

Introduction
The development of efficient approaches for delivering therapeutic genes into somatic cells, including T lymphocytes, has fostered applications in human cancer therapy based on administering T cells that are engineered to recognize molecules expressed by malignant cells. Genetic modification of T cells to confer tumor specificity can circumvent the local and systemic tolerance mechanisms that limit endogenous antitumor T cells, overcome the logistical difficulties in isolating and expanding often exceedingly rare tumor-reactive T cells from patients, and theoretically provide for control of the magnitude, specificity, avidity, and function of the antitumor response. Definitive evidence for in vivo therapeutic activity of genetically modified T cells has been obtained in small clinical trials in specific malignancies, and these successes have renewed optimism that immunotherapy will become an effective modality for a broad range of human cancers [1,2]. Here, we review recent progress and discuss the challenges and future prospects for this developing field of cancer therapy.
Engineering T cells to express MHC-restricted T cell receptors (TCRs) for adoptive therapy
The adoptive transfer of tumor-infiltrating T lymphocytes (TIL), expanded from resected melanoma specimens and selected for reactivity with tumor associated peptides displayed by MHC molecules on cancer cells, can mediate durable tumor regression in a subset of patients with advanced metastatic melanoma [3,4]. A major advance was the recognition that administering lymphodepleting chemotherapy to patients prior to TIL infusions improved the persistence of transferred T cells and antitumor activity [3,5], and lymphodepletion is now employed in most clinical trials that utilize genetically modified T cells. Because tumor-reactive TIL cannot be derived from many patients with melanoma, or from most other solid tumors, it was anticipated that TCR genes could be isolated from tumor-reactive T cell clones in TIL that mediated tumor regression, assembled in gene transfer vectors, and introduced into T cells from any patient of the appropriate MHC type to confer tumor specificity. While conceptually straightforward, TCR gene transfer presented several obstacles.
The first obstacle is the need to isolate a high avidity T cell clone for a defined target antigen, ideally one presented by a common HLA allele, selectively expressed in tumors from many individuals, and not in normal cells. Dissection of the specificity of TIL from melanoma revealed a low frequency of T cells that recognized many known self-antigens such as MART-1, gp100, the cancer-testes antigens, and overexpressed proteins, but unfortunately failed to identify a clear correlation between any single antigen specificity and tumor regression [6]. A concern with targeting self-antigens is that T cell responses are often of low avidity because the highest avidity T cells are deleted during thymic selection to avoid autoimmunity. Mice that express both human MHC and TCR genes have been developed and can be vaccinated to elicit T cell responses (and TCRs) from a repertoire that is not tolerant to human self or tumor associated determinants [7]. It is also possible by mutation to improve the affinity of TCRs obtained from low avidity T cells specific for self-proteins [8]. However, a challenge is determining what TCR affinity for a self-antigen is necessary for therapeutic activity without toxicity to normal tissues or unexpected off-target toxicity, and this issue is difficult to address for human TCRs in vitro or in animal models. It is apparent from animal models that the introduction of TCRs that are of too high affinity can result in the deletion of transferred T cells in vivo [9], and as described below, clinical trials of high affinity receptors specific for self antigens have shown significant on-target toxicity to normal tissues [10,11]. Ideally, TCRs that recognize tumor-specific mutated proteins that are shared in many tumors would be utilized to avoid autoimmunity. This possibility may be exploited in the future for melanoma and other tumors, as exome sequencing uncovers shared mutations in coding sequences that can be examined for immunogenicity [12,13].
A second obstacle is the development of safe and efficient vectors for transferring and stably expressing high levels of introduced TCR genes in T cells. Preclinical evidence suggested that the oncogenic risk of integrating retroviral or lentiviral vectors in T cells may be lower than in hematopoietic stem cells, and long term follow-up of a clinical trial in which T cells were modified with an integrating retroviral vector demonstrated that integration sites did not favor oncogenic sites [14,15]. Thus, both retroviral and lentiviral vectors are now commonly used to deliver codon-optimized TCR genes to T cells. Expression of the introduced TCR alpha and beta chains that confers the desired tumor specificity is diminished by cross pairing o with endogenous TCR chains, which also inadvertently creates novel specificities that underlie the autoimmune toxicities observed in preclinical mouse models of TCR gene therapy [16]. Such unwanted pairing is reduced by modifications of the sequences of introduced TCR chains to promote their selective association [17,18], or by knocking down the expression of endogenous TCR chains using inhibitory RNA [19]. An elegant approach that eliminates the problem of mispairing of TCR chains is to abolish expression of the endogenous receptors using specific zinc-finger nucleases to introduce double strand breaks in the TCR alpha and beta gene loci, which when repaired by error-prone non-homologous end joining results in nucleotide insertions or deletions that disrupt gene expression [20].
Clinical trials using TCR gene transfer to target differentiation antigens (MART-1, gp100, CEA) and the cancer-testes antigen NY-ESO-1 have been reported [10,21,22]. The first study used a retroviral vector to express an HLA A2-restricted MART-1-specific TCR in peripheral blood T cells from melanoma patients. The MART-1 TCR engineered cells were re-infused after lymphodepleting chemotherapy and transient antitumor responses were observed in a minority of patients [21]. In a subsequent trial, 36 melanoma patients received T cells modified with either a MART-1 or a gp100 TCR that was selected for higher affinity, and one complete remission and 8 transient responses were observed. However, on-target toxicity to normal melanocytes in the skin, eye and ear that required local corticosteroid therapy was observed in a significant fraction of patients that received the high avidity TCR [10]. Similarly, the introduction of a high affinity carcinoembryonic antigen (CEA)-specific TCR into autologous T cells to treat colorectal cancer resulted in severe colitis presumably from recognition of normal epithelial cells that express CEA, and the trial was halted after only 3 patients [11]. The on-target toxicity to normal tissues observed with high affinity TCRs specific for differentiation antigens suggested that targeting antigens with more restricted expression on tumor cells would be safer. A TCR specific for an HLA A*0201 restricted NY-ESO-1 epitope, which is expressed in many human cancers but not in normal tissues apart from testes, was used to engineer autologous T cells for treatment of metastatic melanoma and synovial sarcoma (SS). Objective responses were observed in 5 of 11 melanoma patients and 4 of 6 SS patients without toxicity to normal tissues [22]. These clinical results of therapy with TCR engineered T cells targeting a single antigen in patients with advanced refractory malignancies are encouraging, and additional investigation of this approach is warranted, particularly with TCRs that specifically target malignant and not normal cells. Off-target autoimmune toxicities resulting from mispairing of introduced and endogenous TCRs were not seen in these initial clinical trials, however this remains a serious concern and it is anticipated that future trials will employ selective knock-down or knock-out of endogenous TCR gene expression to avoid this complication [16,19,20].
Engineering T cells to express chimeric receptors that target cell surface molecules on cancer cells
The genetic modification of T cells with vectors that encode chimeric antigen receptors (CARs) specific for cell surface molecules overcomes the constraint of TCR recognition of peptide antigens presented by only certain MHC molecules, and avoids tumor escape through impairments in antigen presentation or HLA expression [23]. CARs typically consist of an scFV, derived from the VH and VL sequences of a monoclonal antibody specific for a tumor cell surface molecule that is fused to the CD3ζ intracellular signaling domain alone, or with one or more costimulatory signaling modules, such as CD28, 4-1BB, OX40, or CD27 [24,25]. CARs have been designed with scFvs that target molecules expressed on many different malignancies, and have the attraction that a single receptor construct can be used to treat all patients that express the target molecule. The tumor recognition domain need not be an scFv; for example a construct that encodes a cytokine (IL-13) as the tumor-targeting moiety is in clinical trials for patients with glioblastoma that expresses the IL-13Rα2 [26]. There are potential disadvantages of CARs including the immunogenicity of murine monoclonal antibodies and/or the fusion sites between components of the CAR, and the uncertainty as to how T cells will respond in vivo to signals that do not fully recapitulate those mediated through physiologic interactions of T cells with antigens presented by MHC molecules. Several aspects of CARs are the subject of intensive research including the density of target molecules required for effective T cell signaling in vivo, the affinity of the scFv and optimal costimulatory domains, and potential steric constraints imposed by the location of the target epitope that may dictate the required extracellular spacer length. Modeling of receptor and target ligand structures may provide insights into the optimal design of CARs specific for individual target molecules.
CAR engineered T cells have been tested for antitumor activity in animal models, and the results of pilot clinical trials of T cells modified to express CARs specific for CD19, CD20, ERBB2, and GD2 have been reported [27-33]. The initial clinical studies used first generation CARs that lacked costimulatory domains and/or introduced the CAR gene by electroporation, which was inefficient and required long-term culture to generate T cell products, and significant antitumor activity was not observed [34,35]. Recent studies using efficient retroviral or lentiviral transduction to introduce CAR transgenes that contain costimulatory domains into T cells have provided striking evidence of in vivo antitumor activity in humans, and also revealed the potential for serious toxicity. A fatal toxicity was observed after infusion of a single high dose of ERBB2-specific CAR T cells that contained both CD28 and 4-1BB costimulatory domains, and was attributed to cytokine release and recognition of normal lung epithelium that expresses low amounts of ERBB2 [33]. Encouraging clinical results have been obtained targeting the B-cell lineage restricted CD19 molecule that is expressed on B-cell leukemias and lymphomas with CD19-specific CAR T cells [27-30,36]. June and colleagues reported dramatic and durable remissions in two of three patients with B cell chronic lymphocytic leukemia after the infusion of autologous T cells transduced to express a CD19-specific CAR that contained a 4-1BB costimulatory domain. In this study, the infusion of low doses of T cells led to in vivo expansion of CAR T cells, subsequent tumor lysis, and a sustained deficiency of normal CD19+ B cells [27]. Significant antitumor activity, depletion of normal B cells, and side effects related to tumor lysis and cytokine release have also been observed in patients with CLL and lymphoma who received T cells modified to express CD19 CARs that contain a CD28 costimulatory domain [28,30]. That such durable responses can be achieved in patients with advanced chemotherapy refractory B cell malignancies illustrates the potency of CAR-directed T cell therapy, and suggests that future work to define optimal CD19 CAR constructs and composition of cell products, and to integrate T cell therapy earlier for patients with poor prognostic features may lessen the immediate risks of therapy.
Whether CAR-modified T cells can be effective for solid tumors remains an intense area of investigation. An important issue is to define candidate surface molecules on solid tumors that can be effectively targeted without serious or unremitting toxicity to normal tissues. Mesothelin, L1CAM, and GD2 are being actively pursued as targets, but a significant obstacle is the lack of suitable animal models for preclinical toxicity studies. Significant antitumor activity without toxicity was observed in patients with neuroblastoma treated with GD2 CAR T cells, however the persistence of the transferred cells was relatively short, perhaps due to the lack of a costimulatory domain in the CAR design, and it is uncertain if a sustained T cells response to GD2 would be tolerated [32].
Modification of T cells to enhance safety and efficacy of therapy
Genetic modifications can also potentially enhance the safety and/or therapeutic efficacy of T cells engineered with TCRs or CARs. Strategies to improve safety have included the incorporation of a conditional suicide gene or regulated (on-off) expression of the tumor-targeting receptor. A conditional suicide gene has been developed that encodes human caspase 9 fused to a modified human FK-binding protein. Dimerizing the fusion protein by exposing cells to a cell permeable synthetic small molecule activates caspase 9 and induces apoptosis. This approach was effective in ablating alloreactive T cells in vivo after human hematopoietic cell transplant, and may prove useful to abrogate on-target or off-target toxicities of TCR or CAR engineered T cells [37].
Genetic strategies to overcome local tumor immunosuppression mediated by tumor stromal cells, cytokines, and negative signaling pathways in T cells have also been devised. An intriguing approach is to express a single chain IL-12 molecule in tumor-specific T cells, which in murine tumor models enhanced the efficacy of low doses of MHC-restricted tumor-reactive T cells by re-programming tumor associated myeloid cells to cross present tumor antigens [38]; and eliminated the need for lymphodepleting chemotherapy prior to CAR-directed T cell therapy[39]. The development of vectors that incorporate a T cell activation dependent promoter to drive IL-12 expression may further enhance the safety of this approach for clinical applications [40].
Composition of gene engineered T cell products
The question of what types of T cells should be genetically modified for cancer therapy is beginning to receive close scrutiny. The first trials of genetically engineered T cells used unselected polyclonal T cells obtained by leukapheresis for gene transfer. Thus, the composition of T cell products administered to individual patients varied widely, and the consequence of this variation for interpreting product potency, safety, and efficacy is uncertain. The peripheral T cell pool is composed of multiple phenotypically distinct CD4+ and CD8+ subsets with divergent functions, transcriptional profiles and epigenetic programs, and the phenotypic distribution differs substantially in individuals because of age, pathogen exposure and prior chemotherapy [41,42]. Recent studies in which individual CD8+ T cell subsets were purified prior to genetic modification and adoptive transfer demonstrates remarkably different capacities for long-term persistence and antitumor activity. In animal models, both CD62L+ central memory T cells (TCM) and the infrequent memory subset (termed memory stem cell; TSCM) that has an intermediate phenotype between naïve (TN) and TCM cells, exhibited superior survival in vivo and/or mediated superior antitumor activity after adoptive transfer compared with more differentiated CD62L- effector memory (TEM) cells [43-45]. Analysis of human T cell products used in adoptive therapy has similarly suggested a correlation between retention of TCM markers and persistence in vivo [32]. CD8+ TN cells are undifferentiated and have substantial proliferative capacity, and as a consequence might be directed by cytokines or small molecules during in vitro expansion and transduction to have enhanced therapeutic activity [2]. The CD4+ T cell compartment is also heterogeneous, with defined naïve and memory subsets, and FoxP3+ cells with regulatory function [46]. CD4+ T cells alone have been shown to exhibit antitumor activity after adoptive transfer in animal models and in humans, at least in part by providing help for CD8+ T cells [47,48], and the potential to formulate gene-modified T cell products that contain defined proportions of CD8 and CD4 T cells from distinct subsets is beginning to be explored.
Challenges and Future Directions
Advances in the development and application of immunotherapy for cancer have been impressive in recent years, fueling optimism that this modality will soon have a meaningful impact in patient care. The ability to rapidly derive tumor-reactive T cells from any patient by gene transfer is a significant step for broad application. Many challenges remain, including the identification of target molecules for the most common solid tumors, and it is anticipated that initiatives such as The Cancer Genome Atlas and others that profile and characterize the genes expressed in tumor cells will identify potential candidates both for TCR and CAR directed therapy. Some molecules such as ROR1, which was initially described as a candidate for CAR T cell therapy in chronic lymphocytic leukemia, are also expressed on a variety of solid tumors, including lung and breast cancer [49-51]. A significant obstacle remains the relative lack of animal models for evaluating safety of individual targets, necessitating careful cell dose escalation in clinical trials to assess toxicity to normal tissues. The optimal design of receptor constructs to avoid autoimmunity in the case of TCRs and to enhance antitumor efficacy in the case of CARs requires additional investigation, and may differ for individual target molecules. Similarly, the T cell subsets that should be genetically modified requires further study, and may depend on the type and location of the malignancy being treated. The potential to integrate TCR or CAR modified T cells into combinations that include immune modulating agents such as the anti CTLA-4 or PD-1 checkpoint inhibitors, or antitumor drugs that delay tumor growth remains to be fully explored. The field is energized by the prospect of meeting these challenges and establishing cellular immunotherapy as a broadly effective modality.
Figure 1. Schema for adoptive immunotherapy with genetically modified T cells.
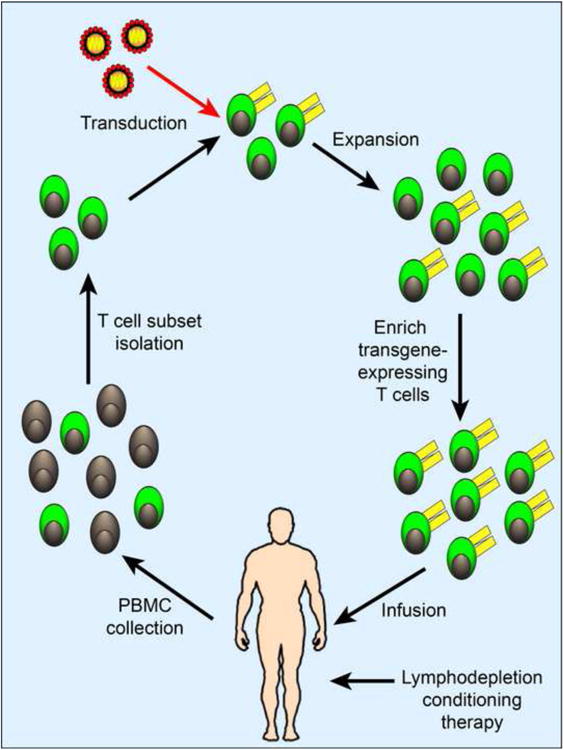
T cells or defined T cell subsets with distinct functional attributes (green) can be isolated from patient blood and genetically modified to express a transgene ecoding a tumor-targeting TCR or CAR (yellow). The genetically modified T cells are then expanded in vitro and if necessary, enriched before infusion into the patient. Conditioning chemoradiotherapy may be administered to lymphodeplete the patient prior to T cell infusion to enhance persistence of the infused T cells. The figure depicts an example of a general strategy for engineering autologous T cells for adoptive immunotherapy.
Figure 2. Factors to consider in the design of tumor-targeting CARs.
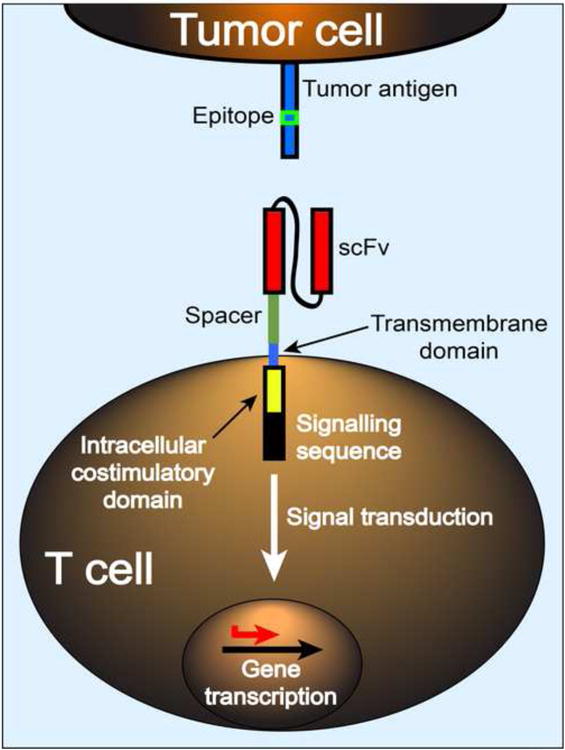
CAR specificity is governed by the selection of scFv but variation in target epitope density and location, scFv affinity, VH/VL orientation, spacer length and costimulatory signaling may influence the nature of synapse formation between the T cell and target cell, the functional efficacy of a given CAR, and effects on T cell gene transcription and epigenetic programming. Evaluation of the variables in CAR design remains empiric, and relies on in vitro assays and in vivo antitumor activity in animal models that are of uncertain value for predicting clinical efficacy.
Highlights.
Gene insertion into T cells overcomes obstacles to effective cancer immunotherapy
TCR and CAR genes enable precise targeting of tumors by distinct T cell subsets
Tumor regressions occur in patients after therapy with gene engineered T cells
T cells engineered to express high avidity receptors may cause on-target toxicity
Conditional suicide genes may improve the safety of engineered T cells
Acknowledgments
The authors acknowledge support from National Institutes of Health grants CA154608, CA13655, and CA138293, and from the Leukemia and Lymphoma Society.
Footnotes
Conflict of Interest: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480:480–489. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer:harnessing the T cell response. Nature reviews Immunology. 2012;12:269–281. doi: 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clinical cancer research an official journal of the American Association for Cancer Research. 2011;17:4550–4557. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, Robbins PF, Huang J, Citrin DE, Leitman SF, et al. Adoptive cell therapy for patients with metastatic melanomaevaluation of intensive myeloablative chemoradiation preparative regimens. Journal of clinical oncology official journal of the American Society of Clinical Oncology. 2008;26:5233–5239. doi: 10.1200/JCO.2008.16.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andersen RS, Thrue CA, Junker N, Lyngaa R, Donia M, Ellebaek E, Svane IM, Schumacher TN, Thor Straten P, Hadrup SR. Dissection of T-cell antigen specificity in human melanoma. Cancer research. 2012;72:1642–1650. doi: 10.1158/0008-5472.CAN-11-2614. [DOI] [PubMed] [Google Scholar]
- 7.Li LP, Lampert JC, Chen X, Leitao C, Popovic J, Muller W, Blankenstein T. Transgenic mice with a diverse human T cell antigen receptor repertoire. Nature medicine. 2010;16:1029–1034. doi: 10.1038/nm.2197. [DOI] [PubMed] [Google Scholar]
- 8.Stone JD, Chervin AS, Aggen DH, Kranz DM. T cell receptor engineering. Methods in enzymology. 2012;503:189–222. doi: 10.1016/B978-0-12-396962-0.00008-2. [DOI] [PubMed] [Google Scholar]
- 9.Engels B, Chervin AS, Sant AJ, Kranz DM, Schreiber H. Long-term persistence of CD4(+) but rapid disappearance of CD8(+) T cells expressing an MHC class I-restricted TCR of nanomolar affinity. Molecular therapy the journal of the American Society of Gene Therapy. 2012;20:652–660. doi: 10.1038/mt.2011.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, Kammula US, Royal RE, Sherry RM, Wunderlich JR, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535–546. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, Davis JL, Morgan RA, Merino MJ, Sherry RM, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Molecular therapy the journal of the American Society of Gene Therapy. 2011;19:620–626. doi: 10.1038/mt.2010.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wei X, Walia V, Lin JC, Teer JK, Prickett TD, Gartner J, Davis S, Stemke-Hale K, Davies MA, Gershenwald JE, et al. Exome sequencing identifies GRIN2A as frequently mutated in melanoma. Nature genetics. 2011;43:442–446. doi: 10.1038/ng.810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, Varela I, Lin ML, Ordonez GR, Bignell GR, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191–196. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *14.Scholler J, Brady TL, Binder-Scholl G, Hwang WT, Plesa G, Hege KM, Vogel AN, Kalos M, Riley JL, Deeks SG, et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Science translational medicine. 2012;4:132ra153. doi: 10.1126/scitranslmed.3003761. This manuscript demonstrates long-term persistence of T cells modified with a retrovirally encoded chimeric antigen receptor comprised of CD4 and CD3ζ and administered to patients with HIV. Encouragingly, retroviral integration site analysis showed no evidence of persistent clonal expansion or enrichment of integration sites near oncogenes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Newrzela S, Cornils K, Li Z, Baum C, Brugman MH, Hartmann M, Meyer J, Hartmann S, Hansmann ML, Fehse B, et al. Resistance of mature T cells to oncogene transformation. Blood. 2008;112:2278–2286. doi: 10.1182/blood-2007-12-128751. [DOI] [PubMed] [Google Scholar]
- 16.Bendle GM, Linnemann C, Hooijkaas AI, Bies L, de Witte MA, Jorritsma A, Kaiser AD, Pouw N, Debets R, Kieback E, et al. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nature medicine. 2010;16:565–570. doi: 10.1038/nm.2128. 561p following 570. [DOI] [PubMed] [Google Scholar]
- 17.Sommermeyer D, Uckert W. Minimal amino acid exchange in human TCR constant regions fosters improved function of TCR gene-modified T cells. Journal of immunology. 2010;184:6223–6231. doi: 10.4049/jimmunol.0902055. [DOI] [PubMed] [Google Scholar]
- 18.Kuball J, Dossett ML, Wolfl M, Ho WY, Voss RH, Fowler C, Greenberg PD. Facilitating matched pairing and expression of TCR chains introduced into human T cells. Blood. 2007;109:2331–2338. doi: 10.1182/blood-2006-05-023069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ochi T, Fujiwara H, Okamoto S, An J, Nagai K, Shirakata T, Mineno J, Kuzushima K, Shiku H, Yasukawa M. Novel adoptive T-cell immunotherapy using a WT1-specific TCR vector encoding silencers for endogenous TCRs shows marked antileukemia reactivity and safety. Blood. 2011;118:1495–1503. doi: 10.1182/blood-2011-02-337089. [DOI] [PubMed] [Google Scholar]
- *20.Provasi E, Genovese P, Lombardo A, Magnani Z, Liu PQ, Reik A, Chu V, Paschon DE, Zhang L, Kuball J, et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nature medicine. 2012 doi: 10.1038/nm.2700. Provasi et al demonstrate that mispairing of introduced and endogenous TCR α and β chains can be prevented by targeted disruption of the TRBC and TRAC genes with zinc finger nucleases. The strategy has the potential to improve the safety profile of T cells engineered to express a tumor-reactive TCR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, Wunderlich JR, Nahvi AV, Helman LJ, Mackall CL, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. Journal of clinical oncology official journal of the American Society of Clinical Oncology. 2011;29:917–924. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drake CG, Jaffee E, Pardoll DM. Mechanisms of immune evasion by tumors. Advances in immunology. 2006;90:51–81. doi: 10.1016/S0065-2776(06)90002-9. [DOI] [PubMed] [Google Scholar]
- 24.Song DG, Ye Q, Poussin M, Harms GM, Figini M, Powell DJ., Jr CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood. 2012;119:696–706. doi: 10.1182/blood-2011-03-344275. [DOI] [PubMed] [Google Scholar]
- 25.Sadelain M, Riviere I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat Rev Cancer. 2003;3:35–45. doi: 10.1038/nrc971. [DOI] [PubMed] [Google Scholar]
- 26.Brown CE, Starr R, Aguilar B, Shami AF, Martinez C, D'Apuzzo M, Barish ME, Forman SJ, Jensen MC. Stem-like Tumor-Initiating Cells Isolated from IL13Ralpha2 Expressing Gliomas Are Targeted and Killed by IL13-Zetakine-Redirected T Cells. Clinical cancer research an official journal of the American Association for Cancer Research. 2012;18:2199–2209. doi: 10.1158/1078-0432.CCR-11-1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **27.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Science translational medicine. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. The manuscripts from Kalos et al and Porter et al report impressive clinical responses in a limited cohort of patients with chronic lymphocytic leukemia who were treated with T cells modified to express a chimeric antigen receptor targeting CD19. Clinical responses and toxicity due to tumor lysis syndrome were delayed after a period of in vivo proliferation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, Stetler-Stevenson M, Phan GQ, Hughes MS, Sherry RM, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **29.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. The New England journal of medicine. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. See Kalos et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, Taylor C, Yeh R, Bartido S, Borquez-Ojeda O, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Till BG, Jensen MC, Wang J, Qian X, Gopal AK, Maloney DG, Lindgren CG, Lin Y, Pagel JM, Budde LE, et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domainspilot clinical trial results. Blood. 2012;119:3940–3950. doi: 10.1182/blood-2011-10-387969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, Rossig C, Russell HV, Diouf O, Liu E, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118:6050–6056. doi: 10.1182/blood-2011-05-354449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Molecular therapy the journal of the American Society of Gene Therapy. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Till BG, Jensen MC, Wang J, Chen EY, Wood BL, Greisman HA, Qian X, James SE, Raubitschek A, Forman SJ, et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood. 2008;112:2261–2271. doi: 10.1182/blood-2007-12-128843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park JR, Digiusto DL, Slovak M, Wright C, Naranjo A, Wagner J, Meechoovet HB, Bautista C, Chang WC, Ostberg JR, et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther. 2007;15:825–833. doi: 10.1038/sj.mt.6300104. [DOI] [PubMed] [Google Scholar]
- 36.Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, Maric I, Raffeld M, Nathan DA, Lanier BJ, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099–4102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **37.Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, Straathof K, Liu E, Durett AG, Grilley B, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. The New England journal of medicine. 2011;365:1673–1683. doi: 10.1056/NEJMoa1106152. The authors show that the safety of infusion of donor T cells into allogeneic transplant recipients can be improved by modification of the infused T cells to express an inducible caspase 9. Systemic adminstration of a small molecule dimerizing agent, AP1903, results in apoptosis of the engineered T cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kerkar SP, Goldszmid RS, Muranski P, Chinnasamy D, Yu Z, Reger RN, Leonardi AJ, Morgan RA, Wang E, Marincola FM, et al. IL-12 triggers a programmatic change in dysfunctional myeloid-derived cells within mouse tumors. The Journal of clinical investigation. 2011;121:4746–4757. doi: 10.1172/JCI58814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, Brentjens RJ. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012;119:4133–4141. doi: 10.1182/blood-2011-12-400044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang L, Feldman SA, Zheng Z, Chinnasamy N, Xu H, Nahvi AV, Dudley ME, Rosenberg SA, Morgan RA. Evaluation of gamma-Retroviral Vectors That Mediate the Inducible Expression of IL-12 for Clinical Application. Journal of immunotherapy. 2012;35:430–439. doi: 10.1097/CJI.0b013e31825898e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mackall CL, Fleisher TA, Brown MR, Andrich MP, Chen CC, Feuerstein IM, Horowitz ME, Magrath IT, Shad AT, Steinberg SM, et al. Age, thymopoiesis, and CD4+ T-lymphocyte regeneration after intensive chemotherapy. The New England journal of medicine. 1995;332:143–149. doi: 10.1056/NEJM199501193320303. [DOI] [PubMed] [Google Scholar]
- 42.Turtle CJ, Swanson HM, Fujii N, Estey EH, Riddell SR. A distinct subset of self- renewing human memory CD8+ T cells survives cytotoxic chemotherapy. Immunity. 2009;31:834–844. doi: 10.1016/j.immuni.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. The Journal of clinical investigation. 2008;118:294–305. doi: 10.1172/JCI32103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang X, Berger C, Wong CW, Forman SJ, Riddell SR, Jensen MC. Engraftment of human central memory-derived effector CD8+ T cells in immunodeficient mice. Blood. 2011;117:1888–1898. doi: 10.1182/blood-2010-10-310599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **45.Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, Almeida JR, Gostick E, Yu Z, Carpenito C, et al. A human memory T cell subset with stem cell-like properties. Nature medicine. 2011;17:1290–1297. doi: 10.1038/nm.2446. The authors identify a memory CD8+ T cell subset with stem cell characteristics and the capacity to persist after in vitro expansion and adoptive transfer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zygmunt B, Veldhoen M. T helper cell differentiation more than justcytokines. Advances in immunology. 2011;109:159–196. doi: 10.1016/B978-0-12-387664-5.00005-4. [DOI] [PubMed] [Google Scholar]
- 47.Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R, Jungbluth A, Gnjatic S, Thompson JA, Yee C. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. The New England journal of medicine. 2008;358:2698–2703. doi: 10.1056/NEJMoa0800251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martin-Orozco N, Muranski P, Chung Y, Yang XO, Yamazaki T, Lu S, Hwu P, Restifo NP, Overwijk WW, Dong C. T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity. 2009;31:787–798. doi: 10.1016/j.immuni.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hudecek M, Schmitt TM, Baskar S, Lupo-Stanghellini MT, Nishida T, Yamamoto TN, Bleakley M, Turtle CJ, Chang WC, Greisman HA, et al. The B-cell tumor-associated antigen ROR1 can be targeted with T cells modified to express a ROR1-specific chimeric antigen receptor. Blood. 2010;116:4532–4541. doi: 10.1182/blood-2010-05-283309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yamaguchi T, Yanagisawa K, Sugiyama R, Hosono Y, Shimada Y, Arima C, Kato S, Tomida S, Suzuki M, Osada H, et al. NKX2-1/TITF1/TTF-1-Induced ROR1 is required to sustain EGFR survival signaling in lung adenocarcinoma. Cancer cell. 2012;21:348–361. doi: 10.1016/j.ccr.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 51.Zhang S, Chen L, Cui B, Chuang HY, Yu J, Wang-Rodriguez J, Tang L, Chen G, BasakGW, Kipps TJ. ROR1 is expressed in human breast cancer and associated with enhanced tumor-cell growth. PloS one. 2012;7:e31127. doi: 10.1371/journal.pone.0031127. [DOI] [PMC free article] [PubMed] [Google Scholar]