

Abstract
Chronic alcohol consumption leads to inflammation and cirrhosis of the liver. In this study, we observed that liver sinusoidal endothelial cells (LSEC) derived from ethanol-fed rats showed several fold increases in the mRNA expression of endothelin-1 (ET-1), hypoxia-inducible factor-1α (HIF-1α), and inflammatory cytochemokines compared with control rat LSEC. We also observed the same results in acute ethanol-treated LSEC from control rats and human dermal microvascular endothelial cells. Ethanol-mediated ET-1 expression involved NADPH oxidase and HIF-1α activation. Furthermore, ethanol increased the expression of the ET-1 cognate receptor ET-BR in Kupffer cells and THP-1 monocytic cells, which also involved HIF-1α activation. Promoter analysis and chromatin immunoprecipitation showed that hypoxia response element sites in the proximal promoter of ET-1 and ET-BR were required for the binding of HIF-1α to up-regulate their expression. We showed that microRNAs, miR-199 among several microRNAs, attenuated HIF-1α and ET-1 expression, while anti-miR-199 reversed the effects, suggesting that ethanol-induced miR-199 down-regulation may contribute to augmented HIF-1α and ET-1 expression. Our studies, for the first time to our knowledge, show that ethanol-mediated ET-1 and ET-BR expression involve HIF-1α, independent of hypoxia. Additionally, ethanol-induced ET-1 expression in rat LSEC is regulated by miR-199, while in human endothelial cells, ET-1 expression is regulated by miR-199 and miR-155, indicating that these microRNAs may function as novel negative regulators to control ET-1 transcription and, thus, homeostatic levels of ET-1 to maintain microcirculatory tone.
Alcoholic liver disease is a common medical consequence of chronic alcohol abuse. The earliest stage of liver injury is fatty liver (steatosis), followed by liver inflammation (steatohepatitis), cirrhosis, and hepatocellular carcinoma (1– 4). Inflammation is recognized as being important in alcoholic liver disease pathogenesis (1, 2). It has been suggested that inflammatory reactions associated with alcoholic liver disease are due to alcohol-induced increase in gut-derived endotoxin absorption, which activates Kupffer cells to release proinflammatory cytokines (TNF-α), chemokines (IL-8), eicosanoids, reactive oxygen species, and NO (1, 4 –7). Among the various cytokines, TNF-α has emerged as one of the key factors in liver disease (4, 8). However, enteral alcohol fed rats develop necroinflammation and increased TNF-α expression, even in the absence of increased plasma levels of endotoxin, suggesting the involvement of other mechanisms by which inflammatory mediators may be produced, culminating in liver injury. Additionally, relatively less is known about how ethanol may activate other liver cells to generate inflammatory molecules, which can then activate infiltrated neutrophils and T cells, leading to liver injury (2).
Among several mechanisms by which alcohol causes liver injury, accentuated hypoxia in the pericentral region (zone 3) in which oxygen tension is physiologically lowest (9) has long been considered important. It is hypothesized that injurious effect at this site (zone 3) by ethanol is mediated at least in part by hypoxia, presumably due to enhanced oxygen utilization by hepatocytes during ethanol metabolism. This is supported by studies of French and his coworkers (10), who showed that high blood alcohol levels cause hypoxia in the liver. Specifically, these investigators utilized an intragastric ethanol-feeding model (11), initially developed by Tsukamoto and his coworkers (12, 13) to show (10) that high urinary blood alcohol levels were accompanied by activation of hypoxia-inducible factor-1α (HIF-1α),3 an indicator of hypoxia. Concomitantly, at the peak of blood alcohol levels, there was a several fold increase in the expression of genes, such as adrenomedullin and erythropoietin, compared with trough blood alcohol levels (10). Moreover, oxygen measurements at the liver surface revealed lower pO2 at the peaks compared with the control (10). These findings suggest that ethanol causes hypoxia in the liver to up-regulate HIF-regulated genes.
In cirrhosis and portal hypertension, the liver sinusoids experience increased resistance (14), which has been suggested to be due to reduced intrahepatic NO production by liver sinusoidal endothelial cells (LSEC) (15). However, the contribution of ET-1 as a vasoconstrictor in microcirculation has been suggested to occur via binding to the ET-A receptor in vascular smooth muscle cells (14). Studies have shown that ethanol perfusion into isolated rat livers via the portal vein increases portal pressure in the early phase (2–5 min), which gradually decreases over the period of ethanol infusion (16). The increase in ethanol-induced portal pressure was attenuated by sodium nitroprusside, a vasodilator, as well as by antiserum to ET-1. Under these conditions, serum levels of IL-8 were high and there was increased infiltration of PMN into the liver (16). Additionally, studies have revealed that under hepatic ischemia/reperfusion conditions (after transplantation or hemorrhagic shock), ET-1 fails to activate endothelial NO synthase in LSEC (17), thus contributing to hepatic microcirculatory failure. However, relatively little is known about how ethanol causes increased ET-1 expression in liver sinusoidal endothelium, which likely affects hepatic microcirculation.
In this report, we show that LSEC derived from ethanol-fed rats exhibit several fold increases in the mRNA expression of MCP-1, MIP-2, ET-1, and HIF-1α, compared with control rats. Since liver-derived SEC do not adapt well in culture and maintain fenestrae (18) relative to endothelial cells from other vascular beds, we utilized human dermal microvascular endothelial cell line (HMEC-1) as a model system for mechanistic studies. Acute treatment of HMEC-1 with ethanol (25–100 mM) resulted in the expression of the same genes as was observed in LSEC derived from ethanol-fed rats. We show that ethanol-mediated signaling leading to increased ET-1 mRNA expression in endothelial cells involves activation of NADPH oxidase and HIF-1α. In Kupffer cells and THP-1 monocytic cells, ethanol increased the mRNA expression of endothelin-B receptor (ET-BR), a cognate receptor for ET-1, via HIF-1α. We also examined the potential involvement of microRNAs (miRNA) in the regulation of ethanol-mediated expression of HIF-1α mRNA and its co-regulated gene, ET-1. We show that miR-199 was specifically involved in regulating ethanol-induced HIF-1α and ET-1 mRNA expression in rat (r)LSEC and human endothelial cells, while miR-155 was primarily involved in ET-1 expression in human endothelial cells. These studies, for the first time to the best of our knowledge, suggest that HIF-1α and ET-1-related miRNAs may function as novel negative regulators to control ET-1 transcription and, thus, homeostatic levels of ET-1 to maintain microcirculatory tone. Moreover, our studies also demonstrate that ethanol mediated up-regulation of ET-1 and ET-BR involves activation of HIF-1α, independent of hypoxia. We show that ethanol-induced ET-1 release from either LSEC or HMEC-1 acts on ethanol-activated Kupffer cells via an ET-BR loop to augment chemokines (MCP-1) expression, with the latter contributing to infiltration of PMN/monocytes from circulation into the liver, contributing to liver injury.
Materials and Methods
Animal experiments
The use of animals for this study was approved by the Institutional Animal Care and Use Committee of the University of Southern California. For chronic alcohol administration, male Wistar rats were implanted with gastrostomy catheters as previously described (19). These rats were given a high-fat diet (35% calories as corn oil; Dyets) and infused with an increasing dose of ethanol (9 –16 g/kg/day) or isocaloric dextrose solutions (20), and weekly enteral LPS (5 mg/kg) for 9 wk (Alcoholic Liver and Pancreatic Diseases Animal Core).
Isolation of LSEC
Sinusoidal endothelial cells from the liver of control and ethanol-fed rats were isolated by procedure as previously described (21). Briefly, liver was perfused with 0.05% type 1a collagenase, meshed, pressed through polypropylene mesh, and centrifuged. SEC were isolated from the liver digest by density gradient centrifugation and centrifugal elutriation as described (21, 22). These LSEC exhibited >95% purity as determined by positive staining for acetylated low density lipoprotein. The absence of contaminating Kupffer cells was ascertained by peroxidase staining. The viability of LSEC was >95% as determined by trypan blue. These cells displayed fenestrae and sieve plates (21), characteristic of sinusoidal endothelial cells in vivo (23). It is pertinent to mention that LSEC in culture do not display fenestrae but show expression of PECAM-1 (CD31) (18), a characteristic marker of endothelial cells (24, 25), showing that LSEC in culture become defenestrated and capillarized.
Endothelial cell culture
LSEC were cultured in DMEM media containing 10% FBS for 2 days. These cells exhibited cobblestone morphology, characteristic of endothelial cells. The immortalized HMEC-1, originally developed by Dr. Edwin Ades and Francisco J. Candall of the Centers for Disease Control and Prevention, and Dr. Thomas Lawley of Emory University, was obtained from the Centers for Disease Control and Prevention. HMEC-1 were cultured in RPMI 1640 containing 10% FBS, 1 mM glutamine, 1 mM sodium pyruvate, 5 mM HEPES, MEM vitamins and nonessential amino acids (1×), 50 μg/ml endothelial cell mitogen (Biomedical Technologies), and heparin (20 U/ml). Cells were cultured overnight in serum-free conditions before the start of experiments.
Isolation and culture of Kupffer cells
Animal use for this study was approved by the Institutional Animal Care and Use Committee of the University of Southern California. The Non-Parenchymal Liver Cell Core of the Research Center for Alcoholic Liver and Pancreatic Diseases and Cirrhosis isolated Kupffer cells from normal Wistar rats, alcohol fed (E-rKC) and pair fed control rats (C-rKC) by in situ digestion of the liver with pronase and then collagenase, followed by arabinogalactan gradient ultracentrifugation and adherence purification as described previously (20). The cells were cultured in low glucose DMEM containing 5% FBS for 2 days before experiments. Kupffer cells isolated from alcohol-fed and pair-fed animals were cultured for 3 h in serum-free conditions before the start of experiments.
THP-1 cell culture
THP-1 cells were cultured in RPMI 1640 containing 10% FBS, 1 mM glutamine, 1 mM sodium pyruvate, 5 mM HEPES, 4.5 μg/ml glucose, 1.5 μg/ml sodium bicarbonate, and 0.05 mM 2-ME (26). Cells were cultured overnight in serum-free conditions before the start of experiments.
Reagents
Diphenylene iodonium (DPI) was obtained from Sigma-Aldrich. LY294002, ascorbate, R59949, and BQ788 were obtained from Tocris Bioscience. These pharmacological inhibitors were used at the indicated concentrations: DPI (10 μM), LY294002 (15 μM), ascorbate (25 μM), R59949 (30 μM), and BQ788 (20 μM) as deemed optimal from literature. Primary Abs for HIF-1α and β-actin, and secondary Abs conjugated to HRP, were purchased from Santa Cruz Biotechnology. All other reagents, unless otherwise specified, were purchased from Sigma-Aldrich. HIF-1α small interfering RNA (siRNA), HIF-1α scrambled RNA (scRNA), prolylhydroxylase (PHD)-2 siRNA, and PHD-2 scRNA were synthesized at the microchemical core facility of the University of Southern California Comprehensive Cancer Center as previously described (27). Endothelin-1 promoter-luciferase plasmids (−669 and −176 bp) with mutation in HIF-1α binding were provided by Dr. K. Webster (University of Miami, Miami, FL) (28).
Isolation of RNA and quantitative RT-PCR
LSEC isolated from control (C-rLSEC) and ethanol-fed rats (E-rLSEC) were extracted for total RNA with TRIzol reagent (Invitrogen). C-rLSEC in culture were treated with ethanol (50 –100 mM) for the indicated time period followed by extraction for RNA. HMEC-1 was treated with ethanol in the absence and presence of indicated pharmacological inhibitors followed by extraction of RNA. mRNA expression was determined and quantified using specific mRNA primers (Table I). Real-time quantitative PCR of mRNA templates was done using the iScript One-Step RT-PCR kit with SYBR Green (Bio-Rad). PCR amplification of 100 ng of RNA was performed for 40 cycles under the following conditions: cDNA synthesis at 50°C for 10 min, iScript reverse transcriptase inactivation at 95°C for 5 min, and PCR cycling and detection at 95°C for 10 s, followed by 60°C for 30 s. Values are expressed as relative expression of mRNA normalized to housekeeping GAPDH mRNA.
Table I.
Oligonucleotide primers used in this studya
| Organism/Gene | Method | Forward Sequence | Reverse Sequence |
|---|---|---|---|
| R, GAPDH | PCR | ttcaatggcacagtcaaggc | tcaccccatttgatgttagcg |
| R, MCP-1 | PCR | cagatctctcttcctccaccactat | caggcagcaactgtgaacaac |
| R, MIP-2 | PCR | cgggatccactgcacctctgggcct | gctctagaggactcgagaattcgatgct |
| R, ET-1 | PCR | atggattattttcccgtgat | gggagtgttgacccagatga |
| R, inducible NO synthase | PCR | gcatcccaagtacgagtggt | gaaggcgtagctgaacaagg |
| R, HIF-1α | PCR | cgagctgcctcttcgacaag | cccagccgctggagcta |
| R, ET-BR | PCR | ttccaactccagtctgatgcg | ctgtcttgtaaaactgcatgaag |
| H, GAPDH | PCR | aacctgccaagtacgatgacatc | gtagcccaggatgcccttga |
| H, MCP-1 | PCR | cgcctccagcatgaaaagtct | atgaaggtggctgctatgagc |
| H, ET-1 | PCR | tgattttctctctgctgtttgtg | caatgtgctcggttgtgggtca |
| H, inducible NO synthase | PCR | tccaacctgcaggtcttcgatgc | ggaccagccaaatccagtctgc |
| H, HIF-1α | PCR | ctcaaagtcggacagcctca | ccctgcagtaggtttctgct |
| H, ET-BR | PCR | cttcacctcagcaggattctg | ggcccagccttaaagcagtt |
| H, ET-BR mutant HRE1 | SDM | cttgagttacttttgagcagggatactggcgaagaggctg | cagcctcttcgccagtatccctgctcaaaagtaactcaag |
| H, ET-BR mutant HRE2 | SDM | ctgtattcctcccgttacaggaaagagttcggagctttg | caaagctccgaactctttcctgtaacgggaggaatacag |
| H, ET-1 wild-type HRE consensus oligo | EMSA | ctccggctgcacgttgcctg | caggcaacgtgcagccggag |
| H, ET-1 mutant HRE consensus oligo | EMSA | ctccggcttaccgttgcctg | caggcaacggtaagccggag |
| H, ET-1 | ChIP | cctcacggatctttctccgat | gtctgacttggacagctctct |
R indicates rat; H, human; SDM, site-directed mutagenesis. Site-specific mutations are depicted in boldface type.
Preparation of cytosolic and nuclear extracts
Cytosolic and nuclear extracts were prepared from HMEC-1 according to the modified procedure of Dignam et al. (29) as previously described (26, 30). Briefly, 5 × 106 cells were washed with cold PBS, lysed in cell lyses buffer (100 mM KCl, 1.5 mM MgCl2, 0.1 mM EGTA, 0.5 mM DTT, 0.5 mM PMSF, 10 mM HEPES (pH 8.0), 0.5% Nonidet P-40, and 1 μl/ml protease inhibitor cocktail) followed by centrifugation at 10,000 × for 1 min. Supernatant was collected (cytosolic extract) and the pellet was re-suspended in 50 μl of nuclear extraction buffer (10 mM HEPES (pH 7.9), 1.5 mM MgCl2, 420 mM NaCl, 0.1 mM EGTA, 0.5 mM DTT, 5% glycerol, 0.5 mM PMSF, and 1 μl/ml protease inhibitor cocktail) and mixed intermittently for 60 min. The nuclear extracts were obtained by centrifugation at 10,000 × g for 10 min at 4°C.
Western blot analysis
HMEC-1 (5 × 106 cells) in serum-free medium were treated with ethanol (100 mM) for the indicated time period. Nuclear and cytosolic extracts were prepared and subjected to electrophoresis, followed by transfer to nitrocellulose membrane. Membranes were probed with Ab to HIF-1α (1/250) as described (27). Blots were stripped and reprobed with β-actin Ab (1/250) to monitor equal protein loading. The protein bands were detected using Immunobilon Western reagents (Millipore).
Transient transfection of HMEC-1 with promoter constructs and siRNA
HMEC-1 (1 × 106 cells) was grown to 80% confluence in culture media containing 5% FCS. The cells were resuspended in 100 μl of Nucleofector solution (Amaxa Kit-T) containing 1 μg of expression plasmid and 0.5 μg of β-galactosidase reporter plasmid or siRNA (50 nM) followed by nucleofection according to the supplier’s protocol using program S-005 (30). Cells were transferred to 6-well plates containing complete media for overnight growth followed by incubation in serum-free medium for 3 h, before stimulation with ethanol for the indicated time period. After treatment, cells were rinsed once with PBS and either RNA was extracted or cells were lysed with reporter lysis buffer (Promega) and centrifuged at 8000 rpm for 5 min. The supernatants were collected and assayed for luciferase activity using a luminometer (Lumat LB 9501; Berthold Technologies) while β-galactosidase activity was measured using the Promega kit. Firefly luciferase values were normalized to β-galactosidase values for transfection efficiency. Data are expressed as values relative to the promoterless PGL3 basic vector.
EMSA for transcription factor HIF-α
The double-stranded oligonucleotides of wild-type ET-1 and ET-1 containing a mutation in its hypoxia response element (HRE) site (Table I) were labeled with biotin using a light shift chemiluminescent EMSA kit (Pierce Chemical). The nuclear extract (10 μ g), 5% glycerol, 5 mM MgCl2, 50 ng/μl poly(dI:dC), 0.05% Nonidet P-40, and 0.5 ng of biotinylated ET-1 oligonucleotide were incubated at room temperature for 20 min. The samples were subjected to nondenaturing 6% PAGE followed by transfer to Hybond-N+ nylon membrane (Amersham). The bands were detected with streptavidin-HRP/chemiluminescence as per the supplier’s instructions. The specificity of DNA-protein interaction was determined using 50-fold excess of unlabeled probe.
Chromatin immunoprecipitation (ChIP) assays
HMEC-1 (5 × 106 cells) in serum-free medium was treated with ethanol (100 mM) in the presence or absence of inhibitors for the indicated time period. ChIP analysis was performed utilizing Ab to HIF-1α as described (30). Briefly, 5 μl of DNA sample was subjected to PCR amplification utilizing primers for the ET-1 promoter region (Table I). PCR was performed for 35 cycles under the following conditions: denaturation at 95°C for 30 s, annealing at 55°C for 60 s, and extension at 72°C for 2 min. The PCR products were subjected to electrophoresis on a 2% agarose gel and visualized by ethidium bromide staining.
Isolation and quantification of miRNAs
Total RNA was isolated from C-rLSEC, E-rLSEC, or HMEC-1 by using the mirVana miRNA isolation kit (Ambion/Applied Biosystems). miRNA expression was determined and quantified utilizing specific miRNA primers (Table I). cDNA was synthesized from 200 ng of isolated miRNA using the reverse transcription system (Promega), according to the manufacturer’s protocol. Quantitative PCR amplification was performed using iQ SYBR Green Supermix (Bio-Rad) for 40 cycles under the following conditions: hot-start iTaq DNA polymerase at 95°C for 2 min, denaturation at 95°C for 15 s, and annealing and data collection at 60°C for 60 s. miRNA quantitative PCR was normalized to constitutively expressed 5S miRNA levels.
Transfection of LSEC and HMEC-1 with anti-miRNA
LSEC and HMEC-1 (1 × 106 cells) were grown to 80% confluence in culture media. The cells were resuspended in 100 μl of Nucleofector solution (Amaxa Kit-T) containing 60 pmol of antisense miRNA followed by nucleofection according to the supplier’s protocol using program S-005. LSEC and HMEC-1 in culture were treated with ethanol (100 mM) for the indicated time period followed by extraction for RNA. mRNA expression was determined and quantified using specific mRNA primers (Table I). Real-time quantitative PCR of mRNA templates was done using the iScript One-Step RT-PCR kit with SYBR Green (Bio-Rad). Values are expressed as relative mRNA expression, normalized to housekeeping GAPDH mRNA.
Quantification of ET-1 release
HMEC-1 and rLSEC (1 × 106 cells/ml) in serum-free medium were treated with ethanol (100 mM) in the presence or absence of inhibitors for 48 and 24 h, respectively. ET-1 release was assayed from the supernatants from these cells using an ELISA kit (American Research Products).
Statistical analysis
Data are presented as means ± SD. Differences in significance of mean values between multiple groups were analyzed with parametric one-way ANOVA followed by a Tukey-Kramer test using the Instat 2 software program (GraphPad Software). Ethanol-treated samples were used for multiple comparisons, and Student’s t test was used to evaluate differences in significance between untreated and ethanol-treated samples.
Results
Rat LSEC show increased mRNA expression of chemokines, ET-1, and HIF-1α
To determine whether feeding of ethanol to rats affects expression of inflammatory molecules, LSEC were isolated from ethanol fed-rats (E-rLSEC) and control rats (C-rLSEC). There was a 3-fold increase in MCP-1, a 2-fold increase in MIP-2, and a 4.5-fold increase in endothelin-1 (ET-1) mRNA expression, as determined by quantitative RT-PCR, in E-rLSEC compared with rLSEC (Fig. 1A). Furthermore, there was no difference in inducible NO synthase mRNA levels in E-rLSEC vs C-rLSEC. However, there was a ~3.5-fold increase in HIF-1α mRNA expression in E-rLSEC compared with C-rLSEC (Fig. 1A). Treatment of rLSEC with ethanol (100 mM) showed a 3-fold increase in both ET-1 and HIF-1α mRNAs at 24 h (Fig. 1B), as was observed in E-rLSEC, indicating that acute treatment with ethanol of isolated rLSEC recapitulates the induction of the same chemokines, ET-1, and HIF-1α seen in vivo. A lower concentration of ethanol (25 and 50 mM) treatment for 24 h also up-regulated the expression of the above-indicated genes in rLSEC, although to a lower extent (data not shown).
FIGURE 1.
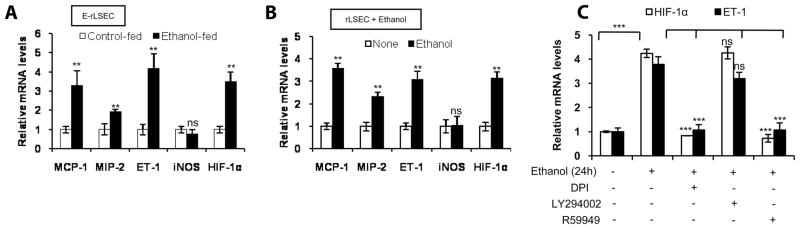
Ethanol augments chemokine gene expression in rLSEC via the NADPH oxidase pathway and HIF-1α. A, Ethanol-induced gene expression in LSEC derived from ethanol-fed rats (E-rLSEC), compared with control-fed rats (C-rLSEC). The data represent fold increases in mRNA expression following ethanol feeding compared with control. B, Ethanol-induced gene expression in ethanol-treated rLSEC, compared with untreated rLSEC. C, Ethanol-induced HIF-1α and ET-1 expression in ethanol-treated rLSEC that were preincubated with the inhibitors DPI (flavin oxidase inhibitor), LY294002 (PI3K inhibitor), or R59949 (HIF-1α inhibitor) for 30 min before ethanol treatment. Quantitative RT-PCR data of ethanol-treated rLSEC represent fold increases in mRNA expression following stimulation with ethanol (100 mM) for 24 h compared with no ethanol. All mRNA expressions were normalized to GAPDH mRNA levels, and the data shown represent three independent experiments (means ± SD). ***, p < 0.001;**, p < 0.01; ns, p > 0.05.
Ethanol-induced ET-1 expression in rat LSEC involves activation of NADPH oxidase and HIF-1α, but not PI3K
Treatment of rLSEC with pharmacologic inhibitors of NADPH oxidase (DPI) and HIF-1α (R59949) before ethanol stimulation (100 mM for 24 h) reduced ET-1 and HIF-1α mRNA expression by 98 ± 2.5% and 108 ± 1.3%, respectively (Fig. 1C). However, PI3K inhibitor (LY294002) did not affect ethanol-induced ET-1 expression (Fig. 1C). Ethanol-induced HIF-1α mRNA expression was reduced completely and below basal levels by both DPI and R59949, while LY294002 had no significant effect (Fig. 1C).
Ethanol treatment of HMEC-1 augments mRNA expression of chemokines, ET-1, and HIF-1α
Since we observed that ethanol in vivo and in vitro up-regulated expression of chemokines and ET-1 in rLSEC, we examined whether human endothelial cells from vascular beds other than liver sinusoids responded to ethanol treatment in a similar manner. Treatment of HMEC-1 with ethanol (100 mM) for 24 h, compared with control HMEC-1, showed an increase in mRNA expression of MCP-1 by 4.5-fold, ET-1 by 6-fold, and HIF-1α by 3-fold (Fig. 2A), similar to that observed in rLSEC acutely treated with ethanol. These results indicate that ethanol exhibited its optimal effect at 100 mM, which corresponds to a 0.46% concentration of alcohol. We utilized HMEC-1 cells as a model system for LSEC in delineating ethanol-induced cell signaling pathways for ease of culture, transfection efficiency, and availability of human promoter constructs.
FIGURE 2.

Ethanol promotes chemokine gene expression in HMEC-1 via the NADPH oxidase pathway and HIF-1α, where ethanol metabolism is required. A, Gene expression levels in HMEC-1 stimulated with ethanol, compared with untreated cells. B, Ethanol-induced HIF-1α and ET-1 mRNA expression in ethanol-treated HMEC-1 that were preincubated with the inhibitors DPI, LY294002, or ascorbate (HIF-1α inhibitor) for 30 min before ethanol treatment. C, Ethanol-induced HIF-1α and ET-1 expression in ethanol-treated HMEC-1 that were transiently transfected with p47phox (NADPH oxidase inhibitor), using scrambled p47phox as a control, and dominant-negative PI3K before stimulation with ethanol. D, HIF-1α and ET-1 expression in HMEC-1 that were treated with ethanol, acetaldehyde, or isopropanol at the indicated concentrations and durations. Where indicated, HMEC-1 were also preincubated with 4-methylpyrazole (1.0 mM) for 30 min before ethanol treatment. E, Ethanol-induced HIF-1α and ET-1 expression in ethanol-treated HMEC-1 that were transiently transfected with HIF-1α siRNA, using scrambled RNA as a control, and HIF-2α siRNA, and then treated with ethanol. F, Ethanol-induced ET-1 expression in rLSEC that were transiently transfected with HIF-1α siRNA and scRNA before ethanol treatment for the indicated times. Quantitative RT-PCR data represent fold increases in mRNA expression following stimulation with ethanol (100 mM) for 24 h compared with no ethanol. All mRNA expressions were normalized to GAPDH mRNA levels, and the data shown represent three independent experiments (means ± SD). ***, p < 0.001; **, p < 0.01; *, p < 0.05; ns, p > 0.05.
Ethanol-induced ET-1 expression in HMEC-1 involves activation of NADPH oxidase and HIF-1α, but not PI3K
Treatment of HMEC-1 with pharmacologic inhibitors of NADPH oxidase (DPI) and HIF-1α (ascorbate) reduced ET-1 mRNA expression by 58 ± 4.3% and 67 ± 3.7%, respectively (Fig. 2B). However, PI3K inhibitor (LY294002) did not affect ethanol-induced ET-1 expression (Fig. 2B). Ethanol-induced HIF-1α mRNA expression was reduced below basal levels by both DPI and ascorbate, while LY294002 had no significant effect (Fig. 2B). Since pharmacological inhibitors can act in a nonspecific manner, HMEC-1 were transfected with siRNA for p47phox (a subunit of NADPH oxidase) and a dominant mutant of PI3K. siRNA for p47phox, but not scRNA for p47phox, completely attenuated ethanol-mediated ET-1 mRNA expression, while dominant PI3K had no effect on ET-1 expression (Fig. 2C). Since ethanol is metabolized to acetaldehyde by alcohol dehydrogenase in cells, we examined whether metabolism of ethanol is required for ET-1 induction. Pretreatment of HMEC-1 with 4-methylpyrazole (1.0 mM) before ethanol stimulation attenuated ET-1 expression by 127 ± 4.2% (Fig. 2D). Moreover, acetaldehyde (0.5–1.0 mM) dose-dependently augmented ET-1 expression to the same level as was observed with equivalent concentrations of ethanol (Fig. 2D). The expression of ET-1 was specific for ethanol, as nonmetabolizing alcohol such as isopropanol did not augment ET-1 expression (Fig. 2D). It is pertinent to mention that a lower concentration of ethanol (50 mM) increased ET-1 and HIF-1α mRNA expression by 2.5-and 2.8-fold, respectively (see Fig. 2D), while a further lower concentration of ethanol (25 mM) also up-regulated the expression of the above-indicated genes, although to a lesser extent (data not shown). Although these concentrations of ethanol and acetaldehyde may seem high, cell viability remained unaffected, and, under chronic alcohol conditions, such a situation may exist. Taken together, these results indicate that ethanol-mediated ET-1 expression requires metabolism of ethanol and activation of NADPH oxidase.
Ethanol-induced ET-1 expression in HMEC-1 and rLSEC involves HIF-1α
To determine whether HIF-1α played a role in ethanol-mediated ET-1 expression, HMEC-1 was transfected with siRNA for HIF-1α. As shown Fig. 2E, transfection with HIF-1α siRNA reduced ET-1 and HIF-1α mRNA expression by 110 ± 5% and 88 ± 2.3%, respectively. However, transfection with scHIF-1α siRNA had no effect on the ethanol-induced expression of HIF-1α and ET-1 mRNA (Fig. 2E). Moreover, transfection with siRNA for HIF-2α had no effect on ethanol-mediated ET-1 mRNA expression (Fig. 2E). To determine whether ethanol-mediated ET-1 expression in rLSEC also involved HIF-1α, rLSEC were transfected with HIF-1α siRNA. As shown in Fig. 2F, rLSEC transfected with HIF-1α siRNA completely (>90 ± 4.3%) attenuated ET-1 expression, while scHIF-1α siRNA had a modest effect (<23 ± 2.5%). These results indicate that, in both rLSEC and human endothelial cells, ethanol-mediated ET-1 expression is silenced by HIF-1α siRNA. Taken together, these data indicate that ethanol-induced ET-1 expression involves HIF-1α but not HIF-2α.
Ethanol-mediated up-regulation of ET-1 gene involves a proximal HRE in the ET-1 promoter
Previous studies have shown that hypoxia-mediated up-regulation of ET-1 involved a proximal HRE site in its promoter (31). As shown in Fig. 3A, treatment of HMEC-1 with ethanol (100 mM) resulted in a ~5-fold increase in both full-length (−669 bp) and truncated (−176 bp) ET-1 promoter luciferase activity. However, a mutation (−124 to −121 bp) of the HRE site in the truncated promoter (−176 bp) resulted in complete attenuation of ethanol-induced ET-1 promoter luciferase activity (Fig. 3A). Next, we determined whether pharmacologic inhibitors, which attenuated ethanol-mediated ET-1 mRNA expression, also affected ET-1 promoter activity. As shown in Fig. 3B, ethanol-induced ET-1 promoter activity was completely reduced to basal level by inhibitors of NADPH oxidase (DPI) and HIF-1α (R59949), but not by an inhibitor of PI3K (LY294002), as was observed for the expression of ET-1 mRNA. Pretreatment of HMEC-1 with 4-methylpyrazole (an inhibitor of alcohol dehydrogenase) before ethanol stimulation completely abrogated ethanol-induced ET-1 promoter activity (Fig. 3B). Next, we examined whether ethanol metabolite acetaldehyde itself affected ET-1 promoter activity. As shown in Fig. 3C, treatment of HMEC-1 with acetaldehyde (1.0 mM), corresponding to 1/100th molar concentration of ethanol, resulted in a 4.5-fold increase in ET-1 promoter luciferase activity, which was completely attenuated by the inhibitors of NADPH oxidase (DPI) and HIF-1α (R59949), but not by inhibitors of PI3K (LY294002) and alcohol dehydrogenase (4-methylpyrazole). These data indicate that ethanol or its metabolite mediates HIF-1α binding to HRE sites in the ET-1 promoter to increase ET-1 promoter activity. Furthermore, metabolism of ethanol to acetaldehyde is required for ethanol-mediated increase in ET-1 promoter activity. These results indicate that the HRE site in the ET-1 proximal promoter is essential for ethanol-mediated ET- 1 promoter activity.
FIGURE 3.

Ethanol augments the promoter activity of ET-1 in HMEC-1 through the NADPH oxidase pathway and HIF-1α, the effects of which require ethanol metabolism. A, Ethanol-induced ET-1 wild-type full-length promoter (−669 bp) and minimal promoter (−176 bp) activities in response to ethanol (100 mM) in HMEC-1. The ET-1 mutant HRE (−176 bp) luciferase construct contains a mutation in its HRE at −124bp to −121 bp. B, Ethanol-induced ET-1 minimal promoter (−176 bp) activity, in response to ethanol (100 mM) of HMEC-1, where cells were preincubated with the inhibitors DPI, LY294002, R59949, or 4-methylpyrazole (1.0 mM) for 30 min before ethanol treatment where indicated. C, Acetaldehyde (1.0 mM) induced ET-1 promoter (−176 bp) activity. Where indicated, cells were preincubated with the inhibitors DPI, LY294002, R59949, or 4-methylpyrazole (1.0 mM) for 30 min before acetaldehyde treatment. Luciferase assay data are expressed as fold change and have been normalized relative to the change in luciferase activity of the untreated controls and to the transfection efficiency with β-galactosidase. The data shown represent two independent experiments (means ± SE). ***, p < 0.001; ns, p > 0.05. D, Time course of ethanol-induced HIF-1α protein expression in nuclear extracts of HMEC-1 for the indicated times. Densitometric analysis was used to determine fold change of HIF-1α protein expression. The data were normalized to β-actin as a loading control. E, The HRE site in the ET-1 promoter was used as a probe, which shows increased binding of HIF-1α in nuclear extracts from HMEC-1 in response to ethanol (100 mM for 24 h), which was attenuated when the HRE site was mutated, as seen by EMSA. Where indicated, HMEC-1 were preincubated with DPI for 30 min before ethanol treatment. Twenty-five nanograms of biotin-labeled ET-1 probe was used. Where indicated, 50-fold excess of cold probe was added to the nuclear extracts. F, Ethanol-induced association of HIF-1α with the HRE site on the ET-1 promoter of HMEC-1, as assessed by ChIP analysis. Where indicated, cells were pretreated with DPI and LY294002 for 30 min before stimulation with ethanol. Data are representative of two independent experiments. Vertical lines indicate repositioned gel lanes.
Ethanol augments HIF-1α binding to HRE consensus region in ET-1 promoter
Treatment of HMEC-1 with ethanol showed a time-dependent (6 –24 h) increase in the expression of HIF-1α protein (Fig. 3D). Next, we performed EMSA to determine whether HIF-1α in nuclear extracts bound to the HRE consensus region of the ET-1 promoter. As shown in Fig. 3E, ethanol increased by 3-fold the binding of HIF-1α to oligonucleotides corresponding to the HRE sequence in the ET-1 promoter. HIF-1α DNA binding was reduced by DPI to the basal level (Fig. 3E, lane 3). Additionally, 50-fold excess of unbiotinylated probe completely displaced binding of biotinylated probe to HIF-1α in nuclear extracts (Fig. 3E, lane 4). Furthermore, biotinylated HRE probe with a mutation in its HRE sites showed no binding to HIF-1α in the nuclear extracts from HMEC-1 treated with ethanol (Fig. 3E, lane 5). These results indicate that HIF-1α in the nuclear extracts binds to bona fide HRE in the ET-1 promoter, and the binding is augmented in response to ethanol.
Ethanol augments HIF-1α binding to HRE sites in ET-1 promoter in chromatin
To determine whether HIF-1α binds to the ET-1 promoter in chromatin in response to ethanol, we performed ChIP analysis. Chromatin was isolated from ethanol-treated HMEC-1 and immunoprecipitated with an Ab to either HIF-1α or normal rabbit IgG (control). As shown in Fig. 3F, ethanol treatment of HMEC-1 resulted in a 3-fold increase in the expected 611-bp product corresponding to −606 to +5 bp of the ET-1 promoter region containing the HRE site, as measured by densitometry. The binding of HIF-1α to the promoter region in chromatin was >90% reduced by DPI (Fig. 3F, lane 4), but not by LY294002 (Fig. 3F, lane 3). As expected, no PCR product bands were observed in control IgG immunoprecipitates (Fig. 3F, lower panel). As shown in Fig. 3F, middle panel, the amplification of input DNA before immunoprecipitation was equal in all of the samples. Taken together, these results indicate that ethanol promotes the binding of HIF-1α to the proximal HRE site in ET-1 promoter both in vitro and in vivo to augment the transcription of ET-1 in human endothelial cells (HMEC-1).
Identification of miRNAs involved in ethanol-mediated HIF-1α and ET-1 mRNA expression
Since ethanol induced the mRNA expression of HIF-1α and its co-regulated ET-1 gene, we examined the miRNAs that may be involved in stabilization and degradation. Previous studies (32) have identified several miRNAs regulated by hypoxia and hypoxia-inducible factor in cancer cells. Analysis of the miRNA database revealed the presence of miR-135, miR-155, and miR-199 binding sites in the proximal 5′- to 3′-untranslated region (UTR) of HIF-1α mRNA, and the presence of the same miRNAs in the 3′-UTR of ET-1 mRNA as depicted in schematics of Fig. 4A. Thus, we analyzed the expression of these putative miRNAs in response to ethanol in endothelial cells. As shown in Fig. 4B, there was a diminution in miR-135 and miR-199 expression in E-rLSEC compared with C-rLSEC. Similarly, ethanol-treated LSEC showed reduced expression of miR-135 and miR-199 (Fig. 4C). The expression of miR-20 and miR-203 were high in E-rLSEC and in ethanol-treated C-rLSEC compared with control (Fig. 4, B and C). Fig. 4D shows a decrease in miR-155 and miR-199 expression in HMEC-1 treated with ethanol compared with untreated HMEC-1. However, the expression of miR-135 in these cells was at the basal level, while expression of miR-20 increased by 3-fold. We used miR-20 as a control, as its level consistently increased in response to ethanol. To determine which of the above miRNAs regulated mRNA expression of HIF-1α, ET-1, or both, we transfected HMEC-1 with miR-135, miR-155, and miR-199 overexpression plasmids (Fig. 4E). As shown in Fig. 4E, overexpression of miR-135 abrogated HIF-1α mRNA to the basal level, and the expression of ET-1 mRNA was also significantly affected (94 ± 4.8%). Additionally, overexpression of either miR-155 or miR-199 completely abrogated ethanol-induced mRNA expression of HIF-1α and ET-1 (Fig. 4E). Next, we determined the effect of overexpression of anti-miR oligonucleotides on ethanol-induced ET-1 and HIF-1α mRNA expression. As shown in Fig. 4F, ethanol induced the expression of HIF-1α and ET-1 mRNAs by 4- and 5-fold, respectively. Transfection with anti-miR-20 (control) reduced ethanol-mediated HIF-1α and ET-1 mRNA expression (Fig. 4F, lane 3). However, transfection with anti-miR-135 did not significantly affect HIF-1α mRNA, while it reduced ET-1 mRNA expression (Fig. 4F, lane 4). Transfection with anti-miR-155 increased the expression of both HIF-1α and ET-1 mRNA levels by 2-fold compared with HMEC-1 treated with ethanol alone (Fig. 4F, lane 5). However, anti-miR-199 augmented ethanol-induced HIF-1α mRNA expression by ~15-fold and ET-1 by ~17–fold (Fig. 4F, lane 6). Taken together, these results indicate that miR-155 and miR-199 affect ethanol-mediated HIF-1α and ET-1 mRNA expression in cultured HMEC-1, although anti-miR-199 had a pronounced effect.
FIGURE 4.

Ethanol-induced HIF-1α and ET-1 mRNA expressions in endothelial cells are regulated by HIF-1α-related miRNAs. A, Schematics of partial complementary binding sites of miRNAs in the 3′-UTRs of HIF-1α and ET-1 mRNA. B, Ethanol-induced expression of HIF-1α-related miRNA levels in E-rLSEC. The data represent fold changes in miRNA expression in E-rLSEC relative to C-rLSEC. C, Ethanol-induced expression of HIF-1α-related miRNA levels in ethanol-treated rLSEC. D, Ethanol-induced expression of HIF-1α-related miRNA levels in ethanol-treated HMEC-1. All miRNA expression levels were normalized to miR-5S levels, and the data shown represent three independent experiments (means ± SD). E, Ethanol-induced abrogation of HIF-1a and ET-1 mRNA expression in HMEC-1 when transfected with miR-135, miR-155, and miR-199 overexpression plasmids. F, HMEC-1 were transiently transfected with anti-miRNAs (60 pmol) followed by ethanol stimulation for 24 h, and then HIF-1α and ET-1 mRNA levels were assessed. G, rLSEC were transiently transfected with anti-miRNAs (60 pmol) before ethanol stimulation, and HIF-1α and ET-1 mRNA expressions were assessed. All mRNA expressions were normalized to GAPDH mRNA levels, and the data shown represent three experiments (means ± SD). ***, p < 0.001; **, p < 0.01; *. p < 0.05. H, Ethanol-mediated increase in ET-1 protein expression in cytosolic extracts of HMEC-1. Where indicated, cells were transfected with an overexpression plasmid for either anti-miR-199 or miR-199. Densitometric analysis was used to determine fold change of ET-1 protein expression. The data were normalized to β-actin as a loading control.
Next, we examined whether the same miRNAs were involved in ethanol-mediated expression of HIF-1α and ET-1 in rLSEC (Fig. 4G). As shown in Fig. 4G, ethanol treatment of rLSEC resulted in a 3.5-fold increase in HIF-1α mRNA and a 5-fold increase in ET-1 mRNA. Overexpression of anti-miR-20 (control) reduced the mRNA levels of HIF-1α and ET-1 to the basal level. Although overexpression of anti-miR-135 and anti-miR-199 increased HIF-1α mRNA expression by 2- and 3-fold, respectively, anti-miR-199 overexpression, but not anti-miR-135, resulted in a 2-fold increase in ET-1 mRNA expression. Furthermore, overexpression of anti-miR-199 augmented ethanol-induced ET-1 protein expression by ~2.5-fold in HMEC-1 (Fig. 4H). However, overexpression of miR-199 caused a ~1.2-fold change in ET-1 protein expression, indicating the role of miR-199 in regulating ET-1 mRNA and protein levels (Fig. 4H). Taken together, these results indicate that miR-135 and miR-199 affect HIF-1α mRNA expression, while only miR-199 regulates ET-1 mRNA expression in rLSEC. Thus, miR-199 is a common mediator of HIF-1α and ET-1 mRNA expression in both rLSEC and HMEC-1.
Ethanol augments expression of ET-1 cognate receptor ET-BR in E-rLSEC, rKC, HMEC-1, and THP-1 monocytic cells
ET-1 mediates its physiological effect in various cells via binding to either ET-A receptor or ET-BR. Thus, we determined whether ethanol in vitro and in vivo affected the expression of the ET-1 receptor in Kupffer cells isolated from ethanol-fed rats (E-rKC) and pair-fed control animals (C-rKC). As shown in Fig. 5A, E-rKC showed a 3.5-fold increase in ET-BR mRNA expression, while treatment of C-rKC with ethanol resulted in a 5.5-fold increase in ET-BR mRNA expression (Fig. 5B). Additionally, human monocytic cells (THP-1 cells), used as a surrogate model system for KCs, when treated with ethanol showed a 4-fold increase in ET-BR expression (Fig. 5C). Our recent studies (30) had shown that placenta growth factor (PlGF)-induced ET-BR expression involved HIF-1α, and thus we examined the role of HIF-1α in ethanol-induced ET-BR expression. As shown in Fig. 5D, transfection of THP-1 with HIF-1α siRNA, but not scrambled HIF-1α siRNA, attenuated ethanol-mediated ET-BR expression. Since the proximal promoter region (−392 to +1 bp) of the ET-BR has two HRE sites (30), we examined whether ethanol induced binding of HIF-1α to the HRE in the ET-BR promoter. Since both Kupffer cells and THP-1 cells exhibited a similar response to ethanol in the expression of ET-BR, we used THP-1 cells as a model system for ease of transfection. As shown in Fig. 5E, treatment of THP-1 cells transfected with the ET-BR promoter construct (−392 to +493 bp) resulted in a 3.5-fold increase above the control. However, mutation of either the HRE-1 (−155 to −152 bp) or HRE-2 (−371 to −368 bp) sites, relative to the transcription start site in the proximal promoter region of ET-BR, completely abrogated ethanol-mediated ET-BR promoter luciferase activity (Fig. 5E). These results indicate that ethanol-mediated activation and binding of HIF-1α to either HRE site in the ET-BR promoter leads to an increase in ET-BR gene expression.
FIGURE 5.

Ethanol induces ET-BR expression in rKC and THP-1 monocytic cells via HIF-1α. A, Ethanol-induced ET-BR expression in Kupffer cells derived from ethanol-fed rats. The data represent fold increases in mRNA expression following ethanol feeding compared with control. B, Ethanol-induced ET-BR expression in ethanol-treated rKC. C, Ethanol-induced ET-BR expression in ethanol-treated THP-1 cells. D, Transient transfection of THP-1 cells with HIF-1α siRNA attenuates ethanol-induced ET-BR expression in ethanol-treated THP-1 cells. HIF-1α scrambled RNA was used as a control. Quantitative RT-PCR data for ethanol-treated rKC and THP-1 represent fold increases in mRNA expression following stimulation with ethanol (100 mM for 2 h) compared with no ethanol. All mRNA expressions were normalized to GAPDH mRNA levels, and the data shown represent three independent experiments (means ± SD). E, Ethanol-induced ET-BR promoter activity in THP-1 cells stimulated with ethanol, which was attenuated when two HRE sites were individually mutated. The data are expressed as fold change and have been normalized relative to the change in luciferase activity of the untreated controls and to the transfection efficiency with β-galactosidase. The data shown represent two independent experiments in duplicate (means ± D). ***, p < 0.001; ns, p > 0.05.
Ethanol augments ET-1-induced MCP-1 expression in THP-1 and Kupffer cells through a positive feedback ET-BR loop involving HIF-1α activation
Since we observed that ethanol caused an increase in the expression of ET-1 in rLSEC and its cognate receptor ET-BR in Kupffer cells, we hypothesized that the interaction of ET-1 with ET-BR could result in an increased expression of chemokines. As shown in Fig. 6A, ethanol treatment of HMEC-1 for 48 h resulted in 1.5-fold increase in ET-1 release, which was attenuated to basal level when HMEC-1 were pretreated with DPI and R59949, but not with LY294002. Thus, the release of ET-1 protein and ET-1 mRNA expression utilizes the same signaling pathway in HMEC-1. Next, we investigated whether ET-1 released from ethanol-treated HMEC-1 activated THP-1 cells to augment MCP-1 expression. The conditioned media from ethanol-treated HMEC-1 for 48 h, corresponding to a concentration of 250 nM of ET-1, resulted in an ~12-fold increase in MCP-1 mRNA expression in THP-1 monocytes (Fig. 6B, lane 2) compared with untreated THP-1 cells (Fig. 6B, lane 1). However, THP-1 cells pretreated with ethanol for 2 h, which express increased levels of ET-BR, showed a 24-fold increase in MCP-1 mRNA with the same amount of conditioned medium from HMEC-1 cells (Fig. 6B, lane 4) compared with THP-1 cells not treated with supernatant (Fig. 6B, lane 3). Furthermore, THP-1 preincubated with BQ788, an ET-BR antagonist, before ethanol stimulation did not show MCP-1 mRNA expression upon incubation with the conditioned medium from ethanol-treated MHEC-1 (Fig. 6B, lane 5). The addition of conditioned medium from untreated HMEC-1 did not augment MCP-1 expression in THP-1 cells treated with ethanol over and above that observed with ethanol alone (data not shown). Taken together, these results indicate that ethanol-induced ET-BR expression in monocytes leads to an additive effect of ET-1-induced MCP-1 expression from monocytes.
FIGURE 6.

Ethanol promotes ET-1-induced MCP-1 expression in THP-1 and Kupffer cells through an endothelial cell-induced positive feedback loop of HIF-1α activation. A, ET-1 release from HMEC-1 in response to ethanol treatment (100 mM for 48 h) expressed as fold change, compared with no treatment. Where indicated, cells were pretreated with DPI, LY294002, and R59949. The data represent two independent experiments in duplicate (means ± SD). B, mRNA expression of MCP-1, ET-BR, and HIF-1α in THP-1 cells incubated with conditioned medium from ethanol-treated HMEC-1. Where indicated, THP-1 cells were treated with ethanol for 2 h in the absence or presence of BQ788. All mRNA expressions were normalized to GAPDH mRNA levels, and the data shown represent two independent experiments in duplicate (means ± SD). C, ET-1 released in rLSEC treated with ethanol (100 mM for 24 h) is mediated through NADPH oxidase and HIF-1α. The data represent two independent experiments in duplicate (means ± SD). D, mRNA expression of MCP-1, ET-BR, and HIF-1α in rKC incubated with conditioned medium from ethanol-treated rLSEC. Where indicated, rKC were treated with ethanol for 12 h in the absence or presence of BQ788, or treated with synthetic ET-1 (250 nm). All mRNA expressions were normalized to GAPDH mRNA levels, and the data shown represent two independent experiments in duplicate (means ± SD). ***, p < 0.001; **, p < 0.01; *, p < 0.05; ns, p > 0.05.
To determine whether ethanol-mediated ET-1 release from rLSEC could activate rKCs in vivo to generate chemokines, MCP-1, we utilized an in vitro assay to mimic this situation. Ethanol treatment of rLSEC for 24 h showed a 1.4-fold increase in the release of ET-1 (Fig. 6C), which was attenuated by the same pharmacological inhibitors, namely DPI and R59949, but not with LY294002, indicating similarities in the expression of ET-1 in both rLSEC and HMEC-1 in response to ethanol. When the conditioned medium from ethanol-treated rLSEC, corresponding to a concentration of 250 nM of ET-1, was added to KCs, there was an ~6-fold increased expression of MCP-1 mRNA, but levels of HIF-1α and ET-BR mRNA remained similar to those of control (Fig. 6D, lane 2). However, rKC treated with ethanol for 12 h showed 6- and 8-fold increases in ET-BR and MCP-1 mRNA expression, respectively (Fig. 6D, lane 3). Additionally, when conditioned medium from ethanol-treated rLSEC was added to ethanol-treated rKC, there were 14- and 12-fold increases in ET-BR and MCP-1 expression, respectively (Fig. 6D, lane 4), indicating an additive effect of ET-1 and ethanol. Furthermore, the expression of MCP-1 was attenuated when ET-BR receptor antagonist (BQ788) was added to rKC treated with ethanol followed by addition of conditioned medium from ethanol-treated rLSEC (Fig. 6D, lane 5). These results indicated that ET-1 released from ethanol-treated rLSEC augmented MCP-1 expression in rKC via ET-BR. These results are similar to those seen in alcohol-treated rKC in response to synthetic ET-1 (Fig. 6D, lane 6), wherein we observed a 14-fold increase in MCP-1 mRNA expression with ET-1 (250 nM). The increase in ET-BR expression seen in ethanol-treated KCs with conditioned medium from rLSEC was unexpected, and thus could be due to soluble factors in conditioned medium such as cytokines, which could have an additive effect with ethanol on ET-BR expression. The addition of conditioned medium from ethanol-treated THP-1 cells, which do not produce ET-1, failed to augment MCP-1 expression in KCs treated with ethanol (data not shown). Taken together, these results indicate that ethanol primes rKC for the expression of ET-BR, which leads to exaggerated response to ET-1 in the formation of chemokines, MCP-1.
Discussion
Our studies show that LSEC from ethanol-fed rats are highly activated and show increased expression of ET-1 and chemokines compared with LSEC isolated from control rats. We focused our studies on how ethanol regulates the expression of ET-1, a potent vasoconstrictor, which likely plays a role in microcirculatory tone and portal hypertension in patients with cirrhosis. In this report, we show that ethanol or its metabolite acetaldehyde induced ET-1 expression in LSEC and HMEC-1, as well as the expression of ET-1 cognate receptor (ET-BR) in Kupffer cells and THP-1 monocytic cells. Our studies show that ethanol-induced up-regulation of ET-1 in endothelial cells involves activation of NADPH oxidase and HIF-1α, but not PI3K. We have recently observed that PlGF, elaborated from erythroid cells, increases the expression of ET-1 in human pulmonary microvascular endothelial cells, which involves activation of both the PI3K and NADPH oxidase pathways, and in HIF-1α (30). These results indicate different signaling events among ligands, that is, between ethanol and PlGF for the expression of ET-1 in endothelial cells.
The ET-1 promoter contains an inverted HIF-1α binding element (GCAC) at −118 bp upstream of the transcription start site, which binds to HIF-1α and HIF-1β, and is essential for the promoter response to hypoxia (28). Additional studies indicated that GATA-2, AP-1, and HIF-1α transcriptional factors in the minimal ET-1 promoter (−176 bp from transcriptional start site) formed a complex to regulate ET-1 gene expression in response to hypoxia (31). In the present study, the role of HIF-1 in the ethanol-mediated expression of ET-1 was established by a number of approaches. Ethanol causes a time-dependent increase in HIF-1α protein levels, which is attenuated by a NADPH oxidase inhibitor but not by a PI3K inhibitor, indicating the role of reactive oxygen species in stabilization of HIF-1α protein, independent of hypoxia. It is well established that under hypoxic conditions HIF-1α protein is stabilized and does not undergo degradation, as PHD-1–3 do not cause hydroxylation of proline residues in HIF-1α for subsequent degradation by the ubiquitin-proteosome pathway (33–35). Studies have shown that specific silencing of PHD-2 is able to stabilize HIF-1α steady-state levels under normoxia in mammalian cells (35). Our studies show that silencing with PHD-2 siRNA resulted in increased levels of HIF-1α and concomitant increased expression of ET-1. Furthermore, we observed that silencing with HIF-1α siRNA resulted in attenuation of ethanol-induced ET-1 expression. Mutation of the HRE site (−124 to −121 bp) resulted in complete attenuation of ethanol-induced ET-1 promoter luciferase activity. The role of HIF-1α in the regulation of ET-1 gene expression is further supported by studies showing the binding of HIF-1α to the HRE site in the ET-1 promoter as demonstrated by EMSA and ChIP analysis.
Since ethanol induced expression of HIF-1α mRNA and co-dependent expression of ET-1 in LSEC, we identified the miRNAs that likely regulate HIF-1α mRNA and ET-1 gene expression. Among several signature miRNAs regulated by hypoxia, which have been characterized and target HIF-1α (32), we have identified miR-199 as a target for ethanol-induced HIF-1α mRNA expression. In silico analysis of the 3′-UTR of HIF-1α shows the presence of miR-199 complementary binding sites at 162 to 184 bp, along with complementary sites for miR-135 (775 to 797 bp) and miR-155 (1152 to 1173 bp), while analysis of the 3′-UTR of ET-1 revealed the presence of complementary binding sites for miR-199 (20 to 40 bp), miR-155 (168 to 190), and miR-135 (8 to 30 bp), indicating the presence of the same complementary binding sites for miRNAs in the 3′-UTRs of both HIF-1α and ET-1 mRNA. Our studies show that miR-199 is a common mediator of HIF-1α and ET-1 mRNA expression in both rLSEC and HMEC-1. However, miR-155, present in human cells, also regulates HIF-1α and ET-1 mRNA expression. MicroRNA-155-mediated translational repression of angiotensin II type 1 receptor (AT-1R) occurs only in humans, chimpanzeesm and dogs, but not in mouse and rat AT-1R genes (36), suggesting that miR-155 is species specific. Since miR-155 is not expressed in rat cells, we suggest that ethanol-mediated expression of HIF-1α and ET-1 mRNA in both rat liver sinusoidal endothelial cells and human microvascular endothelial cells is regulated by miR-199. However, miR-155 also regulates HIF-1α and ET-1 expression in human endothelial cells. Our studies demonstrate, for the first time, that translational repression by miR-155 and miR-199 provides another avenue by which ET-1 expression, mediated by ethanol, can be modulated. Recently, miRNAs have been identified that target sites in the 3′-UTRs of selected genes (37, 38) and specifically affect innate immune response (39), expression of TLRs (40), and erythropoiesis (41).
We also observed that ethanol increased the expression of ET-1 cognate receptor ET-BR in Kupffer cells and LSEC derived from chronic ethanol-fed rats. Moreover, acute treatment of human THP-1 monocytic cells with ethanol showed an increase in the mRNA expression of ET-BR. Ethanol-induced mRNA expression of ET-BR was also shown to be regulated by HIF-1α. An in silico analysis of the ET-BR promoter element (−1259 to −1 bp) showed the presence of several hypoxia-response elements (RCGTG) and inverted HRE elements (GCAC), along with a potential SP-1 site, GATA motif, acute phase regulatory elements, and E-box (42). Our results show that mutation of either the HRE-1 (−155 to −152 bp) or HRE-2 (−371 to −368 bp) sites among several HREs in the proximal promoter region of ET-BR completely abrogated ethanol-mediated ET-BR promoter luciferase activity, indicating that ethanol-mediated binding of HIF-1α to either HRE in ET-BR promoter leads to an increase in ET-BR gene expression. Note that PlGF-induced ET-BR expression only required HRE-1 site (−155 to −152 bp) for the binding of HIF-1α to its promoter (30).
Our studies also show that ET-1 released from either rLSEC or HMEC-1, in response to ethanol treatment, augmented the expression of the chemokine MCP-1 from Kupffer cells and THP-1 monocytic cells, as illustrated in the schematics of Fig. 7. This involved the binding of ET-1 to its cognate receptor ET-BR, as the antagonist for ET-BR (BQ788) abrogated MCP-1 expression. Although ethanol itself augments the expression of MCP-1 in KCs, ET-1-mediated expression of MCP-1 is additive, but not synergistic, with the effect of ethanol alone. We propose that, in response to ethanol, with the up-regulation of ET-1, there will be exaggerated expression of chemokines from KCs (Fig. 7), which will likely promote the infiltration of monocytes, PMN, and T cells from circulation via sinusoids into the liver, contributing to liver injury.
FIGURE 7.

Working model of ethanol-induced ET-1-mediated MCP-1 expression in rKC through a positive feedback loop of HIF-1α activation in rLSEC. Ethanol enters rLSEC and activates the NADPH oxidase pathway, generating reactive oxygen species. Increased reactive oxygen species stabilize HIF-1α protein and activate its translocation into the nucleus, where it heterodimerizes with HIF-1β. This complex binds to the HRE on the ET-1 promoter, causing augmented transcription of ET-1. ET-1 released from rLSEC binds to its cognate receptor ET-BR, which is also up-regulated by ethanol-induced HIF-1α activation in rKC. The binding of ET-1 to ET-BR in rKC leads to increased MCP-1 expression, which acts as a chemoattractant to mediate infiltration of neutrophils from the circulation into the sinusoids and then into the liver, perpetuating ethanol-induced liver injury.
Overall, these studies show that ethanol, its metabolism or metabolite acetaldehyde, augments the expression of ET-1 and its receptor ET-BR via activation of HIF-1α, independent of hypoxia. The translational repression by miR-199 and miR-155 in human endothelial cells and miR-199 in rLSEC provides another novel mechanism by which expression of ET-1 can be modulated. The up-regulation of ET-1 in LSEC and ET-BR in Kupffer cells by ethanol leads to additive effects on ethanol-mediated expression of inflammatory mediators, such as MCP-1. Additionally, ET-1 released from LSEC can cause hepatic vascular dysregulation by interacting with endothelin receptors expressed on LSEC, Kupffer cells, and adjacent stellate cells in activating endothelial NO synthase and formation of NO, as has been shown in LSEC from rats exposed to ischemic-reperfusion injury (17). Thus, ethanol likely causes inflammatory response, as seen by increased expression of inflammatory cytochemokines, but it may induce vasoconstrictive responses via ET-1 and ET-BR up-regulation. Previous studies have shown that hepatocytes (43) and hepatic stellate cells, in addition to LSEC (44), show increased ET-BR expression in liver tissues from cirrhotic animals. Note that, in the liver, hepatic stellate cells, which are pericytes, are known to control the vascular tone at the sinusoidal level. Therefore, ethanol-induced ET-1 by LSEC may cause contraction of hepatic stellate cells via ET-BR and consequent sinusoidal constriction. We suggest that ethanol-mediated ET-1 expression likely contributes to inflammation as seen in patients with cirrhosis. These studies provide a new therapeutic approach, based on HIF-1α target sites, to ameliorate ethanol-induced inflammation.
Acknowledgments
We thank Dr. Kinji Asahina and Jiaohong Wang for kindly isolating and providing rat liver sinusoidal endothelial cells and Kupffer cells. We also thank Dr. Keith Webster at the University of Miami (Miami, FL) for providing the ET-1 luciferase constructs.
Footnotes
This work was supported by pilot project funding (V.K.K.) from National Institutes of Health Grant P50-AA011999 (to H.T.) and its animal and morphological core facilities, Grants R24-AA012885 (Non-Parenchymal Liver Cell Core; to H.T.) and T32-AA07578 (Predoctoral Fellowship to S.Y.).
Abbreviations used in this paper: HIF-1α, hypoxia-inducible factor-1α; ChIP, chromatin immunoprecipitation; DPI, diphenylene iodonium; ET-1, endothelin-1; ET-BR, endothelin-B receptor; HMEC-1, human dermal microvascular endothelial cell line; HRE, hypoxia response element; KC, Kupffer cell; LSEC, liver sinusoidal endothelial cell; miRNA, microRNA; PHD, prolylhydroxylase; PlGF, placenta growth factor; scRNA, scrambled RNA; siRNA, small interfering RNA; UTR, untranslated region; PMN, polymorphonuclear leukocyte.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Vidali M, Stewart SF, Albano E. Interplay between oxidative stress and immunity in the progression of alcohol-mediated liver injury. Trends Mol Med. 2008;14:63–71. doi: 10.1016/j.molmed.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 2.Ramaiah SK, Jaeschke H. Role of neutrophils in the pathogenesis of acute inflammatory liver injury. Toxicol Pathol. 2007;35:757–766. doi: 10.1080/01926230701584163. [DOI] [PubMed] [Google Scholar]
- 3.Tsukamoto H, Kaplowitz N. Pathogenesis of alcoholic liver disease. In: Kaplowitz N, editor. Liver and Biliary diseases. Williams & Wilkins; Baltimore: 1996. pp. 121–137. [Google Scholar]
- 4.Pritchard MT, Nagy LE. Ethanol-induced liver injury: potential roles for Egr-1. Alcohol Clin Exp Res. 2005;29(Suppl 11):146S–150S. doi: 10.1097/01.alc.0000189286.81943.51. [DOI] [PubMed] [Google Scholar]
- 5.Lalor P, Faint J, Aarbodem Y, Hubscher S, Adams D. The role of cytokines and chemokines in the development of steatohepatitis. Semin Liver Dis. 2007;27:173–193. doi: 10.1055/s-2007-979470. [DOI] [PubMed] [Google Scholar]
- 6.McVicker BL, Tuma DJ, Kharbanda KK, Kubik JL, Casey CA. Effect of chronic ethanol administration on the in vitro production of proinflammatory cytokines by rat Kupffer cells in the presence of apoptotic cells. Alcohol Clin Exp Res. 2007;31:122–129. doi: 10.1111/j.1530-0277.2006.00270.x. [DOI] [PubMed] [Google Scholar]
- 7.McClain CJ, Song Z, Barve SS, Hill DB, Deaciuc I. Recent advances in alcoholic liver disease, IV: Dysregulated cytokine metabolism in alcoholic liver disease. Am J Physiol. 2004;287:G497–G502. doi: 10.1152/ajpgi.00171.2004. [DOI] [PubMed] [Google Scholar]
- 8.Tilg H, Diehl AM. Cytokines in alcoholic and nonalcoholic steatohepatitis. N Engl J Med. 2000;343:1467–1476. doi: 10.1056/NEJM200011163432007. [DOI] [PubMed] [Google Scholar]
- 9.Okanoue T, Sakamoto S, Mori T, Sawa Y, Kanaoka H, Nishioji K, Itoh Y. Role of sinusoidal endothelial cells in alcoholic liver disease. In: Tanikawa K, Ueno T, editors. Liver Disease and Hepatic Sinusoidal Cells. Springer; Tokyo: 1999. pp. 190–198. [Google Scholar]
- 10.Li J, French B, Wu Y, Venkatesh R, Montgomery R, Bardag-Gorce F, Kitto J, French SW. Liver hypoxia and lack of recovery after reperfusion at high blood alcohol levels in the intragastric feeding model of alcohol liver disease. Exp Mol Pathol. 2004;77:184–192. doi: 10.1016/j.yexmp.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 11.French SW. Intragastric ethanol infusion model for cellular and molecular studies of alcoholic liver disease. J Biomed Sci. 2001;8:20–27. doi: 10.1007/BF02255967. [DOI] [PubMed] [Google Scholar]
- 12.Tsukamoto H, French SW, Reidelberger RD, Largman C. Cyclical pattern of blood alcohol levels during continuous ethanol infusion in rats. Alcohol Clin Exp Res. 1985;9:31–37. doi: 10.1111/j.1530-0277.1985.tb05046.x. [DOI] [PubMed] [Google Scholar]
- 13.Mathurin P, Deng QG, Keshavarzian A, Choudhary S, Holmes EW, Tsukamoto H. Exacerbation of alcoholic liver injury by enteral endotoxins in rats. Hepatology. 2000;32:1008–1017. doi: 10.1053/jhep.2000.19621. [DOI] [PubMed] [Google Scholar]
- 14.Dunn W, Shah V. Hepatic microcirculation. In: Aird WC, editor. Endothelial Biomedicine. Cambrige Univ. Press; New York: 2007. pp. 1239–1247. [Google Scholar]
- 15.Shah V, Haddad FG, Garcia-Cardena G, Fangos JA, Mennone A, Groszmann RJ. Liver sinusoidal endothelial cells are responsible for nitric oxide modulation of resistance in the hepatic sinusoids. J Clin Invest. 1997:2923–2930. doi: 10.1172/JCI119842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takei Y, Kawano S, Oshita M, Hijioka T, Kamada T, Sao N. Ethanol-induced perturbation of hepatic microcirculation: role of endothelin-1 and nitric oxide in regulation of sinusoidal tone. In: Tanikawa K, Ueno H, editors. Liver Disease and Hepatic Sinusoidal Cells. Springer; Tokyo: 1999. pp. 209–218. [Google Scholar]
- 17.Lee SH, Culberson C, Korneszczuk K, Clemens MG. Differential mechanisms of hepatic vascular dysregulation with mild vs. moderate ischemia-reperfusion. Am J Physiol. 2008;294:G1219–G1226. doi: 10.1152/ajpgi.00527.2007. [DOI] [PubMed] [Google Scholar]
- 18.DeLeve LD, Wang X, Hu L, McCuskey MK, McCuskey RS. Rat liver sinusoidal endothelial cell phenotype is maintained by paracrine and autocrine regulation. Am J Physiol. 2004;287:G757–G763. doi: 10.1152/ajpgi.00017.2004. [DOI] [PubMed] [Google Scholar]
- 19.Tsukamoto H, Hasmik M, Alla D. Intragastric ethanol infusion model in rodents. Methods Mol Biol. 2008;447:33– 48. doi: 10.1007/978-1-59745-242-7_3. [DOI] [PubMed] [Google Scholar]
- 20.Xiong S, She H, Zhang AS, Wang J, Mkrtchyan H, Dynnyk A, Gordeuk VR, French SW, Enns CA, Tsukamoto H. Hepatic macrophage iron aggravates experimental alcoholic steatohepatitis. Am J Physiol. 2008;295:G512–G521. doi: 10.1152/ajpgi.90327.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeLeve LD, Wang X, McCuskey MK, McCuskey RS. Rat liver endothelial cells isolated by anti-CD31 immunomagnetic separation lack fenestrae and sieve plates. Am J Physiol. 2006;291:G1187–G1189. doi: 10.1152/ajpgi.00229.2006. [DOI] [PubMed] [Google Scholar]
- 22.Deleve LD. Dacarbazine toxicity in murine liver cells: a model of hepatic endothelial injury and glutathione defense. J Pharmacol Exp Ther. 1994;268:1261–1270. [PubMed] [Google Scholar]
- 23.Braet F, Wisse E. Structural and functional aspects of liver sinusoidal endothelial cell fenestrae: a review. Comp Hepatol. 2006;1:1–17. doi: 10.1186/1476-5926-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jaffe S, Oliver PD, Farooqui SM, Novak PL, Sorgente N, Kalra VK. Separation of luminal and abluminal membrane enriched domains from cultured bovine aortic endothelial cells: monoclonal antibodies specific for endothelial cell plasma membranes. Biochim Biophys Acta. 1987;898:37–52. doi: 10.1016/0005-2736(87)90107-6. [DOI] [PubMed] [Google Scholar]
- 25.Newman PJ, Albelda SM. Cellular and molecular aspects of PECAM-1. Nouv Rev Fr Hematol. 1992;34(Suppl):S9–S13. [PubMed] [Google Scholar]
- 26.Giri RK, Selvaraj SK, Kalra VK. Amyloid peptide-induced cytokine and chemokine expression in THP-1 monocytes is blocked by small inhibitory RNA duplexes for early growth response-1 messenger RNA. J Immunol. 2003;170:5281–5294. doi: 10.4049/jimmunol.170.10.5281. [DOI] [PubMed] [Google Scholar]
- 27.Kim KS, Rajagopal V, Gonsalves C, Johnson C, Kalra VK. A novel role of hypoxia-inducible factor in cobalt chloride- and hypoxia-mediated expression of IL-8 chemokine in human endothelial cells. J Immunol. 2006;177:7211–7224. doi: 10.4049/jimmunol.177.10.7211. [DOI] [PubMed] [Google Scholar]
- 28.Hu J, Discher DJ, Bishopric NH, Webster KA. Hypoxia regulates expression of the endothelin-1 gene through a proximal hypoxia-inducible factor-1 binding site on the antisense strand. Biochem Biophys Res Commun. 1998;245:894– 899. doi: 10.1006/bbrc.1998.8543. [DOI] [PubMed] [Google Scholar]
- 29.Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patel N, Gonsalves CS, Malik P, Kalra VK. Placenta growth factor augments endothelin-1 and endothelin-B receptor expression via hypoxia-inducible factor-1α. Blood. 2008;112:856– 865. doi: 10.1182/blood-2007-12-130567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamashita K, Discher DJ, Hu J, Bishopric NH, Webster KA. Molecular regulation of the endothelin-1 gene by hypoxia: contributions of hypoxia-inducible factor-1, activator protein-1, GATA-2, and p300/CBP. J Biol Chem. 2001;276:12645–12653. doi: 10.1074/jbc.M011344200. [DOI] [PubMed] [Google Scholar]
- 32.Dalmay T, Edwards DR. MicroRNAs and the hallmarks of cancer. Oncogene. 2006;25:6170– 6175. doi: 10.1038/sj.onc.1209911. [DOI] [PubMed] [Google Scholar]
- 33.Huang LE, Gu J, Schau M, Bunn HF. Regulation of hypoxia-inducible factor 1α is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci USA. 1998;95:7987–7992. doi: 10.1073/pnas.95.14.7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berra E, Benizri E, Ginouves A, Volmat V, Roux D, Pouyssegur J. HIF prolyl hydroxylase 2 is the key oxygen sensor setting low steady state levels of HIF-1α in normoxia. EMBO J. 2008;22:4082– 4090. doi: 10.1093/emboj/cdg392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martin MM, Lee EJ, Buckenberger JA, Schmittgen TD, Elton TS. MicroRNA-155 regulates human angiotensin II type 1 receptor expression in fibroblasts. J Biol Chem. 2006;281:18277–18284. doi: 10.1074/jbc.M601496200. [DOI] [PubMed] [Google Scholar]
- 37.Bino J, Enright AJ, Aravim A, Tuschl T, Sander C, Marks DS. Human nicroRNa targets. PLoS Biol. 2008;2:1862–1879. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Doench JG, Sharp PA. Specificity of microRNA target selection in translational repression. Genes Dev. 2004;18:504–511. doi: 10.1101/gad.1184404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA. 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen XM, Splinter PL, O’Hara SP, LaRusso NF. A cellular micro-RNA, let-7i, regulates Toll-like receptor 4 expression and contributes to cholangiocyte immune responses against Cryptosporidium parvum infection. J Biol Chem. 2007;282:28929–28938. doi: 10.1074/jbc.M702633200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Q, Huang Z, Xue H, Jin C, Ju XL, Han JD, Chen YG. MicroRNA miR-24 inhibits erythropoiesis by targeting activin type I receptor ALK4. Blood. 2008;111:588–595. doi: 10.1182/blood-2007-05-092718. [DOI] [PubMed] [Google Scholar]
- 42.Arai H, Nakao K, Takaya K, Hosoda K, Ogawa Y, Nakanishi S, Imura H. The human endothelin-B receptor gene: structural organization and chromosomal assignment. J Biol Chem. 1993;268:3463–3470. [PubMed] [Google Scholar]
- 43.Kuddus RH, Nalesnik MA, Subbotin VM, Rao AS, Gandhi CR. Enhanced synthesis and reduced metabolism of endothelin-1 (ET-1) by hepatocytes: an important mechanism of increased endogenous levels of ET-1 in liver cirrhosis. J Hepatol. 2000;33:725–732. doi: 10.1016/s0168-8278(00)80302-5. [DOI] [PubMed] [Google Scholar]
- 44.Yokomori H, Oda M, Yasogawa Y, Nishi Y, Ogi M, Takahashi M, Ishii H. Enhanced expression of endothelin B receptor at protein and gene levels in human cirrhotic liver. Am J Pathol. 2001;159:1353–1362. doi: 10.1016/S0002-9440(10)62522-9. [DOI] [PMC free article] [PubMed] [Google Scholar]