

Abstract
Even in healthy individuals there is an inexorable agerelated decline in cognitive function. This is due, in large part, to reduced synaptic plasticity caused by changes in the molecular composition of the postsynaptic membrane. AMPA receptors (AMPARs) are glutamate-gated cation channels that mediate the overwhelming majority of fast excitatory transmission in the brain. Changes in AMPAR number and/or function are a core feature of synaptic plasticity and age-related cognitive decline, AMPARs are highly dynamic proteins that are subject to highly controlled trafficking, recycling, and/or degradation and replacement. This active regulation of AMPAR synthesis, targeting, synaptic dwell time, and degradation is fundamentally important for memory formation and storage. Further, aberrant AMPAR trafficking and consequent detrimental changes in synapses are strongly implicated in many brain diseases, which represent a vast social and economic burden. The purpose of this article is to provide an overview of the molecular and cellular AMPA receptor trafficking events that control synaptic responsiveness and plasticity, and highlight what is known currently known about how these processes change with age and disease.
Keywords: AMPA receptor, glutamate receptor, synaptic plasticity, LTP, LTD, protein trafficking, AMPA receptor trafficking
Abstract
Incluso en los sujetos sanos existe una inexorable declinación de la función cognitiva relacionada con la edad. Esta es debida, en gran parte, a una reducción de la plasticidad sináptica causada por cambios en la composición molecular de la membrana post-sináptica. Los receptores AMPA (AMPARs) son canales catiónicos dependientes de glutamato que median la mayor parte de la transmisión excitatoria rápida en el cerebro. Los cambios en el número ylo función de los receptores AMPAR constituyen una característica central de la plasticidad sináptica y de la declinación cognitiva relacionada con la edad. Los AMPARs son proteínas altamente dinámicas que están sujetas a un alto control respecto al transporte, reciclado ylo degradación y reemplazo. Esta regulación activa de la síntesis, localización, tiempo de permanencia en la sinapsis y degradación de AMPAR es de fundamental importancia para la formación y almacenamiento de la memoria. Además, el transporte aberrante de AMPAR y los consecuentes cambios dañinos en las sinapsis se asocian fuertemente con muchas enfermedades cerebrales, las que representan un gran costo social y económico. El propósito de este artículo es aportar una perspectiva de los acontecimientos moleculares y celulares del transporte del receptor AMPA que controlan la respuesta sináptica y la plasticidad, y destacar lo que actualmente se sabe acerca de cómo estos procesos cambian con la edad y la enfermedad.
Abstract
Le déclin cognitif lié a l'âge, inexorable même chez les individus sains, est dû en grande partie à une diminution de la plasticité synaptique causée par des changements de la composition moléculaire de la membrane post-synaptique. Les récepteurs AMPA (AMPAR) sont des canaux de cations contrôlés par le glutamate qui assurent la médiation de la grande majorité de l'excitation rapide du cerveau. Les modifications en nombre et/ou en fonction des AMPAR sont au coeur de la plasticité synaptique et du déclin cognitif lié á l'age. Les AMPAR sont des protéines extrêmement dynamiques sujettes á une circulation, un recyclage et/ou une dégradation et à une substitution très contrôlés. Cette régulation active de la synthèse, du ciblage, du temps de maintien synaptique et de la dégradation des AMPAR est fondamentalement importante pour le stockage et la formation de la mémoire. De plus, une circulation aberrante des AMPAR et les modifications préjudiciables qui s'en suivent dans les synapses sont forte-ment impliquées dans de nombreuses maladies cérébrales, ce qui représente un lourd fardeau économique et social. Cet article a pour but de présenter la circulation du récepteur moléculaire et cellulaire AMPA qui contrôle la plasticité et la réactivité synaptiques et de souligner les connaissances actuelles sur les changements de ces processus avec l'age et la maladie.
Introduction
Aging is characterized by a progressive multisystemic deterioration of biological processes that inevitably leads to death. In much of the developed world, improvements in public heath have led to significantly extended average life expectancy. In consequence, a major aim of biomedical science is to reduce or prevent the negative consequences of aging to allow individuals to remain productive, healthy, and fulfilled for as long as possible into old age.
Age-dependent decline in cognitive capacity is one of most challenging aspects of aging research. Even in otherwise healthy individuals, the ability to learn new information and to retrieve existing memory becomes compromised and limits intellectual ability. In neurodegenerative diseases such as Alzheimer's disease (AD) and other dementias, the impact on the quality of life for affected individuals, carers, and families is devastating, and these diseases constitute a huge and growing economic burden on society, with an estimated cost in 2010 in Europe of € 477 billion.1
Surprisingly, although some studies have reported the loss of neurons between adolescence an d old age,2 this appears not to significantly contribute to age-related cognitive impairments. Rather, small, region-specific changes in neuronal morphology and structural plasticity such as dendritic branching and spine density appear to be much more important indicators of age-related memory decline.3,4
What is synaptic plasticity?
In the 1940s the Canadian neuroscientist Donald Hebb proposed that neurons strengthen their communication if the presynaptic cell persistently stimulates the postsynaptic cell. This is often restated as “Neurons that fire together, wire together.” Applied to multiple synapses across a group of neurons, it gave rise to the concept that memories are encoded as engrams, which are biophysical changes to a neuronal network.5 Experimental proof of experience-dependent Hebbian plasticity was first obtained in 1973 when it was shown that repeated stimulation of presynaptic perforant path cells in the hippocampus caused lasting increases in postsynaptic responses in dentate gyrus neurons in anesthetized rabbits.6
A diverse range of Hebbian and non-Hebbian types of plasticity have since been discovered, but can generally be divided into four main classes:
Short-term synaptic plasticity, where activation of a synapse increases or decreases the efficacy of synaptic transmission at that particular synapse for seconds or minutes.
Long-term synaptic plasticity, which is like short-term plasticity but where the synapse-specific changes last from minutes to a lifetime.7
Metaplasticity, where synaptic or cellular activity regulates the capacity of individual synapses to undergo subsequent synaptic plasticity. This is sometimes termed the “plasticity of synaptic plasticity.” 8
Homeostatic plasticity or synaptic scaling, in which a neuron adjusts sensitivity of its excitatory synapses up or down in response to network activity in order to tune synaptic gain and stabilize firing.9
Synaptic plasticity can either potentiate or depress synaptic function, depending on the frequency of activity at that synapse. In general, high-frequency stimulation potentiates synaptic activity, leading to long-term potentiation (LTP), whereas lower-frequency stimulation depresses synaptic activity, leading to long-term depression (LTD). A variety of presynaptic and postsynaptic factors can modulate synaptic strength but, as discussed below, it is widely accepted that synaptic plasticity is predominantly expressed through changes in the number, location, and properties of postsynaptic receptors.
Synaptic plasticity and memory
Both LTP and LTD are cellular mechanisms for learning10,11 and there are pronounced parallels between LTP and memory formation and storage. Both have two mechanistically distinct phases, which take place on very similar time scales. The induction phase of LTP, in which synaptic function is initially enhanced, lasts under an hour. There is then a subsequent maintenance phase, in which the increased synaptic strength is fully established. In memory formation there is also an early phase, corresponding to initial learning, and a mechanistically distinguishable late phase, which corresponds to memory consolidation. The induction phase of LTP and the initial learning process in memory both occur without synthesis of new proteins, relying on post-transiational modifications of proteins already present at sites of potentiation.12 Since these changes are not permanent, and proteins have a limited half-life before they are degraded, the maintenance and consolidation phases of LTP and memory therefore both require de novo protein synthesis.13
Mechanisms of plasticity
The most widely studied forms of plasticity are induced by activation of postsynaptic N-methyl-D-aspartate (NMDA) receptors (NMDARs) and expressed by changes in the number of postsynaptic AMPA receptors (AMPARs).14,15 NMDARs are nonspecific cation channels with a high permeability to Ca2+. Under normal resting membrane potential, however, the channel is blocked by Mg2+ ions and this block is released by membrane depolarization.16,17 This property makes NMDARs coincidence detectors since they require both presynaptic glutamate release and postsynaptic depolarization for activation. The entry of Ca2+ and Na+ ions through the activated NMDAR leads to further depolarization, and when the local intracellular Ca2+ concentration reaches a threshold, signal transduction pathways are initiated that ultimately lead to changes in synaptic responsiveness.
Different patterns of NMDAR activity and spatiotemporal calcium dynamics elicit LTP or LTD. In electrophysiology experiments a train of electrical pulses is generally used to depolarize the neuron with high stimulus frequency to induce a rapid Ca2+ influx for LTP and lower frequency for LTD.18 Strong stimulation of afferent presynaptic neurons in hippocampal slices such as trains of 4 x 100 Hz stimulation with a 200-ms interval between θ bursts causes a rapid and substantial Ca2+ influx at the postsynapse which initiates LTP. This is believed to resemble the physiological activity that takes place in the brain during learning processes.19 In dispersed cultured neurons, it is possible to invoke LTP via activation of synaptic NMDARs with the coagonist, glycine.20 In contrast, a more sustained, lower Ca2+ influx evoked by a high number of low-frequency stimulations, eg, 900 pulses at 0.5-5 Hz, causes LTD.21 In addition, direct activation of NMDARs or Group I metabotropic glutamate (mGlu) receptors can cause LTD.22,23
AMPA receptors
AMPARs mediate the overwhelming majority of fast excitatory neurotransmission in the central nervous system (CNS) and are critically important for nearly all aspects of brain function, including learning, memory, and cognition. They are ligand-gated ion channels composed of combinations of four separate subunits (GluA1-4). AMPARs are highly mobile proteins that undergo constitutive and activity-dependent translocation to; recycling at, and removal from, synapses.24,25 All subunits share a common membrane topology with each other, and with NMDAR and kainate receptor subunits (Figure 1). Complex combinations of signaling pathways regulated by global network activity and by the history of activity at the synapse control the number, synaptic localization, and subunit composition of synaptic AMPARs. Increases in the number as well as changes in the composition and/or properties of synaptic AMPARs mediate LTP and LTD, which occur at synapses throughout the CNS26 (Figure 2). Furthermore, as discussed below, aberrant AMPAR trafficking is implicated in neurodegenerative diseases.
Figure 1. AMPAR subunit topology, interacting partners and diverse intracellular c-termini. A) The membrane topology of an AMPA receptor subunit (AMPAR). AMPAR subunits have large extracellular N-termini, three full transmembrane domains, and a cytoplasmic re-entrant loop, which forms the lining of the channel pore and, in GluA2, contains the RNA editing site that determines calcium permeability. The glutamate binding site is formed by the extracellular N-terminus and the loop between the second and third full transmembrane domains. The intracellular c-terminus differs between subunits and binds numerous proteins required for the trafficking and synaptic expression of AMPARs. B) Summary of GluA1 and GluA2 interacting proteins discussed in the text. See text for details. C) The intracellular c-termini of the predominant isoforms of human AMPAR subunits. Amino acid numbers represent positions in the mature protein lacking the signal peptide. Highlighted in GluA1 and GluA2 are proposed phosphorylation sites (blue) and ubiquitination sites (orange) discussed in the text. Underlined in GluA1 -3 are the c-terminal PDZ ligands required for binding PDZ domain-containing proteins.

Figure 2. Basic principles of AMPAR trafficking and synaptic plasticity. Long-term changes in synaptic function can be induced by activation of postsynaptic N-methyl-D-aspartate (NMDA) receptors, which alter synaptic strength through regulating the number of postsynaptic AMPA receptors (AMPARs). NMDAR activation leads to calcium influx through the receptor, which, depending on the spatiotemporal activation profile, can initiate long-term potentiation (LTP) or long-term depression (LTD). Increased synaptic strength during LTP occurs through an increase in the number of postsynaptic AMPARs, while LTD is characterized by a decrease in postsynaptic AMPAR number. Enhanced AMPAR number during LTP can be mediated through both exocytosis of AMPARs and/or lateral diffusion of AMPARs within the membrane to the synapse. Conversely, LTD leads to AMPAR diffusion away from the synapse and receptor endocytosis.
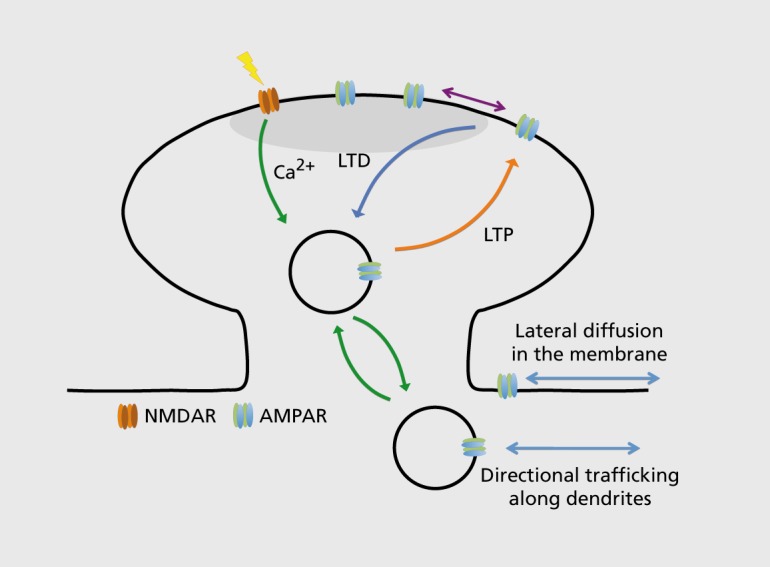
AMPAR subunit composition, assembly, and ER exit
AMPARs assemble in the endoplasmic reticulum (ER) first as dimers, which then come together to form dimers of dimers to make a tetramer.27,28 In adult rat hippocampal neurons AMPARs mainly comprise combinations of GluA1/2 or GluA2/3 subunits,29 and synaptic AMPARs are predominantly combinations of GluA1 and GluA2.30 The GluA2 subunit contains an RNA editing site that replaces the glutamine residue Q607 coded for in the genomic DNA to an arginine residue (Q/R editing) and almost all GluA2 is edited in adult neurons.31 This residue forms part of the channel lining, and the switch to arginine functions both to act as an ER retention motif and to render GluA2-containing AMPARs impermeable to calcium.32-34 GluA1, which lacks this motif, is both calcium permeable and rapidly exported from the ER and trafficked to the plasma membrane.35 Transmembrane AMPAR regulatory proteins (TARPs) which, as discussed below, facilitate correct AMPAR folding and modify channel properties, also participate in export of AMPARs from the ER.36
The intracellular c-terminal domains (tails) of AMPAR subunits can be classified into either long or short tails, which determine their trafficking. GluA1 and GluA4 are long-tailed subunits but GluA4 is expressed mainly during early development and is present only at low levels in adult brain. The trafficking properties of long-tailed AMPAR subunits predominate over those of shorttailed subunits, so receptors containing the GluA1/2 subunit combination exhibit the surface trafficking properties of GluA1. They are rapidly mobilized from the receptor pool in the ER to the surface, as the GluA1 subunit masks the retention sequence in the GluA2 subunit. AMPARs comprising the short-tail subunits GluA2 and GluA3 without GluA1, are trafficked from the ER more slowly.31,37 These receptors also constitu lively recycle to and from the surface to maintain AMPAR numbers.38 In general, GluA1 containing AMPARs are activity-dependently delivered to synapses and are then replaced by GluA2/3, leading to a net increase in synaptic AMPARs in LTP.9-41 (Figure 1).
Calcium-permeable AMPARs and LTP
Q/R edited GluA2-containing AMPARs have negligible Ca2+ permeability.42-44 AMPARs that either lack the GluA2 subunit or contain an unedited version (ie, Ca2+permeable AMPARs; CP- AMPARs, (Figure 3) are initially delivered to perisynaptic sites, and are then translocated to synapses during LTP induction and subsequently replaced by GluA2-containing receptors.45,46 The Ca2+ influx through GluA2-lacking AMPARs appears to drive the insertion of GluA2-containing receptors and this change from Ca2T-permeable to Ca2+ impermeable AMPARs stabilizes LTP.45-47 Until this switch in AMPARs occurs the LTP status of the synapse is labile and susceptible to AMPAR removal by low-frequency stimulation. This early reversible stage in LTP likely corresponds to a fleeting experience that is never laid down as a memory.48
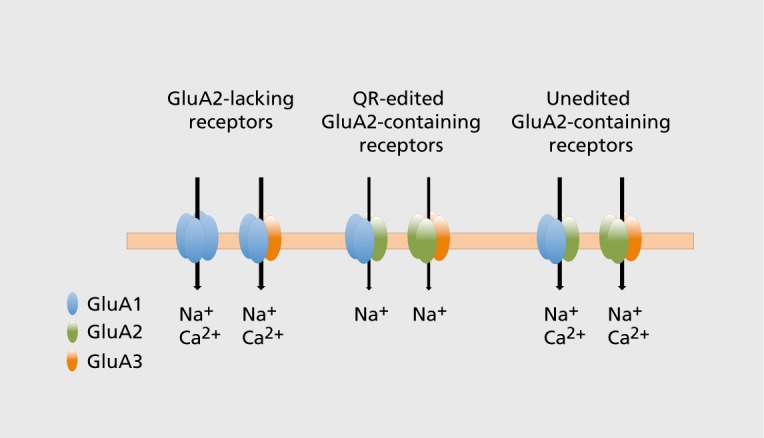
Figure 3. RNA editing of the GluA2 subunit determines calcium permeability of AMPARs. AMPA receptors (AMPARs) lacking the GluA2 subunit, or an unedited GluA2 subunit are calcium-permeable. However, receptors containing an edited GluA2 subunit do not gate calcium. For simplicity, and because their existence in neurons is unclear, GluA3 homomers, which are calcium permeable, and GluA2 homomers, whose calcium permeability depends on the RNA editing state of the GluA2 subunits involved, are not shown. GluA4 (not shown) behaves identically to GluA1.
As with many other aspects of plasticity, the regulation of CP- AMPARs is regulated by phosphorylation. CPAMPARs are incorporated into synapses via a-calciumcalmodulin-dependent protein kinase II (CaMKII)dependent49 and protein kinase C (PKC)-dependent46 mechanisms during early stages of LTP and calcium influx through these receptors is required for the LTPinduced regulation of actin dynamics and spine expansion via activation of the small GTPase Racl and the downstream PAK-LIM kinase pathway.50
Protein phosphorylation in synaptic plasticity
Protein phosphorylation and dephosphorylation is an overarching regulatory mechanism of most cell signal-ing pathways. In neurons in general, and in plasticity in particular, the signaling pathways are especially complex involving multiple kinases and phosphatases. However, despite a wide range of kinases being implicated in LLP and LTD, the core regulatory kinases appear to be CaMKII, mitogen-activated protein kinase (MAPK), protein kinase A (PKA), and isoforms of PKC. Several comprehensive reviews have detailed the roles of AMPAR phosphorylation in plasticity.51-54 Each of the AMPAR subunits GluA1-4 are regulated by phosphorylation. A general rule seems to be that activity-dependent phosphorylation of GluA1 delivers AMPARs to synapses in LLP, whereas GluA1 dephosphorylation is a signal for internalization and LTD. In contrast, PKC phosphorylation of GiuA2 promotes internalization by releasing it from the glutamate receptor anchoring protein (GRIP) and allowing it to bind to the mobilizing protein PICKl.Thus, GluA2 phosphorylation is required for AMPAR internalization and its dephosphorylation is important in synaptic retention.55
Phosphorylation and LTP
CaMKII is necessary and sufficient for LTP.56,57 CaMKII, along with PKC, can phosphorylate the GluA1 subunit at Ser831.58-60 Phosphorylation of Ser831 increases the conductance of homomeric GluA1 and GluA1/2 heteromers in the presence of transmembrane AMPA receptor regulatory proteins (TARPs).61 However, the exact role of Ser831 phosphorylation in vivo is still unclear, since mice lacking phosphorylation at Ser831 still show CaMKII-dependent synaptic insertion and normal hippocampal LTP.62,63
CaMKII also phosphorylates the AMPAR-interacting protein stargazin. Stargazin is one of the TARPs, which are proposed auxiliary AMPAR subunits, and associates with AMPARs, delivering them to, and helping anchor them at, synapses.64 CaMKII phosphorylation of stargazin favors its interaction with the synaptic scaffold protein PSD-95, and this interaction helps anchor AMPARs at synaptic sites.65 Although it remains unclear how CaMKII activation drives the insertion of AMPARs during LTP, it has been reported that the molecular motor protein myosin Va is required for this effect. MyosinVa associates with AMPARs and this interaction is enhanced through activation of the small GTPase Rabll.This mediates the short-range endosomal transport of GluA1-containing receptors from pools in the dendritic shaft, to the spine head where it can be inserted at the synapse during LTP.66
The role of phosphorylation in synaptic plasticity also extends beyond the synapse to enable these changes to persist in the long term. The transcription factor cAMP response element-binding protein (CREB) is important for synthesis of proteins required for LTP consolidation. CREB and other transcription factors are activated via a complex kinase cascade. Calcium entry through NMDARs during the induction stage of LTP increases levels of Ras-GTP, which activates the protein kinase Raf. Activated Raf stimulates MAPK/extraceiiular signal-related kinase (ERK) kinase (MEK), which activates ERK1 and ERK2, which in turn, phosphorylate the transcription factors Eikl and CREB.67 This leads to the synthesis of proteins required for LTP maintenance and memory consolidation.68 The intermediate genes Zif268 and Arc/Arg 3.1 are upregulated by the activation of Elkl and CREB and are specifically connected with the protein synthesis-dependent stage of memory consolidation.69,70
An important aspect of this Ras-ERK signaling pathway is that it is diffusive, allowing downstream effects at locations relatively distant to the initial site of activation. Furthermore, this pathway may be required to recruit AMPARs from distal sites to synapses. AMPAR exocytosis several micrometres away from potentiated synapses is prevented by blocking Ras-ERK signaling, suggesting it initiates AMPAR insertion at relatively distant dendritic regions, ready for incorporation into the synapse.71
The PKC family of serine/threonine kinases participate at different stages in the induction and maintenance of plasticity. LTP expression and memory formation require PKC activity72,73 and activation of PKC can rescue LTP prevented by NMDAR blockade.74 Direct PKC phosphorylation of Ser816 and Ser818 in GluA1 mediates activity-dependent insertion during LTP75 by enhancing binding of GluA1 to the actin cytoskeletal linker protein, 4. IN.76 PKC isoforms generally require both calcium and diacylglycerol for activation, although atypical PKCs (ζ, and ι / λ isoforms) require neither.77 Of these, the constitutively active atypical PKC isoform protein kinase M zeta (PKMζ is of particular interest and has been the focus of intensive research. PKMζ has been dubbed the “memory molecule” since it is proposed to be both necessary and sufficient to maintain potentiated synapses.78,79 In electrophysiology experiments perfusion of PKMζ in a patch pipette has been reported to be sufficient to produce LTP in slices78 and inhibition of PKMζ erases memory and reverses LTP in vivo.80
Intriguingly, inhibition of PKMζ does not block LTP induction. Rather, it prevents maintenance of LTP and can erase established memories without preventing formation of new short-term memories.81 Subsequent studies have suggested that the mechanism of action of PKMζ appears to involve regulation of the GluA2 interacting proteins N-ethymaleimide-sensitive factor (NSF) and PICK1, although the exact mechanisms involved, and the targets of PKMζ which mediate its roles in synaptic plasticity remain unclear.82,83 It should be noted, however that these data remain controversial since they rely mainly on the use of the zeta inhibitory peptide (ZIP) and issues have been raised about the selectivity of ZIP between different PKC isoforms.84
Phosphorylation and LTD
As for AMPAR exocytosis and LTP, the interplay between synaptic phosphorylation and dephosphorylation is central to regulated AMPAR endocytosis and LTD. For example, PKA is located at the postsynaptic density by the anchoring protein AKAP150, which binds directly to PSD-95. Blocking these interactions causes deficits in synaptic transmission85 and inhibits NMDARdependent AMPAR endocytosis and LTD.86 GluA1 is phosphorylated by PKA at Ser845 to regulate the open probability of the channel and promote receptor exocytosis and anchoring at perisynaptic sites.58,87-89 Phosphorylation of Ser845, along with Ser831, appears to “prime” GluA1-containing AMPARs for LTP since, while neither residue appears absolutely required for LTP,63 knock-in mice lacking both of these phosphorylation sites show diminished LTP90 and mice expressing phosphomimetic aspartate residues at these positions show enhanced LTP.91,92 However, dephosphorylation of Ser845 appears important for LTD, since mice lacking phosphorylation at this residue show defects in hippocampal LTD, potentially through phosphorylationmediated regulation of receptor endocytosis.63,89 Another c-terminal GluA1 residue, Thr840 is phosphorylated by PKC93 or p70S6K.94 Dephosphorylation at this site occurs in response to NMDA stimulation94 suggesting a potential role in LTD.
PKC phosphorylation of GluA2 is a major determinant of LTD. Ser880 is located within the GluA2 c-terminal PDZ iigand (see below) responsible for binding to the PDZ domain-containing proteins PICK1 and GRIP. Phosphorylation of Ser880 reduces binding of GRIP1 to GluA2, but leaves PICK1 binding unaffected.95,96 Since GRIP1 binding stabilizes GluA2 at the surface and PICK1 has been proposed to function as a mobilization factor to promote receptor internalization, this differential binding to phosphorylated GluA2 is proposed to underlie GluA2 removal during LTD.97
GluA2 is also phosphorylated by Src family tyrosine kinases at Tyr876, which regulates binding to the guanine-nucleotide exchange factor BRAG2. BRAG2 activates the small GTPase Arf6 and deletion of BRAG2 or inhibition of the GluA2-BRAG2 interaction prevents AMPAR endocytosis and blocks both NMDAR- and mGluR-dependent LTD.98 Phosphorylation of GluA2 at Tyr876 reduces the GluA2-BRAG2 interaction, stabilizing GluA2-containing AMPARs at the surface.
Similarly to LTP, phosphorylation of proteins other than AMPA subunits themselves plays an important role in LTD. For example, the adaptor protein RalBPl promotes receptor endocytosis through binding to the APcomplex and the endocytic proteins epsin and Epsl5. RalBPl binds PSD-95 and the small GTPase RalA, which act in concert to localize RalBPl to dendritic spines. The RalBPl -PSD-95 interaction is negatively regulated by PKA phosphorylation of RalBPl, and NMDA-induced dephosphorylation of RalBPl by protein phosphatase 1 promotes its binding to PSD-95 to recruit RalBPl into spines leading to AMPAR endocytosis.99
Multiple interacting proteins orchestrate AMPAR trafficking
AMPARs are the hub of highly dynamic macromolecular signaling complexes, which consist of a range of direct and indirect interacting proteins that regulate their biosynthesis, trafficking, scaffolding, stability, signaling, and turnover. The core components of the complex vary depending on the location of the AMPAR and the activity of the neuron.
GluA1, 2, and 3 possess a PDZ-binding motif at their extreme c-terminus (Figure 2). These motifs differentially interact with proteins that contain an 80-90 aminoacid PDZ domain (PDZ is an acronym of the first letters of three of the first proteins found to contain this domain).100 PDZ proteins act to bind transmembrane proteins to the cytoskeleton and stabilize signaling complexes.100
GluA1 and GluA2 bind to different subsets of PDZ proteins. Prominent among these are GluA1 binding to synapse-associated protein 97 (SAP97)101 and GluA2 binding to PICK1102 and GRIP.103
The GluA1 interacting protein SAP97 is a member of the membrane-associated guanylate kinase (MAGUK) family of proteins that also includes PSD-95.104 SAP97 links to microtubule-based transport mechanisms via an interaction with the motor protein myosin VI105 and is targeted to spines by CaMKII phosphorylation to deliver GluA1 containing AMPARs.106
PICK1 acts as a Ca2+ sensor and plays important roles in both LTP and LTD. It is involved in the activity-dependent decrease in synaptic GluA2 during NMDARLTD107 and contains a BAR domain that may sense existing membrane curvature, or actively induce the curvature during clathrin-coated pit formation, assisting AMPAR internalization. PICK1 also inhibits Arp2/3mediated actin polymerization to mediate AMPAR internalization during LTD108 and to mediate the decrease in spine size associated with LTD.109 PICK1 shows enhanced localization with Rab5 and early endosomes on induction of NMDAR-LTP,110 and it is involved in mediating the increase in GluA2-lacking CPAMPARs at synapses,111 possibly through the intracellular retention of GiuA2 containing AMPARs.112 Consistent with this, PICK1 knock-down increases the rate of AMPAR recycling to the membrane.113
GRIP also plays an essential role in plasticity. LTD in cerebellar Purkinje cells is abolished in GRIP knockout mice.114 GRIP may have a role in the attachment and anchoring of AMPARs at internal115 and/or surface locations.116 In contrast, PICK1 mobilizes AMPARs and facilitates association with trafficking vesicles. This model explains the importance of these molecules in both forward trafficking to the synapse during LTP, and removal from the synapse during LTD. Additionally, through their interaction with GRIP, AMPARs indirectly bind the heavy chain of the motor protein kinesin117 to direct GluA2-containing AMPARs into dendrites. GRIP also binds to the kinesin KIF1 interacting protein liprin-α118 and to the Arf GTPase-activating protein GIT1.119 These interactions play important roles in AMPAR distribution since inhibiting either reduces AMPAR forward traffic.
AMPAR subunit c-termini also bind to non-PDZ proteins. GluA1 binds to the Ca2+-sensitive actin-based motor protein Myosin Vb120 as well as Myosin Va.66 Myosin Vb transports GluA1-containing AMPAR recycling endosomes to sites of exocytosis. This process couples stimuli that induce LTP to the increased trafficking of cargo necessary for AMPAR insertion and spine enlargement.121
GiuA2 interacts directly with the ATPase NSF at a site upstream from the PDZ motif. The NSF interaction is Ca2+-dependent122 and is required for the maintenance of synaptic AMPARs.123 Blocking NSF binding to GluA2 results in a relatively rapid rundown of AMPAR surface expression under basal non-stimulated conditions with a half-life of around 10 minutes, highlighting the dynamic nature of AMPAR surface expression and recycling.123,124 Mechanisms include the fact that NSF binding blocks the interaction of GiuA2 with the endocytic adaptor protein AP2 to prevent internalization.125 The NSF interaction also disrupts GiuA2/PICKl binding, which prevents PICK1-mediated internalization and intracellular retention of AMPARs to promote their synaptic expression.126
AMPARs are regulated by auxiliary subunits
A growing number of transmembrane proteins have been proposed to associate with AMPAR complexes to function as “auxiliary subunits.” What makes a protein an auxiliary subunit is a matter of debate, but a tentative definition is a protein that forms a stable complex with mature AMPARs.64 TARPs were the first defined family of AMPAR auxiliary subunits and these are critical regulators of several aspects of AMPAR trafficking, pharmacology, and channel kinetics.64,127,128 The prototypic TARP is Stargazin (y-2), which acts as a chaperone protein.128,129 Stargazin mediates AMAPR exit from the ER36,130 stabilizes synaptic AMPARs by binding to the postsynaptic density scaffolding protein PSD-95131 via a process that involves CaMKII phosphorylation,65 and regulates channel properties of surface expressed receptor complexes (for recent reviews on TARP function see refs 64,132).
Subsequent proteomic and homology screens have identified a number of unrelated transmembrane proteins that exhibit similar effects on AMPAR trafficking and are thus putative auxiliary subunits. Cornichon homologs-2 and -3 (CNIH-2 and CNIH-3) have been reported to increase AMPAR surface expression and markedly slow deactivation and desensitization kinetics.133 However, later studies suggest that these proteins act as ER chaperones rather than auxiliary subunits, which associate with the mature, surface-expressed receptor complex.134
Cystine-knot AMPAR modulating protein (CKAMP44) is a brain-specific protein that interacts with ail AMPAR subunits. It is a transmembrane protein with a cysteinerich N-terminai domain.135 It has a widespread distribution in brain but seems to be expressed at relatively low levels. Surprisingly, it seems that CKAMP44 reduces AMPAR currents by extending deactivation and enhancing desensitization. However, the molecular mechanisms that regulate CKAMP44 and its functional consequences on plasticity and memory remain unclear.135
Synapse Differentially Induced Gene 1 (SynDig1) is a transmembrane protein that regulates AMPAR localization at developing hippocampal synapses.136 SynDig1 clusters with GluA2 in cultured neurons and coimmunoprecipitates with GluA2 when expressed in heterologous cells, and with GluA1 and GluA2 from brain extracts. Further, SynDig1 knock-down reduces synapse formation, and surface expression of both GluA1 and GluA2,136 suggesting SynDig1 may represent a potential AMPAR auxiliary subunit with a role in synapse development. However, the relevance of SynDig1 to synaptic plasticity remains to be determined.
AMPAR surface expression and localization at synapses
AMPAR exocytosis and maintenance
The general consensus is that AMPARs are inserted into the plasma membrane close to, but not at, synapses. Once at the surface local lateral diffusion is required for constitutive cycling of AMPARs,137 for the activity-dependent delivery of AMPARs to synapses138 and for the replacement of desensitized AMPARs with functional nondesensitized AMPARs near the synapse to maintain synaptic transmission.139
During LTP induction AMPARs undergo PKA-dependent insertion at perisynaptic sites where they are initially stabilized by actin polymerization and translocate to the synapse on full expression of LTP.48 Following membrane insertion AMPARs can either disperse immediately, increasing the concentration of receptors available for recruitment into spines, or disperse more slowly, contributing to diffuse overall surface pools of receptors.140 Consistent with this, most AMPARs entering spines (70% to 90%) come from receptors already expressed in adjacent areas of dendritic membrane.141,142 One likely method of recruitment is activity-dependent dynamin-mediated endocytosis within spines, which can generate a net inward membrane drift to enhance membrane protein delivery to active spines.143 Even which located at the postsynaptic density AMPARs are highly dynamic and undergo constant recycling. In fact, constant cycles of exocytosis and endocytosis at zones adjacent to the PSD have been proposed to be a major mechanism for retaining AMPARs at synapses.144 AMPARs internalize at endocytic zones (EZs) localized adjacent to the PSD. These EZs are localized through an interaction between the GTPase dynamin-3 and the adaptor protein Homer which, through its interaction with the PSD protein Shank, anchors EZs adjacent to the PSD. Paradoxically, this restricted zone of endocytosis serves to capture AMPARs as they diffuse from the PSD, allowing for them to be locally recycled, thus maintaining synaptic AMPAR number.144 Subsequent work has suggested that localized AMPAR exocytosis occurs at a domain rich in the membrane t-SNARE syntaxin 4 close to the PSD and disruption of syntaxin 4 impairs both spine exocytosis and LTP.145 The combination of localized endo- and exocytosis provides a highly responsive system which allows retention of synaptic AMPARs and provides a dynamic tunable mechanism through which small alterations in the ratio of insertion to internalization can profoundly alter the efficacy of synaptic transmission.
Cell adhesion molecules contribute to anchoring AMPARs at synapses
Trans-synaptic cell adhesion molecules play important roles in the synaptic localization of AMPARs during plasticity.146 N-cadherin is a member of the cadherin family of proteins that mediate Ca2+-dependent adhesion.147 Cadherins rapidly accumulate at points of cell-cell contact prior to synaptic differentiation and disruption of cadherin-based contact inhibits the formation of synapses in primary hippocampai cultures.148 N-cadherin increases surface expression of GluA1149 and a protein complex of N-cadherin, δ-catenin, ABP and GRIP retains GluA2/3 at synapses.150 Additionally, N-cadherin appears to interact with the extracellular N-terminal domain of GiuA2 and disruption of this interaction prevents GluA2-mediated spine enlargement.151
Neurexins and neuroligins are another class of transsynaptic cell-adhesion molecules that play important roles in synapse formation, signaling across the synapse and synaptic function.152 Neuroligin aggregations cluster postsynaptic proteins including GluA2-containing AMPARs153 and disrupting neurexin-neuroligin interactions prevents AMPAR accumulation at synapses.154 Thus, in addition to their structural roles, synaptic adhesion molecules serve to restrict the mobility of AMPARs to regulate synaptic maturation and strength.
AMPAR post-endocytic sorting, degradation pathways, and synaptic plasticity
The sorting events that occur following endocytosis and the regulation of protein degradation are critical aspects of AMPAR trafficking. AMPARs can either be recycled back to the plasma membrane or sorted for lysosomal degradation.155,156 However, the pathways determining whether AMPARs are recycled or degraded have remained elusive. In fact, as outlined below, AMPARs can be degraded by both the ubiquitin-proteasome and ubiquitin-lysosome systems, both of which are strongly implicated in age-related neurodegenerative diseases.
The turnover of many proteins is regulated post-translational modification with the protein ubiquitin. Ubiquitin is conjugated to lysine residues in target proteins through the sequential action of E1, E2, and E3 enzymes. Ubiquitin can target a single lysine in a substrate protein (monoubiquitination) or, through internal lysine residues within ubiquitin itself, form chains (poiyubiquitination), leading to distinct trafficking and degradative pathways.157 It is well established that ubiquitin mediated protein degradation plays a central role in synaptic function and plasticity.158 For example, NMDAR activation can recruit proteasomes to spines and regulate proteasomal function.159 Inhibition or dysfunction of Na+/K+ ATPase causes a rapid decrease in surface expressed and total AMPARs by turnover through proteasome-mediated proteolysis.160 PSD-95 is ubiquitinated in response to NMDAR activation and rapidly degraded by the proteasome. Proteasome inhibitors or mutations that block PSD-95 ubiquitination prevent NMDA-induced AMPAR endocytosis and LTD.161
AMPAR subunits have been reported to be directly ubiquitinated.162-165 Agonist activation induces the Ca2+-sensitive ubiquitination of GluA1 by the E3 ligase Nedd4-1, which specifically binds GluA1, leading to endocytosis and lysosomal degradation.162,164 Ubiquitination occurs primarily at Lys868 and overexpression of Nedd4 enhances GluA1 ubiquitination and decreases AMPAR surface expression.162,164 Knock-down of Nedd4 reduces GluA1 ubiquitination and blocks agonist-induced endocytosis of GiuAl-containing AMPARs.164 Interestingly, GluA1 ubiquitination is specific to agonist stimulation since AMPARs internalized in response to NMDAR activation were not ubiquitinated.162
GluA1 has also been reported to be ubiquitinated in response to EphA4 activation during homeostatic plasticity.166,167 Cdhl, a component of the multi-protein ubiquitin ligase anaphase-promoting complex (APC) binds to and ubiquitinates GluA1 leading to degradation via the ubiquitin/proteasome system.166 Thus, depending on the stimulus and the ligase involved, ubiquitin modification of GluA1 can lead to either endocytosis followed by lysosomal degradation or to degradation by the proteasome.
It has also been reported that GluA2 can be directly and rapidly ubiquitinated in response agonist stimulation or by increasing synaptic activity by antagonizing GABAARs with bicuculline.163 As for GluA1, NMDAR activation does not cause GluA2 ubiquitination but, in contrast to GluA1, ciathrin and dynamin activity is required for GiuA2 ubiquitination suggesting modification occurs after endocytosis.163 Since the currently defined E3s for AMPAR ubiqutination appear to be GiuAl-specific, it will now be important to define the E3s involved in GluA2 ubiquitination and the effects on AMPAR stability, localization and function.
Homeostatic scaling and AMPAR trafficking
Homeostatic scaling is a negative feedback process by which neuronal excitability is adjusted to compensate for changes in network activity.168 Chronically reducing neuronal activity by, for example, preventing action potentials using the sodium channel blocker teterodotoxin (TTX) or blocking NMDA or AMPAR receptors enhances synaptic strength. Conversely, chronic increases in neuronal activity reduce synaptic strength. These homeostatic feedback mechanisms tune neuronal excitability and maintain network activity within a physiologically tractable range. At the postsynaptic membrane homeostatic synaptic scaling is mediated by altering the number of synaptic AMPARs. Many of the trafficking pathways outlined above have been implicated in scaling evoked AMPAR insertion or removal. Importantly, scaling processes are highly relevant to aging and one emerging concept is that inappropriate scaling contributes to the progression of Alzheimer's disease.169
The increase in AMPARs evoked by sustained suppression of synaptic activity exhibits some properties in common with AMPAR increases during LTP. There is an initial insertion of Ca2+-permeable AMPARs and subsequent replacement with GluA2-containing CaCa2+-impermeable AMPARs.170-173 This initial insertion of GluA1 may signal the recruitment of GiuA2 containing receptors since inhibitors of CP- AMPARs block scaling at early, but not at later, timepoints.174 However, some studies have reported the recruitment of both GluA1 and GluA2 in response to suppression of neuronal activity175,176 and GluA2 has been reported to be required for initial synaptic scaling,177 suggesting that the mode of induction of homeostatic scaling, as well as the neuron and synapse type, may determine the AMPAR subunit specificity required.
Various secreted molecules are important for synaptic scaling. Glial cell-derived TNFα increases surface GluA1 followed at later time points by GluA2.178-180 Brainderived neurotrophic factor (BDNF) has differential effects on synaptic scaling depending on the synapse.181,182 Similar to TNFα, BDNF-mediated scaling leads to an initial enhancement of GluA1 surface expression followed by increased GluA2 at later timepoints.183,184 Decreased synaptic activity also increases retinoic acid synthesis and enhances synaptic transmission via increased translation and surface delivery of GluA1 containing AMPARs.172 As with Hebbian plasticity, a complex interplay of kinases and phosphatises contribute to both homeostatic scaling with documented roles for several CaMKII isoforms.185,187
Cell adhesion molecules contribute to the synaptic retention of AMPARs in homeostatic plasticity. Dominant negative N-cadherin reduces TTX-induced upscaling188 and decreased network activity increases surface levels of postsynaptic P-3-integrin, which stabilizes synaptic AMPARs by decreasing GluA2 endocytosis through activation of the GTPase Rapl.189
Homeostatic scaling requires protein synthesis and Arc/Arg3.1 undergoes activity-dependent translation induced by neuronal activity. 9,190 Overexpression or knockdown of Arc respectively up or down regulates basal AMPAR endocytosis via pathways in which Arc interacts with endophilin and dynamin components of the endocytic machinery.191 In vivo levels of Arc control spine density and morphology, and specifically regulate AMPAR trafficking at thin spines.192 As expected of a protein that so intimately controls surface AMPAR number, Arc is also subject to tight post-translational regulation and is modified by both ubiquitin193 and SUMO,194 which act to regulate Arc number and activity, respectively, in order to tune synaptic AMPAR number to neuronal activity.
Synaptic plasticity in normal aging
Cognitive decline, such as mild defects in working or special memory, is an unavoidable consequence of aging. However, while numerous neurodegenerative disorders are characterized by dramatic neuronal cell death, this does not seem to be a characteristic of normal age-related cognitive decline. Rather, it appears that agerelated cognitive decline is mediated through alterations in synaptic number and function in brain regions responsible for memory-related tasks, such as the hippocampus or prefrontal cortex (for reviews see refs 4, 195).
To our knowledge, no studies have directly assessed the trafficking of AMPARs in animal models of normal aging, but the capacity of hippocampal synapses to exhibit plasticity has been investigated. Although the biophysical properties of hippocampal granule or pyramidal neurons seem to be largely unaffected in aging animals,196 depending on the hippocampal synapse examined, aged animals show either a higher threshold for LTP induction197 or a decreased level of LTP induction compared with young animals.198-200 In addition, LTP maintenance is decreased in the dentate gyrus and CA3 of aged rats,201,202 and LTP observed in these animals is more susceptible to depotentiation.203 Thus, while aged animals still exhibit LTP, higher levels of stimulation are required and the potentiation is less stable. Conversely, aged animals show-enhanced induction of LTD at CA3-CA1 synapses, potentially as a result of differences between calcium homeostasis between young and old rats.203
Thus, it seems clear that deficiencies in synaptic plasticity occur during normal aging and these deficits are likely attributable to defects in AMPAR trafficking.
AMPAR trafficking and neural disease
Essentially all age -associated neurological and neurodegenerative disorders involve synaptic abnormalities. A particularly well-studied example of AMPAR dysfunction in disease pathogenesis is Alzheimer's disease (AD). Multiple approaches have been used to model the pathology of AD and common general features of these models are reduced synaptic AMPARs and aberrations in LTP20' and LTD.204,205 Furthermore, disruption of AMPAR trafficking by soluble amyloid beta (Aβ) oligomers is a major causative agent of synaptic dysfunction in AD.206
Aβ treatment of neurons leads to decreased AMPAR surface expression through increased AMPAR endocytosis.207 Interestingly, there are functional similarities between LTD and Aβ-induced AMPAR internalization,208 suggesting these processes may occur through common mechanisms. Synaptic localization of CaMKII is altered in APP transgenic mice and in cultures treated with Aβ oligomers. Knockdown of CaMKII occludes, and CaMKII overexpression blocks the effect of long-term exposure to Aβ on AMPAR surface expression.209 LTD and the Aβ-induced loss of synaptic AMPARs also share other signaling molecules including p38, MAPK, calcineurin (PP2B), and GSK3β.205 Inhibition of calcineurin-mediated AMPAR endocytosis prevents Aβ induced AMPAR internalization and spine loss.207 Similarly, GSK3 inhibition prevents Aβ effects on steady state AMPAR surface expression and delivery of AMPAR into spines following LTP.210
Another route that Aβ interferes AMPAR trafficking appears to be competition with proteolytic maturation of BDNF, which is required for synaptic potentiation associated with classical conditioning.211 The only direct binding partner reported for Aβ oligomers to date is the cellular prion protein (PrP[C]),212 but this accounts for only half of the total oligomer binding. Intriguingly, however, Aβ oligomers preferentially label GluA2-positive spines and crosslinking experiments suggest that the Aβ oligomers bind in close proximity to GiuA2-containing complexes.207 Furthermore, AMPAR antagonists inhibit Aβ oligomer binding and synaptic loss, raising the possibility that Aβ may affect AMPAR trafficking by binding directly to the GluA2 protein complex.
Concluding remarks and future directions
In the last 20 years there has been remarkable progress in the field of AMPAR trafficking. We now understand in increasing molecular detail how AMPARs are inserted into and removed from the plasma membrane, as well as how they diffuse within the membrane to and from the synapses. Impressive though these advances are, much more work is needed before it will be possible to envisage therapeutic strategies for correcting defects in higher brain function associated with aging. For example, it is unclear how memories encoded by synaptic plasticity and network engrains are retained over a lifetime when the synaptic AMPARs that provide the substrate for this information storage have a half-life of about 30 hours. Framed in this way, the surprising fact is that any memories are retained in old age, rather than that there is age-related memory decline. However, recent work has begun to examine the differences in memory formation and synaptic plasticity in various animal models of both normal aging and of neurodegenerative disease. A crucial aspect of future research will therefore involve extending these observations and relating them to what we already know about the trafficking and behaviour of AMPA receptors. Fundamentally, we must seek to define the molecular pathways of AMPAR trafficking that underlie the defects in synaptic plasticity and memory formation associated with cognitive aging and neurodegenerative disease. The challenge of this transition from the observed defects to the unpicking of the molecular detail is not to be underestimated. However, defining the underlying molecular and cellular mechanisms of agedependent alterations in AMPAR trafficking and defining the functional consequences for synaptic transmission represent key long-term goals that hold promise for the development of strategies to combat the memoryloss associated with both normal aging and age-related neurological disorders.
Acknowledgments
We are grateful to the ERC, MRC, and BBSRC for funding.
Selected abbreviations and acronyms
- AMPAR
AMPA receptor
- CaMKII
α-calcium -calmodulin-dependent protein kinase II
- ERK
extracellular signal-related kinase
- Glu
glutamate
- GRIP
glutamate receptor anchoring protein
- LTD
long-term depression
- LTP
long-term potentiation
- MAPK
mitogen-activated protein kinase
- NMDA
N-methyl-D-aspartate
- PKA
protein kinase A
- PKC
protein kinase C
- TARP
transmembrane AMPAR regulatory proteins
Contributor Information
Jeremy M. Henley, School of Biochemistry, MRC Centre for Synaptic Plasticity, University of Bristol, Bristol, UK.
Kevin A. Wilkinson, School of Biochemistry, MRC Centre for Synaptic Plasticity, University of Bristol, Bristol, UK.
REFERENCES
- 1.Gustavsson A., Svensson M., Jacobi F., et al Cost of disorders of the brain in Europe 2010. Eur Neuropsychopharmacol. 2011;21:718–779. doi: 10.1016/j.euroneuro.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 2.Coleman PD., Flood DG. Neuron numbers and dendritic extent in normal aging and Alzheimer's disease. Neurobiol Aging. 1987;8:521–545. doi: 10.1016/0197-4580(87)90127-8. [DOI] [PubMed] [Google Scholar]
- 3.Bergado JA., Almaguer W. Aging and synaptic plasticity: a review. Neural Plast. 2002;9:217–232. doi: 10.1155/NP.2002.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burke SN., Barnes CA. Neural plasticity in the ageing brain. Nat Rev Neurosci. 2006;7:30–40. doi: 10.1038/nrn1809. [DOI] [PubMed] [Google Scholar]
- 5.Hebb DO. The Organization of Behavior: a Neuropsychological Theory. 2002 ed. London, UK: Psychology Press; 1949. [Google Scholar]
- 6.Bliss TV., Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol. 1973;232:331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hughes JR. Post-tetanic potentiation. Physiol Rev. 1958;38:91–113. doi: 10.1152/physrev.1958.38.1.91. [DOI] [PubMed] [Google Scholar]
- 8.Abraham WC., Bear MF. Metaplasticity: the plasticity of synaptic plasticity. Trends Neurosci. 1996;19:126–30. doi: 10.1016/s0166-2236(96)80018-x. [DOI] [PubMed] [Google Scholar]
- 9.Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bliss T., Collingridge G. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 11.Bear MF., Abraham WC. Long-term depression in the hippocampus. Ann Rev Neurosci. 1 996; 1 9:437–462 . doi: 10.1146/annurev.ne.19.030196.002253. [DOI] [PubMed] [Google Scholar]
- 12.Abel T., Lattal KM. Molecular mechanisms of memory acquisition, consolidation and retrieval. CurrOpin Neurobiol. 2001;11:180–187. doi: 10.1016/s0959-4388(00)00194-x. [DOI] [PubMed] [Google Scholar]
- 13.Reymann KG., Frey JU. The late maintenance of hippocampal LTP: Requirements, phases,'synaptic tagging', 'late-associativity' and implications. Neuropharmacology. 2007;52:24–40. doi: 10.1016/j.neuropharm.2006.07.026. [DOI] [PubMed] [Google Scholar]
- 14.Morris RGM., Anderson E., Lynch GS., Baudry M. Selective impairment of learning and blockade of LTP by NMDA receptor antagonist, AP5. Nature. 1986;319:774–776. doi: 10.1038/319774a0. [DOI] [PubMed] [Google Scholar]
- 15.Dudek SM., Bear MF. Homosynaptic long-term depression in area ca1 of hippocampus and effects of N-methyl-D-aspartate receptor blockade. Proc Natl Acad Sci USA. 1992;89:4363–4367. doi: 10.1073/pnas.89.10.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nowak L., Bregestovski P., Ascher P., Herbert A., Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- 17.Mayer ML., Westbrook GL., Guthrie PB. Voltage-dependent block by Mg2+of NMDA responses in spinal cord neurones. Nature. 1984;309:261–263. doi: 10.1038/309261a0. [DOI] [PubMed] [Google Scholar]
- 18.Ismailov I., Kalikulov D., Inoue T., Friedlander MJ. The kinetic profile of intracellular calcium predicts long-term potentiation and long-term depression. J Neurosci. 2004;24:9847–9861. doi: 10.1523/JNEUROSCI.0738-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Larson J., Lynch G. Induction of synaptic potentiation in hippocampus by patterned stimulation involves two events. Science. 1986;232:985–988. doi: 10.1126/science.3704635. [DOI] [PubMed] [Google Scholar]
- 20.Lu W., Man H., Ju W., Trimble WS., MacDonald JF., Wang YT. Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron. 2001;29:243–254. doi: 10.1016/s0896-6273(01)00194-5. [DOI] [PubMed] [Google Scholar]
- 21.Yang SN., Tang YG., Zucker RS. Selective induction of LTP and LTD by postsynaptic [ca2+]i elevation. J Neurophysiol. 1999;81:781–787. doi: 10.1152/jn.1999.81.2.781. [DOI] [PubMed] [Google Scholar]
- 22.Li R., Dozmorov M., Hellberg F., Tian Y., Jilderos B., Wigstrom H. Characterization of NMDA induced depression in rat hippocampus: involvement of AMPA and NMDA receptors. Neurosci Lett. 2004;357:87–90. doi: 10.1016/j.neulet.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 23.Snyder EM., Philpot BD., Huber KM., Dong X., Fallon JR., Bear MF. Internalization of ionotropic glutamate receptors in response to mGluR activation. Nat Neurosci. 2001;4:1079–1085. doi: 10.1038/nn746. [DOI] [PubMed] [Google Scholar]
- 24.Henley JM., Barker EA., Glebov OO. Routes, destinations and delays: recent advances in AMPA receptor trafficking. Trends Neurosci. 2011;34:258–268. doi: 10.1016/j.tins.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anggono V., Huganir RL. Regulation of AMPA receptor trafficking and synaptic plasticity. Curr Opin Neurobiol. 2012;22:461–469. doi: 10.1016/j.conb.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malenka RC., Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 27.Ayalon G., Stern-Bach Y. Functional assembly of AMPA and kainate receptors is mediated by several discrete protein-protein interactions. Neuron. 2001;31:103–113. doi: 10.1016/s0896-6273(01)00333-6. [DOI] [PubMed] [Google Scholar]
- 28.Tichelaar W., Safferling M., Keinanen K., Stark H., Madden DR. The three-dimensional structure of an ionotropic glutamate receptor reveals a dimer-of-dimers assembly. J Mol Biol. 2004;344:435–442. doi: 10.1016/j.jmb.2004.09.048. [DOI] [PubMed] [Google Scholar]
- 29.Wenthold RJ., Petralia RS., Blahos J., II, Niedzielski AS. Evidence for multiple AMPA receptor complexes in hippocampal Ca1/Ca2 neurons. J Neurosci. 1996;16:1982–1989. doi: 10.1523/JNEUROSCI.16-06-01982.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu W., Shi Y., Jackson AC., et al Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuion. 2009;62:254–268. doi: 10.1016/j.neuron.2009.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Greger IH., Khatri L., Kong X., Ziff EB. AMPA receptor tetramerization is mediated by Q/R editing. Neuron. 2003;40:763–774. doi: 10.1016/s0896-6273(03)00668-8. [DOI] [PubMed] [Google Scholar]
- 32.Sommer B., Kohler M., Sprengel R., Seeburg PH. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell. 1991;67:11–19. doi: 10.1016/0092-8674(91)90568-j. [DOI] [PubMed] [Google Scholar]
- 33.Swanson GT., Kamboj SK., Cull-Candy SG. Single-channel properties of recombinant AMPA receptors depend on RNA editing, splice variation, and subunit composition. J Neurosci. 1997;17:58–69. doi: 10.1523/JNEUROSCI.17-01-00058.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Greger IH., Ziff EB., Penn AC. Molecular determinants of AMPA receptor subunit assembly. Trends Neurosci. 2007;30:407–416. doi: 10.1016/j.tins.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 35.Greger IH., Khatri L., Ziff EB. RNA editing at ARG607 controls AMPA receptor exit from the endoplasmic reticulum. Neuron. 2002;34:759–772. doi: 10.1016/s0896-6273(02)00693-1. [DOI] [PubMed] [Google Scholar]
- 36.Vandenberghe W., Nicoll RA., Bredt DS. Interaction with the unfolded protein response reveals a role for stargazin in biosynthetic AMPA receptor transport. J Neurosci. 2005;25:1095–1102. doi: 10.1523/JNEUROSCI.3568-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mah SJ., Cornell E., Mitchell NA., Fleck MW. Glutamate receptor trafficking: endoplasmic reticulum quality control involves ligand binding and receptor function. J Neurosci. 2005;25:221 5–2225. doi: 10.1523/JNEUROSCI.4573-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Araki Y., Lin DT., Huganir RL. Plasma membrane insertion of the AMPA receptor GLUA2 subunit is regulated by NSF binding and Q/R editing of the ion pore. Proc Natl Acad Sci U S A. 2010;107:11080–11085. doi: 10.1073/pnas.1006584107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Passafaro M., Piech V., Sheng M. Subunit-specif ic temporal and spatial patterns of AMPA receptor exocytosis in hippocampal neurons. Nat Neurosci. 2001;4:917–926. doi: 10.1038/nn0901-917. [DOI] [PubMed] [Google Scholar]
- 40.Piccini A., Malinow R. Critical postsynaptic density 95/disc large/zonula occludens-1 interactions by glutamate receptor 1 (GLUR1) and GLUR2 required at different subcellular sites. J Neurosci. 2002;22:5387–5392. doi: 10.1523/JNEUROSCI.22-13-05387.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi S., Hayashi Y., Esteban JA., Malinow R. Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell. 2001;105:331–343. doi: 10.1016/s0092-8674(01)00321-x. [DOI] [PubMed] [Google Scholar]
- 42.Isaac JT., Ashby M., McBain CJ. The role of the GLUR2 subunit in AMPA receptor function and synaptic plasticity. Neuron. 2007;54:859–871. doi: 10.1016/j.neuron.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 43.Liu SJ., Zukin RS. Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 2007;30:126–134. doi: 10.1016/j.tins.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 44.Cull-Candy S., Kelly L., Farrant M. Regulation of Ca2+-permeable AMPA receptors: synaptic plasticity and beyond. Curr Opin Neurobiol. 2006;16:288–297. doi: 10.1016/j.conb.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 45.Plant K., Pelkey KA., Bortolotto ZA., et al Transient incorporation of native glur2-lacking AMPA receptors during hippocampal long-term potentiation. Nat Neurosci. 2006;9:602–604. doi: 10.1038/nn1678. [DOI] [PubMed] [Google Scholar]
- 46.Yang Y., Wang XB., Zhou Q. Perisynaptic GLUR2-lacking AMPA receptors control the reversibility of synaptic and spines modifications. Proc Natl Acad SciUSA. 2010;107:11999–12004. doi: 10.1073/pnas.0913004107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Asrar S., Zhou Z., Ren W., Jia Z. Ca(2+) permeable AMPA receptor induced long-term potentiation requires PI3/MAP kinases but not Ca/cam-dependent kinase II. PLoS One. 2009;4:e4339. doi: 10.1371/journal.pone.0004339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang Y., Wang XB., Frerking M., Zhou Q. Delivery of AMPA receptors to perisynaptic sites precedes the full expression of long-term potentiation. Proc Natl Acad Sci USA. 2008;105:11388–11393. doi: 10.1073/pnas.0802978105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guire ES., Oh MC., Soderling TR., Derkach VA. Recruitment of calciumpermeable AMPA receptors during synaptic potentiation is regulated by CAM-KINASE I. J Neurosci. 2008;28:6000–6009. doi: 10.1523/JNEUROSCI.0384-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fortin DA., Davare MA., Srivastava T., et al Long-term potentiationdependent spine enlargement requires synaptic Ca2+-permeable AMPA receptors recruited by Cam-Kinase I. J Neurosci. 2010;30:11 565–11 575. doi: 10.1523/JNEUROSCI.1746-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang JQ., Arora A., Yang L., Parelkar NK., Zhang G., Liu X., et al Phosphorylation of AMPA receptors: mechanisms and synaptic plasticity. Mol Neurobiol. 2005;32:237–49. doi: 10.1385/MN:32:3:237. [DOI] [PubMed] [Google Scholar]
- 52.Lee HK. Synaptic plasticity and phosphorylation. Pharmacol Ther. 2006;112:810–832. doi: 10.1016/j.pharmthera.2006.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Santos SD., Carvalho AL., Caldeira MV., Duarte CB. Regulation of AMPA receptors and synaptic plasticity. Neuroscience . 2009;158:105–125. doi: 10.1016/j.neuroscience.2008.02.037. [DOI] [PubMed] [Google Scholar]
- 54.Lu W., Roche KW. Posttranslational regulation of AMPA receptor trafficking and function. CurrOpin Neurobiol. 2012;22:470–479. doi: 10.1016/j.conb.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Steinberg JP., Takamiya K., Shen Y., et al Targeted in vivo mutations of the AMPA receptor subunit Glur2 and its interacting protein pickl eliminate cerebellar long-term depression. Neuron. 2006;49:845–860. doi: 10.1016/j.neuron.2006.02.025. [DOI] [PubMed] [Google Scholar]
- 56.Malinow R., Schulman H., Tsien RW. Inhibition of postsynaptic PKC or CAMKII blocks induction but not expression of LTP. Science. 1989;245:862–866. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- 57.Lledo PM., Hjelmstad GO., Mukherji S., Soderling TR., Malenka RC., Nicoll RA. Calcium/calmodulin-dependent kinase II and long-term potentiation enhance synaptic transmission by the same mechanism. Proc Natl Acad Sci USA. 1995;92:11175–11179. doi: 10.1073/pnas.92.24.11175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Roche KW., O'Brien RJ., Mammen AL., Bernhardt J., Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GLUR1 subunit. Neuron. 1996;16:1179–1188. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- 59.Mammen AL., Kameyama K., Roche KW., Huganir RL. Phosphorylation of the alpha-amino-3-hydroxy-5-methylisoxazole4- propionic acid receptor GLUR1 subunit by calcium/calmodulin-dependent kinase II. J Biol Chem. 1997;272:32528–32533. doi: 10.1074/jbc.272.51.32528. [DOI] [PubMed] [Google Scholar]
- 60.Barria A., Muller D., Derkach V., Griffith LC., Soderling TR. Regulatory phosphorylation of AMPA-type glutamate receptors by CAM-KM during long-term potentiation. Science. 1997;276:2042–2045. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- 61.Kristensen AS., Jenkins MA., Banke TG., et al Mechanism of Ca2+/Calmodulin-Dependent Kinase II regulation of AMPA receptor gating. Nat Neurosci. 2011;14:727–735. doi: 10.1038/nn.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hayashi Y., Shi SH., Esteban JA., Piccini A., Poncer JC., Malinow R. Driving AMPA receptors into synapses by LTP and CAMKII: Requirement for glurl and pdz domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- 63.Lee H-K., Takamiya K., He K., Song L., Huganir RL. Specific roles of AMPA receptor subunit GLUR1 (GLUA1) phosphorylation sites in regulating synaptic plasticity in the Ca1 region of hippocampus. J Neurophysiol. 2010;103:479–489. doi: 10.1152/jn.00835.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jackson AC., Nicoll RA. The expanding social network of ionotropic glutamate receptors: TARPS and other transmembrane auxiliary subunits. Neuron. 2011;70:178–199. doi: 10.1016/j.neuron.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Opazo P., Labrecque S., Tigaret CM., et al CAMKII triggers the diffusional trapping of surface AMPARs through phosphorylation of stargazin. Neuron. 2010;67:239–252. doi: 10.1016/j.neuron.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 66.Correia SS., Bassani S., Brown TC., et al Motor protein-dependent transport of AMPA receptors into spines during long-term potentiation. Nat Neurosci. 2008;11:457–466. doi: 10.1038/nn2063. [DOI] [PubMed] [Google Scholar]
- 67.Davis S., Vanhoutte P., Pages C., Caboche J., Laroche S. The mapk/erk cascade targets both ELK-1 and CAMP response element-binding protein to control long-term potentiation-dependent gene expression in the dentate gyrus in vivo. J Neurosci. 2000;20:4563–4572. doi: 10.1523/JNEUROSCI.20-12-04563.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schafe GE., Swank MW., Rodrigues SM., Debiec J., Doyere V. Phosphorylation of ERK/MAP kinase is required for long-term potentiation in anatomically restricted regions of the lateral amygdala in vivo. Learn Mem. 2008;15:55–62. doi: 10.1101/lm.746808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lonergan ME., Gafford GM., Jarome TJ., Helmstetter FJ. Time-dependent expression of ARC and ZIF268 after acquisition of fear conditioning. Neural Plast. 2010;2010:139891. doi: 10.1155/2010/139891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ploski JE., Pierre VJ., Smucny J., et al The activity-regulated cytoskeletalassociated protein (ARC/ARG3.1) is required for memory consolidation of Pavlovian fear conditioning in the lateral amygdala. J Neurosci. 2008;28:12383–12395. doi: 10.1523/JNEUROSCI.1662-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Patterson MA., Szatmari EM., Yasuda R. AMPA receptors are exocytosed in stimulated spines and adjacent dendrites in a RAS-ERK-dependent manner during long-term potentiation. Proc Natl Acad SciUSA. 2010;107:15951–15956. doi: 10.1073/pnas.0913875107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Colley PA., Sheu FS., Routtenberg A. Inhibition of protein kinase C blocks two components of LTP persistence, leaving initial potentiation intact. J Neurosci. 1990;10:3353–3360. doi: 10.1523/JNEUROSCI.10-10-03353.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lovinger DM., Routtenberg A. Synapse-specific protein kinase C activation enhances maintenance of long-term potentiation in rat hippocampus. J Physiol. 1988;400:321–333. doi: 10.1113/jphysiol.1988.sp017122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kleschevnikov AM., Routtenberg A. PKC activation rescues LTP from NMDA receptor blockade. Hippocampus. 2001;11:168–175. doi: 10.1002/hipo.1034. [DOI] [PubMed] [Google Scholar]
- 75.Boehm J., Kang MG., Johnson RC., Esteban J., Huganir RL., Malinow R. Synaptic incorporation of AMPA receptors during LTP is controlled by a PKC phosphorylation site on GLURL. Neuron. 2006;51:213–225. doi: 10.1016/j.neuron.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 76.Lin DT., Makino Y., Sharma K., et al Regulation of AMPA receptor extrasynaptic insertion by 4.1 n, phosphorylation and palmitoylation. Nat Neurosci. 2009;12:879–887. doi: 10.1038/nn.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ohno S., Nishizuka Y. Protein kinase C isotypes and their specific functions: prologue. J Biochem. 2002;132:509–511. doi: 10.1093/oxfordjournals.jbchem.a003249. [DOI] [PubMed] [Google Scholar]
- 78.Ling DSF., Benardo LS., Serrano PA., et al Protein kinase MZETA is necessary and sufficient for LTP maintenance. Nat Neurosci. 2002;5:295–296. doi: 10.1038/nn829. [DOI] [PubMed] [Google Scholar]
- 79.Sacktor TC. PKMZETA, LTP maintenance, and the dynamic molecular biology of memory storage. Prog Brain Res. 2008;169:27–40. doi: 10.1016/S0079-6123(07)00002-7. [DOI] [PubMed] [Google Scholar]
- 80.Shema R., Sacktor TC., Dudai Y. Rapid erasure of long-term memory associations in the cortex by an inhibitor of PKM zeta. Science. 2007;317:951–953. doi: 10.1126/science.1144334. [DOI] [PubMed] [Google Scholar]
- 81.Pastalkova E., Serrano P., Pinkhasova D., Wallace E., Fenton AA., Sacktor TC. Storage of spatial information by the maintenance mechanism of LTP. Science. 2006;313:1141–1144. doi: 10.1126/science.1128657. [DOI] [PubMed] [Google Scholar]
- 82.Yao Y., Kelly MT., Sajikumar S., Serrano P., Tian D., Bergold PJ., et al PKM zeta maintains late long-term potentiation by N-ethylmaleimide-sensitive factor/GLUR2-dependent trafficking of postsynaptic AMPA receptors. J Neurosci. 2008;28:7820–7827. doi: 10.1523/JNEUROSCI.0223-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Migues PV., Hardt O., Wu DC., et al PKMzeta maintains memories by regulating glur2-dependent AMPA receptor trafficking. Nat Neurosci. 2010;13:630–634. doi: 10.1038/nn.2531. [DOI] [PubMed] [Google Scholar]
- 84.Wu-Zhang AX., Schramm CL., Nabavi S., Malinow R., Newton AC. Cellular pharmacology of protein kinase MZETA (PKMZETA) contrasts with its in vitro profile: Implications for pkmzeta as a mediator of memory. J Biol Chem. 2012;287:12879–12895. doi: 10.1074/jbc.M112.357244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tunquist BJ., Hoshi N., Guire ES., Zhang F., Mullendorff K., Langeberg LK., et al Loss of AKAP1 50 perturbs distinct neuronal processes in mice. Proc Natl Acad Sci US A. 2008;105:12557–12562. doi: 10.1073/pnas.0805922105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bhattacharyya S., Biou V., Xu W., Schluter O., Malenka RC. A critical role for Psd-95/Akap interactions in endocytosis of synaptic AMPA receptors. Wat. Neurosci. 2009;12:172–181. doi: 10.1038/nn.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Banke TG., Bowie D., Lee H., Huganir RL., Schousboe A., Traynelis SF. Control of GLUR1 AMPA receptor function by camp-dependent protein kinase. J Neurosci. 2000;20:89–102. doi: 10.1523/JNEUROSCI.20-01-00089.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Oh MC., Derkach VA., Guire ES., Soderling TR. Extrasynaptic membrane trafficking regulated by GLUR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J Biol Chem. 2006;281:752–758. doi: 10.1074/jbc.M509677200. [DOI] [PubMed] [Google Scholar]
- 89.He K., Song L., Cummings LW., Goldman J., Huganir RL., Lee HK. Stabilization of Ca2+-permeable AMPA receptors at perisynaptic sites by GLUR1-S845 phosphorylation. Proc Natl Acad Sci U S A. 2009;106:20033–20038. doi: 10.1073/pnas.0910338106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lee HK., Takamiya K., Han JS., et al Phosphorylation of the AMPA receptor GLUR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell. 2003;112:631–643. doi: 10.1016/s0092-8674(03)00122-3. [DOI] [PubMed] [Google Scholar]
- 91.Makino Y., Johnson RC., Yu Y., Takamiya K., Huganir RL. Enhanced synaptic plasticity in mice with phosphomimetic mutation of the GLUA1 AMPA receptor. Proc Natl Acad Sci USA. 201 1;108:8450–8455. doi: 10.1073/pnas.1105261108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hu XD., Huang Q., Yang X., Xia H. Differential regulation of AMPA receptor trafficking by neurabin-targeted synaptic protein phosphatase-1 in synaptic transmission and long-term depression in hippocampus. J Neurosci. 2007;27:4674–4686. doi: 10.1523/JNEUROSCI.5365-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee HK., Takamiya K., Kameyama K., et al Identification and characterization of a novel phosphorylation site on the glurl subunit of AMPA receptors. Mol Cell Neurosci. 2007;36:86–94. doi: 10.1016/j.mcn.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Delgado JY., Coba M., Anderson CN., et al NMDA receptor activation dephosphorylates AMPA receptor glutamate receptor 1 subunits at threonine 840. J Neurosci. 2007;27:13210–13221. doi: 10.1523/JNEUROSCI.3056-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Perez JL., Khatri L., Chang C., Srivastava S., Osten P., Ziff EB. PICK1 targets activated protein kinase calpha to AMPA receptor clusters in spines of hippocampal neurons and reduces surface levels of the AMPA- type glutamate receptor subunit. 2. J Neurosci. 2001;21:5417–5428. doi: 10.1523/JNEUROSCI.21-15-05417.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Seidenman KJ., Steinberg JP., Huganir R., Malinow R. Glutamate receptor subunit 2 serine 880 phosphorylation modulates synaptic transmission and mediates plasticity in Ca1 pyramidal cells. J Neurosci. 2003;23:9220–9228. doi: 10.1523/JNEUROSCI.23-27-09220.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Palmer CL., Cotton L., Henley JM. The molecular pharmacology and cell biology of alpha-am ino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors. Pharmacol Rev. 2005;57:253–277. doi: 10.1124/pr.57.2.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Scholz R., Berberich S., Rathgeber L., Kolleker A., Kohr G., Kornau HC. AMPA receptor signaling through BRAG2 and ARF6 critical for long-term synaptic depression. Neuron. 2010;66:768–780. doi: 10.1016/j.neuron.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 99.Han K., Kim MH., Seeburg D., et al Regulated RALBP1 binding to RALA and PSD-95 controls AMPA receptor endocytosis and LTD. PLoS Biol. 2009;7:e1000187. doi: 10.1371/journal.pbio.1000187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kim E., Sheng M. PDZ domain proteins of synapses. Wat. Rev Neurosci. 2004;5:771–781. doi: 10.1038/nrn1517. [DOI] [PubMed] [Google Scholar]
- 101.Leonard AS., Davare MA., Home MC., Garner CC., Hell JW. SAP97 is associated with the alpha-amino-3-hydroxy-5-methylisoxazole-4- propionic acid receptor GLUR1 subunit. J BiolChem. 1998;273:19518–19524. doi: 10.1074/jbc.273.31.19518. [DOI] [PubMed] [Google Scholar]
- 102.Staudinger J., Zhou J., Burgess R., Elledge SJ., Olson EN. PICK1: a perinuclear binding protein and substrate for protein kinase c isolated by the yeast two-hybrid system. J Cell Biol. 1995;128:263–271. doi: 10.1083/jcb.128.3.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dong H., O'Brien RJ., Fung ET., Lanahan AA., Worley PF., Huganir RL. GRIP: a synaptic Pdz domain-containing protein that interacts with AMPA receptors [see comments]. Nature. 1997;386:279–284. doi: 10.1038/386279a0. [DOI] [PubMed] [Google Scholar]
- 104.Montgomery JM., Zamorano PL., Garner CC. Maguks in synapse assembly and function: an emerging view. Cell Mol Life Sci. 2004;61:911–929. doi: 10.1007/s00018-003-3364-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wu H., Nash JE., Zamorano P., Garner CC. Interaction of SAP97 with minus-end-directed actin motor myosin vi. Implications for AMPA receptor trafficking. J Biol Chem. 2002;277:30928–30934. doi: 10.1074/jbc.M203735200. [DOI] [PubMed] [Google Scholar]
- 106.auceri D., Cattabeni F., Di Luca M., Gardoni F. Calcium/calmodulindependent protein kinase II phosphorylation drives synapse-associated protein 97 into spines. J Biol Chem. 2004;279:23813–23821. doi: 10.1074/jbc.M402796200. [DOI] [PubMed] [Google Scholar]
- 107.anley JG., Henley JM. Pickl is a calcium-sensor for NMDA-induced AMPA receptor trafficking. Embo J. 2005;24:3266–3278. doi: 10.1038/sj.emboj.7600801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.occa DL., Martin S., Jenkins EL., Hanley JG. Inhibition of ARP2/3-mediated actin polymerization by pickl regulates neuronal morphology and AMPA receptor endocytosis. Nat Cell Biol. 2008;10:259–271. doi: 10.1038/ncb1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nakamura Y., Wood CL., Patton AP., et al Pickl inhibition of the ARP2/3 complex controls dendritic spine size and synaptic plasticity. EMBO J. 2011;30:719–730. doi: 10.1038/emboj.2010.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sossa KG., Court BL., Carroll RC. NMDA receptors mediate calcium-dependent, bidirectional changes in dendritic PICK1 clustering. Mol Cell Neurosci. 2006;31:574–585. doi: 10.1016/j.mcn.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 111.Terashima A., Pelkey KA., Rah JC., et al An essential role for PICK1 in NMDA receptor-dependent bidirectional synaptic plasticity. Neuron. 2008;57:872–882. doi: 10.1016/j.neuron.2008.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jaafari N., Henley JM., Hanley JG. PICK1 mediates transient synaptic expression of GLUA2-lacking AMPArs during LTP. J Neurosci. 201 2;32:11618–11630. doi: 10.1523/JNEUROSCI.5068-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lin DT., Huganir RL. PICK1 and phosphorylation of the glutamate receptor 2 (GLUR2) AMPA receptor subunit regulates GLUR2 recycling after NMDA receptor-induced internalization. J Neurosci. 2007;27:13903–13908. doi: 10.1523/JNEUROSCI.1750-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Takamiya K., Mao L., Huganir RL., Linden DJ. The glutamate receptorinteracting protein family of GLUR2-binding proteins is required for longterm synaptic depression expression in cerebellar Purkinje cells. J Neurosci. 2008;28:5752–5755. doi: 10.1523/JNEUROSCI.0654-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Daw Ml., Chittajallu R., Bortolotto ZA., et al PDZ proteins interacting with C-terminal GLUR2/3 are involved in a Pkc- dependent regulation of AMPA receptors at hippocampal synapses. Neuron. 2000;28:873–886. doi: 10.1016/s0896-6273(00)00160-4. [DOI] [PubMed] [Google Scholar]
- 116.Mao L., Takamiya K., Thomas G., Lin DT., Huganir RL. GRIP1 and 2 regulate activity-dependent AMPA receptor recycling via exocyst complex interactions. Proc Natl Acad Sci U S A. 2011;107:19038–19043. doi: 10.1073/pnas.1013494107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Setou M., Seog DH., Tanaka Y., et al Glutamate-receptor-interacting protein GRIP1 directly steers kinesin to dendrites. Nature. 2002;417:83–87. doi: 10.1038/nature743. [DOI] [PubMed] [Google Scholar]
- 118.Shin H., Wyszynski M., Huh KH., et al Association of the kinesin motor KIF1A with the multimodular protein liprin-alpha. J Biol Chem. 2003;278:11393–11401. doi: 10.1074/jbc.M211874200. [DOI] [PubMed] [Google Scholar]
- 119.Ko J., Kim S., Valtschanoff JG., et al Interaction between liprin-alpha and GIT1 is required for AMPA receptor targeting. J Neurosci. 2003;23:1667–1677. doi: 10.1523/JNEUROSCI.23-05-01667.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lise MF., Wong TP., Trinh A., et al Involvement of myosin VB in glutamate receptor trafficking. J Biol Chem. 2006;28:3669–3678. doi: 10.1074/jbc.M511725200. [DOI] [PubMed] [Google Scholar]
- 121.Wang Z., Edwards JG., Riley N., et al Myosin VB mobilizes recycling endosomes and AMPA receptors for postsynaptic plasticity. Cell. 2008;135:535–548. doi: 10.1016/j.cell.2008.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hanley JG. NSF binds calcium to regulate its interaction with AMPA receptor subunit GLUR2. J Neuron. 2007;101:1644–1650. doi: 10.1111/j.1471-4159.2007.04455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nishimune A., Isaac JT., Molnar E., et al NSF binding to GLUR2 regulates synaptic transmission. Neuron. 1998;21:87–97. doi: 10.1016/s0896-6273(00)80517-6. [DOI] [PubMed] [Google Scholar]
- 124.Noel J., Ralph GS., Pickard L., et al Surface expression of AMPA receptors in hippocampal neurons is regulated by an NSF-dependent mechanism. Neuron. 1999;23:365–376. doi: 10.1016/s0896-6273(00)80786-2. [DOI] [PubMed] [Google Scholar]
- 125.Lee SH., Liu L., Wang YT., Sheng M. Clathrin adaptor AP2 and NSF interact with overlapping sites of GLUR2 and play distinct roles in AMPA receptor trafficking and hippocampal LTD. Neuron. 2002;36:661–674. doi: 10.1016/s0896-6273(02)01024-3. [DOI] [PubMed] [Google Scholar]
- 126.Hanley JG., Khatri L., Hanson PI., Ziff EB. NSF ATPASE and a-/(3-snaps disassemble the AMPA receptor-PICK1 complex. Neuron. 2002;34:53–67. doi: 10.1016/s0896-6273(02)00638-4. [DOI] [PubMed] [Google Scholar]
- 127.Ziff EB. Tarps and the AMPA receptor trafficking paradox. Neuron. 2007;53:627–633. doi: 10.1016/j.neuron.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 128.ato AS., Gill MB., Yu H., Nisenbaum ES., Bredt DS. TARPS differentially decorate AMPA receptors to specify neuropharmacology. Trends Neurosci. 2010;33:241–248. doi: 10.1016/j.tins.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 129.Nicoll RA., Tomita S., Bredt DS. Auxiliary subunits assist AMPA-type glutamate receptors. Science. 2006;311:1253–1256. doi: 10.1126/science.1123339. [DOI] [PubMed] [Google Scholar]
- 130.Vandenberghe W., Nicoll RA., Bredt DS. Stargazin is an AMPA receptor auxiliary subunit. Proc Natl Acad Sci U S A. 2005;102:485–490. doi: 10.1073/pnas.0408269102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bats C., Groc L., Choquet D. The interaction between stargazin and PSD95 regulates AMPA receptor surface trafficking. Neuron. 2007;53:719–734. doi: 10.1016/j.neuron.2007.01.030. [DOI] [PubMed] [Google Scholar]
- 132.Straub C., Tomita S. The regulation of glutamate receptor trafficking and function by TARPS and other transmembrane auxiliary subunits. Curr Opin Neurobiol. 2012;22:488–495. doi: 10.1016/j.conb.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Schwenk J., Harmel N., Zolles G., et al Functional proteomics identify cornichon proteins as auxiliary subunits of AMPA receptors. Science. 2009;323:1313–1319. doi: 10.1126/science.1167852. [DOI] [PubMed] [Google Scholar]
- 134.Shi Y., Suh YH., Milstein AD., et al Functional comparison of the effects of TARPS and cornichons on AMPA receptor trafficking and gating. Proc Natl Acad Sci US A. 2010;107:16315–16319. doi: 10.1073/pnas.1011706107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.von Engelhardt J., Mack V., Sprengel R., et al CKAMP44: a brain-specific protein attenuating short-term synaptic plasticity in the dentate gyrus. Science. 2010;327:1518–2152. doi: 10.1126/science.1184178. [DOI] [PubMed] [Google Scholar]
- 136.Kalashnikova E., Lorca RA., Kaur I., et al SynDig1: an activity-regulated, AMPA- receptor-interacting transmembrane protein that regulates excitatory synapse development. Neuron. 2010;65:80–93. doi: 10.1016/j.neuron.2009.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ashby MC., Maier SR., Nishimune A., Henley JM. Lateral diffusion drives constitutive exchange of AMPA receptors at dendritic spines and is regulated by spine morphology. J Neurosci. 2006;26:7046–7055. doi: 10.1523/JNEUROSCI.1235-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.orgdorff AJ., Choquet D. Regulation of AMPA receptor lateral movements. Nature. 2002;417:649–653. doi: 10.1038/nature00780. [DOI] [PubMed] [Google Scholar]
- 139.Heine M., Groc L., Frischknecht R., et al Surface mobility of postsynaptic AMPARs tunes synaptic transmission. Science. 2008;320:201–205. doi: 10.1126/science.1152089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.udowski GA., Puthenveedu MA., Leonoudakis D., et al Real-time imaging of discrete exocytic events mediating surface delivery of AMPA receptors. J Neurosci. 2007;27:1 1112–11121. doi: 10.1523/JNEUROSCI.2465-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kopec CD., Li B., Wei W., Boehm J., Malinow R. Glutamate receptor exocytosis and spine enlargement during chemically induced long-term potentiation. J Neurosci. 2006;26:2000–2009. doi: 10.1523/JNEUROSCI.3918-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Makino H., Malinow R. AMPA receptor incorporation into synapses during LTP: the role of lateral movement and exocytosis. Neuron. 2009;64:381–390. doi: 10.1016/j.neuron.2009.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Jaskolski F., Mayo-Martin B., Jane D., Henley JM. Dynamin-dependent membrane drift recruits AMPA receptors to dendritic spines. J Biol Chem. 2009;284:12491–12503. doi: 10.1074/jbc.M808401200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lu J., Helton TD., Blanpied TA., et al Postsynaptic positioning of endocytic zones and AMPA receptor cycling by physical coupling of dynamin-3 to homer. Neuron. 2007;55:874–889. doi: 10.1016/j.neuron.2007.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kennedy MJ., Davison IG., Robinson CG., Ehlers MD. Syntaxin-4 defines a domain for activity-dependent exocytosis in dendritic spines. Cell. 2010;141:524–535. doi: 10.1016/j.cell.2010.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Tai CY., Kim SA., Schuman EM. Cadherins and synaptic plasticity. CurrOpin Cell Biol. 2008;20:567–575. doi: 10.1016/j.ceb.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 147.Takeichi M. The cadherins: cell-cell adhesion molecules controlling animal morphogenesis. Development. 1988;102:639–655. doi: 10.1242/dev.102.4.639. [DOI] [PubMed] [Google Scholar]
- 148.Togashi H., Abe K., Mizoguchi A., Takaoka K., Chisaka 0., Takeichi M. Cadherin regulates dendritic spine morphogenesis. Neuron. 2002;35:77–89. doi: 10.1016/s0896-6273(02)00748-1. [DOI] [PubMed] [Google Scholar]
- 149.Nuriya M., Huganir RL. Regulation of AMPA receptor trafficking by Ncadherin. J Neurochem. 2006;97:652–661. doi: 10.1111/j.1471-4159.2006.03740.x. [DOI] [PubMed] [Google Scholar]
- 150.Silverman JB., Restituito S., Lu W., Lee-Edwards L., Khatri L., Ziff EB. Synaptic anchorage of AMPA receptors by cadherins through neural plakophilin-related arm protein AMPA receptor-binding protein complexes. J Neurosci. 2007;27:8505–8516. doi: 10.1523/JNEUROSCI.1395-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Saglietti L., Dequidt C., Kamieniarz K., et al Extracellular interactions between GLUR2 and N-cadherin in spine regulation. Neuron. 2007;54:461–477. doi: 10.1016/j.neuron.2007.04.012. [DOI] [PubMed] [Google Scholar]
- 152.Craig AM., Kang Y. Neurexin-neuroligin signaling in synapse development. Curr Opin Neurobiol. 2007;17:43–52. doi: 10.1016/j.conb.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Heine M., Thoumine 0., Mondin M., Tessier B., Giannone G., Choquet D. Activity-independent and subunit-specific recruitment of functional AMPA receptors at neurexin/neuroligin contacts. Proc Natl Acad Sci USA. 2008;105:20947–20952. doi: 10.1073/pnas.0804007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Thyagarajan A., Ting AY. Imaging activity-dependent regulation of neurexin-neuroligin interactions using trans-synaptic enzymatic biotinylation. Cell. 2010;143:456–469. doi: 10.1016/j.cell.2010.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 155.Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/s0896-6273(00)00129-x. [DOI] [PubMed] [Google Scholar]
- 156.Park M., Penick EC., Edwards JG., Kauer JA., Ehlers MD. Recycling endosomes supply AMPA receptors for LTP. Science. 2004;305:1972–1975. doi: 10.1126/science.1102026. [DOI] [PubMed] [Google Scholar]
- 157.Hurley JH., Stenmark H. Molecular mechanisms of ubiquitin-dependent membrane traffic. Annu Rev Biophys. 2011;40:119–42. doi: 10.1146/annurev-biophys-042910-155404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mabb AM., Ehlers MD. Ubiquitination in postsynaptic function and plasticity. Annu Rev Cell Dev Biol. 2010;26:179–210. doi: 10.1146/annurev-cellbio-100109-104129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Bingol B., Schuman EM. A proteasome-sensitive connection between PSD-95 and GLUR1 endocytosis. Neuropharmacology. 2004;47:755–763. doi: 10.1016/j.neuropharm.2004.07.028. [DOI] [PubMed] [Google Scholar]
- 160.Zhang D., Hou Q., Wang M., et al NA,K-ATPase activity regulates AMPA receptor turnover through proteasome-mediated proteolysis. J Neurosci. 2009;29:4498–4511. doi: 10.1523/JNEUROSCI.6094-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Colledge M., Snyder EM., Crozier RA., et al Ubiquitination regulates PSD95 degradation and AMPA receptor surface expression. Neuron. 2003;40:595–607. doi: 10.1016/s0896-6273(03)00687-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Schwarz LA., Hall BJ., Patrick GN. Activity-dependent ubiquitination of GluA1 mediates a distinct AMPA receptor endocytosis and sorting pathway. J Neurosci. 2010;30:16718–16729. doi: 10.1523/JNEUROSCI.3686-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lussier MP., Nasu-Nishimura Y., Roche KW. Activity-dependent ubiquitination of the AMPA receptor subunit Glua2. J Neurosci. 2011;31:3077–3081. doi: 10.1523/JNEUROSCI.5944-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lin A., Hou Q., Jarzylo L., et al Nedd4-mediated AMPA receptor ubiquitination regulates receptor turnover and trafficking. J Neurochem. 2011;119:27–39. doi: 10.1111/j.1471-4159.2011.07221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Fu AK., Hung KW., Fu WY., et al Apc(cdhl) mediates EPHA4-dependent downregulation of AMPA receptors in homeostatic plasticity. Nat Neurosci. 2011;14:181–119. doi: 10.1038/nn.2715. [DOI] [PubMed] [Google Scholar]
- 166.Fu AKY., Hung K-W., Fu W-Y., Shen C., Chen Y., Xia J., et al Epha4 signaling mediates apccdhl-dependent down-regulation of AMPAr in homeostatic plasticity. Nat Neurosci. 2011;14:181–189. doi: 10.1038/nn.2715. [DOI] [PubMed] [Google Scholar]
- 167.Kantamneni S., Wilkinson KA., Henley JM. Ubiquitin regulation of neuronal excitability. Nat Neurosci. 2011;14:126–128. doi: 10.1038/nn0211-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Pozo K., Goda Y. Unraveling mechanisms of homeostatic synaptic plasticity. Neuron. 2010;66:337–351. doi: 10.1016/j.neuron.2010.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Small DH. Network dysfunction in Alzheimer's disease: does synaptic scaling drive disease progression? Trends Mol Med. 2008;14:103–108. doi: 10.1016/j.molmed.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 170.Sutton MA., Ito HT., Cressy P., Kempf C., Woo JC., Schuman EM. Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell. 2006;125:785–799. doi: 10.1016/j.cell.2006.03.040. [DOI] [PubMed] [Google Scholar]
- 171.Ju W., Morishita W., Tsui J., et al Activity-dependent regulation of dendritic synthesis and trafficking of AMPA receptors. Nat Neurosci. 2004;7:244–253. doi: 10.1038/nn1189. [DOI] [PubMed] [Google Scholar]
- 172.Aoto J., Nam CI., Poon MM., Ting P., Chen L. Synaptic signaling by all-trans retinoic acid in homeostatic synaptic plasticity. Neuron. 2008;60:308–320. doi: 10.1016/j.neuron.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Thiagarajan TC., Lindskog M., Tsien RW. Adaptation to synaptic inactivity in hippocampal neurons. Neuron. 2005;47:725–737. doi: 10.1016/j.neuron.2005.06.037. [DOI] [PubMed] [Google Scholar]
- 174.Hou Q., Zhang D., Jarzylo L., Huganir RL., Man HY. Homeostatic regulation of AMPA receptor expression at single hippocampal synapses. Proc. Natl Acad Sci US A. 2008;105:775–780. doi: 10.1073/pnas.0706447105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.O'Brien RJ., Kamboj S., Ehlers MD., Rosen KR., Fischbach GD., Huganir RL. Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron. 1998;21:1067–1078. doi: 10.1016/s0896-6273(00)80624-8. [DOI] [PubMed] [Google Scholar]
- 176.Wierenga CJ., Ibata K., Turrigiano GG. Postsynaptic expression of homeostatic plasticity at neocortical synapses. J Neurosci. 2005;25:2895–2905. doi: 10.1523/JNEUROSCI.5217-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Gainey MA., Hurvitz-Wolff JR., Lambo ME., Turrigiano GG. Synaptic scaling requires the GLUR2 subunit of the AMPA receptor. J Neurosci. 2009;29:6479–6489. doi: 10.1523/JNEUROSCI.3753-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Beattie EC., Stellwagen D., Morishita W., et al Control of synaptic strength by glial TNF alpha. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- 179.Stellwagen D., Malenka RC. Synaptic scaling mediated by glial TNFalpha. Nature. 2006;440:1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- 180.Steinmetz CC., Turrigiano GG. Tumor necrosis factor-alpha signaling maintains the ability of cortical synapses to express synaptic scaling. J Neurosci. 2010;30:14685–14690. doi: 10.1523/JNEUROSCI.2210-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Rutherford LC., Nelson SB., Turrigiano GG. BDNF has opposite effects on the quantal amplitude of pyramidal neuron and interneuron excitatory synapses. Neuron. 1998;21:521–530. doi: 10.1016/s0896-6273(00)80563-2. [DOI] [PubMed] [Google Scholar]
- 182.Bolton MM., Pittman AJ., Lo DC. Brain-derived neurotrophic factor differentially regulates excitatory and inhibitory synaptic transmission in hippocAMPAl cultures. J Neurosci. 2000;20:3221–3232. doi: 10.1523/JNEUROSCI.20-09-03221.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Caldeira MV., Melo CV., Pereira DB., et al Brain-derived neurotrophic factor regulates the expression and synaptic delivery of alpha-amino-3hydroxy-5-methyl-4-isoxazole propionic acid receptor subunits in hippocampal neurons. J Biol Chem. 2007;282:12619–12628. doi: 10.1074/jbc.M700607200. [DOI] [PubMed] [Google Scholar]
- 184.Li X., Wolf ME. Brain-derived neurotrophic factor rapidly increases AMPA receptor surface expression in rat nucleus accumbens. Eur J Neurosci. 2011;34:190–198. doi: 10.1111/j.1460-9568.2011.07754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Thiagarajan TC., Piedras-Renteria ES., Tsien RW. Alpha- and betaCAMKII. Inverse regulation by neuronal activity and opposing effects on synaptic strength. Neuron. 2002;36:1103–1114. doi: 10.1016/s0896-6273(02)01049-8. [DOI] [PubMed] [Google Scholar]
- 186.Goold CP., Nicoll RA. Single-cell optogenetic excitation drives homeostatic synaptic depression. Neuron. 2010;68:512–528. doi: 10.1016/j.neuron.2010.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Groth RD., Lindskog M., Thiagarajan TC., Li L., Tsien RW. B-Ca2+/camdependent kinase type II triggers upregulation of glua! to coordinate adaptation to synaptic inactivity in hippocAMPAl neurons. Proc Natl Acad Sci USA. 2011;108:828–833. doi: 10.1073/pnas.1018022108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Okuda T., Yu LMY., Cingolani LA., Kemler R., Goda Y. Beta-catenin regulates excitatory postsynaptic strength at hippocampal synapses. Proc Natl Acad Sci US A. 2007;104:13479–13484. doi: 10.1073/pnas.0702334104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Cingolani LA., Thalhammer A., Yu LM., et al Activity-dependent regulation of synaptic AMPA receptor composition and abundance by beta3 integrins. Neuron. 2008;58:749–762. doi: 10.1016/j.neuron.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Bramham CR., Worley PF., Moore MJ., Guzowski JF. The immediate early gene ARC/ARG3.1: Regulation, mechanisms, and function. J Neurosci. 2008;28:11760–11767. doi: 10.1523/JNEUROSCI.3864-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Chowdhury S., Shepherd JD., Okuno H., et al Arc/arg3.1 interacts with the endocytic machinery to regulate AMPA receptor trafficking. Neuron. 2006;52:445–459. doi: 10.1016/j.neuron.2006.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Peebles CL., Yoo J., Thwin MT., Palop JJ., Noebels JL., Finkbeiner S. Arc regulates spine morphology and maintains network stability in vivo. Proc Natl Acad Sci US A. 2010;107:18173–18178. doi: 10.1073/pnas.1006546107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Greer PL., Hanayama R., Bloodgood BL., et al The Angelman syndrome protein UBE3a regulates synapse development by ubiquitinating arc. Cell. 2010;140:704–716. doi: 10.1016/j.cell.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Craig TJ., Jaafari N., Petrovic MM., Rubin PP., Mellor JR., Henley JM. Homeostatic synaptic scaling is regulated by protein sumoylation. J Biol Chem. 2013;8:e52345. doi: 10.1074/jbc.M112.356337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Morrison JH., Baxter MG. The ageing cortical synapse: hallmarks and implications for cognitive decline. Nat Rev Neurosci. 2012;13:240–250. doi: 10.1038/nrn3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Barnes CA., Jung MW., McNaughton BL., Korol DL., Andreasson K., Worley PF. LTP saturation and spatial learning disruption: effects of task variables and saturation levels. J Neurosci. 1994;14:5793–5806. doi: 10.1523/JNEUROSCI.14-10-05793.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Barnes CA., Rao G., Houston FP. LTP induction threshold change in old rats at the perforant path-granule cell synapse. Neurobiol Aging. 2000;21:613–620. doi: 10.1016/s0197-4580(00)00163-9. [DOI] [PubMed] [Google Scholar]
- 198.Tombaugh GC., Rowe WB., Chow AR., Michael TH., Rose GM. Theta-frequency synaptic potentiation in Ca1 in vitro distinguishes cognitively impaired from unimpaired aged Fischer 344. rats. J Neurosci. 2002;22:9932–9940. doi: 10.1523/JNEUROSCI.22-22-09932.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Deupree DL., Turner DA., Watters CL. Spatial performance correlates with in vitro potentiation in young and aged Fischer 344 rats. Brain Res. 1991;554:1–9. doi: 10.1016/0006-8993(91)90164-q. [DOI] [PubMed] [Google Scholar]
- 200.Moore CI., Browning MD., Rose GM. HippocAMPAl plasticity induced by primed burst, but not long-term potentiation, stimulation is impaired in area ca1 of aged Fischer 344 rats. Hippocampus. 1993;3:57–66. doi: 10.1002/hipo.450030106. [DOI] [PubMed] [Google Scholar]
- 201.Barnes CA., McNaughton BL., O'Keefe J. Loss of place specificity in hippocampal complex spike cells of senescent rat. Neurobiol Aging. 1983;4:113–119. doi: 10.1016/0197-4580(83)90034-9. [DOI] [PubMed] [Google Scholar]
- 202.Dieguez D., Barea-Rodriguez EJ. Aging impairs the late phase of longterm potentiation at the medial perforant path-Ca3 synapse in awake rats. Synapse. 2004;52:53–61. doi: 10.1002/syn.20004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Norris CM., Korol DL., Foster TC. Increased susceptibility to induction of long-term depression and long-term potentiation reversal during aging. J. Neurosci. 1996;16:5382–5392. doi: 10.1523/JNEUROSCI.16-17-05382.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Shankar GM., Li S., Mehta TH., Garcia-Munoz A., Shepardson NE., Smith I., et al Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Li S., Hong S., Shepardson NE., Walsh DM., Shankar GM., Selkoe D. Soluble oligomers of amyloid beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62:788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Walsh DM., Selkoe DJ. A beta oligomers - a decade of discovery. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 207.Zhao WQ., Santini F., Breese R., et al Inhibition of calcineurin-mediated endocytosis and alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors prevents amyloid beta oligomer-induced synaptic disruption. J Biol Chem. 2010;285:7619–7632. doi: 10.1074/jbc.M109.057182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Hsieh H., Boehm J., Sato C., et al Ampar removal underlies abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Gu Z., Liu W., Yan Z. {beta}-amyloid impairs AMPA receptor trafficking and function by reducing Ca2+/calmodulin-dependent protein kinase II synaptic distribution. J Biol Chem. 2009;284:10639–10649. doi: 10.1074/jbc.M806508200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Rui Y., Gu J., Yu K., Hartzell HC., Zheng JQ. Inhibition of AMPA receptor trafficking at hippocampal synapses by beta-amyloid oligomers: the mitochondrial contribution. Mol Brain. 2010;3:10. doi: 10.1186/1756-6606-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Zheng Z., Sabirzhanov B., Keifer J. Oligomericamyloid-{beta} inhibits the proteolytic conversion of brain-derived neurotrophic factor (BDNF), AMPA receptor trafficking, and classical conditioning. J Biol Chem. 2010;285:34708–34717. doi: 10.1074/jbc.M110.150821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Lauren J., Gimbel DA., Nygaard HB., Gilbert JW., Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457:1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]