

Abstract
The increased risk for neurodegenerative and neuropsychiatric disorders associated with extended lifespan has long suggested mechanistic links between chronological age and brain-related disorders, including depression, Recent characterizations of age-dependent gene expression changes now show that aging of the human brain engages a specific set of biological pathways along a continuous lifelong trajectory, and that the same genes that are associated with normal brain aging are also frequently and similarly implicated in depression and other brain-related disorders. These correlative observations suggest a model of age-by-disease molecular interactions, in which brain aging promotes biological changes associated with diseases, and additional environmental factors and genetic variability contribute to defining disease risk or resiliency trajectories. Here we review the characteristic features of brain aging in terms of changes in gene function over time, and then focus on evidence supporting accelerated molecular aging in depression. This proposed age-by-disease biological interaction model addresses the current gap in research between “normal” brain aging and its connection to late-life diseases. The implications of this model are profound, as it provides an investigational framework for identifying critical moderating factors, outlines opportunities for early interventions or preventions, and may form the basis for a dimensional definition of diseases that goes beyond the current categorical system.
Keywords: brain molecular aging, age, neuroplasticity, depression, psychiatric, neurological disorder
Abstract
El mayor riesgo para los trastornos neurodegenerativos y neuropsiquiátricos asociados con el aumento de la expectativa de vida ha sugerido hace tiempo asociaciones mecanicistas entre la edad cronológica y los trastornos cerebrates, incluyendo la depresión. Recientes caracterizaciones en los cambios en la expresión génica dependiente de la edad han mostrado que el envejecimiento del cerebro humano requiere de un conjunto específico de vías biológicas en un continuo a través del curso de toda la vida, y que los mismos genes que están asociados con el envejecimiento normal se implican frecuentemente y de igual forma en la depresión como en otros trastornos cerebrates. Estas sucesivas observaciones sugieren un modelo de interacciones moleculares de enfermedades por la edad, en las cuales el envejecimiento cerebral promueve cambios biológicos asociados con las enfermedades, y en forma adicional los factores ambientales y la variabilidad genética contribuyen a definir el riesgo de enfermar o de tener una evolución con resiliencia. En este artículo se revisan las caracteristicas distintivas del envejecimiento cerebral en términos de cambios en la función génica a lo largo del tiempo, y luego se enfoca en la evidencia que sustenta un acelerado envejecimiento molecular en la depresión. Este modelo que propone la interacción biológica de enfermedades por la edad Ie da una dirección a la actual brecha en la investigación entre el envejecimiento cerebral “normal” y su conexión con enfermedades del final de la vida. Las sugerencías de este modelo son profundas y proporcionan un marco de investigación para identificar factores moderadores criticos, delinear oportunidades para intervenciones o prevención precoces y poder formar las bases para una definición dimensional de las enfermedades que vaya más allá del actual sistema de categoríes.
Abstract
L'augmentation du risque de troubles neurodégénératifs et neuropsychiatriques associé à l'allongement de la durée de vie évoque depuis longtemps l'existence de liens mécanistes entre l'âge chronologique et les troubles cérébraux, dont la dépression. Des mises en évidence récentes de modifications d'expression des gènes en fonction de l'âge suggèrent maintenant que le vieillissement cérébral humain met enjeu un ensemble spécifique de voies biologiques sur une trajectoire continue tout au long de la vie. Or, les gènes associés au vieillissement normal du cerveau sont aussi impliqués de façon fréquente et identique dans la dépression ainsi que dans d'autres troubles cérébraux. Ces observations proposent un modèle d'interaction moléculaire âge-maladie, dans lequel le vieillissement cérébral favorise des modifications biologiques associeés aux maladies et dans lequel des facteurs environnementaux supplémentaires et la variabilité génétique contribuent à définir le risque pathologique ou celui des trajectoires de résilience. Nous passons en revue ici les traits caractéristiques du vieillissement cérébral en termes de modification de la fonction des gènes au cours du temps. Puis nous nous interessons aux arguments en favour de l'accélération du vieillissement moléculaire dans la dépression. Cette proposition de modèle d'interaction biologique âge-maladie aborde le décalage actuel dans la recherche entre le vieillissement cérébral normal et ses connexions aux maladies de la vieillesse. Les implications de ce modèle sont importantes, en termes de cadre d'investigation pour l'identification des facteurs de modération déterminants, d'opportunités pour une prévention ou un traitement précoce et de création d'une définition dimensionnelle des maladies allant au-delà du système catégoriel actuel.
Introduction
Improvements in quality and accessibility of public health measures, as well as medical interventions for multiple diseases, have led to dramatic increases in the average human lifespan over the last century.1 Along with this increase in life expectancy comes increased risk for the development of neurodegenerative and neuropsychiatric disorders in late life, including significant increases in symptoms related to depression.2 This report focuses on major depression and its associated symptoms as critical factors and potential modulators of a proposed age-by-disease interaction model. Major depression affects subjects of all ages, increases morbidity in the context of several organ diseases, and overall causes greater disability than all other psychiatric disorders.3 Major depression is a severe mental illness that is defined by specific sets of symptoms, but low mood and anhedonia, the two core symptom dimensions of the illness, are observed across major mental illnesses and neurodegenerative disorders. Importantly, biological pathways associated with depression overlap with those frequently implicated in aging processes (eg, stress, inflammation, immune recruitment, and metabolic syndrome), prompting the hypothesis of accelerated aging in depressed subjects:4 Notably, chronic stress, a common precipitating factor in depression, recruits similar pathways and has been suggested as a factor leading to accelerated aging.5 Conversely, while major depression per se does not increase with older age, a constellation of related symptoms are present in many elderly subjects, even if not categorically diagnosed as depression.2 However, there is also a large variability in individual susceptibility to develop depression and related symptoms with increasing age, and while some dysfunction appears inevitable, successful emotional, physical, and cognitive aging is achievable. This suggests that agerelated biological mechanisms and functional outcomes, including vulnerability to experience depressive symptoms, can be slowed down under certain circumstances, and/or that protective mechanisms may be recruited throughout the lifespan. Hence, simultaneously investigating the biological causes and reciprocal links between brain aging and neuropsychiatric disorders may provide novel perspectives on disease mechanisms.
Accordingly, since evidence suggests that neural networks and biological mechanisms underlying mood regulation are specifically at risk across disorders and during aging,6 our group has focused on major depression and aging of the brain, in order to investigate age-by-disease interactions.
During our investigations of the molecular bases of major depression in the human post-mortem brain, we have uncovered a large and robust effect of age on multiple genes and biological pathways.7,8 Notably, this set of age-dependent genes broadly overlaps with disease-related pathways, and the changes in gene function observed during aging occur for the most part in directions that would otherwise promote neurological disorders, including depression.8 For instance, brain-derived neurotrophic factor (BDNF), somatostatin (SST) and other neuropeptides are decreased in mood-related9-14 and neurodegenerative disorders,15-16 but also lose ~ 50% expression during aging in control subjects.7,17 Similar age and disease changes are observed for numerous other genes,8,10 together suggesting that normal brain aging may in fact promote aspects of disease-related mechanisms. Indeed, major depression is associated with anticipated gene expression changes that occur during normal aging of the brain,18 suggesting that an older molecular age of the brain may represent an early biological event in the disease process, and may serve as a useful marker for risk of developing symptoms of depression.
This review summarizes findings and observations in support of an age-by-disease biological interaction model. This model brings together basic research on normal aging with the investigation of neuropsychiatric and neurodegenerative diseases, and suggests that environment and genetic variability are contributing factors in defining risk and/or resiliency trajectories. Further, identifying age -dependent biological processes and their modulators may inform the development of new interventions for the prevention and treatment of a more broadly-defined depressive syndrome and for related functional outcomes in elderly subjects. Aspects of this model and hypothesis have been previously discussed elsewhere.19,20
Depression and age-related functional outcomes
According to the Diagnostic and Statistical Manual Of Mental Disorders. Fourth Edition (DSM-IV), Major Depression is diagnosed in individuals experiencing low mood and/or anhedonia plus five symptoms that may include changes in sleep, feelings of guilt or worthlessness, low energy, poor concentration, changes in appetite, psychomotor retardation, and thoughts of death or suicide.21 Defined this way, Major Depression affects 10% to 15% of people in the general population in their lifetime.22 The biological bases of depression are complex and likely involve multiple interacting disruptions affecting neurons and glial cells within specific brain areas, giving rise to neural network dysfunctions and depressive symptomatology. At the molecular level there is compelling evidence for the involvement of many biological processes in depression, including, but not limited to, altered monoaminergic neurotransmission, altered stress hormone homeostasis, reduced neurotrophic support, metabolic dysregulation, immune reaction, increased inflammation, oxidative stress, and mitochondrial dysfunction, as well as other aspects of brain plasticity and synaptic functions.23 Notably, similar changes have been reported during aging, prompting the hypothesis that major depression may be associated with “accelerated aging” (See Wolkowicz et al4,24 for reviews).
On the other hand, major depression and other mood disorders per se do not necessarily increase with age, and in fact, only approximately 1% of older individuals meet the criteria for major depression, a prevalence much lower than in younger individuals.25 However, approximately 15% to 25% of individuals over the age of 65 experience depressive symptoms that, while not meeting criteria for major depression, do cause significant distress and interfere with daily functioning.2,26 Low mood symptoms are also common in the presymptomatic phases of neurodegenerative disorders, and are often misattributed to general age-related morbidity.27,28 This discrepancy between formal diagnosis and clinically significant depressive symptoms likely reflects the tendency of older individuals to underreport psychiatric symptoms, the predominance of vegetative and somatic symptoms as part of their clinical presentation, the inability to express depressive symptoms secondary to cognitive impairment,29 and the possibility that depression in older individuals represents a different disease entity with unique clinical presentation and pathophysiology.2,30
Although it is difficult to untangle causal relationships, evidence suggests that proper mood regulation—the capacity to exert homeostatic control on emotions over time—may represent a key component of late-life functional success, and conversely, that mood symptoms may represent not only an early marker, but also a potential contributing factor for subsequent spiraling functional declines.31 Indeed, studies of the functional correlates of aging consistently report increased negative outcomes of low mood,2 motor deficits ranging from decreased fine motor control to impairments in balance and gait, and continuous decline in certain aspects of cognitive functions.32 This suggests that aspects of mood regulatory mechanisms may be selectively vulnerable to early homeostatic changes during normal and pathological aging, or that depressive symptoms may represent a common output for various underlying age-related brain declines.30,33 Conversely, a proportion of older individuals are more resilient to the adverse effects of negative life events and are less likely to feel remorse and guilt,2 underscoring the critical role of individual variability.
Aging of the brain
The number of individuals reaching age 65 in the United States rose 3-fold in the 20th century, from 4.1% in 1900 to 12.4% in the year 2000, and may rise above 20% by the middle of this century. This is equivalent to roughly 85 million people at current growth rates.1 Despite its critical importance to a population growing older, “normal” brain aging and its association with late-life brain disorders is an understudied area of research. This is particularly apparent when compared with the investigation of neurodegenerative disorders, among other fields. The lack of attention given to this important topic may be due to the general belief that aging is inescapable, broad-ranging, and nonspecific.
However, in recent years, the identification of single gene mutations affecting aging and longevity in nematodes, insects, and rodents has demonstrated the presence of a genetic program underlying aging, challenging the above assumptions.34,35 In the mammalian brain, the course of aging parallels that of peripheral tissues, but additional mechanisms reflect the unique features of the brain and post-mitotic differentiated neurons.19,36 For instance, the brain has a higher metabolism level than the rest of the body and utilizes a large proportion of consumed oxygen, hence increasing the potential for producing reactive oxygen species and subsequent oxidative stress. Oxidative stress mediates specific neuronal damage, including modifications to lipids, protein, and DNA, resulting in inflammation, an increase in reactive astrocytes, and altered Ca2+- and mitochondria-mediated neuronal functions, which together may contribute to the deterioration of mental capacities with age.37,38 Further, with rare exceptions, neurons do not divide,39 and thus cellular damage tends to accumulate with increasing age. This is paralleled by a decrease in the capacity for cellular repair.36
Structurally, studies reveal a decrease in neuron volumes, a small loss or no change in cell numbers,40,41 and a progressive thinning of cortical thickness, affecting both gray and white matter.42,43 Functionally, studies indicate a continuous decline with age in certain aspects of cognitive functions (speed of processing, working memory, and long-term memory) beginning in the 20s.44 In contrast, verbal knowledge increases throughout the lifetime.32 This latter observation highlights the point that, while studies often demonstrate a negative conceptual bias towards aging, age-related changes can also be positive, and may represent the recruitment of protective mechanisms against known deleterious effects of aging (ie, oxidative stress) or uncharacterized and beneficial late brain-maturation processes. Based on the above observations, and supported by developments in gene array technology, our group7,8 and others45-48 have investigated the presence of age-dependent gene expression changes in the human brain, as molecular correlates of affected cellular functions.
“Molecular aging” of the human brain
It has been known for some time that robust changes in gene expression occur with aging in peripheral tissues.49 The fact that age-related changes in gene expression extend to the brain may not be surprising, given the body of knowledge about changes in structure and function of the brain with age (described briefly above). Indeed, one might hypothesize that age-related changes in gene expression reflect a general deterioration of the brain and that a preponderance of genes would be affected. This, however, does not appear to be the case. Recent genome-wide studies demonstrate that a relatively small number of genes exhibit age-dependent gene expression changes. Studies in rodent, monkey, and human brains estimate the number of genes exhibiting age-dependent changes to represent less than 10%, and commonly less than 5%, of the entire genome,7,50-55 In a study from our group, the age-related changes of a large number of genes were investigated using gene microarray technology in prefrontal cortex samples from human subjects aged 13 to 79 years (Figure 1).7 The data from this study identified life-long progressive changes in expression with age in approximately 7.5% of genes tested, while expression levels for the large majority of genes were strikingly unchanged throughout adult life. This set of age-dependent genes was also very similar to those observed in other studies, and in fact displayed a high degree of conservation across cohorts and cortical brain regions, despite differences in sample size, expression platforms, and analytical methods.7,8,50-52,54,56,57 Together, this conserved and restricted scope of transcript changes suggests that specific cellular populations and biological processes are selectively affected during aging.
Figure 1. Continuous and progressive gene expression changes in human prefrontal cortex. Age-dependent changes for a core set of exemplary genes (n=588) are presented together for two regions of the prefrontal cortex (Brodmann area (BA9), dorsolateral prefrontal cortex; BA47, orbital ventral prefrontal cortex). Each gene is represented by a row, each array, or brain area per subject, by a column. Samples are organized left to right by brain area and increasing age. Green and red bars indicate decreased and increased gene expression, respectively, versus the averaged signal for these genes across all samples. For example, a horizontal row going from red to green indicates a gene for which expression decreases with age in that brain area. Genes are organized along the y-axis according to similarities in expression profiles across age. This study illustrated several points: (i) similar numbers of genes are downregulated or upregulated throughout the lifetime; (ii) gene changes display continuous and progressive trajectories throughout adult life; (iii) profiles of age-dependent changes are conserved across the two areas; and (iv) downregulated genes are mostly expressed in neurons, whereas upregulated genes include most glial-enriched and some neuronal-enriched genes, as indicated on the right hand of the figure. Columns to the right indicate the distribution of genes with glial-(white matter [WM]- enriched) or neuronal-enriched (gray matter [GM]-enriched) signals. Adapted from ref 7: Erraji-Benchekroun L, Underwood MD, Arango V, et al. Molecular aging in human prefrontal cortex is selective and continuous throughout adult life. Biol Psychiatry. 2005;57:549-558. Copyright © Elsevier 2005.
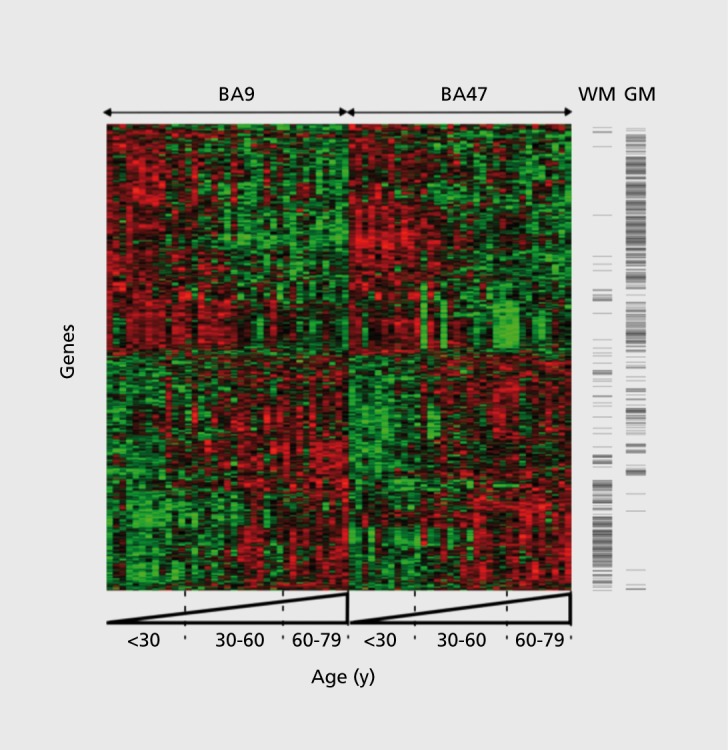
Expression of genes playing a role in glial-mediated inflammation, oxidative stress responses, mitochondrial function, synaptic function and plasticity, and calcium regulation has now consistently been shown to be affected by aging across multiple studies.19,36 Overall, age-upregulated genes are mostly of glial origin and related to inflammation and cellular defenses, while downregulated genes display mostly neuron-enriched transcripts relating to cellular communication and signaling (Figure 1).7 The specificities of genes and cellular functions affected during aging of the brain are briefly summarized in Figure 2, and have been reviewed in detail elsewhere.19,36
Figure 2. Age-dependent biological changes in neurons and glia. Known age-related cellular phenotypes are highlighted for neurons and glia. Blue, pyramidal cells; Purple, interneurons; Orange, astrocyte; Green, microglia; Brown, oligodendrocyte. Not shown are changes in brain white matter track and blood vessel integrity. Many neuronal phenotypes (such as DNA damage) occur in neuron and glia. In parentheses are single representative examples (amongst many) of age-regulated gene expression changes, which may contribute to the particular cellular phenotypes. CRF, Corticotropin-releasing hormone; CALB-1, calbindin 1; SOD2, superoxide dismutase 2; BCL-2, B-cell CLL/lymphoma 2; DRD1, Dopamine receptor D1; SYN2, synapsin II; GFAP, glial fibrillary acidic protein; NF-KB, nuclear factor kappa B; CNP, 2',3'-cyclic nucleotide 3' phosphodiesterase; MHC, myosin heavy chain. Adapted from ref 19: Glorioso C, Sibille E. Between destiny and disease: genetics and molecular pathways of human central nervous system aging. Prog Neurobiol. 2011;93:165-181. Copyright © Pergamon Press 2011.

Together, the consistency and specificity of age-related changes fulfill criteria for aging biomarkers. Accordingly, we have shown that the predicted age for a particular individual, based on regression analysis of age-related trajectories for age-dependent genes, is highly correlated with the chronological age of that individual.7,8 Hence, we have proposed the concept of “molecular age” (ie, predicted age, based on gene expression profile), as a functional assay to measure biological aging of the brain and to assess individual deviation from chronological age and moderators of aging processes.7,8 Using this assay, deviations from expected trajectories were also observed, in which individual subjects displayed greater or lower molecular ages compared with their chronological ages. These deviations did not covary with body mass index, sex, race, or death by cardiovascular accidents. Importantly, one could speculate that individuals with older or younger biological brains may have displayed corresponding functional changes before death. Moreover, identifying factors underlying deviations either in molecular age or in sets of age-dependent genes (See section on BDNF and SST) may provide insight into modulators of age and age-by-disease interactions, hence providing targets for potential therapeutic approaches and preventive strategies. Modulators may include environmental components (diet, disease, exercise, drug exposure, etc), but evidence also suggests a genetic component into functional age trajectories. Here, before reviewing specificities of molecular aging in depression and potential genetic contributions, we review molecular aging and associated genes in the context of disease pathways.
Molecular aging of the brain overlaps with biological pathways implicated in multiple brain disorders
Specific ages of onset are core features of many neuropsychiatric disorders, ranging from late-onset neurodegenerative diseases such as Alzheimer's and Parkinson's diseases58 to earlier onset psychiatric disorders such as schizophrenia and bipolar disorder. Yet, despite their importance, the mechanism(s) underlying age thresholds are largely unknown. Studies have shown that slowing normal aging in model organisms (through genetic or environmental means) results in delayed onset of age-related disorders. For example, mice hypermorphic for the longevity gene, Klotho, live ~ 20% longer and have a corresponding delay in onset of disease59,60 and calorie-restricted primates demonstrated delayed incidence of diabetes, cancer, cardiovascular disease, and brain atrophy.61 Together, these observations suggest an overlap between age- and disease-related biological pathways. Following a broad survey of genes affected during aging and in diseases, we have now reported a large over-representation of neurological-related genes within the human molecular signature of aging.8 In fact, up to a third of genes affected during aging have also been associated in the literature with neuropsychiatric or other brain disorders. Conversely, only 4% of non-age-regulated genes are brain disease-related. For instance, age- and mood disorder-related genes include genes coding for neuropeptides (SSZNPY, CCK, CRF), trophic factors (BDNF, IGF1, FGF), receptors (HTR2A, DRD1, CB1R, GABRAA5, FGF2R) and numerous other genes associated with diseases, including neurodegenerative disorders (MAOB, PER3, CLU, SYN, HTT, NRG1, RLN, TAU, PARK, PINK1, NFKB, SOD2, RGS4, etc).8
This observation that brain disorder-related genes are overrepresented among age-dependent genes, combined with the finding that the observed effects of aging on gene expression are mostly (>90%) in brain disorder-promoting directions, together suggest that the pathways to depression and other brain disorders in late life are aspects of normal molecular aging and may represent one mechanism by which aging precipitates their onset. Notably, these findings came from subjects who were free of neurodegenerative disorders,8 so the observed changes were not sufficient to cause disorders. Instead, normal age-related changes in gene function may represent latent vulnerability factors that are promoted by aging, and that may directly contribute in the disease process (ie, causing or associated with disease) in the context of additional genetic and/or environmental risk factors, which exacerbate age-dependent trajectories. Conversely, moderating factors that delay age-dependent trajectories may promote resiliency not only against age-related declines but also against multiple brainrelated disorders.
Molecular interaction between depression and aging: the cases of BDNF, SST, and dendritic inhibition
An example of a putative interaction between age and disease is provided by the investigation of BDNF and BDNF-dependent genes. BDNF is a signaling neuropeptide that is critical during development and adulthood, specifically in maintaining plasticity and proper functioning of many targeted neuronal cells. Reduced BDNF levels and/or functions have been implicated in multiple brain-related disorders, including major depression,10,13,14 bipolar depression, schizophrenia, Huntington's disease, and Alzheimer's disease,9-16 Interestingly, BDNF is also downregulated with increasing age. A normal non-psychiatric control subject may lose as much as 60% of BDNF expression between the ages of 20 and 60 years.7,17 We have reported evidence of decreased BDNF levels and/or signaling in the amygdala and anterior cingulate cortex of subjects affected with depression compared to controls.10,14,62 We have also reported reduced expression of SST, cortistatin (CORT), and neuropeptide Y (NPY) in the same cohorts. SST, CORT, and NPY are neuropeptides that are expressed in subtypes of γ-aminobutyric acid (GABA) interneurons, which specifically target the dendrites of pyramidal neurons (Figure 3a). SST, CORT, and NPY expressions are dependent on BDNF signaling, as demonstrated by reduced levels in mice with genetically-induced reduction in BDNF functions.14,62,-63 Together, these findings have suggested the presence of a depression-related pathogenic mechanism linking reduced BDNF function to reduced markers of GABA interneurons that provide dendritic inhibition.
Figure 3. Dendritic inhibition, a biological module at the intersection of age and psychiatric disorders. A) Excitatory pyramidal neurons (PYR) are regulated by different types of inhibitory γ-aminobutyric acid (GABA) neurons. Somatostatin (SST)-, neuropeptide Y (NPY)- and cortistatin (CORT)-positive GABA neurons (red) target PYR distal dendrites. Parvalbumin- (PV) and cholecystokinin- (CCK) positive GABA neurons target PYR cell body and axon initial segment (blue). Calretinin-(CR) positive GABA neurons (green) regulate other GABA neurons. B) Markers of interneurons that target PYR dendrites show decreased expression with age, and great effect or statistical significance of changes in subjects with major depression. C) Brain-derived neurotropic factor (BDNF) expression, measured by quantitative polymerase chain reaction (qPCR), is significantly and inversely correlated with chronological age in control and depressed subjects. Values are in arbitrary units of qPCR signal intensity. Respective to age-matched control subjects, subjects with major depression display greater BDNF downregulation (-22%; P<0.05). D) Age-regulation of BDNF- and depression-related genes. The average relative age effects for each individual subject are shown for the set of BDNF-related genes that display significant depression-related effects. Microarray-based gene expression values were normalized to the group means of each gene and averaged per subject. The BDNF-related gene set was split based on the effect of age on those genes in control subjects (top panel, age-upregulated; bottom panel, age-downregulated). The results show that age effects are systematically in the same direction, and of greater effect sizes in subjects with major depression (red squares) compared with controls (blue circles). Values are Pearson correlation factors. *, P<0.05; **, P<0.01 . Data in B is from Erraji et al,7 Glorioso et al,19 and Guilloux et al.14 Figures C and D are adapted from ref 18: Douillard-Guilloux G, Guilloux JP, Lewis DA, Sibille E. Anticipated brain molecular aging in Major Depression. Am J Geriatr Psychiatry. 2012. Oct 31 [epub ahead of print]. Copyright © American Psychiatry Press 2012.

Given that not all elderly subjects develop depression, additional factors must be at play. Indeed, the cross-sectional slope of decrease in BDNF expression in subjects with depression appears to parallel that of control subjects, but at a lower level, demonstrating reduced expression at most ages (Figure 3b). However, the fact that low expression in young depressed subjects overlaps with levels that are reached at older ages in control subjects demonstrates that low BDNF by itself is not sufficient. Rather, this suggests that the molecular context of these reductions are critical contributing factors to developing pathophysiology. Notably, age-related changes for multiple BDNF-dependent genes, including SST, NPY, and to some extent CORT, display increasing rates of change with age compared with BDNF (ie, steeper slopes) and greater overall effect sizes in the context of depression (Figure 3c),18 suggesting an age-by-disease interaction that further and negatively affects gene function in disease-promoting directions, in addition and potentially independently of changes in BDNF function.
Together, these findings have suggested the presence of a BDNF/GABA-related biological module that is responsible for principal neuron dendritic inhibition, and that is positioned at the intersection of age and depression-related mechanisms. In this module, the interaction of both factors may potentially determine if and when decreased function reaches a certain threshold, under which pathophysiological output occurs. These findings also suggest three important aspects of a potential ageby-disease interaction: (i) age-dependent changes in expression of disease-related genes may represent latent vulnerability factors for diseases and associated symptom dimensions; (ii) BDNF and its associated agedependent changes may represent an upstream mediator for age-dependent changes of disease-related genes; and (iii) additional factors must be at play in establishing initial changes in upstream disease-related gene changes (ie, low BDNF) and in moderating the apparent “acceleration” of age-dependent trajectories in disease-promoting directions. Here, we next review additional findings relating to depression and accelerated aging, before discussing a broader age-by-disease interaction model.
Depression is associated with “accelerated” molecular aging
Based on the above-described putative interaction between age- and depression-related mechanisms affecting BDNF and BDNF-dependent genes, and in order to more formally test the hypothesis of accelerated aging in depression, we have investigated changes in broader patterns of age-dependent gene expression in the brains of individuals specifically affected with major depression.18 Results confirmed that affected subjects showed greater changes for BDNF and BDNF-dependent gene expression than the normal age-related changes observed in control subjects. That study also reported that most depression-related genes were frequently age-regulated in both case and control subjects, and that the effects of major depression and age on individual genes were positively correlated. Conversely, most genes that were age-dependent in control subjects displayed greater age effects in subjects with major depression, and the increased prevalence of age effects in depression often corresponded to similar effects in controls, although of reduced magnitude and significance (ie, trend levels). This latter point suggested that the overall increase in number of age-affected genes in depressed subjects may not correspond to de novo age effects, but rather to amplified subthreshold events that were already present in control subjects. So, while that study provided molecular evidence in support of an accelerated brain aging hypothesis in depression, the observed combination of robust (eg, BDNF pathway) and more modest effects on different sets of genes and biological pathways also suggest a heterogeneous impact of age and disease effects on cellular functions. Alternatively, the molecular correlates of aging may represent variable sets of biological age-dependent events, each with their own mediators and modulators and with potential specificities in rates of age-related changes.
Proposed model for age-by-disease molecular interactions
The physiological and functional output of any biological system represents the integration of events occurring at the levels of genes, molecules, cells, microcircuits, and neural networks, and constant feedback across these biological scales contributes to the maintenance of homeostasis in the face of a changing environment. In the context of aging, it is not known which changes represent primary adaptive events that are necessary to maintain homeostasis, and which represent reactive processes and reduced capacity for repair against deleterious events, such as increased oxidative damage, inflammation and accumulation of damaged macromolecules, specifically affecting non-dividing neurons. However, the nature of age-dependent genes and the directions of their expression change with age strongly suggest that the human brain progressively moves with advancing age towards a state that is biologically more consistent with those observed in the context of neuropsychiatric and neurological disorders.20 However, the relative rates of occurrence of psychopathology in elderly subjects also demonstrate that the age-dependent and disease-promoting changes in the expression of disease-related genes are not sufficient to induce overt pathophysiology and associated disease symptoms. For instance, extrapolating for studies in postmortem subjects, reduced BDNF, and markers of dendritic inhibition are probably common in many elderly subjects. One can speculate that these changes may actually be appropriate for the biological landscape of an elderly subject, but similar changes in a younger biological context may induce neural network dysfunctions and deficiencies in information processing and mood regulation, resulting in depression in midlife subjects, for instance. Hence, deviations from predicted trajectories and associated biological context may be more critical than expression changes per se.
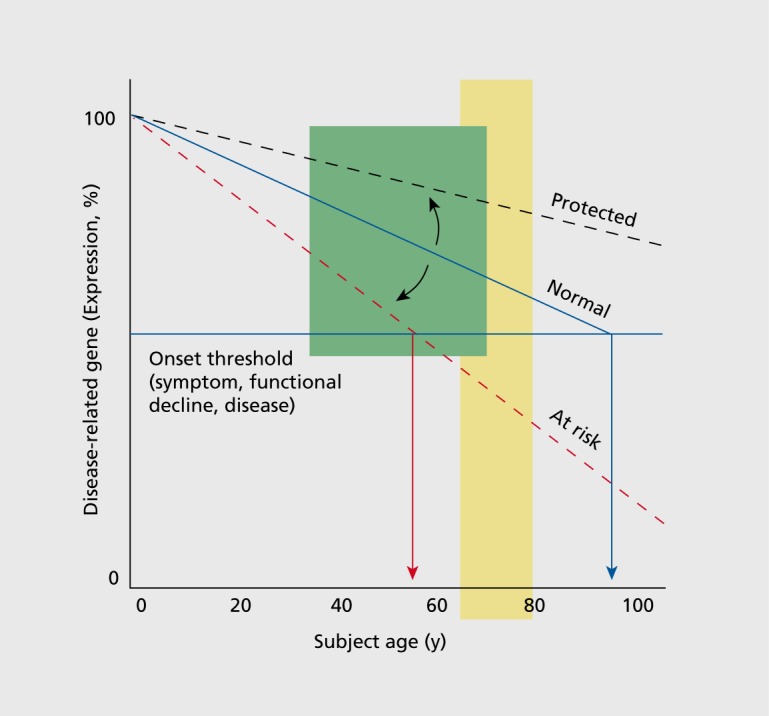
Based on these observations and supported by the fact that gene changes during aging occur in disease-promoting directions, our proposed model is that aging represents an intrinsic vulnerability to pathophysiological changes leading to symptom dimensions (eg, cognitive decline, low mood) and brain disorders, and that additional factors determine if and when critical pathophysiological thresholds are reached (Figure 4). More specifically, loss (or gain) of expression of disease-related genes below or outside expected trajectories and homeostatic range may mark the onset of cellular deficits, leading to disturbances in higher biological scales (microcircuitry, brain region, neural network), in turn promoting the onset of symptoms as the emerging properties of a deregulated system. In this model, factors that affect the trajectory of these age-related changes will determine the timing and potential severity of the initial molecular deficits (Figure 4). The identification of moderators, which place individuals on “at-risk” trajectories, may provide critical information on mechanisms of disease onset. Conversely, factors that delay age-related changes, or that place individuals on “protected” trajectories, may provide critical information on the nature of resiliency, and may offer insight into designing preventive strategies. In short, biological moderators of agedependent trajectories of gene function may represent candidate targets for therapeutic approaches and for promoting resiliency against brain disorders, including psychiatric disorders.
Figure 4. A proposed age-by-disease molecular interaction model. The graph depicts the age-dependent change in expression that is frequently observed for genes that are otherwise implicated in brain-related disorders (a decrease is shown here). Progression below a threshold (horizontal red line) marks the onset of disease symptoms. Changes in the trajectory of agerelated changes in expression of disease-related genes (Y-axis) determine the age (X-axis), or even if, an individual develops disease symptoms (vertical red arrows). Per this model, modulators (black arrows), genetic or environmental, place subjects on an “at risk” or protected trajectory for developing symptoms or brain-related disorders LLD (late-life depression). Adapted from ref 8: Glorioso C, Oh S, Douillard GG, Sibille E. Brain molecular aging, promotion of neurological disease and modulation by Sirtuin5 longevity gene polymorphism. Neurobiol Dis. 2011;41:279-90. Copyright © Blackwell Science 2011; ref 19: Glorioso C, Sibille E. Between destiny and disease:genetics and molecular pathways of human central nervous system aging. Prog Neurobiol. 2011;93:165-181. Copyright © Pergamon Press 2011 .
Implications for future investigations of mechanisms of age and brain-related disorders
Environmental and genetic factors are obvious candidate moderators of an age-by-disease interaction, but identifying their impact on biological aging of the brain may require new experimental strategies. Differences in molecular ages can be assessed in the mid-life range using postmortem brain samples (Figure 4, green shading) since molecular aging displays continuous, life-long, and mostly linear trajectories in adult subjects.7,46 In contrast, when conducting studies to demonstrate associations of biological moderators with functional outcomes in live subjects, it is important to note that brain reserve capacity may buffer functional changes from occurring until years later. The presence of functional declines (emotionality, cognition, health) may be better assessed during later age periods of reduced reserve (ie, over 60 to 65 years of age; Figure 4, yellow shading), where at-risk subjects may start experiencing variable rates of functional declines, while protected individuals may be experiencing more successful aging.
In view of the restricted scope of the molecular profile of aging (ie, less than 10% of expressed genes), studies of genetic factors affecting biological aging are predicted to reduce the number of candidate genes and associated DNA variants to reasonable levels for analytical purposes, hence addressing some of the problems of genome-wide association studies. In a proof-of-concept experiment, we have shown that individual subjects carrying a specific DNA variant located upstream from a candidate gene from the sirtuin family of longevityrelated genes (Sirtuin 5),8 displayed increased molecular ages compared with carriers of the “protective” DNA variant, as measured in anterior cingulate cortex postmortem brain samples. These postmortem genetic studies will need to be followed by studies demonstrating associations of those DNA variants with putative changes in functional trajectories or with altered disease risk ratios in live subjects.
Genetic associations with functional outcomes can be performed using resources from large-scale epidemiological studies, such as the health and body composition, cardiovascular health study or Framingham heart studies, which were specifically designed to investigate critical factors at the vigor-to-frailty age period. These studies may also facilitate the investigation of the moderating effects of the environment (ie, exercise, caloric restriction, nutritional factors such as antioxidants and omega-3 fatty acids, medication, etc), which are more difficult to assess in postmortem conditions due to smaller cohort sizes and reduced antemortem information.
Conclusion
In summary, the considerable overlap between the molecular correlates of brain aging and biological pathways implicated in several neuropsychiatric and neurodegenerative disorders, combined with the potential for a continuum of risk for psychopathology (or conversely resiliency) along life -long trajectories, together suggest a model for age-by-disease molecular interaction in which brain aging promotes biological changes associated with diseases. The implications of a proposed age-by-disease biological interaction model are profound, as it provides an investigational framework for identifying critical moderating factors, outlines opportunities for early interventions or preventions, and finally may form the basis for a dimensional definition of diseases that goes beyond the current categorical system.
Acknowledgments
This work was supported by the National Institute of Mental Health (NIMH) MH084060 and MH093723 grants. The funding agency had no role in the study design, data collection and analysis, decision to publish, and preparation of manuscript. The content is solely the responsibility of the author and does not necessarily represent the official views of the NIMH or the National Institutes of Health. We thank Beverly French for careful comments on the manuscript.
Conflict of Interest: The authors declare no conflicts of interest.
REFERENCES
- 1.Vaupel JW. Biodemography of human ageing. Nature. 2010;464:536–542. doi: 10.1038/nature08984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fiske A., Wetherell JL., Gatz M. Depression in older adults. Annu Rev Clin Psychol. 2009;5:363–389. doi: 10.1146/annurev.clinpsy.032408.153621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Belmaker RH., Again G. Major depressive disorder. N Engl J Med. 2008;358:55–68. doi: 10.1056/NEJMra073096. [DOI] [PubMed] [Google Scholar]
- 4.Wolkowitz OM., Reus VI., Mellon SH. Of sound mind and body: depression, disease, and accelerated aging. Dialogues Clin Neurosci. 2011;13:25–39. doi: 10.31887/DCNS.2011.13.1/owolkowitz. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carroll BJ. Ageing, stress and the brain. Novartis Found Symp. 2002;242:26–36;discussion 36-45. [PubMed] [Google Scholar]
- 6.Butters MA., Young JB., Lopez O., et al Pathways linking late-life depression to persistent cognitive impairment and dementia. Dialogues Clin Neurosci. 2008;10:345–357. doi: 10.31887/DCNS.2008.10.3/mabutters. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Erraji-Benchekroun L., Underwood MD., Arango V., et al Molecular aging in human prefrontal cortex is selective and continuous throughout adult life. Biol Psychiatry. 2005;57:549–558. doi: 10.1016/j.biopsych.2004.10.034. [DOI] [PubMed] [Google Scholar]
- 8.Glorioso C., Oh S., Douillard GG., Sibille E. Brain molecular aging, promotion of neurological disease and modulation by Sirtuin5 longevity gene polymorphism. Neurobiol Dis. 2011;41:279–290. doi: 10.1016/j.nbd.2010.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rakofsky JJ., Ressler KJ., Dunlop BW. BDNF function as a potential mediator of bipolar disorder and post-traumatic stress disorder comorbidity. Mol Psychiatry. 2012;17:22–35. doi: 10.1038/mp.2011.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sibille E., Morris HM., Kota RS., Lewis DA. GABA-related transcripts in the dorsolateral prefrontal cortex in mood disorders. IntJ Neuropsychopharmacol. 2011:1–14. doi: 10.1017/S1461145710001616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Konradi C., Zimmerman El., Yang CK., et al Hippocampal interneurons in bipolar disorder. Arch Gen Psychiatry. 2011;68:340–350. doi: 10.1001/archgenpsychiatry.2010.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Konradi C., Eaton M., MacDonald ML., Walsh J., Benes FM., Heckers S. Molecular evidence for mitochondrial dysfunction in bipolar disorder. Arch Gen Psychiatry. 2004;61:300–308. doi: 10.1001/archpsyc.61.3.300. [DOI] [PubMed] [Google Scholar]
- 13.Tripp A., Kota RS., Lewis DA., Sibille E. Reduced somatostatin in subgenual anterior cingulate cortex in major depression. Neurobiol Dis. 2011;42:116–124. doi: 10.1016/j.nbd.2011.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guilloux JP., Douillard-Guilloux G., Kota R., et al Molecular evidence for BDNF- and GABA-related dysfunctions in the amygdala of female subjects with major depression. Mol Psychiatry. 2012;17:1130–1142. doi: 10.1038/mp.2011.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gahete MD., Rubio A., Duran-Prado M., Avila J., Luque RM., Castano JP. Expression of Somatostatin, cortistatin, and their receptors, as well as dopamine receptors, but not of neprilysin, are reduced in the temporal lobe of Alzheimer's disease patients. J Alzheimers Dis. 2010;20:465–475. doi: 10.3233/JAD-2010-1385. [DOI] [PubMed] [Google Scholar]
- 16.Siegel GJ., Chauhan NB. Neurotrophic factors in Alzheimer's and Parkinson's disease brain. Brain Res Brain Res Rev. 2000;33:199–227. doi: 10.1016/s0165-0173(00)00030-8. [DOI] [PubMed] [Google Scholar]
- 17.Webster MJ., Herman MM., Kleinman JE., Shannon Weickert C. BDNF and trkB mRNA expression in the hippocampus and temporal cortex during the human lifespan. Gene Expr Patterns. 2006;6:941–951. doi: 10.1016/j.modgep.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 18.Douillard-Guilloux G., Guilloux JP., Lewis DA., Sibille E. Anticipated brain molecular aging in Major. Depression. Am J Geriatr Psychiatry. 2012; Oct 31 [Epub ahead of print]. doi: 10.1016/j.jagp.2013.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Glorioso C., Sibille E. Between destiny and disease: genetics and molecular pathways of human central nervous system aging. Prog Neurobiol. 2011;93:165–181. doi: 10.1016/j.pneurobio.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McKinney BC., Sibille E. The age-by-disease interaction hypothesis of late-life depression. Am J Geriatr Psychiatry. In press. doi: 10.1016/j.jagp.2013.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.American Psychiatric Association. Diagnostic criteria from DSM-IV-TR. Washington, DC: American Psychiatric Association;2000 [Google Scholar]
- 22.Kessler RC., Berglund P., Demler O., et al The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA. 2003;289:3095–3105. doi: 10.1001/jama.289.23.3095. [DOI] [PubMed] [Google Scholar]
- 23.Belmaker RH., Agam G. Major depressive disorder. N Engl J Med. 2008;358:55–68. doi: 10.1056/NEJMra073096. [DOI] [PubMed] [Google Scholar]
- 24.Wolkowitz OM., Epel ES., Reus VI., Mellon SH. Depression gets old fast: do stress and depression accelerate cell aging? Depress Anxiety. 2010;27:327–338. doi: 10.1002/da.20686. [DOI] [PubMed] [Google Scholar]
- 25.Weissman MM., Leaf PJ., Tischler GL., et al Affective disorders in five United States communities. Psychol Med. 1988;18:141–153. doi: 10.1017/s0033291700001975. [DOI] [PubMed] [Google Scholar]
- 26.Koenig HG., Blazer DG. Epidemiology of geriatric affective disorders. Clin Geriatr Med. 1992;8:235–251. [PubMed] [Google Scholar]
- 27.Berger AK., Fratiglioni L., Winblad B., Backman L. Alzheimer's disease and depression: preclinical comorbidity effects on cognitive functioning. Cortex. 2005;41:603–612. doi: 10.1016/s0010-9452(08)70200-4. [DOI] [PubMed] [Google Scholar]
- 28.Granholm AC., Boger H., Emborg ME. Mood, memory and movement: an age-related neurodegenerative complex? Curr Aging Sci. 2008;1:133–139. doi: 10.2174/1874609810801020133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wiener P., Alexopoulos GS., Kakuma T., Meyers BS., Rosenthal E., Chester J. The limits of history-taking in geriatric depression. Am J Geriatr Psychiatry. 1997;5:116–125. [PubMed] [Google Scholar]
- 30.Alexopoulos GS. Depression in the elderly. Lancet. 2005;365:1961–1970. doi: 10.1016/S0140-6736(05)66665-2. [DOI] [PubMed] [Google Scholar]
- 31.Lenze EJ., Schulz R., Martire LM., et al The course of functional decline in older people with persistently elevated depressive symptoms: longitudinal findings from the Cardiovascular Health Study. J Am Geriatr Soc. 2005;53:569–575. doi: 10.1111/j.1532-5415.2005.53202.x. [DOI] [PubMed] [Google Scholar]
- 32.Park DC., Lautenschlager G., Hedden T., Davidson NS., Smith AD., Smith PK. Models of visuospatial and verbal memory across the adult life span. Psychol Aging. 2002;17:299–320. [PubMed] [Google Scholar]
- 33.Alexopoulos GS., Schultz SK., Lebowitz BD. Late-life depression: a model for medical classification. Biol Psychiatry. 2005;58:283–289. doi: 10.1016/j.biopsych.2005.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hekimi S., Guarente L. Genetics and the specificity of the aging process. Science. 2003;299:1351–1354. doi: 10.1126/science.1082358. [DOI] [PubMed] [Google Scholar]
- 35.Fontana L., Partridge L., Longo VD. Extending healthy life span — from yeast to humans. Science. 2010;328:321–326. doi: 10.1126/science.1172539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yankner BA., Lu T., Loerch P. The aging brain. Annu Rev Pathol. 2008;3:41–66. doi: 10.1146/annurev.pathmechdis.2.010506.092044. [DOI] [PubMed] [Google Scholar]
- 37.Thibault O., Porter NM., Chen KC., et al Calcium dysregulation in neuronal aging and Alzheimer's disease: history and new directions. Cell Calcium. 1998;24:417–433. doi: 10.1016/s0143-4160(98)90064-1. [DOI] [PubMed] [Google Scholar]
- 38.Thibault O., Hadley R., Landfield PW. Elevated postsynaptic [Ca2+]i and L-type calcium channel activity in aged hippocampal neurons: relationship to impaired synaptic plasticity. J Neurosci. 2001;21:9744–9756. doi: 10.1523/JNEUROSCI.21-24-09744.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhardwaj RD., Curtis MA., Spalding KL., et al Neocortical neurogenesis in humans is restricted to development. Proc Natl Acad Sci U S A. 2006;103:12564–12568. doi: 10.1073/pnas.0605177103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pakkenberg B., Gundersen HJ. Neocortical neuron number in humans: effect of sex and age. J Comp Neurol. 1997;384:312–320. [PubMed] [Google Scholar]
- 41.Morrison JH., Hof PR. Life and death of neurons in the aging brain. Science. 1997;278:412–419. doi: 10.1126/science.278.5337.412. [DOI] [PubMed] [Google Scholar]
- 42.Resnick SM., Pham DL., Kraut MA., Zonderman AB., Davatzikos C. Longitudinal magnetic resonance imaging studies of older adults: a shrinking brain. J Neurosci. 2003;23:3295–3301. doi: 10.1523/JNEUROSCI.23-08-03295.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sowell ER., Peterson BS., Thompson PM., Welcome SE., Henkenius AL., Toga AW. Mapping cortical change across the human life span. Nat Neurosci. 2003;6:309–315. doi: 10.1038/nn1008. [DOI] [PubMed] [Google Scholar]
- 44.Wilson RS., Bienias JL., Berry-Kravis E., Evans DA., Bennett DA. The apolipoprotein E epsilon 2 allele and decline in episodic memory. J Neurol Neurosurg Psychiatry. 2002;73:672–677. doi: 10.1136/jnnp.73.6.672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lu T., Pan Y., Kao SY., et al Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–891. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- 46.Kang HJ., Kawasawa Yl., Cheng F., et al Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–489. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Colantuoni C., Lipska BK., Ye T., et al Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature. 2011;478:519–523. doi: 10.1038/nature10524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Berchtold NC., Cribbs DH., Coleman PD., et al Gene expression changes in the course of normal brain aging are sexually dimorphic. Proc Natl Acad Sci U S A. 2008;105:15605–15610. doi: 10.1073/pnas.0806883105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Finch CE., Tanzi RE. Genetics of aging. Science. 1997;278:407–411. doi: 10.1126/science.278.5337.407. [DOI] [PubMed] [Google Scholar]
- 50.Lee CK., Weindruch R., Prolla TA. Gene-expression profile of the ageing brain in mice. Nat Genet. 2000;25:294–297. doi: 10.1038/77046. [DOI] [PubMed] [Google Scholar]
- 51.Lee CK., Klopp RG., Weindruch R., Prolla TA. Gene expression profile of aging and its retardation by caloric restriction. Science. 1999;285:1390–1393. doi: 10.1126/science.285.5432.1390. [DOI] [PubMed] [Google Scholar]
- 52.Blalock EM., Chen KC., Sharrow K., et al Gene microarrays in hippocampal aging: statistical profiling identifies novel processes correlated with cognitive impairment. J Neurosci. 2003;23:3807–3819. doi: 10.1523/JNEUROSCI.23-09-03807.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jiang CH., Tsien JZ., Schultz PG., Hu Y. The effects of aging on gene expression in the hypothalamus and cortex of mice. Proc Natl Acad Sci U S A. 2001;98:1930–1934. doi: 10.1073/pnas.98.4.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lu T., Pan Y., Kao SY., et al Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–891. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- 55.Avramopoulos D., Szymanski M., Wang R., Bassett S. Gene expression reveals overlap between normal aging and Alzheimer's disease genes. Neurobiol Aging. 2011;32:2319;e2327–2334. doi: 10.1016/j.neurobiolaging.2010.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Blalock EM., Geddes JW., Chen KC., Porter NM., Markesbery WR., Landfield PW. Incipient Alzheimer's disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc Natl Acad Sci U S A. 2004;101:2173–2178. doi: 10.1073/pnas.0308512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miller JA., Horvath S., Geschwind DH. Divergence of human and mouse brain transcriptome highlights Alzheimer disease pathways. Proc Natl Acad Sci U S A. 2010;107:12698–12703. doi: 10.1073/pnas.0914257107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nussbaum RL., Ellis CE. Alzheimer's disease and Parkinson's disease. N Engl J Med. 2003;348:1356–1364. doi: 10.1056/NEJM2003ra020003. [DOI] [PubMed] [Google Scholar]
- 59.Kurosu H., Yamamoto M., Clark JD., et al Suppression of aging in mice by the hormone Klotho. Science. 2005;309:1829–1833. doi: 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Masuda H., Chikuda H., Suga T., Kawaguchi H., Kuro-o M. Regulation of multiple ageing-like phenotypes by inducible klotho gene expression in klotho mutant mice. Mech Ageing Dev. 2005;126:1274–1283. doi: 10.1016/j.mad.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 61.Colman RJ., Anderson RM., Johnson SC., et al Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325:201–204. doi: 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tripp A., Oh H., Guilloux JP., Martinowich K., lewis DA., Sibille E. Brainderived neurotrophic factor signaling and subgenual anterior cingulate cortex dysfunction in major depression. Am J Psychiatry. 2012;169:1194–1202. doi: 10.1176/appi.ajp.2012.12020248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sakata K., Woo NH., Martinowich K., et al Critical role of promoter IV-driven BDNF transcription in GABAergic transmission and synaptic plasticity in the prefrontal cortex. Proc Natl Acad Sci U S A. 2009;106:5942–5947. doi: 10.1073/pnas.0811431106. [DOI] [PMC free article] [PubMed] [Google Scholar]