by Dr. Corrado Spadafora (Italian National Institute of Health, Rome, Italy) and Dr. Christopher Counter (Duke University, Durham, NC), respectively. This work was supported by Bill Walter III Melanoma Research Fund of Melanoma Research Foundation, Harry J. Lloyd Charitable Trust, and Cancer and Leukemia Group B Foundation (to J.D.).
Footnotes
Source of Support: This work was supported by Bill Walter III Melanoma Research Fund of Melanoma Research Foundation, Harry J. Lloyd Charitable Trust, and Cancer and Leukemia Group B Foundation (to J.D.).
Conflict of Interest: None declared.
REFERENCES
- 1.Eggermont AM, Robert C. New drugs in melanoma: It's a whole new world. Eur J Cancer. 2011;47:2150–7. doi: 10.1016/j.ejca.2011.06.052. [DOI] [PubMed] [Google Scholar]
- 2.Flaherty KT. Next generation therapies change the landscape in melanoma. F1000 Med Rep. 2011;3:8. doi: 10.3410/M3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li J, Xu M, Yang Z, Li A, Dong J. Simultaneous inhibition of MEK and CDK4 leads to potent apoptosis in human melanoma cells. Cancer Invest. 2010;28:350–6. doi: 10.3109/07357900903286966. [DOI] [PubMed] [Google Scholar]
- 4.Zhao Y, Zhang Y, Yang Z, Li A, Dong J. Simultaneous knockdown of BRAF and expression of INK4A in melanoma cells leads to potent growth inhibition and apoptosis. Biochem Biophys Res Commun. 2008;370:509–13. doi: 10.1016/j.bbrc.2008.03.148. [DOI] [PubMed] [Google Scholar]
- 5.Li Z, Sheng T, Wan X, Liu T, Wu H, Dong J. Expression of HERV-K correlates with status of MEK-ERK and p16INK4A-CDK4 pathways in melanoma cells. Cancer Invest. 2010;28:1031–7. doi: 10.3109/07357907.2010.512604. [DOI] [PubMed] [Google Scholar]
- 6.de Parseval N, Heidmann T. Human endogenous retroviruses: From infectious elements to human genes. Cytogenet Genome Res. 2005;110:318–32. doi: 10.1159/000084964. [DOI] [PubMed] [Google Scholar]
- 7.Kurth R, Bannert N. Beneficial and detrimental effects of human endogenous retroviruses. Int J Cancer. 2010;126:306–14. doi: 10.1002/ijc.24902. [DOI] [PubMed] [Google Scholar]
- 8.Blomberg J, Benachenhou F, Blikstad V, Sperber G, Mayer J. Classification and nomenclature of endogenous retroviral sequences (ERVs): Problems and recommendations. Gene. 2009;448:115–23. doi: 10.1016/j.gene.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 9.Ahn K, Kim HS. Structural and quantitative expression analyses of HERV gene family in human tissues. Mol Cells. 2009;28:99–103. doi: 10.1007/s10059-009-0107-y. [DOI] [PubMed] [Google Scholar]
- 10.Büscher K, Trefzer U, Hofmann M, Sterry W, Kurth R, Denner J. Expression of human endogenous retrovirus K in melanomas and melanoma cell lines. Cancer Res. 2005;65:4172–80. doi: 10.1158/0008-5472.CAN-04-2983. [DOI] [PubMed] [Google Scholar]
- 11.Muster T, Waltenberger A, Grassauer A, Hirschl S, Caucig P, Romirer I, et al. An endogenous retrovirus derived from human melanoma cells. Cancer Res. 2003;63:8735–41. [PubMed] [Google Scholar]
- 12.Mangeney M, Pothlichet J, Renard M, Ducos B, Heidmann T. Endogenous retrovirus expression is required for murine melanoma tumor growth in vivo. Cancer Res. 2005;65:2588–91. doi: 10.1158/0008-5472.CAN-04-4231. [DOI] [PubMed] [Google Scholar]
- 13.Oricchio E, Sciamanna I, Beraldi R, Tolstonog GV, Schumann GG, Spadafora C. Distinct roles for LINE-1 and HERV-K retroelements in cell proliferation, differentiation and tumor progression. Oncogene. 2007;26:4226–33. doi: 10.1038/sj.onc.1210214. [DOI] [PubMed] [Google Scholar]
- 14.Pothlichet J, Mangeney M, Heidmann T. Mobility and integration sites of a murine C57BL/6 melanoma endogenous retrovirus involved in tumor progression in vivo. Int J Cancer. 2006;119:1869–77. doi: 10.1002/ijc.22066. [DOI] [PubMed] [Google Scholar]
- 15.Serafino A, Balestrieri E, Pierimarchi P, Matteucci C, Moroni G, Oricchio E, et al. The activation of human endogenous retrovirus K (HERV-K) is implicated in melanoma cell malignant transformation. Exp Cell Res. 2009;315:849–62. doi: 10.1016/j.yexcr.2008.12.023. [DOI] [PubMed] [Google Scholar]
- 16.Carter A. Cell fusion theory: Can it explain what triggers metastasis? J Natl Cancer Inst. 2008;100:1279–81. doi: 10.1093/jnci/djn336. [DOI] [PubMed] [Google Scholar]
- 17.Pawelek JM, Chakraborty AK. Fusion of tumour cells with bone marrow-derived cells: A unifying explanation for metastasis. Nat Rev Cancer. 2008;8:377–86. doi: 10.1038/nrc2371. [DOI] [PubMed] [Google Scholar]
- 18.Tchenio T, Heidmann T. Defective retroviruses can disperse in the human genome by intracellular transposition. J Virol. 1991;65:2113–8. doi: 10.1128/jvi.65.4.2113-2118.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ancrile B, Lim KH, Counter CM. Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev. 2007;21:1714–9. doi: 10.1101/gad.1549407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rotolo S, Diotti R, Gordon RE, Qiao RF, Yao Z, Phelps RG, et al. Effects on proliferation and melanogenesis by inhibition of mutant BRAF and expression of wild-type INK4A in melanoma cells. Int J Cancer. 2005;115:164–9. doi: 10.1002/ijc.20865. [DOI] [PubMed] [Google Scholar]
- 21.Dewannieux M, Blaise S, Heidmann T. Identification of a functional envelope protein from the HERV-K family of human endogenous retroviruses. J Virol. 2005;79:15573–7. doi: 10.1128/JVI.79.24.15573-15577.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao P, Zheng J. Oncogenic virus-mediated cell fusion: New insights into initiation and progression of oncogenic viruses – Related cancers. Cancer Lett. 2011;303:1–8. doi: 10.1016/j.canlet.2010.12.021. [DOI] [PubMed] [Google Scholar]
- 23.Parris GE. The role of viruses in cell fusion and its importance to evolution, invasion and metastasis of cancer clones. Med Hypotheses. 2005;64:1011–4. doi: 10.1016/j.mehy.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 24.Büscher K, Hahn S, Hofmann M, Trefzer U, Ozel M, Sterry W, et al. Expression of the human endogenous retrovirus-K transmembrane envelope, Rec and Np9 proteins in melanomas and melanoma cell lines. Melanoma Res. 2006;16:223–34. doi: 10.1097/01.cmr.0000215031.07941.ca. [DOI] [PubMed] [Google Scholar]
- 25.Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: A mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol. 2011;12:385–92. doi: 10.1038/nrm3115. [DOI] [PubMed] [Google Scholar]
- 26.Hufton AL, Panopoulou G. Polyploidy and genome restructuring: A variety of outcomes. Curr Opin Genet Dev. 2009;19:600–6. doi: 10.1016/j.gde.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 27.Davoli T, de Lange T. The causes and consequences of polyploidy in normal development and cancer. Annu Rev Cell Dev Biol. 2011;27:585–610. doi: 10.1146/annurev-cellbio-092910-154234. [DOI] [PubMed] [Google Scholar]
- 28.Ganem NJ, Storchova Z, Pellman D. Tetraploidy, aneuploidy and cancer. Curr Opin Genet Dev. 2007;17:157–62. doi: 10.1016/j.gde.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 29.Neville PJ, Conti DV, Krumroy LM, Catalona WJ, Suarez BK, Witte JS, et al. Prostate cancer aggressiveness locus on chromosome segment 19q12-q13.1 identified by linkage and allelic imbalance studies. Genes Chromosomes Cancer. 2003;36:332–9. doi: 10.1002/gcc.10165. [DOI] [PubMed] [Google Scholar]
- 30.Nakayama K, Takebayashi Y, Hata K, Fujiwaki R, Iida K, Fukumoto M, et al. Allelic loss at 19q12 and Xq11-12 predict an adverse clinical outcome in patients with mucinous ovarian tumours of low malignant potential. Br J Cancer. 2004;90:1204–10. doi: 10.1038/sj.bjc.6601681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barlogie B, Drewinko B, Schumann J, Göhde W, Dosik G, Latreille J, et al. Cellular DNA content as a marker of neoplasia in man. Am J Med. 1980;69:195–203. doi: 10.1016/0002-9343(80)90379-4. [DOI] [PubMed] [Google Scholar]
- 32.Søndergaard K, Larsen JK, Møller U, Christensen IJ, Hou-Jensen K. DNA ploidy-characteristics of human malignant melanoma analysed by flow cytometry and compared with histology and clinical course. Virchows Arch B Cell Pathol Incl Mol Pathol. 1983;42:43–52. doi: 10.1007/BF02890369. [DOI] [PubMed] [Google Scholar]
- 33.Whang-Peng J, Chretien P, Knutsen T. Polyploidy in malignant melanoma. Cancer. 1970;25:1216–23. doi: 10.1002/1097-0142(197005)25:5<1216::aid-cncr2820250529>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 34.Larsson LI, Bjerregaard B, Talts JF. Cell fusions in mammals. Histochem Cell Biol. 2008;129:551–61. doi: 10.1007/s00418-008-0411-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Witze E, Rothman JH. Cell fusion: An EFFicient sculptor. Curr Biol. 2002;12:R467–9. doi: 10.1016/s0960-9822(02)00948-x. [DOI] [PubMed] [Google Scholar]
- 36.Lin EH, Salon C, Brambilla E, Lavillette D, Szecsi J, Cosset FL, et al. Fusogenic membrane glycoproteins induce syncytia formation and death in vitro and in vivo: A potential therapy agent for lung cancer. Cancer Gene Ther. 2010;17:256–65. doi: 10.1038/cgt.2009.74. [DOI] [PubMed] [Google Scholar]
- 37.Cui Y, Borysova MK, Johnson JO, Guadagno TM. Oncogenic B-Raf(V600E) induces spindle abnormalities, supernumerary centrosomes, and aneuploidy in human melanocytic cells. Cancer Res. 2010;70:675–84. doi: 10.1158/0008-5472.CAN-09-1491. [DOI] [PubMed] [Google Scholar]
- 38.Kamata T, Hussain J, Giblett S, Hayward R, Marais R, Pritchard C. BRAF inactivation drives aneuploidy by deregulating CRAF. Cancer Res. 2010;70:8475–86. doi: 10.1158/0008-5472.CAN-10-0603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saavedra HI, Fukasawa K, Conn CW, Stambrook PJ. MAPK mediates RAS-induced chromosome instability. J Biol Chem. 1999;274:38083–90. doi: 10.1074/jbc.274.53.38083. [DOI] [PubMed] [Google Scholar]
- 40.Fuchs NV, Kraft M, Tondera C, Hanschmann KM, Löwer J, Löwer R. Expression of the human endogenous retrovirus (HERV) group HML-2/HERV-K does not depend on canonical promoter elements but is regulated by transcription factors Sp1 and Sp3. J Virol. 2011;85:3436–48. doi: 10.1128/JVI.02539-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reiche J, Pauli G, Ellerbrok H. Differential expression of human endogenous retrovirus K transcripts in primary human melanocytes and melanoma cell lines after UV irradiation. Melanoma Res. 2010;20:435–40. doi: 10.1097/CMR.0b013e32833c1b5d. [DOI] [PubMed] [Google Scholar]
- 42.Schanab O, Humer J, Gleiss A, Mikula M, Sturlan S, Grunt S, et al. Expression of human endogenous retrovirus K is stimulated by ultraviolet radiation in melanoma. Pigment Cell Melanoma Res. 2011;24:656–65. doi: 10.1111/j.1755-148X.2011.00860.x. [DOI] [PubMed] [Google Scholar]
- 43.Stengel S, Fiebig U, Kurth R, Denner J. Regulation of human endogenous retrovirus-K expression in melanomas by CpG methylation. Genes Chromosomes Cancer. 2010;49:401–11. doi: 10.1002/gcc.20751. [DOI] [PubMed] [Google Scholar]