

Abstract
Background
The vestibular system controls the ion composition of its luminal fluid through several epithelial cell transport mechanisms under hormonal regulation. The semicircular canal duct (SCCD) epithelium has been shown to secrete Cl- under β2-adrenergic stimulation. In the current study, we sought to determine the ion transporters involved in Cl- secretion and whether secretion is regulated by PKA and glucocorticoids.
Results
Short circuit current (Isc) from rat SCCD epithelia demonstrated stimulation by forskolin (EC50: 0.8 μM), 8-Br-cAMP (EC50: 180 μM), 8-pCPT-cAMP (100 μM), IBMX (250 μM), and RO-20-1724 (100 μM). The PKA activator N6-BNZ-cAMP (0.1, 0.3 & 1 mM) also stimulated Isc. Partial inhibition of stimulated Isc individually by bumetanide (10 & 50 μM), and [(dihydroindenyl)oxy]alkanoic acid (DIOA, 100 μM) were additive and complete. Stimulated Isc was also partially inhibited by CFTRinh-172 (5 & 30 μM), flufenamic acid (5 μM) and diphenylamine-2,2′-dicarboxylic acid (DPC; 1 mM). Native canals of CFTR+/− mice showed a stimulation of Isc from isoproterenol and forskolin+IBMX but not in the presence of both bumetanide and DIOA, while canals from CFTR−/− mice had no responses. Nonetheless, CFTR−/− mice showed no difference from CFTR+/− mice in their ability to balance (rota-rod). Stimulated Isc was greater after chronic incubation (24 hr) with the glucocorticoids dexamethasone (0.1 & 0.3 μM), prednisolone (0.3, 1 & 3 μM), hydrocortisone (0.01, 0.1 & 1 μM), and corticosterone (0.1 & 1 μM) and mineralocorticoid aldosterone (1 μM). Steroid action was blocked by mifepristone but not by spironolactone, indicating all the steroids activated the glucocorticoid, but not mineralocorticoid, receptor. Expression of transcripts for CFTR; for KCC1, KCC3a, KCC3b and KCC4, but not KCC2; for NKCC1 but not NKCC2 and for WNK1 but only very low WNK4 was determined.
Conclusions
These results are consistent with a model of Cl- secretion whereby Cl- is taken up across the basolateral membrane by a Na+-K+-2Cl- cotransporter (NKCC) and potentially another transporter, is secreted across the apical membrane via a Cl- channel, likely CFTR, and demonstrate the regulation of Cl- secretion by protein kinase A and glucocorticoids.
Keywords: Chloride secretion, Rat, Knockout mouse, Primary culture, Epithelium, Inner ear, Bumetanide, DIOA, Glucocorticoid, NKCC, KCC
Background
The inner ear controls the ion composition of its luminal fluid, endolymph, through a multiplicity of transepithelial transport mechanisms in different cell types bounding the lumen. The high-K+, low Na+, low Ca2+ endolymph composition is needed for proper auditory and vestibular function [1-3]. K+ secretion by both strial marginal cells and vestibular dark cells is stimulated by β-adrenergic receptors via cytosolic cAMP as second messenger [4,5]. Secretion of the primary anion, Cl-, is known to also be under adrenergic control in semicircular canal duct (SCCD) epithelium [6].
Cl- transport by several epithelia has been shown to be under control of a cAMP signal pathway that is mediated by apical CFTR Cl- channels via protein kinase A (PKA) [7]. Vectorial transport in those epithelia depends also on basolateral Na+-K+-ATPase and K+ channels as well as basolateral Cl transporters such as the Na+-K+-2Cl- cotransporter (NKCC1/Slc12a2) [6,8-10]. Cellular cAMP levels and cAMP-mediated processes, including ion transport, depend on cAMP metabolism regulated by the enzymatic activity of anabolic adenylyl cyclase and catabolic phosphodiesterase [11-13].
Glucocorticoids can modify cellular responses via genomic and non-genomic pathways, including regulation of ion transport processes [14-17]. Na+ absorption by SCCD has already been demonstrated to be stimulated by glucocorticoids [17,18]. There is considerable evidence in various tissues, including epithelia, that glucocorticoids have long-term (genomic) effects on cAMP metabolism and potentiate cAMP-mediated responses, including ion transport by activation of the glucocorticoid receptor [19-21].
We therefore sought to determine in SCCD a) whether there is a basal constitutive adenylyl cyclase activity driving cAMP-mediated Cl- secretion, b) whether Cl- secretion is enhanced by glucocorticoid hormones via activation of glucocorticoid receptors, c) whether Cl- secretion is mediated through PKA action and d) whether Cl- secretion is mediated by a basolateral NKCC and/or KCC and an apical CFTR Cl- channel. Our findings establish that the SCCD is a site in the inner ear for PKA-mediated Cl- transport, that this transport depends on NKCC1 and another Cl- uptake process (perhaps KCC), that apical CFTR is likely involved and that it is enhanced by glucocorticoid-receptor activation.
Results
Adenylyl cyclase activator, cAMP analogs, and phosphodiesterase inhibitors stimulate Isc
The SCCD epithelium actively contributes to endolymph homeostasis by Cl- secretion under control of β2-adrenergic receptors via a cAMP pathway [6], like other epithelia that are known to secrete Cl- upon stimulation by β-adrenergic receptor activation [22-26]. Forskolin (adenylyl cyclase activator) [6] (Figure 1A & B), cell permeable cAMP analogs (8-Br-cAMP and 8-pCPT-cAMP [100 μM]) (Figure 2A & B), the non-selective phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (IBMX; 250 μM) (Figure 2C), and the cAMP-specific phosphodiesterase-4 (PDE4) inhibitor RO-20-1724 (100 μM) (Figure 2D) increased Isc in the presence of apical amiloride (10 μM), an inhibitor of the epithelial Na+ channel. The maximal forskolin-stimulated Isc was 0.58 ± 0.06 μA/cm2 (n=38) (Figure 1B). In the present series of experiments (Figures 2B,C,D), amiloride produced no significant changes in Isc in the absence of steroids, although in a previous larger series of experiments there was a small (15%) but significant decrease in Isc[17]. The initial transepithelial resistance (Figure 1B) was 4.7 ± 0.6 kΩ-cm2 (n=3) and decreased significantly with 30 μM forskolin to 3.1 ± 0.3 kΩ-cm2 (n=3).
Figure 1.

Stimulation of equivalent short circuit current (Isc) by the adenylyl cyclase activator forskolin. (A) Representative recording of VT from primary cultures of SCCD epithelium with the effect of forskolin (0.01 to 10 μM) added cumulatively on both sides at arrows. Downward deflections result from transepithelial current pulses and are proportional to transepithelial resistance. B) Summary of concentration-response of Isc to forskolin (FSK) stimulation (n = 3–5), EC50 = 0.8 μM and Hill coefficient 0.9. Summary data are mean ± SEM; error bars are smaller than symbols.
Figure 2.

Membrane-permeable cAMP analogs and phosphodiesterase inhibitors increase Isc. A) Summary of concentration-response of Isc to 8-Br-cAMP (n = 3–4) on both sides after prior application of 10 μM apical amiloride, EC50 = 180 μM and Hill coefficient 3.0. B) Summary of response of Isc to 8-pCPT-cAMP (8-pCPT; 100 μM; n = 4) on both sides in the presence of apical amiloride (10 μM); no further stimulation by subsequent forskolin (FSK, 10 μM). C) Summary of response of Isc to 3-isobutyl-1-methylxanthine (IBMX; 250 μM; n = 3) on both sides and D) RO-20-1720 (RO-20; 100 μM; n = 3) on both sides after prior application of apical amiloride (10 μM). Summary data are mean ± SEM; *, P < 0.05; NS, not significant; compared to bar immediately to the left.
The lipid-soluble drugs forskolin, 8-pCPT-cAMP, RO-20-1724, 3-isobutyl-1-methylxanthine (IBMX), were added to both the apical and basolateral baths. Amiloride was added to only the apical side and bumetanide to the basolateral side. Amiloride had no significant effect on Isc, whereas subsequent addition of both forskolin (Figure 1) and 8-Br-cAMP (Figure 2A) increased Isc in a concentration dependent manner with an EC50 of about 0.8 μM and 180 μM respectively. Forskolin showed no additional effect after prior stimulation by either 8-pCPT-cAMP (100 μM) (Figure 2B) or by RO-20-1724 (100 μM) (Figure 2D), demonstrating constitutive activity of adenylyl cyclase in SCCD epithelium.
Glucocorticoids increase forskolin-stimulated Isc
We investigated whether forskolin-stimulated Cl- secretion was altered by glucocorticoid treatment (24 hr). As in the absence of dexamethasone, increasing intracellular cAMP in dexamethasone-treated epithelia by exposure to 8-pCPT-cAMP in the presence of amiloride (10 μM) led to an increased Isc (representative recording in Figure 3). Similar responses were seen with forskolin (10 μM), 8-Br-cAMP (100 μM) and IBMX (250 μM) (data not shown). The glucocorticoid-stimulated Na+ absorption via apical sodium channels (ENaC) was blocked by amiloride, which decreased Isc by 81 – 92% [17]; the remaining current was due to Cl- secretion [6].
Figure 3.

Stimulation of Cl-secretion by cAMP after exposure to dexamethasone. Representative trace of response of VT to apical amiloride and the membrane-permeable cAMP analog 8-pCPT-cAMP on both sides after incubation with dexamethasone (100 nM, 24 h).
The concentration-dependence of natural and synthetic glucocorticoids was determined (Figure 4). Interestingly, the stimulation by forskolin was significantly greater after treatment with 100 or 300 nM dexamethasone, as observed previously with single concentrations of dexamethasone and forskolin [17]. Similarly, the stimulation of Isc by forskolin was significantly greater after 24 hr treatment with the other glucocorticoids (hydrocortisone, corticosterone, and prednisolone) and the mineralocorticoid aldosterone in the continued presence of amiloride (Figure 4). The transepithelial resistance was significantly reduced by about one third after exposure to effective concentrations of glucocorticoids (ANOVA analysis of Table two in [17]), as would be expected after insertion of a conductive pathway (epithelial sodium channels) in the apical membrane.
Figure 4.

Response of forskolin-stimulated Iscto corticosteroids. Summary data of responses to forskolin (10 μM) after incubation (24 h) in steroids or steroids + receptor antagonists at the concentrations shown. A) dexamethasone (Dex) ± spironolactone (Spi) or mifepristone (mif); B) prednisolone (Pred); C) corticosterone (Cort); D) hydrocortisone (HC) ± spironolactone (Spi) or mifepristone (mif); E) aldosterone (Aldo) ± spironolactone (Spi) or mifepristone (mif); F) progesterone (Prog). The horizontal dashed lines show the reference levels for statistical comparisons.
The natural and synthetic glucocorticoids stimulated within their respective physiologic and therapeutic ranges [17], while aldosterone was only effective at concentrations much higher than found under normal physiologic conditions [17].
Glucocorticoids increase forskolin-stimulated Isc by activation of glucocorticoid receptor
We investigated whether dexamethasone, hydrocortisone, and aldosterone increased FSK-stimulated Isc by activation of glucocorticoid receptors and/or mineralocorticoid receptors. SCCD epithelia were incubated in the presence of dexamethasone (100 nM), hydrocortisone (1 μM) or aldosterone (1 μM) alone or in the presence of receptor antagonists. Mifepristone significantly reduced the effects of dexamethasone, hydrocortisone and aldosterone (Figure 4A,D,E), consistent with action of all of these corticosteroids at the glucocorticoid receptor. Mifepristone is also known to be an antagonist of the progesterone receptor; however, progesterone (10–1000 nM) had no effect on forskolin stimulation (Figure 4F). The mineralocorticoid receptor antagonist spironolactone had no significant effect on dexamethasone-, hydrocortisone- or aldosterone-treated epithelia (Figure 4A,D,E), consistent with a lack of involvement of mineralocorticoid receptor in the increase of the FSK-stimulated Isc in these cells. These findings suggest that corticosteroids increase forskolin-stimulated Isc (cAMP-mediated Cl- secretion) solely by activation of glucocorticoid receptors in SCCD epithelia.
PKA activator stimulates Isc
It is known that cAMP-dependent Cl- secretion in many epithelia is mostly mediated through activation of protein kinase A (PKA) [7]. We therefore investigated whether PKA activation increases Isc in SCCD. Indeed, apical and basolateral addition of PKA activator N6-BNZ-cAMP stimulated Isc at 30, 100, 300 and 1000 μM after prior inhibition of Na+ transport with apical amiloride (10 μM) (Figure 5).
Figure 5.

PKA activation of Isc. Summary of response of Isc to the PKA activator N6-BNZ-cAMP (n = 3) after treatment with dexamethasone in the presence of apical amiloride (10 μM). Summary data are mean ± SEM; *, P < 0.05; compared to bar prior to addition of activator. Note a break in the Y-axis.
Blockers of basolateral Na+-K+-2Cl- and K+-Cl- cotransporters inhibit forskolin and/or forskolin + IBMX-stimulated Isc
We have previously shown that both Ba2+-sensitive K+ channels and ouabain-sensitive Na+-K+-ATPase are involved in Cl- secretion by SCCD [6]. However, the participation of Na+-K+-2Cl- and K+-Cl- cotransporters is not known. Therefore, we investigated whether these transport proteins are involved in cAMP-mediated Cl- secretion. Untreated and dexamethasone (100 nM; 24 hr)-treated SCCD epithelia were stimulated with apical and basolateral forskolin and forskolin + IBMX respectively, followed by application of blockers of ion transporters to the basolateral side. Na+-K+-2Cl- cotransporter inhibitor bumetanide (50 μM) partially decreased the magnitude of forskolin (5 μM) + IBMX (125 μM)-stimulated Isc (Figure 6A). Bumetanide (10 & 50 μM) also decreased the forskolin (10 μM)-stimulated Isc by 20 ± 2% (10 μM; n = 8) and 18 ± 5% (50 μM; n = 3) (data not shown). Similarly, the K+-Cl- cotransporter blocker [(dihydroindenyl)oxy]alkanoic acid (DIOA, 100 μM) partially inhibited the forskolin + IBMX-stimulated Isc (Figure 6B). Interestingly, DIOA and bumetanide, when added to the bath cumulatively, completely inhibited all of the forskolin + IBMX-stimulation, returning Isc to the level observed with amiloride (Figure 6C). These findings are consistent with the presence of Na+-K+-2Cl- and K+-Cl- cotransporters at the basolateral membrane and their participation in Cl- secretion by SCCD epithelium, although DIOA is lipophilic and could cross to a KCC on the apical membrane. However, the observed effect is not likely a result of action of DIOA on an apical KCC since transport of Cl- by an apical KCC would also result in a similar rate of K+ secretion, and no transepithelial K+ (Rb+) flux was detected from cAMP-stimulated SCCD [6].
Figure 6.

Inhibition of forskolin-stimulated Iscby bumetanide and DIOA. A) Summary of inhibition by basolateral bumetanide (BUM) (n = 11) of cAMP-stimulated Isc (forskolin, FSK + IBMX). Epithelia were incubated in dexamethasone and Na+ transport was inhibited with apical amiloride before stimulation by cAMP. Note that Isc does not return after only bumetanide to the basal Cl- secretion rate in amiloride (dotted line). B) Representative recording of VT and the response to apical amiloride forskolin + IBMX, basolateral DIOA and basolateral bumetanide (BUM). C) Summary of response of Isc to DIOA and bumetanide (BUM) (n = 3) after stimulation by forskolin+ IBMX. Na+ transport was inhibited with apical amiloride before stimulation. Note that Isc returns after both DIOA and bumetanide to the basal Cl- secretion rate in amiloride (dotted line). Summary data (A and C) are mean ± SEM; *, P < 0.05 compared to bar immediately to the left; NSA, not significantly different from amiloride.
Expression of KCC and WNK isoforms
Additive inhibition of Isc by bumetanide and DIOA suggested dependence of electrogenic Cl- transport in SCCD on expression of NKCC and KCC. The basolateral isoform of NKCC, (NKCC1/Slc12a2 Affymetrix probe set 1367853_at), was shown to be present in a gene array of SCCD and the apical isoform (NKCC2/Slc12a1 Affymetrix probe set 1368548_at) was absent [27]. Not all isoforms of KCC were identified on the gene chips; we tested for expression of transcripts with RT-PCR and observed expression of KCC1, KCC3a, KCC3b and KCC4; but, KCC2 was absent (Table 1).
Table 1.
RT-PCR demonstration of the presence of KCC1, KCC3a, KCC3b, KCC4 and absence of KCC2 in rat SCCD primary cultures
| Ct, average | ||||||
|---|---|---|---|---|---|---|
| Tissue |
18S |
KCC1 |
KCC2 |
KCC3a |
KCC3b |
KCC4 |
| rSCCD |
|
26.4 |
BT |
25.0 |
27.3 |
26.7 |
| rat kidney |
14.2 |
25.9 |
-- |
23.3 |
24.5 |
26.3 |
| rat brain |
-- |
-- |
22.6 |
-- |
-- |
-- |
| blank | 35.0 | 30.2 | BT | BT | 32.3 | 35.8 |
BT, Below Threshold fluorescence at end of 38 thermocycles. Ct values are based on duplicate runs of 4 samples SCCD and duplicates of control tissue total RNA. The difference between Ct values of duplicates varied by 0.3 ± 0.04 (n=26) with a range from 0.0 to 0.8.
It is known that genetic mutations in the kinase WNK1 and WNK4 cause a disease featuring hypertension and hyperkalemia and the etiology appears to be related to regulation of NKCC and KCC (reviewed in [49]). We observed that SCCD expresses WNK1 but only very low or absent WNK4 (Table 2).
Table 2.
RT-PCR demonstration of the presence of WNK1 and absence of WNK4 in rat SCCD primary cultures
| Tissue | 18S | WNK1 | WNK4 |
|---|---|---|---|
| rSCCD |
13.4 |
24.7 |
31.6 |
| rat kidney |
14.7 |
25.0 |
26.0 |
| blank | 34.4 | BT | 35.2 |
Ct, average. BT, Below Threshold fluorescence at end of 38 thermocycles. Ct values are based on duplicate runs of 4 samples SCCD and duplicates of rat kidney as a control tissue. The difference between Ct values of duplicates varied by 0.6 ± 0.09 (n=36) with a range from 0.0 to 2.1.
Blockers of apical Cl- transport inhibit forskolin and/or forskolin + IBMX-stimulated Isc
Cl- secretion across the apical membrane in many epithelia is mediated by the CFTR Cl- channel [8,10]. Na+ currents were blocked by amiloride (100 μM) in dexamethasone (100 nM, 24 hr)-treated SCCD epithelia. Isc was then stimulated with apical and basolateral forskolin (5 μM) + IBMX (125 μM), followed by apical addition of blockers of Cl- transporters. Non-selective Cl- channel inhibitors DPC (1 mM) and flufenamic acid (5 μM) (Figure 7A) partially inhibited the stimulated Isc. The CFTR Cl- channel blocker CFTRinh-172 (5 μM or 30 μM) [28] partially inhibited the forskolin (10 μM) and forskolin (5 μM) + IBMX (125 μM)-stimulated Isc in both the absence and presence of dexamethasone (100 nM, 24 hr; Figure 7B, left and right panels). Taken together, these results suggest that CFTR Cl- channels are functionally expressed at the apical membrane and account for at least part of the Cl- secretion by SCCD epithelium, although CFTR appears to not be essential for vestibular function under normative conditions (see below). The observation of the presence of mRNA transcripts of CFTR in gene arrays of rat primary cultures (Affymetrix Probe set 1384960_at [27]) is consistent with that interpretation.
Figure 7.

Partial inhibition of Iscin rat primary cultures by blockers of CFTR. A) Diphenylamine-2,2′-dicarboxylic acid (DPC) and flufenamic acid (FFA) significantly reduced cAMP-stimulated Isc. B) CFTRinh-172 significantly reduced cAMP-stimulated Isc in both the absence and presence of dexamethasone. FSK, forskolin; CF, CFTRinh-172. *, P < 0.05.
SCCD from CFTR+/− mice showed an increase in response of Isc to a β2-adrenergic receptor agonist isoproterenol (100 nM) and a mixture of forskolin and IBMX (Figure 8, top panel). Heterozygous CFTR mice are known to have an ion transport profile similar to wild-type mice [29]. A mixture of DIOA (KCC inhibitor) and bumetanide (NKCC inhibitor) completely inhibited the increased Isc by forskolin + IBMX (Figure 8, top panel). This result is consistent with the results shown above in rat SCCD primary cultures (Figure 6C). On the other hand, SCCD from CFTR−/− mice lacked response to both isoproterenol (10 μM) and a mixture of forskolin and IBMX, suggesting that all of the cAMP-stimulated Isc is mediated by, or dependent on, CFTR in mouse SCCD (Figure 8, top panel). The vestibular functional phenotype, as assessed by Rota-Rod, of the CFTR−/− mice did not differ significantly from the CFTR+/− mice (Figure 8, bottom panel), in spite of the profound difference in stimulated Isc (above).
Figure 8.

Short circuit current and balance in CFTR knockout mice. (top panel) Vibrating Probe recordings of Isc from native SCCD from heterozygote (CFTR+/−) and CFTR knockout (CFTR−/−) mice (n=3 each). ISO, Isoproterennol (10 μM); F, Forskolin; I, IBMX; D, DIOA; B, Bumetanide. The control bath solution contained Amiloride (10 μm). Perfusion was returned to control solution after isoproterenol. *, significant change in current by treatment; P < 0.05. NS, not significant. (bottom panel) Balance function of heterozygote (CFTR+/−) and CFTR knockout (CFTR−/−) mice was tested with a rota-rod apparatus. No significant difference was found between the two groups (n=5 each).
Discussion
Sensory transduction of acceleration in the vestibular labyrinth is mediated by modulation of K+ currents through sensory cells, where the K+ originates from the high-[K+] luminal fluid. Secretion of K+ is controlled by β-adrenergic stimulation (among other signal pathways) [30,31] and it is to be expected that secretion of the primary anion would also be regulated by the same agonists. Movements of anions would be expected to be transcellular since the paracellular pathway must be extremely tight in this epithelium to support the large ion concentration gradients. Cl- secretion by the semicircular canal duct (SCCD) in the vestibular labyrinth is stimulated by cAMP as second messenger [6] and similar mechanisms have been proposed in the cochlea [32,33], although the cell types responsible in the cochlea have not yet been unambiguously determined (see [34]).
The SCCD is an epithelial domain that has a high ratio of surface area to endolymph volume and would therefore be a strong candidate for a site of effective ion homeostasis. Indeed, it was recently shown that these cells also absorb Na+ via the epithelial sodium channel (ENaC) under glucocorticoid receptor control [17,18] and absorb Ca2+ via an epithelial Ca2+ channel [35], in addition to their role in Cl- secretion.
We determined in the present study that cAMP acts via PKA, whose target may be an apical CFTR Cl channel [6]. Evidence supporting the involvement of CFTR include inhibition of Cl- secretion (cAMP-stimulated Isc) by the poorly-specific inhibitors flufenamic acid and DPC and by the specific inhibitor CFTRinh-172. Partial inhibition in rSCCD by CFTRinh-172 is consistent with reports of significant but only partial inhibition in avian proximal tubule at 20 μM [36]. In addition, mRNA message for CFTR was found to be present in the rat primary cultures of SCCD (GEO database, Accession GSE6196, [27]). The observation of cAMP-stimulated Isc in mouse canals extends the findings in gerbil and rat [6] to another rodent species. The absence of cAMP-stimulated Isc in CFTR knockout mice is consistent with an essential role of CFTR in canal Cl- secretion, although there is no strict proof ruling out the unlikely occurrence of dissection damage to only the knockout mouse canals.
Interestingly, there is no correlation of deafness in persons with dysfunctional CFTR (cystic fibrosis) [37,38] and no reports of vertigo in this population. Our results with vestibular tests of CFTR knockout mice are consistent with that observation. It may be, however, that this anion transport system is not by itself essential for normal inner ear function, but may be necessary in times of systemic stress when β-adrenergic agonist levels are elevated, but this proposition remains to be tested.
The results here demonstrated increased Isc by stimulation of the cAMP signal pathway in several ways: through increased cAMP production (forskolin stimulation of adenylyl cyclase), addition of exogenous cAMP analogs (8-Br-cAMP and 8-pCPT-cAMP), and inhibition of cAMP catabolism (IBMX and RO-20-1724 inhibition of phosphodiesterase). The action of all of these agents has been well-documented in other cells [11-13,19-21]. Increased Cl- secretion by inhibition of phosphodiesterase implies that the adenylyl cyclase is constitutively active in the SCCD epithelium in the absence of β-adrenergic agonists, and is therefore consistent with the earlier observation of constitutive low-level Cl- secretion [6]. Stimulation of Cl- secretion has been reported in mouse jejunum, guinea pig distal colon, T84 monolayers and human colonic epithelial cells by phosphodiesterase inhibition [39-42]. Of particular interest is the finding that phosphodiesterase inhibition did not elevate Cl- secretion in the absence of exogenous stimulation of adenylyl cyclase in T84 epithelial monolayers [41]. This is in contradistinction to our observations in rat semicircular canal primary cultures (vide infra and [6]).
The responsiveness of adenylyl cyclase and phosphodiesterase to multiple physiological challenges is related to the presence of multiple families of isoforms with tissue-specific localization [11-13]. Ten isoforms of adenylyl cyclase (AC; membranous AC1-9 and soluble AC) have been identified in mammalian cells [11]. In SCCD, the non-selective adenylyl cyclase activator forskolin, which is known to activate all identified adenylyl cyclase isoforms with varying sensitivity [11,43,44], stimulated Cl- secretion. Gene array results showed expression of transcripts for AC2 and AC4 in SCCD among the genes that were tested (AC2, AC3, AC4, AC5, AC6, and AC8) using Affymetrix Rat Genome 230 2.0 Array chips [27]. However, it is not known whether other isoforms that were not represented on the gene array are expressed in SCCD.
Eleven families of phosphodiesterases (PDE1 to PDE11) have been identified in mammalian cells, with each family having several isoforms [13]. Phosphodiesterases show three types of substrate specificity [13]. The PDE4, PDE7, and PDE8 families hydrolyze cAMP specifically. PDE5, PDE6, and PDE9 are cGMP specific and PDE1, PDE2, PDE3, PDE10, and PDE11 are dual substrate phosphodiesterases i.e. they hydrolyze both cAMP and cGMP. In SCCD, both the non-selective phosphodiesterase inhibitor IBMX and the selective PDE4 inhibitor RO-20-1724 [19] stimulated Cl- secretion. Gene array results showed expression of the transcripts for PDE4 and PDE7 isoforms but not PDE8 in SCCD [27]. Expression of PDE4 at the transcript level is consistent with RO-20-1724 stimulation of Isc.
Glucocorticoids are known to affect cAMP metabolism and potentiate cAMP-mediated responses, including salt and water transport by activation of the glucocorticoid receptor [16,19-21]. It is known that glucocorticoids stimulate anion transport, including Cl- in mammalian ileal mucosa, by increasing the concentration of cyclic nucleotides [16]. Glucocorticoids elevate cellular cAMP levels by regulating the activity of both adenylyl cyclases and phosphodiesterases, predominantly by suppressing the activity of phosphodiesterases [19,20]. It is not known if glucocorticoids increase cAMP-mediated Cl- secretion in SCCD epithelium by a similar mechanism.
Glucocorticoids increase cAMP-mediated Cl- secretion in SCCD epithelium by activation of exclusively the glucocorticoid receptor. However, the exact mechanism of potentiation of cAMP response is not known. The stimulation of cAMP-mediated Isc could not have been a result of the increase in glucocorticoid-activated ENaC function [17], since increased Na+ conductance would depolarize the cell, which in turn would reduce the outward driving force for Cl- across the apical membrane, in contrast to observations. In addition, Na+ absorption was blocked by amiloride in these experiments. Another putative mechanism of action of glucocorticoids in SCCD is regulation of NKCC expression, as observed in vascular smooth muscle and trabecular meshwork cells [45,46]. However, there was no change in expression of NKCC1 in SCCD after incubation with 100 nM dexamethasone for 24 h [27]. The molecular mechanism for glucocorticoid-increased cAMP-mediated Cl- secretion in SCCD epithelium has not yet been resolved.
One pathway of corticosteroid action is via the kinases SGK1 and WNK1 [47] among other kinases involved in regulation of ion transport such as SPAK and OSR1 [48]. Our gene array of rat SCCD cultures [27] showed SPAK or Stk39 to be present (1387059_at based on Genbank NM_019362) and minimally down-regulated 1.2-fold by dexamethasone. There were no probe sets identified on the chips for OSR1. WNK1 is downstream from SGK1 and is implicated in the control of both NKCC and KCC transporters and thereby their roles in Cl- transport [49]. The expression of WNK1 in the SCCD may therefore be involved in the regulation of Cl- secretion in SCCD through these two transporters. The transcript expression for WNK4 was nearly absent.
Complete additive inhibition of the cAMP-stimulated Isc by bumetanide and DIOA is consistent with the participation of both Na+-K+-2Cl- and K+-Cl- cotransporter in Cl- cotransport. The concentration of bumetanide used fully inhibits NKCC1 but has no effect on KCC1 and KCC4 [50,51]. Similarly, DIOA is a potent well-established KCC transport inhibitor (IC50 = 10–40 μM) that is without effect on NKCC [52,53] and has been used as a specific KCC inhibitor in the range 30 – 100 μM [53-55]. However, DIOA has also been observed in several epithelia to have diverse effects [56-59].
The canonical interpretation that the effect of DIOA is a result of inhibition of a KCC, however, is problematic. Foremost, the KCC would be operating “backwards” from its most-commonly observed mode and if this were indeed the case it would necessitate an extremely low intracellular [Cl-]. A low intracellular concentration of the secreted ion species (Cl-) would not favor a strong efflux across the apical membrane. Although most cells operate KCC transporters as cellular efflux pathways, Payne calculates that some neuronal cells have sufficiently-low intracellular [Cl-] to thermodynamically drive influx of KCl [60]. The membrane potential, intracellular [K+] and [Cl-] of SCCD epithelial cells is not known, so it is not possible at this time to evaluate whether these cells are able to support concurrent basolateral KCC electroneutral influx and apical electrogenic Cl- secretion. The discussion by Payne [60] shows that at the normal extracellular [K+] of about 4 mM, the intracellular [Cl-] would need to be about 5 mM or less; a highly unlikely constellation. An elevation of extracellular [K+] at the canal epithelium due to hair cell stimulation is also highly unlikely since the hair cells are located remotely from the canals and excess K+ would be removed from perilymph by vestibular dark cells recycling the K+ back into the lumen.
The observation that basolateral bumetanide causes only a partial inhibition of cAMP-stimulated Cl- secretion at 50 μM suggests the existence of a parallel Cl- uptake mechanism. Block of the remaining secretion by DIOA points to a KCC, subject to the caveats given above. An alternative explanation is that DIOA is not a specific inhibitor for KCC in SCCD and either blocks another Cl- uptake mechanism that operates under thermodynamically-favorable conditions or acts broadly on active transport through inhibition of metabolic processes. This last suggestion has been observed with another lipid-soluble weak acid, the Cl- channel blocker NPPB, restricting its use to <100 μM [61]. In addition, DIOA has been shown to interfere with ATP recovery after metabolic insult and to release cytochrome c from isolated mitochondria at the concentration used here [62]. Nonetheless, if this is the basis for action of DIOA in the semicircular canal epithelium, the metabolic interference is not complete (Figure 6) and the likely involvement of a parallel Cl- uptake pathway in rat canal remains.
In summary, our results are consistent with the following cell transport model of regulated Cl- secretion by SCCD (Figure 9). A basolateral Na+-K+-ATPase removes Na+ from the cell and brings in K+, which is energized by ATP. The resulting Na+ concentration gradient drives the basolateral influx of K+ and Cl- along with the recirculating Na+ ions via the bumetanide-sensitive NKCC1. Cl- would be secreted across the apical membrane through Cl- channels that may include CFTR as a channel or transport modulator. The rate of secretion is controlled by glucocorticoids and β2-adrenergic signalling via cAMP and PKA.
Figure 9.
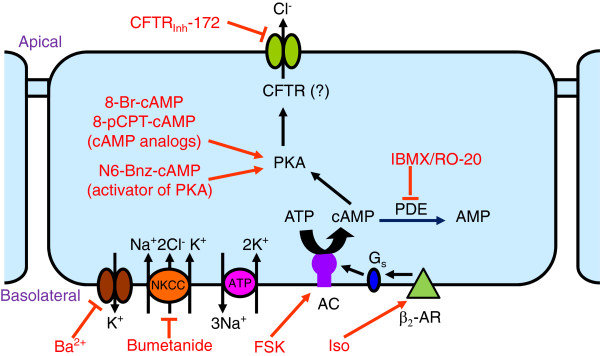
Proposed cell model for cAMP/PKA-mediated Cl- secretion by SCCD epithelium. Cl- is taken up across the basolateral membrane by NKCC (Na+-K+-2Cl- cotransporter). A Na+-K+-ATPase and Ba2+-sensitive K+ conductance in the basolateral membrane is thought to create a negative cell membrane potential that drives Cl- exit across the apical membrane via a Cl- channel, likely CFTR. Cl- secretion is stimulated by PKA from activation of a basolateral β2-adrenergic receptor (β2-AR; activated by agonists such as isoproterenol, ISO) coupled to a cAMP second messenger pathway (Gs-protein activation of adenylyl cyclase (AC) which can also be directly activated by forskolin (FSK). Phosphodiesterase (PDE) catabolizes cAMP and can be inhibited by IBMX and RO-20-1720 (RO-20). The cAMP-dependent Cl- secretion pathway is further stimulated by activation of intracellular glucocorticoid receptors (not shown) after chronic exposure to glucocorticosteroids.
Conclusions
The rat semicircular canal duct epithelium expresses three major ion transport systems, including a β2-adrenergic receptor-stimulated Cl- secretion [6]. We have demonstrated here that Cl- secretion is under control of a PKA-mediated signal pathway that depends on basolateral Cl- uptake via NKCC and (KCC?) transporters and that it is enhanced by activation of glucocorticoid receptors. Interestingly, NKCC does not appear to contribute to ENaC-mediated Na+ absorption [17] in the same cells. The role of the β2-adrenergic receptor/cAMP pathway in inner ear function remains unknown but is likely activated under stress.
Methods
All experiments were performed on primary cultures of rat SCCD, except the measurements of short circuit current from native canals and vestibular function that were performed on CFTR mutant mice.
Cell cultures of rat SCCD epithelium
Primary cultures of neonatal Wistar rat SCCD epithelium were produced as described previously [6]. Briefly, animals were anesthetized and sacrificed according to a protocol approved by the Kansas State University Institutional Animal Care and Use Committee. SCCD epithelial cells were microdissected from isolated temporal bones. Isolated cells, exclusive of connective tissue and common crus, were seeded on 6.5 mm diameter permeable (4 μm) Costar (#3470) Transwell supports and cultured in DMEM/F12 medium (Invitrogen 12500–062; http://www.invitrogen.com/site/us/en/home/support/Product-Technical-Resources/media_formulation.60.html) supplemented with 5% fetal bovine serum, 26.8 mM NaHCO3-, penicillin (100 U/ml) and streptomycin (100 μg/ml). Cultures treated with steroids, in the presence and absence of antagonists, were exposed for 24 h unless otherwise stated.
Reagents
Amiloride (#A-7410, Sigma), forskolin (#F-6886, Sigma), spironolactone (#S-3378, Sigma), mifepristone (#M-8046, Sigma), corticosterone (#C-2505, Sigma), prednisolone (#6004, Sigma), bumetanide (#B-3023, Sigma), RO-20-1724 (#557502, Calbiochem), 3-isobutyl-1-methylxanthine (IBMX; #I-7018, Sigma), flufenamic acid (#F-9005, Sigma), CFTR inh-172 ([28](gift from Dr. Bruce Schultz), and [(dihydroindenyl)oxy]alkanoic acid (DIOA; # D-129, Sigma) were dissolved in DMSO. Progesterone (#P-8783, Sigma), and aldosterone (#215360050, Acros Organics, New Jersey) were dissolved in absolute ethanol. DMSO or ethanol alone had no effect on electrical parameters in Ussing chamber experiments.
Cyclodextrin-encapsulated dexamethasone (#D-2915, Sigma), hydrocortisone (#H-0396, Sigma), isoproterenol (#I-6504, Sigma), 8-Br-cAMP (#203800, Calbiochem), 8-pCPT-cAMP (#C-3912, Sigma), and N6-Benzoyl-Adenosine 3′,5′-cyclic Monophosphate, Sodium Salt (N6-BNZ-cAMP; #116802, Calbiochem) were dissolved in water.
Mice breeding and maintenance
CFTR heterozygous parents in a C57BL/6 background were bred and the offspring were genotyped for CFTR+/+, CFTR+/− or CFTR−/− All the animals were maintained on Colyte in water to avoid intestinal impaction [63].
Dissection of mouse SCCD
CFTR+/− and CFTR−/− mice (34–39 days old) [63] were anesthetized with sodium pentobarbital (50–100 mg/kg; i.p.) and sacrificed under a protocol approved by the Institutional Animal Care and Use Committee of Kansas State University. The temporal bones were removed and SCCD without common crus were dissected in HEPES-buffered saline. The SCCD were mounted in a 200 μl perfusion chamber on the stage of an inverted microscope (Nikon ECLIPSE TE 300) and continuously perfused at 37°C with HEPES-buffed solution at an exchange rate of 180 μl/sec.
Electrophysiological measurements
Isc from Primary cultures of rat SCCD
SCCD epithelia were bathed in HEPES-buffered solution equilibrated with air. The composition was (in mM) 150 NaCl, 3.6 KCl, 1 MgCl2, 0.7 CaCl2, 5 glucose, and 10 HEPES, pH 7.4. Transepithelial voltage (VT) and resistance (RT) were measured from confluent monolayers of SCCD in an Ussing chamber under current clamp (ΔI=1 μA) mode as described previously [17,18]. The equivalent Isc was calculated from Isc = VT/RT. Isc was measured either in the absence of steroids (Figure 1, 2, 7B left) or after 24 h incubation with steroid. Steroid-stimulated Na+ current was blocked with amiloride before measurement of cAMP-stimulated Isc. Cytosolic cAMP levels were raised by stimulation of adenylyl cyclase with forskolin, block of phosphodiesterase with IBMX or RO-20-1724 or a combination of forskolin and IBMX, as indicated. The lipid-soluble drugs dexamethasone, hydrocortisone, aldosterone, forskolin, 8-pCPT-cAMP, N6-BNZ-cAMP, spironolactone, mifepristone, corticosterone, prednisolone, progesterone and [(dihydroindenyl)oxy]alkanoic acid (DIOA) were added to both the apical and basolateral baths. Amiloride was added to only the apical side and bumetanide to the basolateral side.
Isc from mouse native canal ducts; Voltage-sensitive vibrating probe recordings
The vibrating probe technique was identical to that previously described [34,64]. Briefly, the current density was monitored from mouse SCCD by vibrating (200–400 Hz) a platinum-iridium wire microelectrode with a platinum-black tip positioned 20 μm from the basolateral surface of the SCCD with computer-controlled stepper-motor manipulators (Applicable Electronics, Forest Dale, MA) and probe software (ASET version 2.00, Science Wares, East Falmouth, MA). The bath references were 26-gauge Pt-black electrodes. The phase-locked signal from the electrode was detected by phase-sensitive amplifiers, digitized y (0.5 Hz, 16 bit), and the output was expressed as current density at the electrode. HEPES-buffered solution was used as the bath.
Vestibular function test
4 sets of littermate-matched CFTR+/− and CFTR−/− mice (mean age = 56 d) were tested 4 times/day (with a five minute rest between tests) over five days. The RotaRod (Series 8, IITC, Woodland Hills, CA) used a 32 mm diameter rod and was programmed to start at 4 RPM, increasing to 40 RPM over 1 minute. All mice were placed on the rod before rotation was initiated and the time until falling was recorded. The instrument was modified by placing a landing cushion on the platform of the timer stop switch. The daily mean time for each mouse was used to calculate the mean and standard error of each group. After the last run of each day, all homozygous mice were given a 1 ml saline injection to prevent dehydration.
RNA isolation and RT-PCR
Total RNA was extracted from SCCD primary cultures using RNeasy Micro Kit and the quality and quantity were determined as described previously [18]. RT-PCR was performed on total RNA as described previously using a One Step RT-PCR Kit following the manufacturer’s protocol (#210210, Qiagen) [18].
Gene-specific primers for KCC and WNK isoforms were based on GenBank sequences (Table 3) and were verified to amplify the intended targets. Reverse transcription (RT) was performed on 10 ng of total RNA for 30 min at 50°C and 15 min at 95°C. RT was followed by 38 PCR cycles. Each PCR cycle consisted of 95°C for 1 min, 55°C for 1 min and 72°C for 1 min. To exclude the possibility of genomic DNA amplification during the PCR reaction, RT negative controls were performed (−RT). PCR products were run on 2% agarose gels and detected by ethidium bromide. Purified PCR products were sequenced to verify the identity of the RT- PCR products.
Table 3.
Gene-specific primers
| Gene | Primers 5′—3′ | GenBank # (species) | Amplicon size (bp) | Source |
|---|---|---|---|---|
| 18S |
S: GAG GTT CGA AGA CGA TCA GA |
BK000964 (many) |
316 |
[65] |
| AS: TCG CTC CAC CAA CTA AGA AC | ||||
| KCC1 |
S: GTT CGC CTC ACT CTT CCT GGC |
U55815 (rat) |
419 |
[66] |
| AS: TGG GCC ACC ACA TAC AGG GA | ||||
| KCC2 |
S: CAT CAC AGA TGA ATC TCG GG |
U55816 (rat) |
213 |
[66] |
| AS: TTC TCT GGG TCT GTC TCC C | ||||
| KCC3a |
S: CCT CGC CTC CTC ACC TTT GC |
AF211854 (mouse) |
284 |
This study |
| AS: TCA CTC TGA CGC CAG CCA TTG | ||||
| KCC3b |
S: AGT AAA AGC CCG GAT TCA GG |
AF211855 (mouse) |
330 |
[66] |
| AS: ATG AAA GTA CCC ATT TGG GG | ||||
| KCC4 |
S: AGG AAG CTG CTG AGC GCA C |
AF087436 (mouse) |
443 |
[66] |
| AS: CAG CAT TGT ACA GGT GCA GC | ||||
| WNK1 |
S: ACC AGA AAG CCT CAT GTA AGC C |
NM_053794 (rat) |
301 |
This study |
| AS: GTC CGC AGG GAA CGT CAT TG | ||||
| WNK4 |
S: CAC CTC CCG CCG CAA CAG |
NM_175579 (rat) |
347 |
This study |
| AS: TCC ACA CAG CAA AGA GCA CCC |
Primers designed on mouse sequences also specifically recognized and amplified the corresponding rat genes. All primer sets were validated to specifically recognize their targets.
Statistical analyses
Electrophysiology data are presented as original recordings and as mean values ± SE from n observations. Student’s t-test was used to determine statistical significance of paired and unpaired samples. The Hill equation was fitted to concentration-response curves by using individual data points in order to retain appropriate weighting and presented here plotted with the mean and SEM. Differences were considered significant for P < 0.05.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SRP contributed to the design and analysis of the experiments, to performance of many of the electrophysiological measurements and to writing the manuscript. SBK contributed to the design and analysis of the experiments, and to performance of many of the electrophysiological measurements. TW performed the vibrating probe experiments on the CFTR mutant mice. RCD provided the CFTR mutant mice and consulted on their breeding, care and genotyping. NNR designed PCR primers and performed the RT-PCR measurements. DGH contributed to the electrophysiological measurements. DCM contributed to the design and analysis of the experiments and to writing the manuscript. All authors read and approved the final manuscript.
Authors’ information
Current address for SRP: 112 Greene Hall, Department of Anatomy, Physiology and Pharmacology, College of Veterinary Medicine, Auburn University, Auburn, AL 36849.
Contributor Information
Satyanarayana R Pondugula, Email: srp0010@auburn.edu.
Suresh B Kampalli, Email: suresh.kampalli@yahoo.co.in.
Tao Wu, Email: wutao301@gmail.com.
Robert C De Lisle, Email: rdelisle@kumc.edu.
Nithya N Raveendran, Email: nithyanandhini@gmail.com.
Donald G Harbidge, Email: dharbidg@vet.k-state.edu.
Daniel C Marcus, Email: marcus@vet.k-state.edu.
Acknowledgements
This work was supported by NIH-NIDCD grant R01-DC00212 and NIH-NCRR grant P20-RR017686 to DCM and by the Cystic Fibrosis Foundation to RCD. Publication of this article was funded in part by the Kansas State University Open Access Publishing Fund. We thank Dr. Bruce Schultz for the gift of CFTRinh-172.
References
- Marcus DC, Acoustic Transduction. Acoustic Transduction. San Diego: Academic Press: Essentials of Membrane Biophysics. Edited by Sperelakis N; 2012. pp. 649–668. [Google Scholar]
- Marcus DC, Wangemann P, In The Oxford Handbook of Auditory Science. Inner ear fluid homeostasis. Oxford: Oxford University Press: The Ear. Edited by Fuchs PA; 2010. pp. 213–230. [Google Scholar]
- Marcus DC, Wangemann P, In Physiology and Pathology of Chloride Transporters and Channels in the Nervous System--From molecules to diseases. Cochlear and Vestibular Function and Dysfunction. New York: Elsevier: Edited by Alvarez-Leefmans FJ, Delpire E; 2009. pp. 425–437. [Google Scholar]
- Sunose H, Liu J, Shen Z, Marcus DC. cAMP increases apical IsK channel current and K+ secretion in vestibular dark cells. J Membr Biol. 1997;156:25–35. doi: 10.1007/s002329900184. [DOI] [PubMed] [Google Scholar]
- Sunose H, Liu J, Shen Z, Marcus DC. cAMP increases K+ secretion via activation of apical IsK/KvLQT1 channels in strial marginal cells. Hear Res. 1997;114:107–116. doi: 10.1016/S0378-5955(97)00152-4. [DOI] [PubMed] [Google Scholar]
- Milhaud PG, Pondugula SR, Lee JH, Herzog M, Lehouelleur J, Wangemann P, Sans A, Marcus DC. Chloride secretion by semicircular canal duct epithelium is stimulated via b2-adrenergic receptors. Am J Physiol, Cell Physiol. 2002;283:C1752–C1760. doi: 10.1152/ajpcell.00283.2002. [DOI] [PubMed] [Google Scholar]
- Bradbury NA. cAMP signaling cascades and CFTR: is there more to learn? Pflugers Arch. 2001;443(Suppl 1):S85–S91. doi: 10.1007/s004240100651. [DOI] [PubMed] [Google Scholar]
- Hryciw DH, Guggino WB. Cystic fibrosis transmembrane conductance regulator and the outwardly rectifying chloride channel: a relationship between two chloride channels expressed in epithelial cells. Clin Exp Pharmacol Physiol. 2000;27:892–895. doi: 10.1046/j.1440-1681.2000.03356.x. [DOI] [PubMed] [Google Scholar]
- Schultz BD, Singh AK, Devor DC, Bridges RJ. Pharmacology of CFTR chloride channel activity. Physiol Rev. 1999;79:S109–S144. doi: 10.1152/physrev.1999.79.1.S109. [DOI] [PubMed] [Google Scholar]
- Kurihara K, Nakanishi N, Moore-Hoon ML, Turner RJ. Phosphorylation of the salivary Na+-K+-2Cl- cotransporter. Am J Physiol, Cell Physiol. 2002;282:C817–C823. doi: 10.1152/ajpcell.00352.2001. [DOI] [PubMed] [Google Scholar]
- Sunahara RK, Dessauer CW, Gilman AG. Complexity and diversity of mammalian adenylyl cyclases. Annu Rev Pharmacol Toxicol. 1996;36:461–480. doi: 10.1146/annurev.pa.36.040196.002333. [DOI] [PubMed] [Google Scholar]
- Cooper DM. Regulation and organization of adenylyl cyclases and cAMP. Biochem J. 2003;375:517–529. doi: 10.1042/BJ20031061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler MA, Ayyagari RR, Wheeler GL, Weiss RM. Regulation of cyclic nucleotides in the urinary tract. J Smooth Muscle Res. 2005;41:1–21. doi: 10.1540/jsmr.41.1. [DOI] [PubMed] [Google Scholar]
- Dagenais A, Denis C, Vives MF, Girouard S, Masse C, Nguyen T, Yamagata T, Grygorczyk C, Kothary R, Berthiaume Y. Modulation of alpha-ENaC and alpha1-Na+-K+-ATPase by cAMP and dexamethasone in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2001;281:L217–L230. doi: 10.1152/ajplung.2001.281.1.L217. [DOI] [PubMed] [Google Scholar]
- Wehling M. Specific, nongenomic actions of steroid hormones. Annu Rev Physiol. 1997;59:365–393. doi: 10.1146/annurev.physiol.59.1.365. [DOI] [PubMed] [Google Scholar]
- Sellin JH, Field M. Physiologic and pharmacologic effects of glucocorticoids on ion transport across rabbit ileal mucosa in vitro. J Clin Invest. 1981;67:770–778. doi: 10.1172/JCI110094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pondugula SR, Sanneman JD, Wangemann P, Milhaud PG, Marcus DC. Glucocorticoids stimulate cation absorption by semicircular canal duct epithelium via epithelial sodium channel. Am J Physiol Renal Physiol. 2004;286:F1127–F1135. doi: 10.1152/ajprenal.00387.2003. [DOI] [PubMed] [Google Scholar]
- Pondugula SR, Raveendran NN, Ergonul Z, Deng Y, Chen J, Sanneman JD, Palmer LG, Marcus DC. Glucocorticoid Regulation of Genes in the Amiloride-Sensitive Sodium Transport Pathway by Semicircular Canal Duct Epithelium of Neonatal Rat. Physiol Genomics. 2006;24:114–123. doi: 10.1152/physiolgenomics.00006.2005. [DOI] [PubMed] [Google Scholar]
- Yingling JD, Fuller LZ, Jackson BA. Modulation of cyclic AMP metabolism by glucocorticoids in PC18 cells. J Neurochem. 1994;63:1271–1276. doi: 10.1046/j.1471-4159.1994.63041271.x. [DOI] [PubMed] [Google Scholar]
- Jackson BA, Braun-Werness JL, Kusano E, Dousa TP. Concentrating defect in the adrenalectomized rat. Abnormal vasopressin-sensitive cyclic adenosine monophosphate metabolism in the papillary collecting duct. J Clin Invest. 1983;72:997–1004. doi: 10.1172/JCI111072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plee-Gautier E, Grober J, Duplus E, Langin D, Forest C. Inhibition of hormone-sensitive lipase gene expression by cAMP and phorbol esters in 3T3-F442A and BFC-1 adipocytes. Biochem J. 1996;318(Pt 3):1057–1063. doi: 10.1042/bj3181057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy MM, Bell CL. Distinct cellular mechanisms of cholinergic and beta-adrenergic sweat secretion. Am J Physiol. 1996;271:C486–C494. doi: 10.1152/ajpcell.1996.271.2.C486. [DOI] [PubMed] [Google Scholar]
- Chan HC, Liu CQ, Fong SK, Law SH, Wu LJ, So E, Chung YW, Ko WH, Wong PY. Regulation of Cl- secretion by extracellular ATP in cultured mouse endometrial epithelium. J Membr Biol. 1997;156:45–52. doi: 10.1007/s002329900186. [DOI] [PubMed] [Google Scholar]
- Barker PM, Brigman KK, Paradiso AM, Boucher RC, Gatzy JT. Cl- secretion by trachea of CFTR (+/−) and (−/−) fetal mouse. Am J Respir Cell Mol Biol. 1995;13:307–313. doi: 10.1165/ajrcmb.13.3.7544595. [DOI] [PubMed] [Google Scholar]
- Liu W, Sato Y, Hosoda Y, Hirasawa K, Hanai H. Effects of higenamine on regulation of ion transport in guinea pig distal colon. Jpn J Pharmacol. 2000;84:244–251. doi: 10.1254/jjp.84.244. [DOI] [PubMed] [Google Scholar]
- Reinach P, Holmberg N. Inhibition of calcium of beta adrenoceptor mediated cAMP responses in isolated bovine corneal epithelial cells. Curr Eye Res. 1989;8:85–90. doi: 10.3109/02713688909013897. [DOI] [PubMed] [Google Scholar]
- Pondugula SR, Raveendran NN, Marcus DC. Ion transport regulation by P2Y receptors, protein kinase C and phosphatidylinositol 3-kinase within the semicircular canal duct epithelium. BMC Res Notes. 2010;3:100. doi: 10.1186/1756-0500-3-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Thiagarajah JR, Yang H, Sonawane ND, Folli C, Galietta LJ, Verkman AS. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J Clin Invest. 2002;110:1651–1658. doi: 10.1172/JCI16112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke LL, Harline MC. CFTR is required for cAMP inhibition of intestinal Na+ absorption in a cystic fibrosis mouse model. Am J Physiol. 1996;270:G259–G267. doi: 10.1152/ajpgi.1996.270.2.G259. [DOI] [PubMed] [Google Scholar]
- Wangemann P, Liu J, Shimozono M, Scofield MA. b1-adrenergic receptors but not b2-adrenergic or vasopressin receptors regulate K+ secretion in vestibular dark cells of the inner ear. J Membr Biol. 1999;170:67–77. doi: 10.1007/s002329900538. [DOI] [PubMed] [Google Scholar]
- Wangemann P, Liu J, Shimozono M, Schimanski S, Scofield MA. K+ secretion in strial marginal cells is stimulated via b1-adrenergic receptors but not via b2-adrenergic or vasopressin receptors. J Membr Biol. 2000;175:191–202. doi: 10.1007/s002320001067. [DOI] [PubMed] [Google Scholar]
- Doi K, Mori N, Matsunaga T. Effects of adenylate cyclase activation on electrical resistance of scala media. Acta Otolaryngol Suppl. 1993;501:76–79. doi: 10.3109/00016489309126220. [DOI] [PubMed] [Google Scholar]
- Kitano I, Doi K, Mori N, Umemoto M, Sakagami M, Fukazawa K, Matsunaga T. Failure of forskolin to elevate the endocochlear potential in kanamycin-poisoned animals. Hear Res. 1994;78:58–64. doi: 10.1016/0378-5955(94)90044-2. [DOI] [PubMed] [Google Scholar]
- Lee JH, Marcus DC. Endolymphatic sodium homeostasis by Reissner’s membrane. Neuroscience. 2003;119:3–8. doi: 10.1016/S0306-4522(03)00104-0. [DOI] [PubMed] [Google Scholar]
- Yamauchi D, Raveendran NN, Pondugula SR, Kampalli SB, Sanneman JD, Harbidge DG, Marcus DC. Vitamin D upregulates expression of ECaC1 mRNA in semicircular canal. Biochem Biophys Res Commun. 2005;331:1353–1357. doi: 10.1016/j.bbrc.2005.04.053. [DOI] [PubMed] [Google Scholar]
- Laverty G, Anttila A, Carty J, Reddy V, Yum J, Arnason SS. CFTR mediated chloride secretion in the avian renal proximal tubule. Comp Biochem Physiol A Mol Integr Physiol. 2012;161:53–60. doi: 10.1016/j.cbpa.2011.09.005. [DOI] [PubMed] [Google Scholar]
- Cipolli M, Canciani M, Cavazzani M, Uras P, Zampieri P, Mastella G. Ear disease is not a common complication in cystic fibrosis. Eur J Pediatr. 1993;152:265–266. doi: 10.1007/BF01956160. [DOI] [PubMed] [Google Scholar]
- Forman-Franco B, Abramson AL, Gorvoy JD, Stein T. Cystic fibrosis and hearing loss. Arch Otolaryngol. 1979;105:338–342. doi: 10.1001/archotol.1979.00790180036007. [DOI] [PubMed] [Google Scholar]
- Chao PC, Hamilton KL. Genistein stimulates electrogenic Cl- secretion via phosphodiesterase modulation in the mouse jejunum. Am J Physiol, Cell Physiol. 2009;297:C688–C698. doi: 10.1152/ajpcell.00152.2009. [DOI] [PubMed] [Google Scholar]
- Halm ST, Zhang J, Halm DR. beta-Adrenergic activation of electrogenic K+ and Cl- secretion in guinea pig distal colonic epithelium proceeds via separate cAMP signaling pathways. Am J Physiol Gastrointest Liver Physiol. 2010;299:G81–G95. doi: 10.1152/ajpgi.00035.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Veilleux A, Zhang L, Young A, Kwok E, Laliberte F, Chung C, Tota MR, Dube D, Friesen RW, Huang Z. Dynamic activation of cystic fibrosis transmembrane conductance regulator by type 3 and type 4D phosphodiesterase inhibitors. J Pharmacol Exp Ther. 2005;314:846–854. doi: 10.1124/jpet.105.083519. [DOI] [PubMed] [Google Scholar]
- O’Grady SM, Jiang X, Maniak PJ, Birmachu W, Scribner LR, Bulbulian B, Gullikson GW. Cyclic AMP-dependent Cl secretion is regulated by multiple phosphodiesterase subtypes in human colonic epithelial cells. J Membr Biol. 2002;185:137–144. doi: 10.1007/s00232-001-0120-3. [DOI] [PubMed] [Google Scholar]
- Seifert R, Lushington GH, Mou TC, Gille A, Sprang SR. Inhibitors of membranous adenylyl cyclases. Trends Pharmacol Sci. 2012;33:64–78. doi: 10.1016/j.tips.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumbay MG, Watts VJ. Novel regulatory properties of human type 9 adenylate cyclase. J Pharmacol Exp Ther. 2004;310:108–115. doi: 10.1124/jpet.104.065748. [DOI] [PubMed] [Google Scholar]
- Jiang G, Cobbs S, Klein JD, O’Neill WC. Aldosterone regulates the Na-K-2Cl cotransporter in vascular smooth muscle. Hypertension. 2003;41:1131–1135. doi: 10.1161/01.HYP.0000066128.04083.CA. [DOI] [PubMed] [Google Scholar]
- Putney LK, Brandt JD, O’Donnell ME. Effects of dexamethasone on sodium-potassium-chloride cotransport in trabecular meshwork cells. Invest Ophthalmol Vis Sci. 1997;38:1229–1240. [PubMed] [Google Scholar]
- Kim SH, Kim KX, Raveendran NN, Wu T, Pondugula SR, Marcus DC. Regulation of ENaC-mediated sodium transport by glucocorticoids in Reissner’s membrane epithelium. Am J Physiol, Cell Physiol. 2009;296:C544–C557. doi: 10.1152/ajpcell.00338.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon KB, Delpire E. Molecular physiology of SPAK and OSR1: two Ste20-related protein kinases regulating ion transport. Physiol Rev. 2012;92:1577–1617. doi: 10.1152/physrev.00009.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle KT, Rinehart J, Ring A, Gimenez I, Gamba G, Hebert SC, Lifton RP. WNK protein kinases modulate cellular Cl- flux by altering the phosphorylation state of the Na-K-Cl and K-Cl cotransporters. Physiology (Bethesda) 2006;21:326–335. doi: 10.1152/physiol.00015.2006. [DOI] [PubMed] [Google Scholar]
- Marcus DC, Marcus NY, Greger R. Sidedness of action of loop diuretics and ouabain on nonsensory cells of utricle: a micro-Ussing chamber for inner ear tissues. Hear Res. 1987;30:55–64. doi: 10.1016/0378-5955(87)90183-3. [DOI] [PubMed] [Google Scholar]
- Mercado A, Song L, Vazquez N, Mount DB, Gamba G. Functional comparison of the K+-Cl- cotransporters KCC1 and KCC4. J Biol Chem. 2000;275:30326–30334. doi: 10.1074/jbc.M003112200. [DOI] [PubMed] [Google Scholar]
- Garay RP, Nazaret C, Hannaert PA, Cragoe EJ Jr. Demonstration of a [K+, Cl-]-cotransport system in human red cells by its sensitivity to [(dihydroindenyl)oxy]alkanoic acids: regulation of cell swelling and distinction from the bumetanide-sensitive [Na+, K+, Cl-]-cotransport system. Mol Pharmacol. 1988;33:696–701. [PubMed] [Google Scholar]
- Saitta M, Cavalier S, Garay R, Cragoe E Jr, Hannaert P. Evidence for a DIOA-sensitive [K+, Cl-]-cotransport system in cultured vascular smooth muscle cells. Am J Hypertens. 1990;3:939–942. doi: 10.1093/ajh/3.12.939. [DOI] [PubMed] [Google Scholar]
- Capo-Aponte JE, Iserovich P, Reinach PS. Characterization of regulatory volume behavior by fluorescence quenching in human corneal epithelial cells. J Membr Biol. 2005;207:11–22. doi: 10.1007/s00232-005-0800-5. [DOI] [PubMed] [Google Scholar]
- Coull JA, Boudreau D, Bachand K, Prescott SA, Nault F, Sik A, De Koninck P, De Koninck Y. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature. 2003;424:938–942. doi: 10.1038/nature01868. [DOI] [PubMed] [Google Scholar]
- Anfinogenova YJ, Rodriguez X, Grygorczyk R, Adragna NC, Lauf PK, Hamet P, Orlov SN. Swelling-induced K(+) fluxes in vascular smooth muscle cells are mediated by charybdotoxin-sensitive K(+) channels. Cell Physiol Biochem. 2001;11:295–310. doi: 10.1159/000047816. [DOI] [PubMed] [Google Scholar]
- Koltsova SV, Luneva OG, Lavoie JL, Tremblay J, Maksimov GV, Hamet P, Orlov SN. HCO3-dependent impact of Na+, K+,2Cl- cotransport in vascular smooth muscle excitation-contraction coupling. Cell Physiol Biochem. 2009;23:407–414. doi: 10.1159/000218187. [DOI] [PubMed] [Google Scholar]
- Kurbannazarova RS, Bessonova SV, Okada Y, Sabirov RZ. Swelling-activated anion channels are essential for volume regulation of mouse thymocytes. Int J Mol Sci. 2011;12:9125–9137. doi: 10.3390/ijms12129125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauf PK, Misri S, Chimote AA, Adragna NC. Apparent intermediate K conductance channel hyposmotic activation in human lens epithelial cells. Am J Physiol, Cell Physiol. 2008;294:C820–C832. doi: 10.1152/ajpcell.00375.2007. [DOI] [PubMed] [Google Scholar]
- Payne JA. Functional characterization of the neuronal-specific K-Cl cotransporter: implications for [K+]o regulation. Am J Physiol. 1997;273:C1516–C1525. doi: 10.1152/ajpcell.1997.273.5.C1516. [DOI] [PubMed] [Google Scholar]
- Wangemann P, Wittner M, Di SA, Englert HC, Lang HJ, Schlatter E, Greger R. Cl(−)-channel blockers in the thick ascending limb of the loop of Henle. Structure activity relationship. Pflugers Arch. 1986;407(Suppl 2):S128–S141. doi: 10.1007/BF00584942. [DOI] [PubMed] [Google Scholar]
- Pond BB, Galeffi F, Ahrens R, Schwartz-Bloom RD. Chloride transport inhibitors influence recovery from oxygen-glucose deprivation-induced cellular injury in adult hippocampus. Neuropharmacology. 2004;47:253–262. doi: 10.1016/j.neuropharm.2004.04.002. [DOI] [PubMed] [Google Scholar]
- De Lisle RC, Isom KS, Ziemer D, Cotton CU. Changes in the exocrine pancreas secondary to altered small intestinal function in the CF mouse. Am J Physiol Gastrointest Liver Physiol. 2001;281:G899–G906. doi: 10.1152/ajpgi.2001.281.4.G899. [DOI] [PubMed] [Google Scholar]
- Lee JH, Marcus DC. Nongenomic effects of corticosteroids on ion transport by stria vascularis. Audiol Neurootol. 2002;7:100–106. doi: 10.1159/000057657. [DOI] [PubMed] [Google Scholar]
- Wangemann P, Itza EM, Albrecht B, Wu T, Jabba SV, Maganti RJ, Lee JH, Everett LA, Wall SM, Royaux IE, Green ED, Marcus DC. Loss of KCNJ10 protein expression abolishes endocochlear potential and causes deafness in Pendred syndrome mouse model. BMC Med. 2004;2:30. doi: 10.1186/1741-7015-2-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SL, Roussa E, Le Rouzic P, Thevenod F, Alper SL, Best L, Brown PD. Expression of K+-Cl- cotransporters in the alpha-cells of rat endocrine pancreas. Biochim Biophys Acta. 2004;1667:7–14. doi: 10.1016/j.bbamem.2004.08.005. [DOI] [PubMed] [Google Scholar]