

Summary
The dsRNA-dependent protein kinase (PKR) is a key mediator of the anti-viral and anti-proliferative effects of interferon. Unphosphorylated PKR is characterized by inhibitory interactions between the kinase and RNA binding domains (RBDs), but the structural details of the latent state and its unraveling during activation are not well understood. To study PKR regulation by NMR we assigned a large portion of the backbone resonances of the catalytically inactive K296R kinase domain, and performed 15N-HSQC titrations of this kinase domain with the RBDs. Chemical shift perturbations in the kinase indicate that RBD2 binds to the substrate eIF2α docking site in the kinase C-lobe. Consistent with these results, a mutation in the eIF2α docking site, F495A displays weaker interactions with the RBD. The full-length RBD1+2 binds more strongly to the kinase domain than RBD2 alone. The observed chemical shift changes extend from the eIF2α binding site into the kinase N-lobe and inside the active site, consistent with weak interactions between the N-terminal part of the RBD and the kinase.
Introduction
The interferon-inducible dsRNA-dependent protein kinase (PKR) is a key mediator of the cellular antiviral and anitproliferative defense.1–3 PKR belongs to a group of kinases that phosphorylate the α–subunit of translation initiation factor eIF2 and block protein synthesis in response to stress.4 In addition to exerting translational control, PKR mediates the activation of a number of transcription factors, including NF-κB, STATs and p53.5–10 Normally PKR exists in an latent state that is activated or inhibited by non-covalent interactions with various cellular and viral proteins and double-stranded RNAs (Fig. 1), and by phosphorylation/dephosphorylation events at multiple Ser, Thr and Tyr sites along the entire protein.11,12 The structural details of the auto-inhibited state and a myriad of activation and inhibition scenarios are not well understood.
Figure 1.
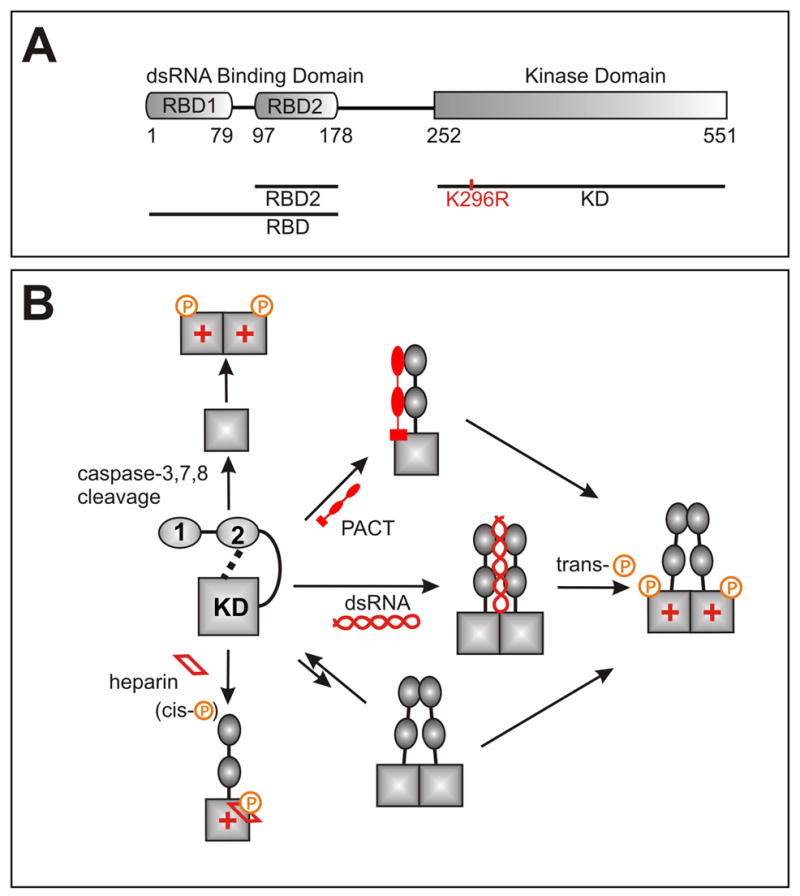
a) PKR domain structure and constructs used in this work. b) Some of the known mechanisms of PKR activation. Kinase catalytic activity is denoted with ‘+’. PKR exists in an autoinhibited state characterized by interactions between the kinase and regulatory domains. Prior to this work only RBD2-KD interactions have been observed, and the surfaces involved have not been identified. Activation requires sequestering of the RBD from the kinase, phosphorylation at mutliple sites, and dimerization. Dimerization is mediated by RBD1 and the N-terminus of the KD1,15,18–20,30 and is enhanced by phosphorylation of the activation loop of the kinase,15,21,30 but the precise details and relationship between activator binding, dimerization and auto-phosphorylation are not completely understood.
Full length PKR (Fig. 1) contains two highly conserved domains (RBD1 and RBD2) which make up the dsRNA binding domain (RBD),13 a linker region of unknown function, and a kinase domain (KD). The structure of the RBD was solved by NMR14 and the X-ray crystal structure of the kinase domain was recently elucidated.15 Numerous biochemical studies have shown that in the latent PKR state the kinase domain is inhibited by interactions with the regulatory domains(reviewed in 1). Binding of RBD2 but not RBD1 to the kinase was demonstrated by NMR16 but the domain surfaces involved in the interaction are not known. In the best known activation scenario, dsRNA binding releases the RBD from the kinase domain and promotes protein dimerization and trans-autophosphorylation in the activation loop.11,17–21 While some viral dsRNAs transcripts activate PKR, others are known to inhibit it, probably via direct interactions with the kinase domain.22 The cellular protein PACT activates PKR in the absence of dsRNA.23 PACT forms a heterodimer with PKR, extending across the RBD and onto the linker or the N-terminal region of the kinase domain.24 PKR is also activated by the polyanion heparin.25 Heparin binds the KD near the active site, as suggested by deletion experiments26 and triggers cis-autophosphorylation.27 PKR is also activated during apoptosis by proteolytic cleavage of the regulatory domain by several caspases,28 and is the only known kinase that is inhibited by viral proteins and RNAs (reviewed in 3).
Here we report the NMR backbone assignments of the kinase domain that will facilitate characterization of the multiple regulatory interactions of this domain. We perform NMR titrations of the KD and RBD to map the auto-inhibitory interactions of PKR. The results suggest that the entire regulatory domain participates in inhibition, by masking the eIF2α recognition site, and by allosterically or directly blocking the catalytic active site.
Results
The K296R mutant used in this study is catalytically inactive and lacks the toxicity of the wild-type protein to host expression systems. The K296 side chain extends into the ATP-binding pocket and replacement with Arg inhibits phosphate transfer without affecting ATP binding.29 The full length K296R mutant binds and is phosphorylated by the wild-type enzyme, and has been widely used to study PKR autophosphorylation and dimerization.1,11,17,19,20,21,25,26,30 Important for the NMR studies, this mutant can not autophoshorylate during bacterial expression, and therefore does not dimerize.21,30 Limited proteolysis experiments on the full-length protein identified a domain coinciding with the product of caspase cleavage during apoptosis, D251-C551.28 The KD construct used in the NMR, M252-C551 (K296R) was cloned with no affinity tags. Expression in Rosetta DE3 (Novagen) cells yielded a large quantity of inclusion body protein which was refolded with 30% yield. The samples had maximum achievable concentrations of 250 uM which remained stable for 2 days at room temperature.
Backbone assignments of the kinase domain
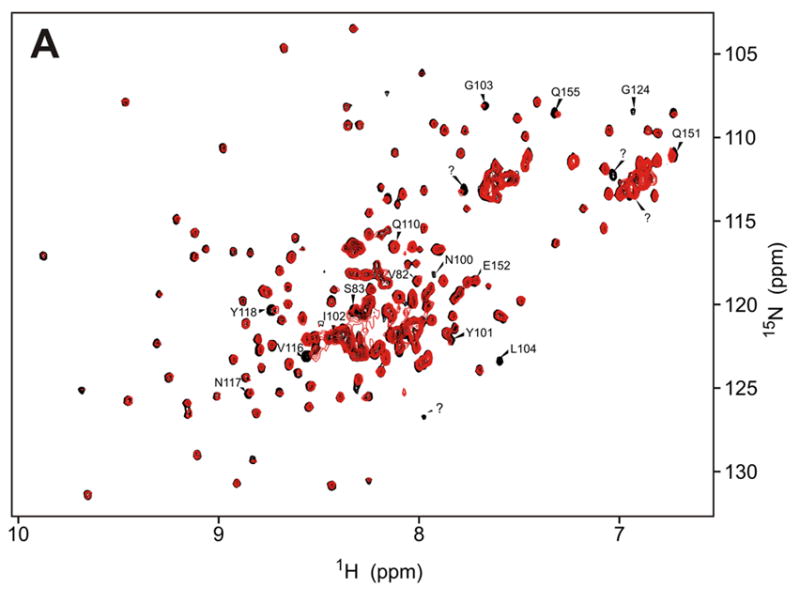
NMR assignment of the protein backbone (Figure 2) was performed using TROSY triple resonance experiments,31 3D-15N-NOESY-HSQC, and 2D 15N-HSQC spectra of amino acid-specific 15N-labeled samples (labeled at Leu, Val, Lys, Phe and Tyr). HNCACB and HNCOCACB triple-resonance spectra were acquired using non-linear sampling32,33 to reduce acquisition time to within the limits imposed by sample stability. The assignments were performed manually, and with the program MARS.34 286 out of the expected 292 residues (98%) of the kinase are observed in the HNCO spectrum. The HN, N, Cα and Cβ resonances of 224 (75%) of the amino acids were assigned (Figure 2b). The remaining resonances are heavily overlapped in the center of the 15N-HSQC spectrum (Fig. 2a) and could not be assigned. Unassigned residues are concentrated mainly in two regions of the protein that appear to be disordered under the studied conditions. Namely, the kinase activation loop is unassigned, as is a serine-rich, acidic region (P334-S357) of unknown function, which contains a repeat of the sequence DYDPEXSXXSXXS. This motif is unique among the eIF2α kinases and is only present once in PKR from other species. The region S337-S350 was deleted in the protein construct used to solve the X-ray crystal structure by Dar et al., who found this region largely responsible for the low solubility of the protein.15 The obtained NMR assignments are sufficient to perform binding studies of the kinase domain.
Figure 2.
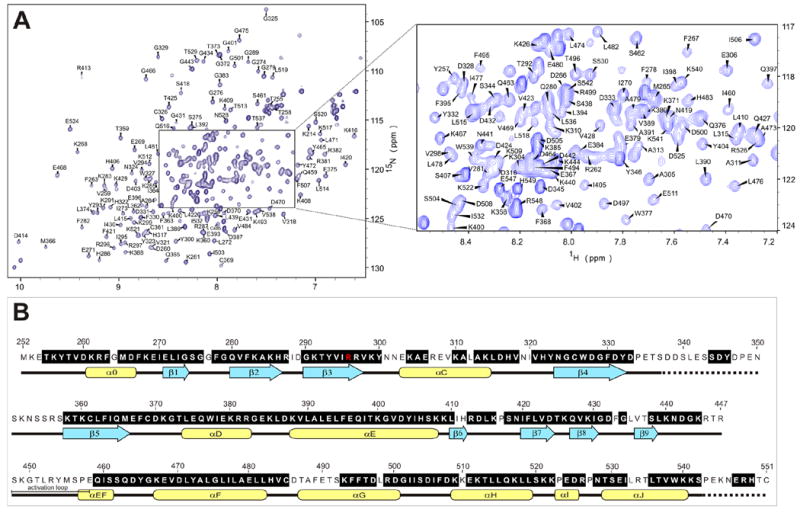
a)Trosy-15N-HSQC spectrum of the PKR K296R kinase domain (residues 252-551, 250μM in 50 mM sodium phosphate, 25 mM ammonium sulfate, pH 6.70); b) PKR kinase primary sequence showing in black the assigned amino acids. With the exception of the K296R mutation (red) the sequence used is that of the wild-type protein. Secondary structure motifs are taken from the X-ray crystal structure.15 Regions with unknown structure are shown with dashed lines.
Kinase - dsRNA binding domain interactions
To map the interaction surface between the RBD and the kinase domain we performed NMR binding studies of full-length RBD (1-178), the second dsRNA binding domain RBD2 (97-178) and the KD (252-551, K296R) (Fig. 1a). An unlabeled sample of one domain was titrated into an 15N-labeled sample of the other domain, monitoring changes in the 15N-HSQC spectra of the labeled protein (Figures 3–5). Addition of either RBD construct significantly affected the spectra of the kinase domain. We observed concentration-dependent shifts consistent with a fast exchange binding regime and, at high RBD concentrations, broadening and the appearance of new peaks (Figs. 3a and 4a). The changes observed in the kinase upon addition of the full RBD are more pronounced than those from the RBD2 indicating that both RNA binding motifs interact with the catalytic domain.
Figure 3.
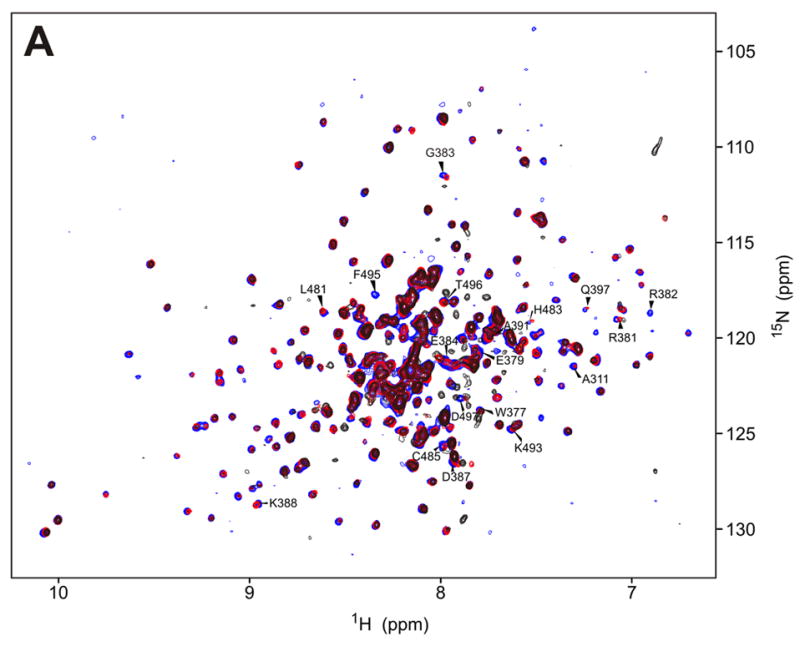
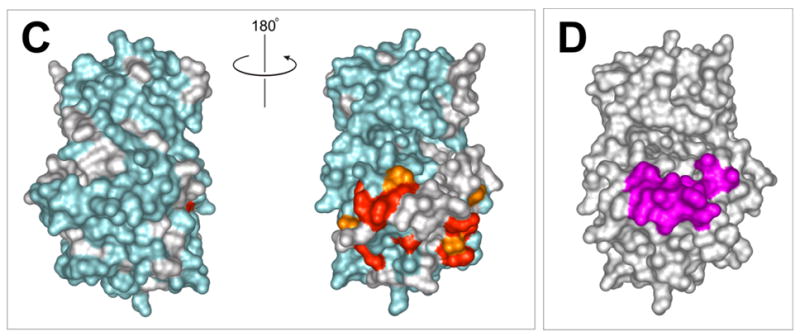
a) 15N-HSQC spectra of the kinase domain alone (0.1 mM, blue trace) and in the presence of 0.2 mM (red) and 0.7 mM (black) RBD2 (97-178). Amide residues that display the most significant changes are labeled; b) Structure of the PKR KD15 (PDB 2A1A) showing in red the locations of the affected amide labeled in (a) (orange: 0.03ppm > δδ > 0.01ppm; red: δδ > 0.03ppm). Unassigned and ambiguous residues are shown in gray, while unaffected residues are in cyan; c) Surface representation of the results shown in (b). The view on the right corresponds to the orientation of the middle structure in (b) and is identical to (d); d) Structure of the KD showing the eIF2α docking site. Residues within 5 angstroms of resolved eIF2α atoms are colored purple.
Figure 5.
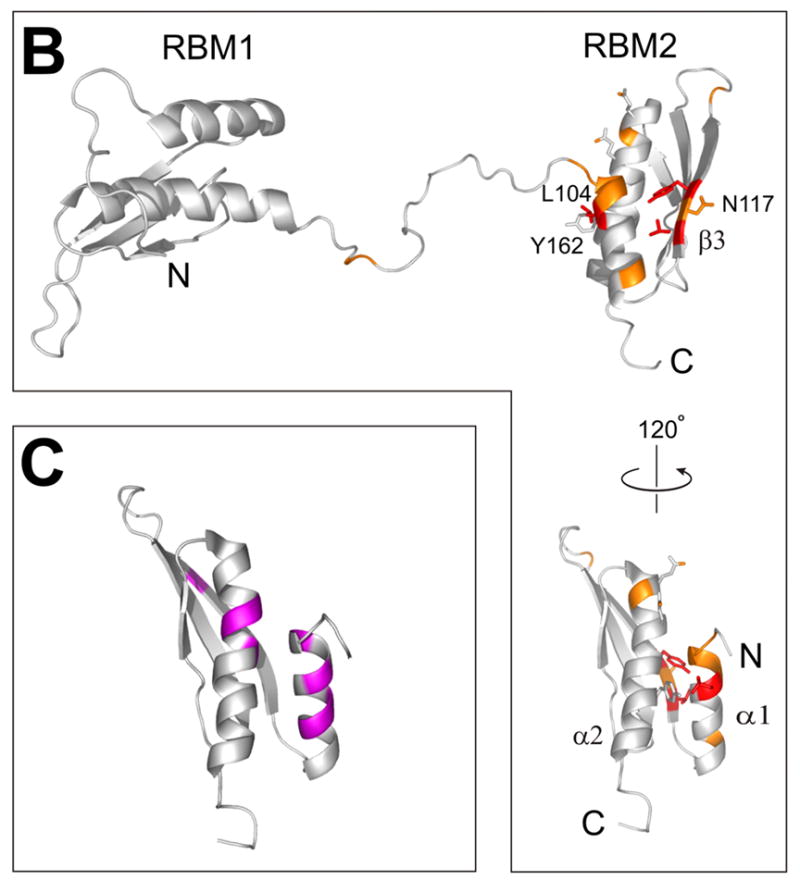
a) 15N-HSQC spectrum of the full-length dsRNA binding domain, RBD (1-178) alone (black trace) and in the presence of two-fold excess KD (K296R 252-551, red). Assignments are based on published data14; Residues that display reproducible changes (δδ > 0.01ppm) in the titration are labeled; b) Structure of the PKR RBD14 (PDB 1QU6) showing the locations of the amide residues affected by addition of KD (orange: δδ > 0.01ppm, red: broadening and δδ >0.03ppm); c) Structure of the PKR RBD showing NMR chemical shift changes observed in titrations with various viral dsRNAs.22,47
Figure 4.
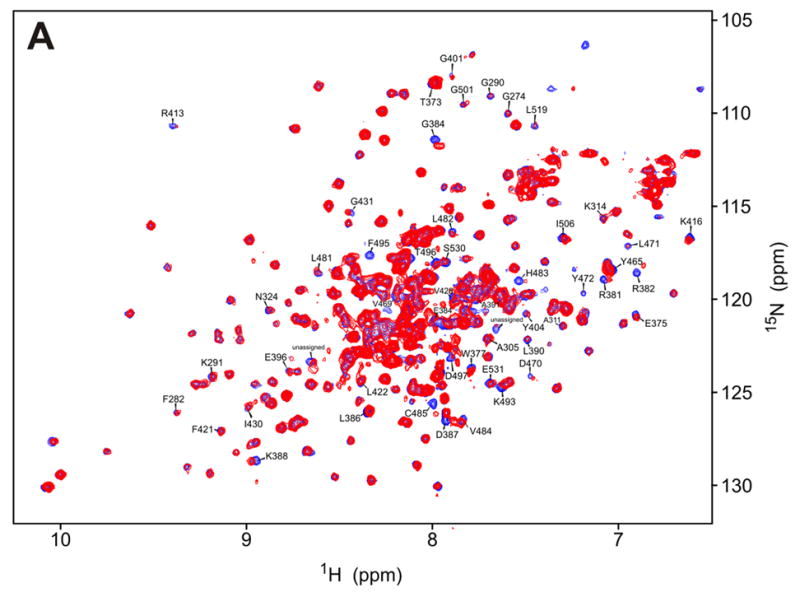
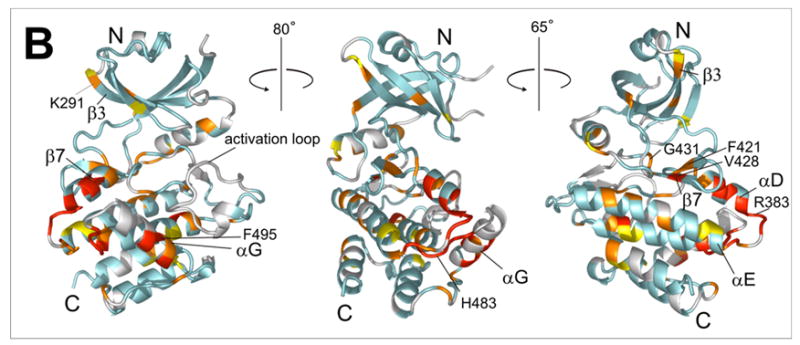
a) 15N-HSQC spectrum of the kinase domain (K296R) alone (0.2 mM, blue trace) and in the presence of 0.6 mM (red) RBD (1-178). Amide residues that display the most significant changes are labeled; b) Structure of the PKR KD15 (PDB 2A1A) showing the locations of the affected amide groups labeled in (a) (orange: δδ > 0.01ppm, red: δδ > 0.03ppm and broadening).
The HSQC spectra of the bound KD have significantly reduced resolution and signal to noise ratios, compared to the kinase domain alone. To eliminate artifacts the titrations were performed in duplicate, at slightly different pH. The observed perturbations are relatively small (δδ < 0.1ppm, Supporting Information) but sufficient to clearly map the interaction surfaces. For clarity the peaks were grouped as unaffected (δδ < 0.01 ppm), weakly affected (δδ > 0.01ppm), strongly affected (δδ > 0.03ppm), and ambiguous or unassigned. The results were mapped on the X-ray structure of kinase domain15 and the NMR structure of the RBD14 (Figs. 3b, 4b and 5b).
Kinase domain - RBD2 interaction
The chemical shift mapping of the kinase surface involved in interactions with the dsRNA binding domain 2 (RBD2) is shown in Figure 3. Kinase residues that exhibit significant changes upon addition of RBD2 (97-179) are clustered in the C-terminal lobe, mainly in a charged region between helices αD and αE (G381-D387), and helix αG and flanking regions (Leu481-Ile506). The rest of the protein, especially the N-lobe, is conspicuously unperturbed. As shown in Figures 3c and d, the data define a kinase-RBD2 binding surface that coincides with the substrate eIF2α docking site identified in the X-ray crystal structure and accompanying mutagenesis studies.15,30 The importance of helix αG for RBD2 binding was confirmed in mutagenesis studies (see below).
Kinase domain - RBD1+2 interaction
To study the interactions of the kinase with the N-terminal part of the RBD, we titrated full-length RBD1+2 (1-178) to the labeled kinase domain (Fig. 4a). Chemical shift changes are observed in the same region that responds to RBD2, and additionally, smaller but reproducible changes occur in the kinase N-lobe. Changes in the KD throughout the protein hint at a conformational change in the kinase upon RBD binding. At low kinase concentration that allows the use of a large excess RBD, more pronounced changes occur in the N-terminal lobe of the kinase, in contiguous regions along the β3-edge of the β–barrel (K291-K296), the hinge region β7–β8, and the kinase active site (Supporting information). However, the low signal to noise ratio of the spectra precluded reliable analysis of that data and it is not presented here. Regardless, comparison of the RBD2 and RBD1+2 titrations shown in Figures 3a and 4a suggest that the entire RBD interacts with the kinase. The data are consistent with weak interactions of the N-terminal part of the RBD with the N-lobe of the kinase.
RBD interaction with kinase domain
In order to map the RBD surface involved in the interaction with the kinase domain, the 15N-HSQC titrations were repeated using 15N-labeled RBD (1-178) and unlabeled kinase. The spectra of the RBD were analyzed using the published NMR assignments of the RBD.14 The limited solubility of the kinase construct precluded the use of large excess unlabeled KD needed to produce large chemical shift changes in the RBD. The spectra of RBD (Fig. 5a) indicate very little change in RBD1 (1-80) but reproducible changes in RBD2 (97-178) and the linker connecting the two subunits. This pattern again indicates stronger interactions of RBD2 than the rest of the RBD with the kinase. Three amide residues in RBD2 stand out due to a larger change in chemical shift (~ 0.03ppm) and significant broadening on addition of KD (Fig. 5). Amino acids V116 and Y118 in the β3 strand and L104 in helix α1 are located on adjacent faces of the RBD2. Recent NMR studies of the interactions of PKR RBD with various viral dsRNAs identified helix α1 as a mediator of the interactions with dsRNA22,47 (Fig. 5c) and it is reasonable to expect at least partial overlap between the KD and dsRNA binding sites on the RBD2 if dsRNA binding is to sequester the RBD from the kinase. Mutagenesis studies of the RBD2 did not confirm the importance of β3 for KD binding (see below), implying that helix α1 containing L104 may be directly involved in binding to the kinase. Y162, a residue in van der Waals contact with Leu104 (Figure 5b) was recently identified as an autophosphorylation site important for PKR activation.12 It is possible that a charged pY162 serves to “eject” the RBD2 from the kinase.
NMR studies of KD and RBD mutants
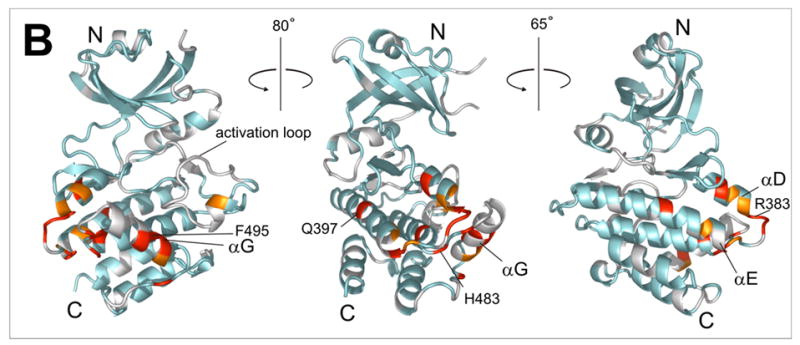
In order to independently verify the location of RBD2 binding on the kinase35 we mutated several of the amino acids implicated in the chemical shift mapping and repeated the 15N-HSQC titrations with mutant proteins. Mutations in the charged region, RR381/382GD and D387R produced misfolded or aggregated protein, as judged by the 15N-HSQC spectra (not shown). The mutants F495A and H483D (both K296R) yielded 15N-HSQC spectra that looked very similar to the original K296R construct, with changes mostly in the vicinity of the mutated residue. H483D kinase aggregated upon addition of RBD preventing further analysis. Addition of excess unlabeled RBD (1-178) to F495A KD (K296R) produced only slight changes in the spectrum, compared to the original K296R construct (Fig. 6a). Notably, residues affected by the F495A mutation and those affected by addition of RBD are clustered in distinct regions of the KD. Most of the difference between the original K296R and the mutated F495A kinase are in residues in the vicinity of F495, and those amide signals do not change significantly upon addition of RBD. The majority of the changes in F495A upon addition of RBD are located primarily in the N-lobe, in the putative RBD1-binding region. The data strongly support the idea that KD-RBD interaction occurs in two distinct regions of the kinase. F495 is located in the αG helix of the kinase, confirming the importance of helix αG for RBD2 binding.
Figure 6.
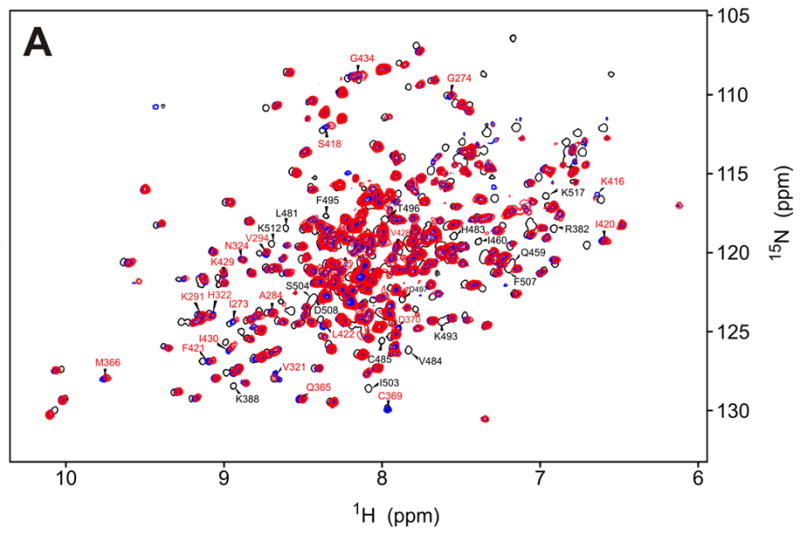
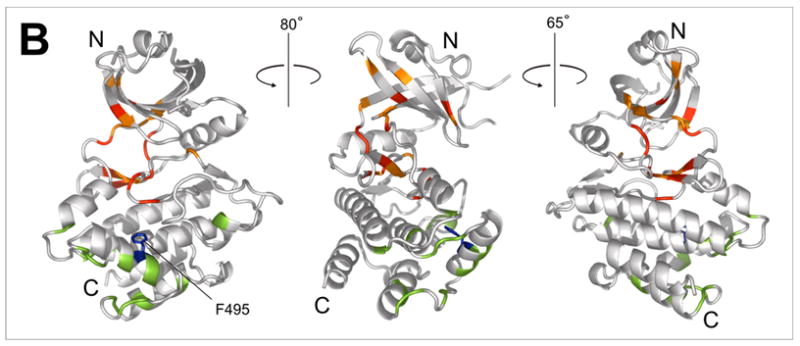
a)15N-HSQC spectrum of the F495A/K296R kinase alone (blue) and in the presence of 5-fold excess RBD (1-178) (red). Affected residues (red labels) are assigned based on the corresponding peaks in the K296R KD spectrum (black trace). Major differences between the K296R and the K296R/F495A spectra are observed in the vicinity of F495 (labeled in black letters); b) Mapping of the residues indicated in (a) on the kinase structure. Chemical shift changes upon addition of RBD are indicated in red (δδ > 0.03ppm) and orange (0.03ppm > δδ > 0.01ppm). Differences in the spectra of K296R and K296R/F495A labeled in (a) are colored green. The mutated F495 is shown in blue.
To verify the RBD2 binding surfaces implicated in the titrations as important for kinase binding, we tested a N117R RBD (1-178) mutant. We chose N117 because it is between the strongly affected amide residues Y118 and V116, and its side chain is exposed, whereas the side chains of V116 and Y118 are buried and mutations at those positions could affect the proper folding of the domain. However, the changes of the 15N-HSQC of the N117R spectrum that occur on addition of unlabeled KD are identical to the changes in the wild-type RBD (data not shown), suggesting that the N117 side chain and the corresponding surface of the β3 strand are not directly involved in KD binding. The observed changes in the V116 and Y118 signals may instead be due to indirect effects on the NH’s of residues 116 and 118, transmitted via the amino acid side chains, which are packed against helix α1.
Discussion
Figure 7 depicts some of the known PKR activation scenarios in the context of our results.The regulatory domain is “wrapped” around the kinase domain, and both RBDs interact with the kinase. DsRNA binding domain 2 blocks access to the substrate eIF2α by binding to the kinase C-terminal lobe, contacting helix αG and the adjacent loop R381-D387. This surface is clearly outlined by the NMR chemical shift data, and is further supported by the weakened F495A interaction with the RBD. Incidentally, Dey and coworkers found that F495P mutation of the KD PKR impaired eIF2α phosphorylation, but not autophosphorylation or phosphorylation of substrates that do not require docking to helix αG.30 This implies that RBD2 alone would inhibit phosphorylation of eIF2α, but not autophosphorylation or phosphorylation of other substrates. Auto-inhibition by masking of the substrate binding site has been previously observed, e. g. in the c-AMP-dependent kinase PKA,36 and has also been proposed for PKR. Sharp et al. found that the vaccinia virus protein e3l, a PKR inhibitor, interacted with the C-terminal lobe of the kinase (amino acids 367-523) and competed with binding of eIF2α. It was proposed that PKR e3I inhibition as well as PKR auto-inhibition occurred via masking of the substrate binding site.37
Figure 7.
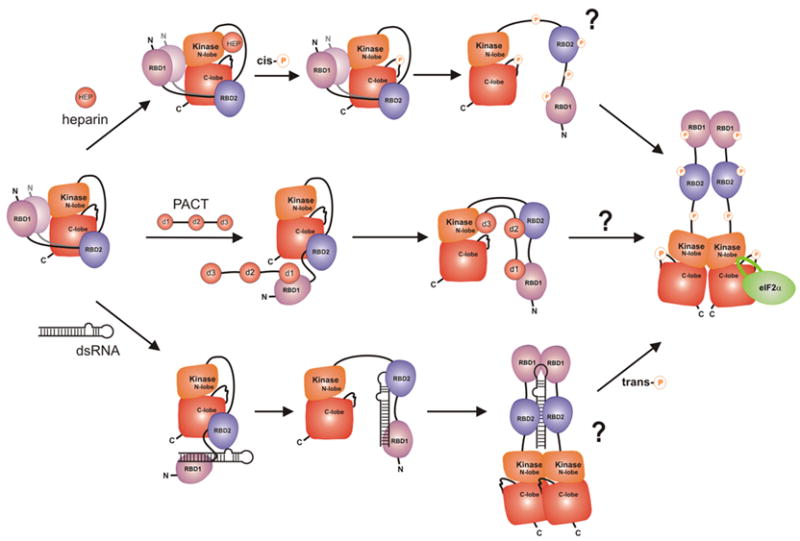
A detailed model of PKR regulation showing previously known activation scenarios in the context of the present work. According to our data, RBD2 masks the substrate eIF2a docking site, while the N-terminal region of the RBD weakly interacts with the kinase N-lobe. The precise location of the RBD1-KD interaction is unknown, indicated by the “blurry” RBD1. Access to the active site may be hindered by the activation loop, allosterically, or by the linker connecting the kinase and RBD2. Activation by heparin occurs via interactions with the kinase N-lobe26 and subsequent cis-autophosphorylation.27 PKR activation by PACT occurs via sequestering of the regulatory domain from the kinase and requires interactions of domain d3 with the kinase N-lobe.24,48 DsRNA releases the RBD from the kinase domain and stimulates dimerization and trans-autophosphorylation.11,17–21 Activation loop phosphorylation stimulates dimerization and is required for activity.1,15,21,30 In addition, multiple phosphorylation sites along the protein may serve to prevent the RBD from re-associating with the kinase, to fine-tune enzymatic activity, or to mediate various non-covalent interactions of PKR. A major challenge that remains is to elucidate the sequence and mechanism of autophosphorylation events that occur during PKR activation.
Based on the NMR data we estimate that the kinase-RBD2 (97-178) interaction has a Kd ~ 200 μM. This number represents the lowest RBD2 concentration that affects the spectrum of the kinase at 50 μM. Using the same criteria, the Kd of the full-length RBD construct (1-178) containing both RBD1 and RBD2 appears to be smaller than 100 μM. However, the observed fast exchange regime is consistent with an interaction that is weaker than the previously reported Kd ~ 500 nM measured by surface plasmon resonance.16 The lack of changes in RBD1 upon addition of unlabeled kinase (Figure 5) indicates that the interactions of RBD with the kinase are mainly due to RBD2. It is therefore unlikely that full length RBD binds with a dramatically higher affinity to the kinase than RBD2 alone.
The location of the N-terminal region of the RBD (RBD1 and the RBD1-RBD2 linker) on the kinase is not clearly defined by the NMR data. The lack of changes in the spectrum of RBD1 (Fig. 5) upon addition of unlabeled kinase confirms that the RBD1-kinase interaction is very weak. This is consistent with previous NMR studies that found no kinase-RBD1 interactions.16 Nevertheless, a comparison of the KD-RBD2 (97-178) and KD-RBD (1-178) titrations (Figs. 3a and 4a) clearly indicates the presence of additional RBD-kinase interactions beyond RBD2. Binding of the N-terminal region of the RBD to both K296R and the mutant F495A/K296R causes changes in the N-lobe and the active site of the kinase (Figures 4b and 6b). These data are consistent with RBD1 and/or the RDB1-RBD2 linker binding to the β3/β7 face of the kinase (Fig. 4b), but are not conclusive.
Experimental evidence suggests that the catalytic site of PKR is inaccessible in the latent protein. For example, it has been observed that full length PKR only binds ATP after addition of dsRNA.38,17 Moreover, a free PKR kinase domain lacking the RBD autophosphorylates (albeit weakly) at T446 and T451 in the activation loop, but does not trans-phosphorylate full-length PKR at these positions.17 Our data does not indicate direct blockage of the active site by RBD-kinase interactions, leaving two possibilities.39 The active site could be restricted by conformational changes in the kinase, e.g. in the activation loop, induced by RBD-KD binding, analogous to the Src tyrosine kinase.40 Alternatively, the linker connecting the kinase domain and the RBD could act as a “pseudo-substrate”, analogous to the PKA-RIα interaction.36 Both possibilities are allowed in our model, but the former seems more plausible. The NMR analysis did not yield information on the activation loop, but the numerous chemical shift perturbations in the hinge region between the N- and C-terminal lobes support the possibility of allosteric regulation. Besides participation of the activation loop, which is likely given the importance of T446 phosphorylation, a kinase domain “squeezed” between the two RBDs (Fig. 7) may be inhibited via movement of the N- and C-lobes.
Very little is known about the role of the RBD-kinase linker. Positioning of the RBD2 on the substrate docking site naturally places the linker in front of the active site (Fig. 7), but to date there is no evidence for KD-linker interactions. Support for the idea that the unphosphorylated activation loop blocks the active site while the kinase-RBD linker does not participate in autoinhibition comes from the activators PACT and heparin. PACT has three domains that parallel the PKR domain structure. The N-terminal PACT domains heterodimerize with the PKR RBDs but alone are not sufficient for PKR activation. A third 66 amino acid domain, d3, interacts with an unknown region of PKR mapped between the RBD and the N-terminus of the KD and is necessary for PKR activation. A PACT-PKR fusion that contains PKR-RBD tethered to d3 via a 50 residue spacer (RBD-spacer-d3) also activates PKR. Based on these observations it was proposed that the entire PKR regulatory domain (1~250), including the RBD and the linker that connects it to the kinase needs to be sequestered to achieve full activation.24 The requirement for a spacer in the artificial PACT construct (RBD-spacer-d3) suggests that the RBD-KD linker of PKR is displaced by PACT-KD interactions rather than sequestered by direct binding to PACT d3. Analogous to PACT d3 domain, heparin has been shown to activate PKR by binding directly to the N-lobe of the KD. PKR deletion studies identified a heparin binding motif located at the entrance of the active site and partially overlapping the activation loop.26 Both heparin and PACT d3 binding could affect the conformation of the activation loop and trigger PKR cis-autophosphorylation, as observed for heparin.27 This has to occur without removing the RBD from the kinase, explaining the weakness of d3 activation compared to full-length PACT.
In summary, we have completed a large portion of the backbone assignments of the 300 residue catalytic domain of PKR. The assignments allowed mapping of the autoinhibitory interactions between the kinase and the dsRNA binding domain, and will facilitate further characterization of the intricate details of PKR inhibition and activation.
Note added in proof
Following completion of this work Li et al.published a report in which the interaction of the PKR kinase and PACT domain d3 was mapped to residues 326-337 on the kinase.48 Convincing NMR titration, immunoprecipitation and mutagenesis data was presented to show that this region in the N-lobe of the kinase is responsible for the autoinhibitory kinase-RBD2 interaction. In contrast, we observe only minor changes in the implicated region and attribute these to RBD1 binding. The assignments reported here will allow further studies to clarify this apparent contradiction.
Materials and Methods
DNA cloning
pBi vectors containing the wild-type and K296P PKR were kindly provided by Monique Davis of the Genetic Institute and the K296R mutant was prepared using the QuickChange mutagenesis kit (Stratagene). To prepare the different deletion constructs the following primers were used. RBD1&2 (Met1 - Ser178) forward: 5′-GGTGGTTCATATGGCTGGTGATCTTTCA-3′; RBD2 (Met97-Ser179) forward: 5′-GGTGGTTCATATGGGGAATTACATAGGCCTT-3′; RBD1&2 and RBD2 reverse: 5′-GGGATCCTCAAGAGGACAGGTAGTCA-3′; KD (Met252-Cys551) forward: 5′-GGTGGTTCATATGAAAGAAACAAAGTATACTG-3′. The PCR products were digested with NdeI (3′) and BamHI (5′) and inserted into plasmid pET15b (Novagen, RBD1&2 and RBD2) or pTWIN2 (New England Biolabs, KD), resulting in a His-tagged RBDs and non-tagged KD.
Protein expression and purification
All proteins were expressed in Rosetta(DE3) cells (Novagen) transformed by the corresponding expression plasmids. The cells were grown in the appropriate LB or M9 medium and induced at 0.8 OD600 with 1 mM (RBD and RBD2) or 3 mM (kinase domain) IPTG for 4 h (LB medium), 6 h (M9/H2O) or 12 h ( M9/D2O medium. Amino acid-specific 15N-labeled samples were prepared by growing the cells in M9 medium lacking nitrogen source and supplemented with nucleosides (200 mg/l each), unlabeled amino acids (200 mg/l each) and the α-15N-labeled amino acid (125 mg/l).
RBD2 and full-length RBD
The cell pellets were lysed in 50 mM sodium phosphate, pH 7.2, 500 mM NaCl, 5 mM β-mercaptoethanol, and the soluble fraction was loaded on Ni-NTA resin (Quiagen), eluted with 150 mM imidazole, dialyzed in 20 mM sodium phosphate, 150 mM NaCl, 5 mM DTT (pH 6.7) and concentrated to 0.8 mM.
Kinase domain
The cells were lysed in 50 ml (per 1L culture) 50 mM sodium phosphate, pH 7.2, 500 mM NaCl, 20 mM EDTA The insoluble fraction was sonicated and centifuged (15,000 g, 20 min) once in lysis buffer, containing 1% Triton X, and twice in water. The resulting inclusion body pellets were suspended in 10 ml 7M Gdm.HCl, 100 mM DTT with sonication, heated at 60°C for 30 min and added dropwise to 500 ml rapidly stirring cold refolding buffer (100 mM sodium phosphate, 100 mM ammonium sulfate, 100 mM arginine hydrochloride, 5mM DTT, pH 7.2). The refolding was allowed to proceed overnight at 4°C without stirring and the solution was filtered through 0.2 μm Express Millipore filter. The protein was precipitated by addition of 0.6 g/ml ammonium sulfate and centrifuged at 8,000 g (30 min). The pellet was suspended in 25 ml 50 mM Na phosphate, 100 mM ammonium sulfate, 5 mM DTT (pH 7.2), dialyzed against the same buffer, and precipitated once more with ammonium sulfate. The pellets were dissolved in minimal volume of the above buffer and loaded on a Superdex 200 gel filtration column (Amersham). The collected fractions were concentrated by ammonium sulfate precipitation and dialyzed against cold NMR buffer (20 mM sodium phosphate, 50 mM ammonium sulfate, 3 mM DTT, pH 6.70). Typically, the yield from 1L of bacterial culture was 10 mg of purified protein (~10%). The kinase was only soluble to 250 μM in cold buffer and gradually precipitated to 100μM at room temperature, in the course of 3 days. Precipitated protein was recycled by a second refolding.
Nuclear Magnetic Resonance
All NMR experiments were acquired at 25°C on Bruker Avance and Varian Inova 600 MHz instruments equipped with cryogenic probes. All protein samples were extensively deuterated to reduce proton-proton relaxation, except in the case of amino acid-specific labeling, where the protocol does not allow inexpensive deuteration. Trosy-HNCO, Trosy-HNCA/HNCOCA, Trosy-HNCACB/HNCOCACB and 3D 15N-NOESY-HSQC were acquired on fully deuterated, 13C/15N-labeled protein in 93%H2O/7%D2O buffer. 15N-HSQC experiments were acquired on protonated protein that was specifically 15N-labeled at Leu, Val, Phe, Tyr or Lys residues. Due to the poor solubility of the samples, the HNCACB/HNCOCACB pair of experiments was acquired using a 700 complex point non-linear schedule.32 The non-linear data was processed by maximum entropy reconstruction in the Rowland NMR toolkit33 (http://www.rowland.harvard.edu/rnmrtk/toolkit.html). All other data was processed by FFT in nmrPipe.41 NMR titrations were performed on 100–200 μM samples of 15N-labeled, perdeuterated protein with gradual addition of a concentrated stock of unlabeled binding partner followed by concentration in a spin-filter (Amicon) at 4°C. Chemical shift changes were calculated as √[(ΔH)2 + (0.2ΔN)2].
Backbone resonance assignment
Spectral analysis was performed in Sparky42 (http://www.cgl.ucsf.edu/home/sparky/). The spin systems were assigned manually and by iterative runs of the program MARS34 which uses triple resonance data, amino acid-specific assignments and chemical shift predictions. The manual analysis included 3D 15N-NOESY-HSQC data in conjunction with a homology model of the kinase generated in SwissModel. The program ShiftX43 was used to generate chemical shift predictions for the homology model of the kinase. In-house perl scripts were written to generate candidate assignments based the ShiftX predictions, as described in detail by others.44
Supplementary Material
Acknowledgments
We thank Drs. Mallika Sastry, Dmitri Ivanov and Songhyouk Park for advice and technical assistance. This work was supported by NIH-NCI grant U19-CA87427 and NIH grants GM47467 and CA 68262.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kaufman R. The double stranded RNA-activated protein kinase PKR. In: Sonenberg N, Hershey JWB, Mathews MB, editors. Translational Control of Gene Expression. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2000. pp. 503–28. [Google Scholar]
- 2.Williams BR. PKR: A sentinel kinase for cellular stress. Oncogene. 1999;18:6112–20. doi: 10.1038/sj.onc.1203127. [DOI] [PubMed] [Google Scholar]
- 3.Gale M, Jr, Katze MG. Molecular mechanisms of interferon resistance mediated by viral-directed inhibition of PKR, the interferon-induced protein kinase. Pharmacol Ther. 1998;78:29–46. doi: 10.1016/s0163-7258(97)00165-4. [DOI] [PubMed] [Google Scholar]
- 4.Sonenberg N, Hershey JWB, Mathews MB, editors. Translational control of gene expression. Cold Spring Harbor Laboratory Press 2000; Cold Spring Harbor, NY: [Google Scholar]
- 5.Gil J, Esteban M. Induction of apoptosis by the dsRNA-dependent protein kinase (PKR): Mechanism of action. Apoptosis. 2000;5:107–14. doi: 10.1023/a:1009664109241. [DOI] [PubMed] [Google Scholar]
- 6.Clemens MJ. Targets and mechanisms for the regulation of translation in malignant transformation. Oncogene. 2004;23:3180–8. doi: 10.1038/sj.onc.1207544. [DOI] [PubMed] [Google Scholar]
- 7.Malmgaard L. Induction and regulation of IFNs during viral infections. J Interferon Cytokine Res. 2004;24:439–54. doi: 10.1089/1079990041689665. [DOI] [PubMed] [Google Scholar]
- 8.Peel AL. PKR activation in neurodegenerative disease. J Neuropathol Exp Neurol. 2004;63:97–105. doi: 10.1093/jnen/63.2.97. [DOI] [PubMed] [Google Scholar]
- 9.D’Acquisto F, Ghosh S. PACT and PKR: Turning on NF-kappa B in the absence of virus. Sci STKE 2001. 2001:RE1. doi: 10.1126/stke.2001.89.re1. [DOI] [PubMed] [Google Scholar]
- 10.Williams BR. Signal integration via PKR. Sci STKE 2001. 2001:RE2. doi: 10.1126/stke.2001.89.re2. [DOI] [PubMed] [Google Scholar]
- 11.Zhang F, Romano P, Nagamura-Inoue T, Tian B, Dever T, Mathews M, Ozato K, Hinnebusch A. Binding of double-stranded RNA to protein kinase PKR is required for dimerization and promotes critical autophosphorylation events in the activation loop. J Biol Chem. 2001;276:24946–58. doi: 10.1074/jbc.M102108200. [DOI] [PubMed] [Google Scholar]
- 12.Su Q, Wang S, Baltzis D, Qu LK, Wong A, Koromillas A. Proc Natl Acd Sci USA. 2006;103:63–68. doi: 10.1073/pnas.0508207103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saunders L, Barber G. The dsRNA binding protein family: critical roles, diverse cellular functions. FASEB J. 2003;17:961–983. doi: 10.1096/fj.02-0958rev. [DOI] [PubMed] [Google Scholar]
- 14.Nanduri S, Carpick BW, Yang Y, Williams BR, Qin J. Structure of the double-stranded RNA-binding domain of the protein kinase PKR reveals the molecular basis of its dsRNA-mediated activation. EMBO J. 1998;17:5458–65. doi: 10.1093/emboj/17.18.5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dar A, Dever T, Sicheri F. Higher-order substrate recognition of eIF2α by the RNA-dependent protein kinase PKR. Cell. 2005;122:887–900. doi: 10.1016/j.cell.2005.06.044. [DOI] [PubMed] [Google Scholar]
- 16.Nanduri S, Rahman F, Williams BR, Qin J. A dynamically tuned double-stranded RNA binding mechanism for the activation of antiviral kinase PKR. EMBO J. 2000;19:5567–74. doi: 10.1093/emboj/19.20.5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu S, Kaufman RJ. Trans-autophosphorylation by the isolated kinase domain is not sufficient for dimerization or activation of the dsRNA-activated protein kinase R. Biochemistry. 2004;43:1107. doi: 10.1021/bi0360105. [DOI] [PubMed] [Google Scholar]
- 18.Vattem KM, Staschke KA, Wek RC. Mechanism of activation of the dsRNA-dependent protein kinase, PKR: role of dimerization and cellular localization in the stimulation of PKR phosphorylation of eukaryotic initiation factor-2 (eIF2) Eur J Biochem. 2001;268:3674–84. doi: 10.1046/j.1432-1327.2001.02273.x. [DOI] [PubMed] [Google Scholar]
- 19.Patel RC, Sen GC. Requirement of PKR dimerization mediated by specific hydrophobic residues for its activation by double stranded RNA and its anti growth effects in yeast. Mol Cell Biol. 1998;18:7009–19. doi: 10.1128/mcb.18.12.7009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu S, Kaufman RJ. A model for the double-stranded DNA (dsRNA)-dependent dimerization and activation of the dsRNA-activated protein kinase PKR. J Biol Chem. 1997;272:1291–1296. doi: 10.1074/jbc.272.2.1291. [DOI] [PubMed] [Google Scholar]
- 21.Lemaire P, Lary J, Cole JL. Mechanism of PKR activation: Dimerization and kinase activation in the absence of double-stranded RNA. J Mol Biol. 2005;345:81–90. doi: 10.1016/j.jmb.2004.10.031. [DOI] [PubMed] [Google Scholar]
- 22.McKenna S, Kim I, Liu CW, Puglisi J. Uncoupling of RNA binding and kinase activation by viral inhibitor RNAs. J Mol Biol. 2006;358:1270–85. doi: 10.1016/j.jmb.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 23.Patel RC, Sen GC. PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J. 1998;17:4379–90. doi: 10.1093/emboj/17.15.4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peters GA, Hartmann R, Qin J, Sen GC. Modular Structure of PACT: Distinct Domains for Binding and Activating PKR. Mol Cell Biology. 2001;21:1908–20. doi: 10.1128/MCB.21.6.1908-1920.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hovanessian AG, Galabru J. The double-stranded RNA-dependent protein kinase is also activated by heparin. Eur J Biochem. 1987;167:467–473. doi: 10.1111/j.1432-1033.1987.tb13360.x. [DOI] [PubMed] [Google Scholar]
- 26.Fasciano S, Hutchins B, Handy I, Patel RC. Identification of the heparin-binding domains of the interferon-induced protein kinase, PKR. FEBS J. 272:1425–39. doi: 10.1111/j.1742-4658.2005.04575.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.George C, Thomis D, McCormack S, Svahn C, Samuel C. Characterization of the heparin-mediated activation of PKR, the interferon-inducible RNA-dependent protein kinase. Virology. 1996;221:180–8. doi: 10.1006/viro.1996.0364. [DOI] [PubMed] [Google Scholar]
- 28.Saelens X, Kalai M, Vandenabeele P. Translation inhibition in apoptosis. Caspase-dependent PKR activation and eIF2-alpha phosphorylation. J Biol Chem. 2001;276:41620–28. doi: 10.1074/jbc.M103674200. [DOI] [PubMed] [Google Scholar]
- 29.Carrera AC, Alexandrov K, Roberts TM. The conserved lysine of the catalytic domain of protein kinases is actively involved in the phosphotransfer reaction and not required for anchoring ATP. Proc Natl Acad Sci USA. 1993;90:442–6. doi: 10.1073/pnas.90.2.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dey M, Cao C, Dar A, Tamura T, Ozato K, Sicheri F, Dever T. Mechanistic link between PKR dimerization, autophosphorylation and eIF2α substrate recognition. Cell. 2005;122:901–13. doi: 10.1016/j.cell.2005.06.041. [DOI] [PubMed] [Google Scholar]
- 31.Salzmann M, Pervushin K, Wider G, Senn H, Wuthrich K. TROSY in triple-resonance experiments: new perspectives for sequential NMR assignment of large proteins. Proc Natl Acad Sci USA. 1998;95:13585–90. doi: 10.1073/pnas.95.23.13585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rovnyak D, Frueh D, Sastry M, Sun Z, Stern A, Hoch J, Wagner G. Accelerated acquisition of high resolution triple-resonance spectra using non-uniform sampling and maximum entropy reconstruction. J Magn Reson. 2004;170:15–21. doi: 10.1016/j.jmr.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 33.Hoch J, Stern A. Maximum entropy reconstruction, spectrum analysis and deconvolution in multidimensional nuclear magnetic resonance. Methods Enzymol. 2001;338:159–78. doi: 10.1016/s0076-6879(02)38219-3. [DOI] [PubMed] [Google Scholar]
- 34.Jung YS, Zweckstetter M. Mars - robust automatic backbone assignment of proteins. J Biomol NMR. 2004;30:11–23. doi: 10.1023/B:JNMR.0000042954.99056.ad. [DOI] [PubMed] [Google Scholar]
- 35.To confirm the chemical shift perturbation data we attempted NOE-based NMR experiments designed for the elucidation of weak intermolecular complexes. In the cross-saturation scheme45 we saturated the fully protonated RBD and looked for intensity changes in the 15N-HSQC spectrum of the perdeuterated kinase. In the ‘driven NOE’ experiment46 single Ile-methyl or Leu/Val-methyl proton signals were selectively saturated in the perdeuterated (Ile/Leu/Val-methyl protonated) RBD and the spectra of the 15N-HSQC of the kinase were monitored for intensity changes. We did not observe any intensity changes in either experiment. In the former case we attribute this to inefficient spin diffusion in the relatively small RBDs. In the latter case, where the intensity changes are expected to be much smaller, the problem lies in the low signal to noise ratio, resulting from the low concentration and stability of the protein.
- 36.Kim C, Xuong NH, Taylor SS. Crystal Structure of a Complex Between the Catalytic and Regulatory (RIa) Subunits of PKA. Science. 2005;307:690. doi: 10.1126/science.1104607. [DOI] [PubMed] [Google Scholar]
- 37.Sharp T, Moonan F, Romashko A, Joshi B, Barber G, Jagus R. The vaccinia virus E3L gene product interacts with both the regulatory and the substrate binding regions of PKR: implications for PKR autoregulation. Virology. 1998;250:302–315. doi: 10.1006/viro.1998.9365. [DOI] [PubMed] [Google Scholar]
- 38.Galabru J, Hovanessian AG. Autophosphorylation of the protein kinase dependent on double-stranded RNA. J Biol Chem. 1987;262:15538–15544. [PubMed] [Google Scholar]
- 39.Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109:275–82. doi: 10.1016/s0092-8674(02)00741-9. [DOI] [PubMed] [Google Scholar]
- 40.Xu W, Doshi A, Lei M, Eck MJ, Harrison SC. Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol Cell. 1999;3:629–38. doi: 10.1016/s1097-2765(00)80356-1. [DOI] [PubMed] [Google Scholar]
- 41.Delaglio F, Grzesiek S, Vuister G, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–93. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 42.Goddard TD, Kneller DG. SPARKY 3. University of California; San Francisco: [Google Scholar]
- 43.Neal S, Nip AM, Zhang H, Wishart DS. Rapid and accurate calculation of protein 1H, 13C and 15N chemical shifts. J Biomol NMR. 2003;26:215–240. doi: 10.1023/a:1023812930288. [DOI] [PubMed] [Google Scholar]
- 44.Langer T, Vogtherr M, Elshorst B, Betz M, Schieborr U, Saxena K, Schwalbe H. NMR backbone assignment of a protein kinase catalytic domain by a combination of several approaches: application to the catalytic subunit of cAMP-dependent protein kinase. Chembiochem. 2004;5:1508–16. doi: 10.1002/cbic.200400129. [DOI] [PubMed] [Google Scholar]
- 45.Takahashi H, Nakanishi T, Kami K, Arata Y, Shimada I. A novel method for determining the interfaces of large protein-protein complexes. Nature Str Biol. 2000;7:220–23. doi: 10.1038/73331. [DOI] [PubMed] [Google Scholar]
- 46.Reibarkh M, Malia T, Wagner G. NMR distinction of single- and multiple-mode binding of small-molecule protein ligands. J Am Chem Soc. 2006;128:2160–61. doi: 10.1021/ja055971z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim I, Liu CW, Puglisi JD. Specific recognition of HIV TAR RNA by the ds RNA binding domains (dsRBD1-dsRBD2) of PKR. J Mol Biol. 2006;358:430–42. doi: 10.1016/j.jmb.2006.01.099. [DOI] [PubMed] [Google Scholar]
- 48.Li S, Peters GA, Ding K, Zhang X, Qin J, Sen GC. Molecular basis for PKR activation by PACT or dsRNA. Proc Natl Acad Sci USA. 2006;103:10005–10. doi: 10.1073/pnas.0602317103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.